


Abstract
Long non-coding RNAs (lncRNAs) can act as scaffolds that promote the interaction of proteins, RNA, and DNA. There is increasing evidence of sequence-specific interactions of lncRNAs with DNA via triple-helix (triplex) formation. This process allows lncRNAs to recruit protein complexes to specific genomic regions and regulate gene expression. Here we propose a computational method called Triplex Domain Finder (TDF) to detect triplexes and characterize DNA-binding domains and DNA targets statistically. Case studies showed that this approach can detect the known domains of lncRNAs Fendrr, HOTAIR and MEG3. Moreover, we validated a novel DNA-binding domain in MEG3 by a genome-wide sequencing method. We used TDF to perform a systematic analysis of the triplex-forming potential of lncRNAs relevant to human cardiac differentiation. We demonstrated that the lncRNA with the highest triplex-forming potential, GATA6-AS, forms triple helices in the promoter of genes relevant to cardiac development. Moreover, down-regulation of GATA6-AS impairs GATA6 expression and cardiac development. These data indicate the unique ability of our computational tool to identify novel triplex-forming lncRNAs and their target genes.
INTRODUCTION
A significant portion of the human genome encodes genes that express long non-coding RNAs (lncRNAs). Nuclear lncRNAs participate in several biological processes, including chromatin organization and transcriptional regulation, and act as structural scaffolds of nuclear domains. Their size allows lncRNAs to facilitate simultaneous interactions of several molecules (1,2). Of particular interest is the interaction of lncRNAs with DNA. The advent of new techniques, including chromatin isolation by RNA purification (ChIRP), capture hybridization analysis of RNA targets (CHART), chromatin oligo affinity precipitation (ChOP), and RNA antisense purification (RAP), has helped to decipher the features of some nuclear lncRNAs and their interactions at the chromatin level (3–6). For example, in human cancer cells, lncRNA HOTAIR has been found to interact with more than 900 genomic regions close to the binding sites of EZH2 and SUZ12 (3). Similarly, thousands of interaction loci have been identified for other lncRNAs, such as MALAT1, NEAT1 (4) and MEG3 (5). Recent protocols go one step further and capture all potential RNA–DNA interactions in a given cell type (GRID-Seq (7), ChAR-Seq (8) and SPRITE (9)). Nevertheless, all these protocols capture both direct RNA–DNA interactions as well as protein mediated RNA–DNA interactions. Therefore, they are not able to reveal molecular mechanisms underlying the interaction of particular RNAs with specific DNA loci and further functional characterization of the RNAs is required.
One mechanism facilitating direct RNA–DNA interactions are triple helices. Double-stranded DNA can form triple-helical structures by accommodating a third single-stranded nucleic acid in its major groove. The third strand binds to duplex DNA by forming Hoogsteen or reverse Hoogsteen hydrogen bonds with a purine-rich (adenine-and-guanine–rich) strand of DNA in either the parallel orientation (both 5′ to 3′) or anti-parallel orientation (5′ to 3′ and 3′ to 5′) (10). Only specific combinations of bases promote the formation of stable triple-helical structures (Figure 1A).
Figure 1.

The computational framework of TRIPLEXES and TDF. (A) Triplexes are formed by binding of single-stranded RNA (blue) with a purine-rich strand (green) of a double-stranded DNA via Hoogsteen base pairing. To form a triplex in the parallel orientation, a pyrimidine or mixed motifs are required, but the anti-parallel orientation requires a purine or mixed motifs. (B) For a given RNA and DNA sequence, TRIPLEXES identifies candidate triple helices with a minimum size and maximum number of mismatches following one of the canonical codes. Each triplex is formed by one RNA sequence (triplex forming oligo – TFO) and a DNA region (triple target sites – TTS). We introduce here the concept of DNA binding domains (DBD) based on the fact that TFOs (orange) usually group in particular regions of a RNA. Contiguous regions with overlapping TFOs (marked in red) define a DNA-binding domain. (C) TDF performs statistical tests by combing predictions from TRIPLEXES to answer the following questions: (1) which regions of a RNA (DBD) are more likely to form triple helices with particular DNA target regions? (2) Which DNA regions (target genes) are more likely to be targeted by the RNA? and (3) which lncRNAs are more likely to form triple helices in a set of target DNA regions?
