


Abstract
P53-binding protein 1 (53BP1) mediates DNA repair pathway choice and promotes checkpoint activation. Chromatin marks induced by DNA double-strand breaks and recognized by 53BP1 enable focal accumulation of this multifunctional repair factor at damaged chromatin. Here, we unveil an additional level of regulation of 53BP1 outside repair foci. 53BP1 movements are constrained throughout the nucleoplasm and increase in response to DNA damage. 53BP1 interacts with the structural protein NuMA, which controls 53BP1 diffusion. This interaction, and colocalization between the two proteins in vitro and in breast tissues, is reduced after DNA damage. In cell lines and breast carcinoma NuMA prevents 53BP1 accumulation at DNA breaks, and high NuMA expression predicts better patient outcomes. Manipulating NuMA expression alters PARP inhibitor sensitivity of BRCA1-null cells, end-joining activity, and immunoglobulin class switching that rely on 53BP1. We propose a mechanism involving the sequestration of 53BP1 by NuMA in the absence of DNA damage. Such a mechanism may have evolved to disable repair functions and may be a decisive factor for tumor responses to genotoxic treatments.
INTRODUCTION
DNA double-strand breaks (DSB) trigger a rapid and comprehensive DNA damage response (DDR) that leads to checkpoint signaling and cell cycle arrest, repair factor recruitment to the damage sites, and DNA repair. The precise orchestration of this response is critical for cell and organism survival (1). Most DDR factors are permanent residents of the nucleoplasm that are not synthesized de novo during the DDR. Rather, repair foci formation relies on posttranslational modifications of histones and DDR factors. DSB are processed predominantly by two competing pathways: Error-prone nonhomologous end-joining (NHEJ) and homologous recombination (HR). HR restores the genetic information from the sister chromatids and the committing step for this pathway is DNA end resection.
53BP1 is a multifunctional DDR protein that plays an important role in repair pathway choice: 53BP1 and its effector RIF1 compete with BRCA1 to prevent CtIP-mediated resection and, as a consequence, antagonize HR in favor of NHEJ (2–5). Additionally, RIF1 recruits the shielding complex that suppresses resection (6–9). This effect is fine-tuned by SCAI, which progressively associates with 53BP1, thereby displacing RIF1 and enabling BRCA1-mediated repair (10). For DNA lesions undergoing HR repair, 53BP1 prevents excessive resection and favors gene conversion over mutagenic single-strand annealing (11). In the absence of functional BRCA1, the balance between HR and NHEJ is tilted and DSB are improperly repaired by the NHEJ pathway, leading to deleterious chromosomal aberrations. This effect is exploited in anticancer therapies with PARP inhibitors (PARPi) (12). Acquired resistance limits clinical efficacy of PARPi, and loss of 53BP1 function is one of the mechanisms conferring PARPi tolerance in cancer cells (13–15). With the exception of BRCA-null tumors, 53BP1 functions as a tumor suppressor, the loss of which radiosensitizes human (16) and mouse cells (17).
53BP1 is continuously expressed in the nucleus and rapidly accumulates at ionizing radiation-induced foci (IRIF) (18,19). The recruitment of 53BP1 to IRIF depends on constitutive H4K20Me2 and damage-induced H2AK15Ub marks recognized by the tudor and ubiquitin-dependent recruitment (UDR) domains of the protein (20–22). In the absence of DNA damage, the demethylase JMJD2A and the Polycomb protein L3MBTL1 compete with 53BP1 for H4K20Me2 binding sites; JMJD2A degradation and L3MBTL1 eviction during the DDR facilitate 53BP1 binding to damaged chromatin (23,24). In addition, the TIP60 acetyltransferase reduces 53BP1 binding to the chromatin, tilting the repair balance towards HR: Acetylation of H4K16 decreases 53BP1’s affinity for H4K20Me2 (25), whereas H2AK15Ac prevents ubiquitination of the same residue and 53BP1 UDR binding (26). Sustained 53BP1 function at IRIF also depends on 53BP1’s BRCT domain binding to ATM-phosphorylated H2AX (27,28).
Less is known about the regulation of 53BP1 spatial distribution and function outside of repair foci. More generally, the mechanisms regulating the access of repair factors to chromatin in the absence of DNA damage remain largely unexplored. Yet such mechanisms may be key to prevent undue activation of the DDR. Here, we show that 53BP1 has a slow nucleoplasmic diffusion behavior that accelerates in response to DNA damage. We identify a novel interaction between 53BP1 and the structural nuclear protein NuMA, which regulates the mobility, IRIF formation, and function of 53BP1.
MATERIALS AND METHODS
Cell culture, transfection and genotoxic treatments
Osteosarcoma U2OS cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS, Sigma). U2OS Lac-ISceI-Tet cells were obtained from T. Misteli (NCI). Non-neoplastic breast epithelial cells (HMT-3522 S1) were cultured in H14 medium (29); HMT-3522 T4-2 breast cancer cells were cultured in H14 without EGF. SUM149PT breast cancer cells (obtained from E. Alli, WFU) were cultured in DMEM supplemented with 10% FBS and with 10 mM HEPES buffer, hydrocortisone (5 μg/ml) and insulin (5 μg/ml). CH12F3-2 cells were obtained from T. Honjo (Kyoto University) and were cultured in RPMI 1640 containing 2 mM l-glutamine, 10% FBS and 50 μM 2-mercaptoethanol in vertically positioned T25 flasks. Their density was kept below 105 cells/ml. Mycoplasma testing was performed yearly and results were systematically negative. Lipofectamine 3000 (ThermoFisher) was used for siRNA (ON-TARGETplus, Dharmacon) and for plasmid DNA transfection. The following expression vectors were used for this study: GFP-53BP1 and GFP-53BP1ct (encoding full length 53BP1 and residues 1200–1711 of 53BP1 fused to GFP, respectively) (30); mCherry-53BP1ct (Addgene plasmid # 19835) (31); GFP-Lac-NLS (32); GFP-MeCP2 (33); GFP-PCNA (34); GFP-MDC1 (Addgene plasmid #26285); and mCherry-NuMA, cloned by replacing GFP in GFP-NuMA (35) by mCherry using KpnI and BsrG1 restriction sites. GFP-NuMA(S395A) was cloned by overlap PCR using the 5′ CAG CTG GAA GAA CAC CTT gCg CAG CTG CAG GAT AAC CCA C 3′ and 5′ GTG GGT TAT CCT GCA GCT GCG CAA GGT GTT CTT CCA GCT G 3′ primer pair. The overlap PCR product was digested with EcoRV and AflII and the fragment (1166 bp) was ligated into the corresponding sites of pcDNA GFP-NuMA. Clones were verified using restriction for presence of the FspI site (introduced by silent mutagenesis) and by DNA sequencing. The shRNA vectors targeting human NuMA were purchased from Origene (TR311065). The shRNA vectors for murine proteins were purchased from the Dharmacon RNAi Consortium and included shNuMA-1 (TRCN0000072130), shNuMA-2 (TRCN0000072132), and sh53BP1 (TRCN0000081778). A shRNA scramble pLKO.1 plasmid (Addgene # 1864) (36) was used as negative control. Lentiviral particles generated using HIV packaging were used for transduction. Stably silenced cell lines were generated after selection with puromycin (0.6 μg/ml). DNA damage was induced by gamma irradiation (3 Gy and 10 Gy for S1 and U2OS cells, respectively; Gammacell 220 irradiator from Nordion), with the radiomimetic drug bleomycin (20 mU/ml for 1 h), with mitomycin C (MMC; 2.6 μM, 18 h), or with hydrogen peroxide (1 mM, 10 min).
Modeling of protein diffusion
Simulations of protein diffusion in the nucleus were implemented in MATLAB (MathWorks). All simulations were performed at 37°C. Macromolecules in solution undergo random collisions from surrounding molecules, resulting in a 3D translational or rotational random walk. 3D translational dynamics in medium satisfies the diffusion equation:
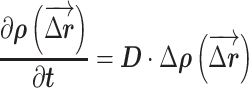 |
(1) |
where 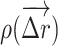 is the displacement distribution of the molecule, D is the diffusion coefficient, and Δ is the 3D Laplace operator. Theoretically, the diffusion coefficient has the following relationship with the size, d, of the macromolecules:
is the displacement distribution of the molecule, D is the diffusion coefficient, and Δ is the 3D Laplace operator. Theoretically, the diffusion coefficient has the following relationship with the size, d, of the macromolecules:
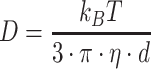 |
(2) |
with 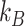 , the Boltzmann constant, T the temperature, and η the viscosity of the medium (1.5 × 10−3 Pa·s in the nucleus (37). The diffusion coefficient can also be obtained from mean squared displacement (MSD) calculations,
, the Boltzmann constant, T the temperature, and η the viscosity of the medium (1.5 × 10−3 Pa·s in the nucleus (37). The diffusion coefficient can also be obtained from mean squared displacement (MSD) calculations, 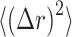 . In our model, we only considered the Brownian motion of the protein in a 2D plane. Therefore,
. In our model, we only considered the Brownian motion of the protein in a 2D plane. Therefore,
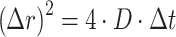 |
(3) |
With Δt, the lag time between each position Δr. We simulated the 2D Brownian motions of the proteins in time windows of 1000 s and the trajectories (xi, yi) per second (Δt) were registered for calculation. The displacements were then calculated as:
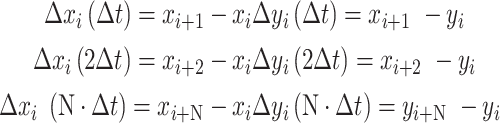 |
(4) |
And the square displacement (Δr)2 was summarized as:
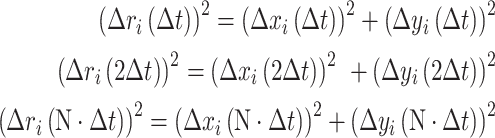 |
(5) |
The diffusion coefficients D depend on the hydrodiameter (hence the shape) of the molecules (equation 2). GFP has a beta barrel structure of 4.2 nm in length (38), used for modeling. To simulate GFP-53BP1 diffusion, we considered two extreme possibilities, a single globular domain (sphere) and a highly elongated molecule (rod). The corresponding physical sizes were predicted using a sedimentation model described in (39). A sphere the size of GPF-53BP1 would have a diameter of 8.22 nm, whereas the calculated length of the rod would be 32.88 nm (using a sedimentation coefficient Smax/S = 4). When predicting the sizes of 53BP1 dimers, we considered combinations of spheres with minimum surface (16.44 nm), as well as concatenated rods (65.56 nm). The diffusion coefficients were obtained by fitting MSD curves using equation (3).
Measurement of protein diffusion
Fluorescence correlation spectroscopy (FCS) measurements were performed on a customized scanning confocal microscope (Microtime 200, PicoQuant), as described previously (40). Cells were maintained at 37°C and 5% CO2 using a stage-top incubator (Tokai Hit). GFP-53BP1 autocorrelation curves were fitted using a two-components model, which was validated by a maximum entropy method-based fitting routine (MEM-FCS) (41). Optimal curve fitting was achieved using a two-component Brownian model.
Continuous photobleaching (CP) measurements were done using a Zeiss LSM880 scanning confocal microscope equipped with a 63× water immersion objective (NA = 1.2) and by point scanning of specific regions in the cell nucleus. Points were continuously scanned at moderate laser power (5%) over 7.8 s and fluorescence intensity was simultaneously recorded. Fluorescence intensity decay curves are characterized by the dynamic equilibrium of photobleaching and diffusion. CP curves for GFP-53BP1 and GFP-MDC1 were fitted to a multi-exponential decay model (42–44) as follows:
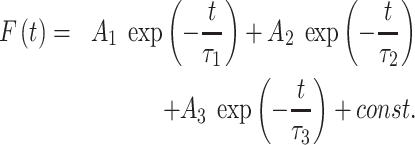 |
(6) |
The fast decay (first term in equation 6) represents the fast initial photobleaching of the fluorescent molecules in the focal volume, which was not considered for our analyses. The second term represents the association/dissociation behavior. We used τ2 to derive the dissociation constant Kd (42). Kd was normalized to the control condition since not all CP constant parameters could be precisely estimated. The third term characterizes the mobility of free-diffusing molecules. Values of τ3 were not affected by DNA damage. For nontagged GFP, CP curves were best fitted using a two-component model (τ1, fast initial bleaching [not considered] and τ2, free diffusion).
Fluorescence loss in photobleaching (FLIP) assays were performed using a Zeiss LSM710 confocal microscope, with a 40× oil immersion lens (NA = 1.3). The 488 laser line was used at 100% intensity to bleach regions of interests (ROIs) corresponding to the upper half of the cell nuclei. This region was initially bleached using 2–3 scanning iterations. Time-lapses were recorded (50 images over 1.5 min), alternating bleaching of the upper-half of the nucleus and imaging of the whole nucleus. GFP intensity was measured as a function of time in elongated vertical rectangular ROIs below the bleaching region. A single component decay curve was used to fit the FLIP curves for the characteristic decay time of the molecules (45).
Mass spectrometry
GFP-fused to 53BP1 containing amino acids 1–34 and 1220–1711 (30) was stably expressed in HEK293T cells under the Tet-On promoter. Cells were exposed to doxycycline (1 μg/ml) for 72 h to induce expression of the GFP-53BP1 construct. Cell lysates were immunoprecipitated using GFP antibodies, resolved by SDS-PAGE, and subjected to trypsin digestion. Peptides were separated by HPLC and analyzed by LC-MS/MS using a LTQ Orbitrap XL (Thermo). Spectra were extracted by ProteoWizard and were analyzed using Mascot (Matrix Science). Scaffold (Proteome Software Inc.) was used to validate MS/MS based peptide and protein identifications. Mass spectrometry data are deposited in the MassIVE repository (https://massive.ucsd.edu; accession MSV000083293).
Immunoprecipitation and western blot analysis
For coimmunoprecipitation, nuclear extracts were prepared using the Universal Magnetic Co-IP kit (Active Motif) in the presence of protease inhibitors (aprotinin, 10 μg/ml, Sigma; Pefabloc, 1 mM, Roche Applied Science), phosphatase inhibitor (sodium fluoride, 250 μM), and PARG inhibitor (DEA, 10 μM, Trevigen). Nuclear extracts (0.5 mg) were immunoprecipitated with NuMA antibodies or nonspecific IgG (1.5 μg) overnight at 4°C, and analyzed by immunoblotting. Similar results were obtained with two different NuMA antibodies (Calbiochem, ab-2 and Bethyl Laboratories, A301-509A). Antibodies used for immunoblot were 53BP1 (Abcam, Ab36823, 1 μg/ml), γH2AX (Ser139; Millipore, clone JBW301, 1 μg/ml), Histone H2B (Abcam, Ab1790, 0.1 μg/ml), lamin B (Abcam, Ab16048, 60 ng/ml), NuMA (B1C11, 1:2, a gift from Dr Jeffrey Nickerson, UMass, Worcester, USA), P-NuMA (S395; Cell Signaling, 3429, 1:1000), and PAR (Trevigen, 4336-APC-050, 1:1000).
Immunostaining
Immunostaining was performed as described (40). Antibodies used for immunofluorescence were 53BP1 (Abcam, 5 μg/ml), γH2AX (Millipore, 2 μg/ml), NuMA (B1C11, 1:2 or Abcam, clone EP3976, 1:250), P-NuMA (Cell Signaling, 1:200), and SNF2h (Abcam Ab3749, 20 μg/ml). Fluorescent signals were imaged with a Zeiss LSM710 confocal microscope using a 63× oil (NA = 1.4) objective. Imaging fields were randomly selected based on DAPI signals. Repair foci were quantified using a custom macro in ImageJ (http://rsbweb.nih.gov/ij/). For experiments comparing HMT-3522 S1 cells transfected with nontargeting and NuMA-targeting siRNA, the macro included a step to subtract NuMA-derived masks from DAPI-derived masks of the nuclei, in order to exclude cells retaining NuMA expression in the siNuMA conditions.
FRET and laser microirradiation
Acceptor photobleaching FRET experiments were performed using a Zeiss LSM710 confocal microscope using a 63x water objective (NA = 1.3). mCherry (FRET acceptor) was bleached in spot areas of the cell nucleus using three pulse iterations (561 diode laser at 100% power and 177 μs/pixel dwell time). FRET efficiency was calculated as E = ((I[post-bleach] – I[pre-bleach])/I[post-bleach]) x 100. To induce DNA damage by laser microirradiation, the 405 nm laser line of the LSM710 was used at maximum power to draw lines across nuclei (100 μs dwelling time; ∼1 s scan/line). Cells were sensitized with BrdU (10 μM) 48 h prior to microirradiation and were irradiated and imaged with the 63x water immersion lens. In all assays involving live cell imaging, cells in glass-bottom dishes (MatTek) were maintained at 37°C and 5% CO2 with a stage-top incubator (Pecon).