Regarding the functional relevance of RNA–DNA triplexes, research has shown that a nucleolar lncRNA (‘pRNA’) directly interacts with a ribosomal DNA (rDNA) promoter, forming a triple-helical structure. This structure is recognized by DNA methyltransferase DNMT3B, which methylates rDNA promoters and represses rDNA transcription (11). Later, several studies have revealed that lncRNAs, including Fendrr (12), MEG3 (5), KHPS1 (13), PARTICLE (14,15) and HOTAIR (16), directly interact with DNA in a sequence-specific manner, forming RNA–DNA triple helices. Such lncRNAs have been shown to bind to proximal (12,13) or distal (5,14–16) genomic regions and to activate (5,13) or repress transcription (12,16) through recruitment of coactivator or corepressor proteins. Besides, some lncRNAs form triple helices in cis (auto-binding) (12,13,17), that is, at the exact location where they are transcribed.
Computational methods are crucial for identification of triple helices. Initial methods were based on the search of purine rich DNA regions, but did not characterize triplex forming RNA regions (18,19). Later, an efficient algorithm for detection of triple helices of a RNA in large DNA sequences named TRIPLEXATOR (20) was proposed. It enumerates all regions of RNA and DNA sequences that are likely to engage in the formation of triple helices with size larger than l bp and with k maximum mismatches. This widely used computational tool will list tens of thousands of triple helices for a single lncRNA, but it offers few statistics to select relevant triple helices. This makes the selection of triplexes for subsequent functional studies a cumbersome manual task. LongTarget is another computational method for prediction of triple helices (21). However, this web based method only evaluates a single DNA region at a time and was recently shown to be significantly slower than TRIPLEXATOR (22). This precludes its use in the analysis of large number of RNA or DNA regions.
Our previous in silico analysis has shown that only particular regions of the lncRNA HOTAIR are likely to form triple helices with DNA (16). These regions were close to but did not overlap with known HOTAIR domains that interact with PRC2 and LSD1 complexes. One of these RNA regions was confirmed to form triple helices in two target genes distal to HOTAIR (16). This indicates the importance of computational methods for statistical characterization of triplex-forming regions within lncRNAs. We denote these regions as DNA binding domains (DBD), i.e. small regions (20–50 bp) within potentially long RNAs (>1000 bp) forming triple helices with specific DNA regions (Figure 1B). Moreover, RNA sequencing enables the discovery of hundreds of lncRNAs with cell type–specific expression and unknown function (23,24). There is a need for computational approaches to rank lncRNAs by their probability to bind to distinct DNA regions via triple-helix formation (25).
Our approach
Here, we present computational methods for identifying RNA–DNA triple helices and for characterizing lncRNAs and their respective DNA targets. First, we describe TRIPLEXES (Figure 1B) a method that improves the computational time for triplex identification in comparison to TRIPLEXATOR by using an efficient bit-parallel algorithm (26). TRIPLEXES also provides an efficient algorithm for genome-wide detection of auto-binding triple helices. Second, we introduce Triplex Domain Finder (TDF) — a computational tool for statistical characterization of the triplex-forming potential of lncRNAs (Figure 1C). TDF uses concepts similar to motif over-representation analysis, which is commonly used to find transcription factors regulating particular genes or genomic regions (27).
That is, TDF detects regions within RNA (DNA binding domains), which are more likely to form triple helices in particular target DNA regions (ChOP-Seq peaks or promoters of particular genes) than in background genomic regions (random genomic regions or all gene promoters). Moreover, TDF ranks DNA target regions or RNAs according to the triplex-forming statistics. These computational tasks addressed by TDF are crucial in the identification of RNA and DNA target regions for further biological validation and, for the first time, allows exploratory analysis evaluating several lncRNAs at a time. We are not aware of any computational method providing the same functionality as TDF.