GFP-based repair assays and cell cycle analysis
NHEJ was assessed in U2OS cells with a stable integration of the NHEJ-I reporter cassette, as described (46). The NHEJ reporter is composed of the GFP coding sequence interrupted by an exogenous exon flanked by ISceI recognition sites. NHEJ reconstitutes the GFP coding sequence after ISceI cleavage. GFP-positive cells were quantified by flow cytometry 24 h after ISceI transfection. 10 000–100 000 cells were analyzed in each sample. Cells were costained with DRAQ5 (Biostatus) to collect DNA content values for cell cycle determination.
Class switch recombination (CSR)
CH12F3-2 cells were nucleofected with shRNA vectors and overexpression plasmids (Lonza Nucleofector™ I device; solution L; program T20; 106 cells and 2 μg DNA per reaction). CSR to IgA was induced by stimulating the cells for 72 h with 5 μg/ml agonist anti-CD40 (clone HM40-3; BD), 5 ng/ml IL-4 (R&D Systems) and 2.5 ng/ml TGF-β1 (R&D Systems). Surface expression of IgA was analyzed by flow cytometry using PE-conjugated goat antimouse IgA antibodies (1:400; SouthernBiotech).
Quantification of chromosomal aberrations
SUM149PT cells were seeded in 6-well plates (one plate per condition) and cultured until they reached 60% confluency for transfection. GFP and GFP-NuMA overexpressing cells were selected with geneticin (200 μg/ml). To quantify chromosomal aberrations, the cells were transferred onto 22 × 22 mm coverslips in 35 mm dishes 48 h after transfection. Cells were then treated with PARP inhibitor (olaparib; 0.5 μM) or vehicle. After 24h incubation, colcemid (10 μg/ml) was added to the cells for 1h. Metaphase spreads were prepared by hypotonic bursting in (2:1) (75 mM KCl: 0.8% sodium citrate) and fixation in (3:1) (methanol: acetic acid). Subsequently, coverslips were air dried, mounted on microscope slides, stained with 2% Giemsa for 3 min and washed in Gurr buffer. Slides were allowed to dry and were analyzed with an Olympus IX83 microscope using a 60× oil objective and CMOS camera (ORCA-Flash4.0; Hamamatsu).
Human breast tissues
Breast invasive ductal carcinoma tissues were obtained from mastectomies and normal breast tissue was obtained from reduction mammoplasties, after obtaining patient consent (Purdue Institutional Review Board approval 1206012467). The excision specimen were resected, minced to approx. 4 mm fragments, and placed in RPMI within 30 min of surgery. Tissue explants were rinsed with, and transported in RPMI. After irradiation (3 Gy; Gammacell 220 irradiator) and recovery in a tissue culture incubator (37°C; 5% CO2), tissue explants were incubated in PBS containing 18% sucrose on ice for 15 min, then with PBS with 30% sucrose on ice for 15 min, and frozen in optimal cutting temperature (OCT) compound on dry ice mixed with 70% ethanol. Controls were mock-irradiated. Tissues were stored at −80°C until sectioning of 12 μm thick sections with a cryostat set at −20°C. Tissue slices were collected on Superfrost plus slides and used for immunostaining.
Analysis of tumor expression profiles
Breast tumor expression profiles generated by The Cancer Genome Atlas (TCGA) Research Network (http://cancergenome.nih.gov/) and the microarray dataset of Nagalla et al. (47) were utilized in this study. The TCGA clinical data and RNA-seq data (level 3-processed) were downloaded from the Broad's FireBrowse website (http://firebrowse.org/; TCGA data version 2016_01_28). The RNA-seq breast tumor data comprising 1100 tumor expression profiles was filtered to exclude male and gender ‘unknown’ samples (n = 13), metastatic tissue samples (n = 7) and one errant skin cancer sample yielding 1079 female primary breast tumor samples. OncoLnc (http://www.oncolnc.org) was used to visualize overall survival (48).
Statistical analysis
Statistical analyses were done using GraphPad Prism 6.0. Data are presented as mean ± SEM or mean ± SD, as indicated in the figure legends. The D’Agostino & Pearson omnibus normality test was used to test for normality before computing either t-test or one-way ANOVA and post-hoc tests. Nonparametric tests were used if the data did not pass the normality test (at alpha = 0.05). Statistical tests are indicated in the figure legends, whereas exact p-values are shown in the figures. All statistical tests were two-sided.
RESULTS AND DISCUSSION
53BP1 movements are restrained in the nucleoplasm
The mechanisms that govern the spatial kinetics of 53BP1 in the absence of DNA damage are largely unknown. Therefore, we measured diffusion of GFP-tagged 53BP1 in regions of the nucleoplasm devoid of DNA breaks using fluorescence correlation spectroscopy, and compared the values with predictions generated with a diffusion model (Figure 1A). Theoretical diffusion values for GFP alone and for a fluorescent protein tandem concurred with FCS measurements, which validated our model. For GFP-53BP1, we used hydrodiameters corresponding to sphere or rod structures to predict diffusion because the full structure of 53BP1 is unknown. The FCS curves for GFP-53BP1 were best fitted with a two-component model interpreted as slow movements (0.5 μm2/s) coexisting with rapid Brownian motions (23.2 μm2/s), although we cannot exclude some contribution of directed motions. The ‘Brownian’ diffusion values matched well with the predicted diffusion value assuming a spherical structure of 53BP1, indicating that 53BP1 is likely to adopt a compact native conformation. In contrast, the second component of GFP-53BP1 diffusion was significantly slower than the predictions, even when considering 53BP1 dimers in the model (Supplementary Table S1). We conclude that 53BP1 diffusion is constrained in the cell nucleus.
Figure 1.

NuMA interacts with 53BP1 and regulates 53BP1 kinetics. (A) Modeling of GFP-53BP1 diffusion, assuming either a spherical or a rod-shaped structure of the protein. GFP and a fluorescent protein tandem (GFP-mCherry) were used as controls. Time trace simulations are shown with the predicted diffusion times and experimental FCS values (mean ± SEM; n ≥ 15). (B) Diffusion of GFP-53BP1 and GFP measured by FCS. U2OS cells were untreated (control), treated with bleomycin (20 mU/ml, 1 h), or exposed to ionizing radiations (IR, 10 Gy and 30 min recovery). n = 50–60 cells from 2 biological replicates; Mann–Whitney test. (C) Immunoprecipitation of NuMA from U2OS nuclear extracts (N.E.). Nonspecific immunoglobulins (IgGs) were used as controls. Blots were probed for NuMA and 53BP1. Cells were exposed to IR (10 Gy, and 30 min recovery) or treated with H2O2 (1 mM, 10 min) prior IP. Densitometric quantification of 53BP1 pull down is shown on the graph (mean ± SEM; one sample t-test; n = 4 (IR) or 3 (H2O2)). In the right panel (H2O2 treatment), all lanes are from the same immunoblot membrane and were taken with the same exposure. Lanes were reassembled for clarity. (D) FRET efficacy in U2OS cells expressing NuMA fused to mCherry and either GFP (used as control), GFP-53BP1, or GFP-53BP1ct (mean ± SD; n = 20–25 cells from at least two experiments; Kruskal–Wallis and Dunn's multiple comparison test). (E) Colocalization (arrowheads) between 53BP1 foci and bright NuMA features in immunostaining images of S1 cells treated with bleomycin or untreated (control). Scale bar, 5 μm. Overlap is quantified on the graph (mean ± SEM; n = 10 images from two experiments, corresponding to 300 nuclei per condition; Student's t-test). (F) FCS analysis of GFP-53BP1, GFP, and GFP-MeCP2 diffusion. Cells were transfected with nontargeting (NT) or with NuMA-targeting siRNA and were treated with bleomycin as indicated. Statistical analysis with one-way ANOVA and Tukey (GFP-53BP1) or Mann–Whitney tests (GFP and GFP-MeCP2). NuMA silencing was verified by western blot, with lamin B as loading control.
A previous study determined the steady state kinetics of two other NHEJ factors, KU80 (14 μm2/s) and DNA-PKc (6 μm2/s) (49). These measurements are similar to the theoretical values for KU80 and DNA-PKc computed with our diffusion model (Supplementary Table S1). Hence, the concept of restricted diffusion that we put forward for 53BP1 might apply to some but not all DDR proteins.
DNA damage increases 53BP1 mobility
To test whether the constraints on 53BP1 are modulated during the DDR, diffusion of GFP-53BP1 was measured by FCS in the absence and presence of DNA damage induced with ionizing radiations (IR) or with the radiomimetic drug bleomycin. Association of 53BP1 with the chromatin flanking DNA breaks strongly reduces 53BP1 mobility (50). To avoid this effect, we selected regions in-between repair foci for FCS point measurements. Exposures to IR and bleomycin both lead to a significant increase in GFP-53BP1 mobility outside repair foci (Figure 1B). This increase could not be explained by altered nucleoplasm viscosity since the kinetics of GFP alone remained unchanged after DNA damage. We used two independent approaches to further assess 53BP1 motions in response to DNA damage, continuous photobleaching (CP) and fluorescence loss in photobleaching (FLIP). The dissociation constant Kd was extracted from CP measurements. In cells treated with bleomycin, Kd for GFP-53BP1 was significantly increased compared to control (Supplementary Figure S1A). We interpret this increased dissociation rate as the release of 53BP1 from an immobile binding site. No differences in CP measurements were measured for GFP alone. We also performed CP measurements for MDC1, a DDR factor with a molecular size similar to that of 53BP1. Unlike GFP-53BP1, Kd for GFP-MDC1 was decreased in cells with DNA damage (albeit not significantly), possibly reflecting an overall increased association with chromatin (Supplementary Figure S1A). In FLIP experiments, diffusion times for GFP-53BP1 significantly decreased in cells treated with bleomycin (Supplementary Figure S1B), meaning that 53BP1 became more mobile in cells with DNA damage, as measured with FCS. Our biophysical results are consistent with biochemical fractionation experiments that detected 53BP1 in the insoluble nuclear fraction in the absence of DNA damage and in the chromatin fraction after IR (51). These findings suggest a mechanism increasing 53BP1 nucleoplasmic mobility in response to DNA damage.
53BP1 interacts with the nucleoskeletal protein NuMA
We used a proteomics approach to identify factors interacting with 53BP1 and regulating its dynamics in the nucleus. We short-listed 53BP1 binding candidates identified in this study and in previous analyses of the 53BP1 interactome deposited in PubMed or in the BioGrid database (http://thebiogrid.org/) (Supplementary Tables S2 and S3). Among these proteins, the nuclear mitotic apparatus protein (NuMA) was identified with the highest confidence in our analysis.
NuMA is a structural nuclear protein with a well-established function in spindle pole assembly and maintenance (52,53). In addition to mitotic functions, NuMA is essential for the establishment of higher-order chromatin organization during epithelial cell differentiation (54,55) and for DNA repair by HR (40). Co-immunoprecipitation (IP) experiments using NuMA antibodies confirmed the interaction between endogenous 53BP1 and NuMA in U2OS osteosarcoma cells, in HMT-3522 T4-2 breast cancer cells, and in non-neoplastic HMT-3522 S1 mammary epithelial cells (Figure 1C and Supplementary Figure S2A). Interaction between the two proteins decreased after DSB induction with ionizing radiations, but not after a short exposure to hydrogen peroxide used as a source of oxidative DNA damage (Figure 1C and Supplementary Figure S2B). Fluorescence resonance energy transfer (FRET) experiments showed that mCherry-tagged NuMA interacts with GFP-53BP1 and that NuMA-53BP1 interaction decreases in cells with DNA damage (Figure 1D). FRET experiments also indicated interaction between NuMA and the minimal focus-forming region of 53BP1 (residues 1200–1711; GFP-53BP1ct) that contains 53BP1’s oligomerization, tudor, and UDR domains. These results suggest that NuMA interacts with a region of 53BP1 that is critical for chromatin binding. We can however not exclude the possibility that NuMA interacts with another portion of 53BP1 (or that the interaction is indirect). Indeed, the oligomarization domain in GFP-53BP1ct may allow the formation of heterodimers with full-length endogenous 53BP1 in these experiments.
To further address the link between NuMA and 53BP1, the relative distribution of the two proteins was quantified by immunostaining. A subset of 53BP1 nuclear bodies overlapped with bright NuMA signals in untreated cells. In bleomycin-treated cells, this colocalization decreased significantly, as revealed by visual scoring (Figure 1E) and by computing Mander's colocalization coefficients (Supplementary Figure S2C). 53BP1 nuclear bodies in nondamaged cells correspond to dysfunctional telomeres (56) as well as ‘shielded’ mitotic lesions (57). The latter are generally found in G1 cells and may contain complex DNA lesions that require the HR pathway for repair. 53BP1 bodies have been proposed to provide a protective environment inhibiting transcription and delaying repair until the appropriate cellular context arises, such sister chromatid synthesis during S-phase (58). Hence, 53BP1 bodies are distinct from 53BP1 IRIF induced by drugs and radiations. The loss of colocalization between NuMA and 53BP1 nuclear bodies and the loss of interaction between NuMA and 53BP1 after DNA damage induction suggest that NuMA regulates 53BP1 function during the initial response to DNA damage.
NuMA reduces 53BP1 mobility outside repair foci
The DNA damage-dependent interaction and colocalization between 53BP1 and the structural protein NuMA prompted us to test if NuMA regulates the dynamics of 53BP1 in the cell nucleus. Mobility of GFP-53BP1 at nuclear regions lacking damage foci was significantly higher in cells silencing NuMA compared to controls (Figure 1F). This increase was similar to the increased diffusion resulting from DNA damage induction with bleomycin, and both treatments had no additive effect. Silencing NuMA did not alter the dynamics of GFP or GFP fused to the methyl CpG binding protein 2 (GFP-MeCP2), nor cause DNA damage (see next section). FLIP experiments confirmed the increased 53BP1 diffusion in cells silencing NuMA compared to controls (Supplementary Figure S1B). Hence, NuMA may reduce 53BP1 diffusion in the absence of DNA damage.
NuMA depletion does not alter 53BP1 expression
NuMA is structurally related to nuclear lamins, which have been shown to stabilize 53BP1 (59). In contrast to laminA/C depletion, NuMA silencing did not alter 53BP1 levels (Figure 2A). Silencing NuMA had also no effect on 53BP1 levels in cells with DNA damage (Supplementary Figure S2D). Hence, NuMA does not regulate 53BP1 expression but may rather sequestrate 53BP1 in the absence of damage to keep this master DDR effector in check. We note that other studies have shown degradation of 53BP1 by the proteasome after DNA damage (60,61). This effect was not observed in U2OS or HMT-3522 S1 cells in our experimental conditions, possibly due to cell line-specific expression and compartmentalization of the protein.
Figure 2.

NuMA antagonizes 53BP1 accumulation at DNA damage sites. (A) Left: NuMA silencing and 53BP1 expression analyzed by western blot in HMT-3522 S1 cells transfected with nontargeting (NT) or NuMA-targeting siRNA. Right: IR-induced focal accumulation of 53BP1 in S1 cells transfected with siNT and siNuMA. Confocal images show NuMA and 53BP1 immunostaining in irradiated cells (3 Gy, 2h recovery). The arrowhead points to a cell retaining NuMA expression in contrast to its neighbors. Quantification of 53BP1 foci is presented in the box-and-whisker plot. *P < 0.05 and ***P < 0.0001 (one-way ANOVA and Bonferroni's test; n = 4; 100–300 cells analyzed per condition for each replicate). The inset shows the average number of 53BP1 foci per nucleus in cells that lost (–) or retained (+) NuMA expression within the siNuMA transfection condition. #P < 0.05 (Student's t-test). (B) Cell cycle distribution in S1 cells transfected with NT and NuMA siRNA determined by flow cytometry (mean ± SEM; n = 4; Student's t-test). (C) Accumulation of 53BP1 at ISceI sites in U2OS cells transfected with NT and NuMA siRNA. Cleavage sites were identified using the Lac arrays flanking the ISceI site and 53BP1 was detected by immunostaining. n > 40 cells from three replicates; Student's t-test. (D) Accumulation of GFP-53BP1ct at laser-microirradiated tracks (arrowheads) in U2OS cells expressing mCherry or mCherry-NuMA (insets) or transfected with NT and NuMA-targeting siRNA. The fraction of GFP signal at the tracks is shown on the bar graphs (mean ± SEM, Mann-Whitney test). (E) NuMA phosphorylation at Ser395 (P-NuMA) after DNA damage. Left: western blot analysis. Total NuMA and lamin B were used as loading controls. Right: immunostaining of cells transfected with NT or NuMA siRNA. The arrow points to P-NuMA signals in a cells expressing NuMA. (F) Accumulation of mCherry-53BP1ct after laser-microirradiation in cells expressing GFP, GFP-NuMA, or a nonphosphorytable NuMA mutant [GFP-NuMA(S395A)] (mean ± SEM, Mann-Whitney test). Arrowheads indicate the position of the laser microirradiation tracks. Scale bars, 10 μm.