To access the power of TDF to find novel DNA binding domains and triple helices, we evaluated the ability of TDF to identify previously reported DNA binding domains and triple helices of Fendrr, HOTAIR and MEG3 by analyzing genome-wide data that contain their potential DNA targets (5,12,28). Furthermore, we applied a new sequencing approach to validate a new DBD of MEG3 predicted by TDF. To evaluate the ability of TDF for de novo detection of triplex-forming RNAs, we performed an unbiased evaluation of 75 lncRNAs expressed during cardiac differentiation. We could confirm the triple-helix formation of the top-ranked lncRNA GATA6-AS.
MATERIALS and METHODS
Methods
First, we describe TRIPLEXES and its algorithm for enumerating all triple helices between the one (or more) provided RNA and DNA sequences following the canonical codes described in Figure 1A. Next, we describe the Triplex Domain Finder, which is a statistical framework to evaluate large numbers of triple helices predicted by TRIPLEXES (or TRIPLEXATOR) on several RNA and DNA sequences. TDF is based on two statistical tests: the promoter test, which evaluates triple-helix formation in the promoters of genes; and the genomic region test, which evaluates triple-helix formation in arbitrary genomic regions.
TRIPLEXES
For a given RNA sequence, P = p1p2⋅⋅⋅pn, and DNA sequence T = t1t2⋅⋅⋅to, the triplex detection problem is the search for (maximum length) substrings pi...pj and tu..tv from P and T with minimum length l. Triple helices follow the matching code from Figure 1A. For the case of an anti-parallel motif, matching is evaluated with the inverted RNA substring (pj⋅⋅⋅pi). In practice, we work with the relaxed version of this problem, which allows up to k mismatches to occur but no indels.
As TRIPLEXATOR, we adopt error rate e for definition of k, such that k = ⌈e × L⌉, where L is the total length of the triplex. Each triplex is represented as tuple t = (rP, rT), where rP = (i, j) represents the location in the RNA forming a triplex, and rT = (u, v) represents the location in the DNA forming a triplex. Here, we denote rP as a triplex forming oligonucleotide (TFO) and rT as a triplex target site (TTS). TRIPLEXES works by first searching for triple-helix seeds of fixed size l. This problem can be loosely formulated as a k-mismatch pattern search and is efficiently solved with the bit-parallel algorithm of Myers (26). Initially, a suffix array of all substrings of length l in P is built to have a unique set of l-grams. Subsequently, this set of l-grams is approximately matched against each DNA sequence T separately by Myers’ bit-parallel algorithm, yielding a set of seeds with length l that represent putative triple helices between P and T.
At the second step, to identify the maximal triplexes for the initial matches (seeds), these regions are extended while ensuring that all constraints, such as the maximum error rate, minimum rate of guanines, and maximum number of consecutive errors, are still satisfied. The extension of each seed is based on a heuristic algorithm: first, we precompute the positions of more distant mismatches on either side of a seed, up until a maximum number of mismatches. This process gives us an interval within which the seed should be maximally extended while satisfying all the constraints. To do so, we start with a window that includes the seed and extends to the left as much as possible. Then, we probe the next mismatch position to the right of the seed and verify if all constraints are still satisfied. If yes, we extend the window and proceed to the next mismatch to the right. If not, we resize the window from the left and continue to the right. During this process, windows of various sizes may be valid, but only the largest one is stored. Such a search is necessary because the guanine and error rates depend on the window length and must be recomputed for each shift (extension). Finally, the extended seeds that overlap are merged to build a single continuous triplex. As a result, TRIPLEXES reports a superset of all maximal triplexes satisfying all the constraints. By definition, both TRIPLEXES and TRIPLEXATOR are exact algorithms returning the same TFO-TTS pairs when presented to the same RNA/DNA sequences and parameters.
A novel feature of TRIPLEXES is the detection of auto-binding RNA–DNA triple helices. In short, in a given RNA (or DNA) sequence, for candidate string pi...pj, TRIPLEXES searches for triple helices in string pu...pv, where |i − u| = |j − v| < g for small g values (default is 3). This problem can be efficiently solved by means of an interval tree to limit the search space. This approach allows us to find all auto-binding sites in the whole genome for the given parametrization within minutes (23 minutes for human hg19).