NuMA antagonizes 53BP1 recruitment at DNA breaks
To test if NuMA influences 53BP1 accumulation at DNA repair foci, HMT-3522 S1 mammary epithelial cells were transfected with NuMA-targeting or with nontargeting siRNA and exposed to IR. Increased 53BP1 foci density was measured in cells silencing NuMA compared to the nontargeting siRNA controls (Figure 2A). We also compared NuMA-positive and NuMA-negative cells within the NuMA siRNA transfections and found that irradiated cells with NuMA expression had significantly less 53BP1 foci compared to cells with no detectable NuMA expression (Figure 2A). We excluded that NuMA silencing sensitized the cells to IR because similar amounts of DSB were detected using the comet assay in siNuMA transfectants and controls (Supplementary Figure S3A). Moreover, cell cycle distribution - an important factor in repair pathway choices - was not affected by NuMA depletion in S1 cells (Figure 2B). Increased 53BP1 foci numbers were also measured in bleomycin-treated S1 cells expressing NuMA-targeting shRNA (Supplementary Figure S3B) and a similar effect was measured for GFP-53BP1 in U2OS cells, where siRNA-mediated NuMA depletion lead to increased GFP-53BP1 foci formation in response to bleomycin or mitomycin C (Supplementary Figure S3C).
Next, we asked if NuMA affects 53BP1 accumulation at single DSB using a cell system with a stable integration of the ISceI cleavage site flanking lac arrays. 53BP1 foci at cleaved arrays were brighter in cells with NuMA silenced compared to controls (Figure 2C). In addition, NuMA overexpression decreased GFP-53BP1 and GFP-53BP1ct accumulation at laser-microirradiated tracks and silencing NuMA lead to increased GFP-53BP1ct line accumulation in microirradiated cells (Figure 2D and Supplementary Figure S3D). Altering NuMA expression did not perturb recruitment of GFP-PCNA and GFP-MDC1 to the laser tracks (Supplementary Figure S3D–E), ruling out nonspecific effects and indicating that NuMA’s effect on 53BP1 is likely independent from the MDC1-RNF8-RNF168 cascade that mediates chromatin recognition by 53BP1 (5).
Pan-nuclear NuMA phosphorylation regulates 53BP1 accumulation at sites of DNA damage
We showed previously that NuMA regulates SNF2h accumulation at DNA breaks (40). Yet silencing SNF2h did not alter 53BP1 line formation in microirradiation assays (Supplementary Figure S3F), suggesting that SNF2h does not mediate the spatial regulation of 53BP1 by NuMA. These results are in agreement with a previous report showing no defect in 53BP1 IRIF formation in SNF2h-depleted cells (62).
NuMA is an ATM substrate that is rapidly phosphorylated at serine 395 in response to IR (40,63) (Figure 2E). Immunostaining with P-NuMA antibodies after bleomycin treatment or laser microirradiation revealed pan-nuclear NuMA phosphorylation in response to DNA damage (Figure 2E and Supplementary Figure S3G). To determine if NuMA phosphorylation may regulate 53BP1 accumulation at damaged chromatin, we first compared GFP-53BP1ct accumulation at laser microirradiated sites in cells treated with an ATM inhibitor (ATMi) versus vehicle. The ATMi treatment significantly reduced GFP-53BP1ct line formation (Supplementary Figure S3H). We could however not exclude pleiotropic effects of ATMi on 53BP1ct accumulation to DNA damage sites and therefore compared 53BP1 line formation in cells expressing GFP, GFP-NuMA, or GFP fused to a nonphosphorylatable NuMA mutant [GFP-NuMA(S395A)]. Whereas GFP-NuMA expression reduced 53BP1 line accumulation by approximately 50%, expression of the nonphosphorylatable mutant almost completely abrogated 53BP1 recruitment to DNA damage sites (Figure 2F). Together, the data suggest that NuMA acts as a barrier preventing 53BP1 accumulation at damaged chromatin. NuMA phosphorylation by ATM may serve as a release mechanism.
NuMA negatively regulates 53BP1 function
53BP1 is essential for immunoglobulin class switch recombination (CSR) during B cell maturation (64,65), a process where DSB induced in the immunoblobulin heavy chain gene are re-ligated by long-range NHEJ. We confirmed that silencing 53BP1 in CH12F3-2 murine B cells reduces immunoglobulin switching to the IgA class (Figure 3A). Next, we found that GFP-NuMA overexpression markedly reduced CSR and expression of the phospho-null NuMA mutant further exacerbated the CSR defect (Figure 3A).
Figure 3.

NuMA negatively regulates 53BP1 function. (A) CSR in CH12F3-2 B cells stably expressing scrambled and 53BP1 shRNA, or nucleofected to express GFP, GFP-NuMA, and GFP-NuMA(S395A) (mean ± SEM; n ≥ 3, ANOVA and Tukey). Representative flow cytometry contour plots are shown for cell nucleofected with GFP and GFP-NuMA constructs. 53BP1 silencing was verified by western blot (right). (B) Chromosomal aberrations (radials + fusions) in BRCA1-null SUM149 cells transfected with nontargeting or 53BP1-targeting siRNA, GFP, or GFP-NuMA. Cells were treated with olaparib (0.5 μM; 24 h). n = 40–120 cells from ≥3 replicates; statistical analysis with ANOVA and Tukey. Representative images of metaphases are displayed, with arrowheads indicating aberrations. Western blot (right) verified 53BP1 silencing and GFP-NuMA expression (upper band). (C) Quantification of NHEJ in U2OS cells with a stably integrated NHEJ-GFP reporter (left; mean ± SEM; Student's t-test). The fraction of cells expressing mCherry or mCherry-NuMA among GFP-positive cells is shown in the cross-ruled graph, whereas cell cycle distribution is shown on the right.
In breast and ovarian tumors with BRCA1 mutations, loss of 53BP1 was shown to partially restore HR, thereby reducing PARP inhibitor toxicity (13,14). As expected, siRNA-mediated depletion of 53BP1 in BRCA1-null SUM149PT breast cancer cells decreased the frequency of chromosomal aberrations in response to the PARP inhibitor olaparib (Figure 3B). GFP-NuMA overexpression significantly reduced olaparib efficacy, similarly to 53BP1 loss. Moreover, combining NuMA overexpression and 53BP1 silencing did not have an additive effect, suggesting epistasis of the two proteins (Figure 3B).
An important function of the 53BP1-RIF1 complex is to promote NHEJ by preventing DSB end-resection, the committing step for HR. In cells overexpressing mCherry-NuMA, NHEJ activity was decreased by 50% relative to controls expressing mCherry only (Figure 3C). In addition, the proportion of NHEJ-competent (GFP-positive) cells with mCherry signals was significantly reduced in the mCherry-NuMA transfections compared to the mCherry transfection, although the transfection efficiencies for mCherry and for mCherry-NuMA were not different (32 ± 4% versus 34 ± 4%, respectively).
From three independent assays (CSR, PARPi sensitivity of BRCA1-deficient cells, and NHEJ efficacy), we conclude that NuMA negatively regulates 53BP1 function. As such, NuMA may phenocopy the tudor interacting repair regulator (TIRR), a soluble factor binding to 53BP1 and preventing 53BP1 interaction with H4K20Me2 (60,66–69). Similar to 53BP1-NuMA, the interaction between 53BP1 and TIRR was shown to decrease in response to DNA damage, enabling 53BP1 chromatin binding and DDR functions (60). Interestingly, the association between 53BP1 and its previously characterized partners—as well as NuMA—increased in cells depleted from TIRR (60). Whereas TIRR stabilizes 53BP1 in the nuclear-soluble fraction (60), 53BP1 also resides in the insoluble fraction of the cell nucleus (51), where NuMA is found as well (54,70). Hence, distinct negative regulators may control (and possibly compete for) 53BP1 in the different compartments of the nucleus.
NuMA expression predicts survival in breast cancer patients
To address the in vivo relevance of 53BP1 regulation by NuMA, invasive ductal carcinoma (IDC) tissue samples were collected after surgery, irradiated, and left to recover in medium for one or six hours. As expected, the number of 53BP1 IRIF increased in irradiated tissues compared to nonirradiated controls and subsequently decreased after the recovery period (Figure 4A). NuMA was detected in discrete nuclear spots, as shown previously (54). Strikingly, the majority of the large 53BP1 nuclear bodies in nonirradiated cells overlapped with the NuMA spots. As measured in cell culture, colocalization between NuMA and 53BP1 decreased after IR in the tissue samples (Figure 4A and Supplementary Figure S4A). After six hours recovery, the overlap between the two proteins went back to the levels found in the nonirradiated controls, consistent with an initial observation that interaction between 53BP1 and NuMA measured by mass spectrometry was not different at a late time point after IR (24h). We noticed and quantified large variations in NuMA expression within IDC samples (but not in normal breast tissues derived from reduction mammoplasties; Supplementary Figure S4B). This heterogeneity in NuMA levels enabled us to ask if NuMA expression affects 53BP1 foci formation. As observed in vitro, NuMA levels negatively correlated with 53BP1 foci densities in irradiated IDC cells (Figure 4B).
Figure 4.

NuMA expression predicts survival in breast cancer patients. (A) Confocal images of 53BP1 and NuMA immunostaining in invasive ductal carcinoma (IDC). Tissue explants were mock-irradiated (control) or exposed to IR (3 Gy) and left to recover for 1 or 6 h. Arrowheads point to colocalized foci. Scale bar, 10 μm. 53BP1 foci numbers and the overlap between 53BP1 foci and bright NuMA features are quantified in the graphs (mean ± SEM; n = 3 [50–300 foci counted per condition in each sample]; Student's t-test). (B) Correlation between NuMA expression and 53BP1 foci density in irradiated tissues (IDC patients #005–7). Left: representative confocal images. NuMA staining intensity is visualized with a heat map. Center: normalized NuMA intensities are plotted against the densities of 53BP1 foci for each cell nucleus. n = 281; Spearman's p-value is indicated. Right: average foci densities in cells with low NuMA (first quintile) and high NuMA (last quintile) (mean ± SEM; n = 56; Mann–Whitney test). Scale bar, 10 μm. (C) Correlation analysis of NUMA1 and TP53BP1 mRNA expression levels in 1954 breast tumors analyzed by microarray. Data are expressed as log2 normalized signal intensities. Spearman correlation (rs) and the corresponding P-value are shown. (D) Kaplan–Meier plot of distant metastasis-free survival (DMFS) with patients stratified by NUMA1 expression quartiles. The log-rank P-value is shown.
53BP1 repair function in cancer cells may promote cancer cell resistance to IR and chemotherapy (with the notable exception of BRCA1−/− tumors) by enabling repair as well as by fueling genomic instability that results from nonhomologous end-joining. Indeed, increased radiosensitivity has been measured in 53BP1-deficient mice (17) and in glioblastoma tumors with very low levels of 53BP1 (16). A negative regulator of 53BP1 may affect cancer cell survival in the presence of DNA damage and hence, cancer treatment responses. We therefore examined NuMA gene expression levels in the Cancer Genome Atlas (TCGA) cohort and in an independent microarray analysis of breast tumors (47). In both cohorts, we observed a significant but moderate positive correlation between NUMA1 and 53BP1 transcript levels (Figure 4C and Supplementary Figure S4B). It remains to be seen if NuMA and 53BP1 protein levels are correlated as well in patient cohorts.
More importantly, NUMA1 expression was significantly associated with patient distant metastasis-free survival by Kaplan-Meier analysis in the microarray dataset, and stratification of tumors based on NUMA1 messenger levels revealed a significant increase in survival for patients with tumors with high NUMA1 expression (Figure 4D). This effect persisted when only basal-like tumors were analyzed, indicating that the good prognosis associated with high NUMA1 expression was not due to the enrichment of breast cancer subtypes with better outcomes among NUMA1-high cases (Supplementary Figure S4C). Accordingly, high NUMA1 predicted longer overall survival in patients from the TCGA cohort (Supplementary Figure S4E).
CONCLUSION
Little is known about regulation of DNA repair factors outside of the context of DNA repair foci. Here, we present data supporting a sequestration mechanism for 53BP1, which may prevent activation of the DNA damage response in the absence of DNA damage. Using multiple biophysical approaches (FCS, CP, and FLIP), we show that 53BP1 movements are restrained in regions of the nucleus devoid of repair foci, and that 53BP1 mobility increases in these regions after DNA damage induction. We identify interaction between 53BP1 and the structural nuclear protein NuMA that may be key to the proposed sequestration mechanism based on our observations that (i) NuMA depletion increases 53BP1 mobility, (ii) 53BP1 interaction and colocalization with NuMA decrease in response to DSB, (iii) NuMA antagonizes 53BP1 repair foci formation, and (iv) 53BP1 DSB repair functions are negatively regulated by NuMA, which is worthwhile considering in light of the previously established pro-HR function of NuMA (40). It will be interesting to assess if and how NuMA influences additional 53BP1-dependent cellular outcomes; in particular the fusion of dysfunctional telomeres (31). Also, the 53BP1-NuMA release mechanism and the structural basis for the interaction deserve further investigation. Both aspects are likely to involve NuMA phosphorylation by ATM, which we find occurs rapidly throughout the nucleus in response to DSB. NuMA expression and localization is frequently altered in cancer cells (71,72) and our analyses show high intra- and intertumor heterogeneity in NuMA levels, as well as a significant survival gain associated with high NUMA1 gene expression in breast cancer patients. NuMA’s mitotic function may influence therapeutic responses. In addition, the new results presented here suggest that NuMA may suppress the DSB repair function of 53BP1, in particular mutagenic NHEJ linked to 53BP1 overexpression (73), thereby increasing tumor sensitivity to genotoxic anticancer treatments and lowering mutagenic rates that contribute to treatment resistances. These effects may be exploited for therapeutic purposes.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to E. Alli (Wake Forest School of Medicine), C. Cardoso (Technische Universität Darmstadt), M. Coppey-Moisan (Institut Jacques Monod), V. Gorbunova (University of Rochester), T. Halazonetis (university of Geneva), T. Honjo (Kyoto University), T. de Lange (The Rockefeller University), T. Misteli (NCI), D. Sabatini (Whitehead Institute), E. Campeau (University of Massachusetts), and J. Nickerson (University of Massachusetts) for generously sharing cell lines and reagents. We thank M. Pettenati, K. Ragheb, J, and M. Gray for technical assistance and J. Muhlemann for helpful comments on the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [K99/R00CA163957 to P.-A.V., R01CA112017 to S.A.L.]; American Cancer Society [RSG-12-170-01-LIB to K.M.H.]; Wake Forest Center for Molecular Signaling (CMS) via its imaging facility; and the National Cancer Institute's Cancer Center Support Grant [P30CA012197] issued to the Wake Forest University CCC. Funding for open access charge: Department of Cancer Biology from the Wake Forest School of Medicine.
Conflict of interest statement. None declared.