TDF – Triplex Domain Finder
TDF is a framework for statistical characterization of triplex-forming potential of RNAs within particular target DNA regions. Starting from TFO-TTS pairs provided by TRIPLEXES, TDF first defines DNA binding domains (DBDs) by finding contiguous RNA regions with overlapping TFOs (see Figure 1B). Then, it compares the number of TTS formed by a given DBD in target DNA regions, i.e. promoters of the genes differentially expressed after the lncRNA knockdown or regions with ChIRP-Seq peaks. This is contrasted to the number of TTS of the same DBD in background regions, i.e. all promoters or random genomic regions. DBDs with statistically significant higher number of target regions with a TTS than in background regions are regarded as triplex-forming domains of the RNA. Moreover, TDF uses the statistical significance to rank detected DBDs in case more than one DBD is indicated as significantly enriched. See Supplementary Figures S1 and S2 for details on tests.
More formally, TDF executes TRIPLEXES to enumerate all triple helices of the lncRNAs found in target regions and background regions. This returns a set of triple helices (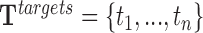 and
and 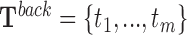 ). From
). From 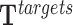 , we can obtain the set of TFOs (
, we can obtain the set of TFOs (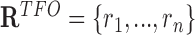 ) and the set of TTSs (
) and the set of TTSs (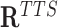 ). Next, TDF defines all candidate DBDs by finding contiguous regions within the RNA with overlapping TFOs (Figure 1B). That is
). Next, TDF defines all candidate DBDs by finding contiguous regions within the RNA with overlapping TFOs (Figure 1B). That is
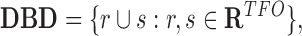 |
(1) |
where for a pair of regions r = (i, j) and s = (u, v), r∪s defines a merge operation for overlapping regions
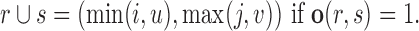 |
(2) |
and 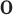 defines the overlap between two regions
defines the overlap between two regions
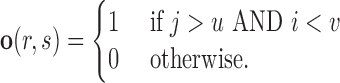 |
(3) |
TDF ignores DBDs with low TFO support (less than 5 TFOs or having TFOs associated to <5% of target DNA regions). Next, for each candidate DBD, we evaluate its triplex-forming potential by testing whether the number of DNA regions with at least one TTS associated to a DBD is greater in target DNA regions as compared to background DNA regions. This operation is currently implemented in two statistical tests described below.
Promoter test
This test evaluates whether the DNA binding domains of a RNA are likely to form triple helices in the promoter regions of candidate target genes, i.e. list of differentially expressed genes in a given biological study. The test compares the events of binding of the lncRNA in the promoters of candidate genes (target regions, 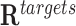 ) with the binding events in the remaining promoters of the genome (background regions,
) with the binding events in the remaining promoters of the genome (background regions, 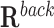 ). First, TDF enumerates all TTSs from a set of triple-helix predictions
). First, TDF enumerates all TTSs from a set of triple-helix predictions 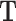 associated with DBD d
associated with DBD d
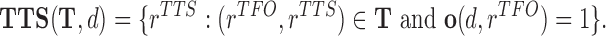 |
(4) |
Then, it counts the number of target regions with at least one TTS for the given DBD d
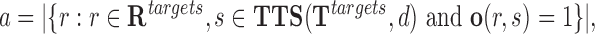 |
(5) |
and non-target regions with at least one TTS for the given DBD d.
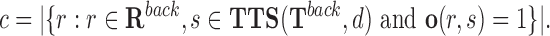 |
(6) |
After that, we define a two-by-two contingency table representing the numbers of target and non-target regions with at least one (or no) TTS for a given DBD as follows:
| with TTS | without TTS | |
|---|---|---|
| Target promoters | a |
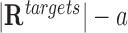
|
| Non-target promoters | c |
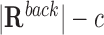
|
Finally, Fisher’s exact test is performed on the above contingency table for each DBD. TDF outputs the corrected P-value (29), an odds ratio, and binding-site statistics a and c for all candidate DBDs (see Supplementary Figure S1 for a schematic of the promoter test).