REFERENCES
- 1. Ciccia A., Elledge S.J.. The DNA damage response: making it safe to play with knives. Mol. Cell. 2010; 40:179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Callen E., Di Virgilio M., Kruhlak M.J., Nieto-Soler M., Wong N., Chen H.T., Faryabi R.B., Polato F., Santos M., Starnes L.M. et al.. 53BP1 mediates productive and mutagenic DNA repair through distinct phosphoprotein interactions. Cell. 2013; 153:1266–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chapman J.R., Barral P., Vannier J.B., Borel V., Steger M., Tomas-Loba A., Sartori A.A., Adams I.R., Batista F.D., Boulton S.J.. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol. Cell. 2013; 49:858–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Escribano-Diaz C., Orthwein A., Fradet-Turcotte A., Xing M., Young J.T., Tkac J., Cook M.A., Rosebrock A.P., Munro M., Canny M.D. et al.. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol. Cell. 2013; 49:872–883. [DOI] [PubMed] [Google Scholar]
- 5. Panier S., Boulton S.J.. Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell Biol. 2014; 15:7–18. [DOI] [PubMed] [Google Scholar]
- 6. Dev H., Chiang T.W., Lescale C., de Krijger I., Martin A.G., Pilger D., Coates J., Sczaniecka-Clift M., Wei W., Ostermaier M. et al.. Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat. Cell Biol. 2018; 20:954–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ghezraoui H., Oliveira C., Becker J.R., Bilham K., Moralli D., Anzilotti C., Fischer R., Deobagkar-Lele M., Sanchiz-Calvo M., Fueyo-Marcos E. et al.. 53BP1 cooperation with the REV7-shieldin complex underpins DNA structure-specific NHEJ. Nature. 2018; 560:122–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gupta R., Somyajit K., Narita T., Maskey E., Stanlie A., Kremer M., Typas D., Lammers M., Mailand N., Nussenzweig A. et al.. DNA repair network analysis reveals shieldin as a key regulator of NHEJ and PARP inhibitor sensitivity. Cell. 2018; 173:972–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Noordermeer S.M., Adam S., Setiaputra D., Barazas M., Pettitt S.J., Ling A.K., Olivieri M., Alvarez-Quilon A., Moatti N., Zimmermann M. et al.. The shieldin complex mediates 53BP1-dependent DNA repair. Nature. 2018; 560:117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Isobe S.Y., Nagao K., Nozaki N., Kimura H., Obuse C.. Inhibition of RIF1 by SCAI Allows BRCA1-Mediated Repair. Cell Rep. 2017; 20:297–307. [DOI] [PubMed] [Google Scholar]
- 11. Ochs F., Somyajit K., Altmeyer M., Rask M.B., Lukas J., Lukas C.. 53BP1 fosters fidelity of homology-directed DNA repair. Nat. Struct. Mol. Biol. 2016; 23:714–721. [DOI] [PubMed] [Google Scholar]
- 12. Zimmermann M., de Lange T.. 53BP1: pro choice in DNA repair. Trends Cell Biol. 2014; 24:108–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bouwman P., Aly A., Escandell J.M., Pieterse M., Bartkova J., van der Gulden H., Hiddingh S., Thanasoula M., Kulkarni A., Yang Q. et al.. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol. 2010; 17:688–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bunting S.F., Callen E., Wong N., Chen H.T., Polato F., Gunn A., Bothmer A., Feldhahn N., Fernandez-Capetillo O., Cao L. et al.. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010; 141:243–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hurley R.M., Wahner Hendrickson A.E., Visscher D.W., Ansell P., Harrell M.I., Wagner J.M., Negron V., Goergen K.M., Maurer M.J., Oberg A.L. et al.. 53BP1 as a potential predictor of response in PARP inhibitor-treated homologous recombination-deficient ovarian cancer. Gynecol. Oncol. 2019; doi:10.1016/j.ygyno.2019.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Squatrito M., Vanoli F., Schultz N., Jasin M., Holland E.C.. 53BP1 is a haploinsufficient tumor suppressor and protects cells from radiation response in glioma. Cancer Res. 2012; 72:5250–5260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ward I.M., Minn K., van Deursen J., Chen J.. p53 Binding protein 53BP1 is required for DNA damage responses and tumor suppression in mice. Mol. Cell Biol. 2003; 23:2556–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schultz L.B., Chehab N.H., Malikzay A., Halazonetis T.D.. p53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double-strand breaks. J. Cell Biol. 2000; 151:1381–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rappold I., Iwabuchi K., Date T., Chen J.. Tumor suppressor p53 binding protein 1 (53BP1) is involved in DNA damage-signaling pathways. J. Cell Biol. 2001; 153:613–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Botuyan M.V., Lee J., Ward I.M., Kim J.E., Thompson J.R., Chen J., Mer G.. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell. 2006; 127:1361–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fradet-Turcotte A., Canny M.D., Escribano-Diaz C., Orthwein A., Leung C.C., Huang H., Landry M.C., Kitevski-LeBlanc J., Noordermeer S.M., Sicheri F. et al.. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature. 2013; 499:50–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mattiroli F., Vissers J.H., van Dijk W.J., Ikpa P., Citterio E., Vermeulen W., Marteijn J.A., Sixma T.K.. RNF168 ubiquitinates K13-15 on H2A/H2AX to drive DNA damage signaling. Cell. 2012; 150:1182–1195. [DOI] [PubMed] [Google Scholar]
- 23. Acs K., Luijsterburg M.S., Ackermann L., Salomons F.A., Hoppe T., Dantuma N.P.. The AAA-ATPase VCP/p97 promotes 53BP1 recruitment by removing L3MBTL1 from DNA double-strand breaks. Nat. Struct. Mol. Biol. 2011; 18:1345–1350. [DOI] [PubMed] [Google Scholar]
- 24. Mallette F.A., Mattiroli F., Cui G., Young L.C., Hendzel M.J., Mer G., Sixma T.K., Richard S.. RNF8- and RNF168-dependent degradation of KDM4A/JMJD2A triggers 53BP1 recruitment to DNA damage sites. EMBO J. 2012; 31:1865–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tang J., Cho N.W., Cui G., Manion E.M., Shanbhag N.M., Botuyan M.V., Mer G., Greenberg R.A.. Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination. Nat. Struct. Mol. Biol. 2013; 20:317–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jacquet K., Fradet-Turcotte A., Avvakumov N., Lambert J.P., Roques C., Pandita R.K., Paquet E., Herst P., Gingras A.C., Pandita T.K. et al.. The TIP60 complex regulates bivalent chromatin recognition by 53BP1 through direct H4K20me binding and H2AK15 acetylation. Mol. Cell. 2016; 62:409–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Baldock R.A., Day M., Wilkinson O.J., Cloney R., Jeggo P.A., Oliver A.W., Watts F.Z., Pearl L.H.. ATM Localization and Heterochromatin Repair Depend on Direct Interaction of the 53BP1-BRCT2 Domain with gammaH2AX. Cell Rep. 2015; 13:2081–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kleiner R.E., Verma P., Molloy K.R., Chait B.T., Kapoor T.M.. Chemical proteomics reveals a gammaH2AX-53BP1 interaction in the DNA damage response. Nat. Chem. Biol. 2015; 11:807–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vidi P.A., Bissell M.J., Lelievre S.. Three-dimensional culture of human breast epithelial cells: the how and the why. Methods Mol. Biol. 2013; 945:193–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Huyen Y., Zgheib O., Ditullio R.A. Jr, Gorgoulis V.G., Zacharatos P., Petty T.J., Sheston E.A., Mellert H.S., Stavridi E.S., Halazonetis T.D.. Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature. 2004; 432:406–411. [DOI] [PubMed] [Google Scholar]
- 31. Dimitrova N., Chen Y.C., Spector D.L., de Lange T.. 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature. 2008; 456:524–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Soutoglou E., Dorn J.F., Sengupta K., Jasin M., Nussenzweig A., Ried T., Danuser G., Misteli T.. Positional stability of single double-strand breaks in mammalian cells. Nat. Cell Biol. 2007; 9:675–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kudo S., Nomura Y., Segawa M., Fujita N., Nakao M., Schanen C., Tamura M.. Heterogeneity in residual function of MeCP2 carrying missense mutations in the methyl CpG binding domain. J. Med. Genet. 2003; 40:487–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Leonhardt H., Rahn H.P., Weinzierl P., Sporbert A., Cremer T., Zink D., Cardoso M.C.. Dynamics of DNA replication factories in living cells. J. Cell Biol. 2000; 149:271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Merdes A., Heald R., Samejima K., Earnshaw W.C., Cleveland D.W.. Formation of spindle poles by dynein/dynactin-dependent transport of NuMA. J. Cell Biol. 2000; 149:851–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sarbassov D.D., Guertin D.A., Ali S.M., Sabatini D.M.. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005; 307:1098–1101. [DOI] [PubMed] [Google Scholar]
- 37. Liang L., Wang X., Xing D., Chen T., Chen W.R.. Noninvasive determination of cell nucleoplasmic viscosity by fluorescence correlation spectroscopy. J. Biomed. Opt. 2009; 14:024013. [DOI] [PubMed] [Google Scholar]