Genomic region test
This test evaluates whether the DNA binding domains of a RNA are likely to form triple helices in particular genomic regions, i.e., peaks from genome-wide essays ChIRP-Seq, ChOP-Seq, or CHART-Seq. As a background region, we generate H random regions by random selection of DNA regions with the same size/length as those of the target region 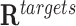 . Then, we carry out an empirical statistical test to determine whether the number of target regions with at least one TTS is greater than the number of ‘random’ non-target regions with one or more TTS for a given DBD. Unless otherwise stated, TDF performs randomization for 1000 times.
. Then, we carry out an empirical statistical test to determine whether the number of target regions with at least one TTS is greater than the number of ‘random’ non-target regions with one or more TTS for a given DBD. Unless otherwise stated, TDF performs randomization for 1000 times.
Formally, we generate H non-target regions by randomly selecting DNA positions with the same size as that of the target regions. After that, we apply TRIPLEXES to the regions and obtain predictions 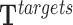 and
and 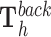 for h = 1, ..., H. Next, we evaluate all potential TTSs from the target regions as described in Equation 4. Then, we estimate the number of target regions (a) with at least one TTS for a given DBD (Equation 5). Similarly, we obtain distribution
for h = 1, ..., H. Next, we evaluate all potential TTSs from the target regions as described in Equation 4. Then, we estimate the number of target regions (a) with at least one TTS for a given DBD (Equation 5). Similarly, we obtain distribution 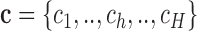 , where ch is the number of non-target regions with at least one TTS per DBD of the hth non-target region. We compute an empirical P-value by counting the values higher than a that are found in
, where ch is the number of non-target regions with at least one TTS per DBD of the hth non-target region. We compute an empirical P-value by counting the values higher than a that are found in 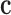 .
.
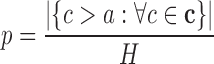 |
(7) |
Finally, we apply the false discovery rate (FDR) (29) as a multiple-test correction method to the P-values (see Supplementary Figure S2 for a schematic of the genomic region test).
Ranking of target DNA regions
TDF provides statistics to rank target regions: the number of TTSs detected in the region normalized by kilobases and the proportion of the base pairs covered by TTSs. For this purpose, TDF considers only TTSs from statistically significant DBDs. TDF also allows for the inclusion of experimental evidence, e.g., gene expression fold change or scores of ChOP-Seq peaks, as an external criterion for ranking. TDF provides a combined statistic involving the sum of ranks on all the available criteria. Moreover, the interface enables the user to select the criteria for ranking candidate target regions.
Ranking of multiple lncRNAs
TDF allows for evaluation of the triple-helix formation of multiple lncRNAs targeting a set of common DNA regions. This approach is useful in an exploratory analysis, i.e., evaluation of all lncRNAs differentially expressed in a particular differentiation process. These lncRNAs are then evaluated in the same set of target regions, i.e. promoter regions of the differentially expressed genes or regions with particular chromatin marks. TDF provides statistics such as the number of enriched DBDs and the number of TTSs to rank candidate lncRNAs. Because the sizes of lncRNAs can vary (from 200 nucleotides to 105 nucleotides in the data analyzed here), statistics are normalized by the number of bases (N = 1000/length) to avoid the bias for larger lncRNAs.
Materials
Fendrr sequence and DNA targets
To define targets of Fendrr (GenBank: JQ973641.2), we obtained RNA-Sequencing of Fendrr shRNA and control conditions deposited in GEO (GSE43078). We calculated the log2 fold change between Fendrr shRNA and control. Genes with a fold change >2 are defined as differentially expressed (1507 genes). We also included genes analysed by Grote et al. (12), which were not present in the differentially expressed gene list. Of those genes, only 1377 are mapped to TDF transcript database (Mouse GENCODE V4). Triple helix binding sites had at least 20 nt with a maximum of four mismatches and 2 consecutive errors. All the sequences were based on mm9 genome.