- 38. Ormo M., Cubitt A.B., Kallio K., Gross L.A., Tsien R.Y., Remington S.J.. Crystal structure of the Aequorea victoria green fluorescent protein. Science. 1996; 273:1392–1395. [DOI] [PubMed] [Google Scholar]
- 39. Erickson H.P. Size and shape of protein molecules at the nanometer level determined by sedimentation, gel filtration, and electron microscopy. Biological Procedures Online. 2009; 11:32–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vidi P.A., Liu J., Salles D., Jayaraman S., Dorfman G., Gray M., Abad P., Moghe P.V., Irudayaraj J.M., Wiesmuller L. et al.. NuMA promotes homologous recombination repair by regulating the accumulation of the ISWI ATPase SNF2h at DNA breaks. Nucleic Acids Res. 2014; 42:6365–6379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sengupta P., Garai K., Balaji J., Periasamy N., Maiti S.. Measuring size distribution in highly heterogeneous systems with fluorescence correlation spectroscopy. Biophys. J. 2003; 84:1977–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wachsmuth M., Weidemann T., Muller G., Hoffmann-Rohrer U.W., Knoch T.A., Waldeck W., Langowski J.. Analyzing intracellular binding and diffusion with continuous fluorescence photobleaching. Biophys. J. 2003; 84:3353–3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Delon A., Usson Y., Derouard J., Biben T., Souchier C.. Continuous photobleaching in vesicles and living cells: a measure of diffusion and compartmentation. Biophys. J. 2006; 90:2548–2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Muller K.P., Erdel F., Caudron-Herger M., Marth C., Fodor B.D., Richter M., Scaranaro M., Beaudouin J., Wachsmuth M., Rippe K.. Multiscale analysis of dynamics and interactions of heterochromatin protein 1 by fluorescence fluctuation microscopy. Biophys. J. 2009; 97:2876–2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cole N.B., Smith C.L., Sciaky N., Terasaki M., Edidin M., Lippincott-Schwartz J.. Diffusional mobility of Golgi proteins in membranes of living cells. Science. 1996; 273:797–801. [DOI] [PubMed] [Google Scholar]
- 46. Mao Z., Bozzella M., Seluanov A., Gorbunova V.. Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair. 2008; 7:1765–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nagalla S., Chou J.W., Willingham M.C., Ruiz J., Vaughn J.P., Dubey P., Lash T.L., Hamilton-Dutoit S.J., Bergh J., Sotiriou C. et al.. Interactions between immunity, proliferation and molecular subtype in breast cancer prognosis. Genome Biol. 2013; 14:R34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Anaya J. OncoLnc: linking TCGA survival data to mRNAs, miRNAs, and lncRNAs. PeerJ Comput. Sci. 2016; 2:e67. [Google Scholar]
- 49. Abdisalaam S., Davis A.J., Chen D.J., Alexandrakis G.. Scanning fluorescence correlation spectroscopy techniques to quantify the kinetics of DNA double strand break repair proteins after gamma-irradiation and bleomycin treatment. Nucleic Acids Res. 2014; 42:e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Asaithamby A., Chen D.J.. Cellular responses to DNA double-strand breaks after low-dose gamma-irradiation. Nucleic Acids Res. 2009; 37:3912–3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gibbs-Seymour I., Markiewicz E., Bekker-Jensen S., Mailand N., Hutchison C.J.. Lamin A/C-dependent interaction with 53BP1 promotes cellular responses to DNA damage. Aging cell. 2015; 14:162–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kisurina-Evgenieva O., Mack G., Du Q., Macara I., Khodjakov A., Compton D.A.. Multiple mechanisms regulate NuMA dynamics at spindle poles. J. Cell Sci. 2004; 117:6391–6400. [DOI] [PubMed] [Google Scholar]
- 53. Silk A.D., Holland A.J., Cleveland D.W.. Requirements for NuMA in maintenance and establishment of mammalian spindle poles. J Cell Biol. 2009; 184:677–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Abad P.C., Lewis J., Mian I.S., Knowles D.W., Sturgis J., Badve S., Xie J., Lelièvre S.A.. NuMA influences higher order chromatin organization in human mammary epithelium. Mol. Biol. Cell. 2007; 18:348–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lelièvre S.A., Weaver V.M., Nickerson J.A., Larabell C.A., Bhaumik A., Petersen O.W., Bissell M.J.. Tissue phenotype depends on reciprocal interactions between the extracellular matrix and the structural organization of the nucleus. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:14711–14716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Denchi E.L., de Lange T.. Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature. 2007; 448:1068–1071. [DOI] [PubMed] [Google Scholar]
- 57. Lukas C., Savic V., Bekker-Jensen S., Doil C., Neumann B., Pedersen R.S., Grofte M., Chan K.L., Hickson I.D., Bartek J. et al.. 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat Cell Biol. 2011; 13:243–253. [DOI] [PubMed] [Google Scholar]
- 58. Fernandez-Vidal A., Vignard J., Mirey G.. Around and beyond 53BP1 nuclear bodies. Int. J. Mol. Sci. 2017; 18:E2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Gonzalez-Suarez I., Redwood A.B., Perkins S.M., Vermolen B., Lichtensztejin D., Grotsky D.A., Morgado-Palacin L., Gapud E.J., Sleckman B.P., Sullivan T. et al.. Novel roles for A-type lamins in telomere biology and the DNA damage response pathway. EMBO J. 2009; 28:2414–2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Drane P., Brault M.E., Cui G., Meghani K., Chaubey S., Detappe A., Parnandi N., He Y., Zheng X.F., Botuyan M.V. et al.. TIRR regulates 53BP1 by masking its histone methyl-lysine binding function. Nature. 2017; 543:211–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mayca Pozo F., Tang J., Bonk K.W., Keri R.A., Yao X., Zhang Y.. Regulatory cross-talk determines the cellular levels of 53BP1 protein, a critical factor in DNA repair. J. Biol. Chem. 2017; 292:5992–6003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Smeenk G., Wiegant W.W., Marteijn J.A., Luijsterburg M.S., Sroczynski N., Costelloe T., Romeijn R.J., Pastink A., Mailand N., Vermeulen W. et al.. Poly(ADP-ribosyl)ation links the chromatin remodeler SMARCA5/SNF2H to RNF168-dependent DNA damage signaling. J. Cell Sci. 2013; 126:889–903. [DOI] [PubMed] [Google Scholar]
- 63. Matsuoka S., Ballif B.A., Smogorzewska A., McDonald E.R. 3rd, Hurov K.E., Luo J., Bakalarski C.E., Zhao Z., Solimini N., Lerenthal Y. et al.. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007; 316:1160–1166. [DOI] [PubMed] [Google Scholar]
- 64. Manis J.P., Morales J.C., Xia Z., Kutok J.L., Alt F.W., Carpenter P.B.. 53BP1 links DNA damage-response pathways to immunoglobulin heavy chain class-switch recombination. Nat. Immunol. 2004; 5:481–487. [DOI] [PubMed] [Google Scholar]
- 65. Ward I.M., Reina-San-Martin B., Olaru A., Minn K., Tamada K., Lau J.S., Cascalho M., Chen L., Nussenzweig A., Livak F. et al.. 53BP1 is required for class switch recombination. J. Cell Biol. 2004; 165:459–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zhang A., Peng B., Huang P., Chen J., Gong Z.. The p53-binding protein 1-Tudor-interacting repair regulator complex participates in the DNA damage response. J. Biol. Chem. 2017; 292:6461–6467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Botuyan M.V., Cui G., Drane P., Oliveira C., Detappe A., Brault M.E., Parnandi N., Chaubey S., Thompson J.R., Bragantini B. et al.. Mechanism of 53BP1 activity regulation by RNA-binding TIRR and a designer protein. Nat. Struct. Mol. Biol. 2018; 25:591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Dai Y., Zhang A., Shan S., Gong Z., Zhou Z.. Structural basis for recognition of 53BP1 tandem Tudor domain by TIRR. Nat. Commun. 2018; 9:2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wang J., Yuan Z., Cui Y., Xie R., Yang G., Kassab M.A., Wang M., Ma Y., Wu C., Yu X. et al.. Molecular basis for the inhibition of the methyl-lysine binding function of 53BP1 by TIRR. Nat. Commun. 2018; 9:2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lydersen B.K., Pettijohn D.E.. Human-specific nuclear protein that associates with the polar region of the mitotic apparatus: distribution in a human/hamster hybrid cell. Cell. 1980; 22:489–499. [DOI] [PubMed] [Google Scholar]
- 71. Bruning-Richardson A., Bond J., Alsiary R., Richardson J., Cairns D.A., McCormac L., Hutson R., Burns P.A., Wilkinson N., Hall G.D. et al.. NuMA overexpression in epithelial ovarian cancer. PLoS One. 2012; 7:e38945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Knowles D.W., Sudar D., Bator-Kelly C., Bissell M.J., Lelièvre S.A.. Automated local bright feature image analysis of nuclear protein distribution identifies changes in tissue phenotype. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:4445–4450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zong D., Callen E., Pegoraro G., Lukas C., Lukas J., Nussenzweig A.. Ectopic expression of RNF168 and 53BP1 increases mutagenic but not physiological non-homologous end joining. Nucleic Acids Res. 2015; 43:4950–4961. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.