MEG3 sequence and DNA targets
DNA target regions of MEG3 (ENST00000451743) were obtained from ChOP-Seq experiments on human breast cancer cell line (BT-549). We used 532 MEG3 ChOP-Seq peaks close to the deregulated genes after MEG3 down-regulation as provided by Mondal et al. (5) We added the peak close to TGFB2 with a validated triple helix, as it was not included in the previous list. Triple helix binding sites had a maximum of three mismatches and two consecutive errors. A minimum triple helix size of 14 nt was used to recover the small triple helices validated in the previous study (5). To get a full list of MEG3 ChOP-Seq peaks, we have also performed peak calling with MACS2 (30) using default parameters (17 953 peaks).
HOTAIR targets
In the knockdown experiment of lncRNA HOTAIR (ENST00000424518) in fibroblasts, 1327 up-regulated genes were identified (28). We applied TDF promoter test on those genes with the parameter l = 15. Five significant DBDs are identified and four of them (I, III, IV and V) coincide with the ones detected with HOTAIR ChiRP-seq data (16). Besides, we also applied TDF on the RNA-DNA interaction data in MDA-MB-231 cell from GRID-seq experiment (GSE82312) (7). We first filtered RNAs reads which overlap with HOTAIR and then obtained reads associated to their DNA targets. Eventually 5,046 reads were identified in replicate 1 and 4,623 for replicate 2. TDF genomic region test was applied to 792 genomics regions with at least three reads. We could not find RNA reads in GRID-seq experiments for any other lncRNA evaluated here.
DBD-Capture-Seq
DBD-Capture-seq (31) experiments were performed with RNA oligos corresponding to MEG3 domain I and domain II, as well as GATA6-AS domain I (see supplementary material for protocol details). Control experiments were based on the same protocol without the inclusion of an oligonucleotide. Experiments were sequenced on a NexSeq500 Illumina platform in duplicates. The reads were aligned to the human genome (hg38) by BWA (32) (version 0.7.15-r1140), filtered according to the blacklisted genomic regions from ENCODE (33). Differential peak calling was performed by RGT-THOR (34) (RGT version 0.11.3) with the P-value cut-off = 10−2.5 by contrasting libraries with oligos (MEG3 domain I) versus control libraries. Peaks with highest signals in control libraries were used as controls.
TDF - Transcript annotation
TDF employs GENCODE annotation (35) for definition of promoters (version 24 for humans and version 4 for mice). The promoter test can be executed with either gene symbols or ENSEMBL IDs as input. Mapping of gene symbols to ENSEMBL is conducted via annotations provided by HGNC for humans (http://www.genenames.org/) and by MGI (version 6.03) for mice (http://www.informatics.jax.org).
See our supplementary material for description of experiments to characterize lncRNAs relevant cardiac differentiation, functional characterization of GATA6-AS and implementation details of TDF and TRIPLEXES.
RESULTS
TDF identifies the known triple helices of Fendrr and HOTAIR
To assess the ability of TDF to find known triple helices, we analyze Fendrr, which was reported to form a triplex in the promoter regions of developmental genes such as Foxf1 and Pitx2 (12). Notably, these triplexes were detected with a computationally expensive [O(n3)] RNA–DNA base-pairing energy model (36) and were therefore restricted to a few selected target genes (<10). To assess the triplex-forming potential of Fendrr in a genome-wide manner, we performed TDF analysis on all genes that are differentially expressed after a knockdown of Fendrr (1,507 genes) (12).
As shown in Figure 2A, TDF detected only one enriched DBD in Fendrr located in the region 1502–1565 (adjusted P-value 0.0069; Supplementary Table S1). Moreover, TDF ranked the Foxf1 promoter as the first target according to combined ranking (the number of TTSs, coverage of TTSs, and fold change in expression), while Pitx2 was ranked as the gene with the second largest number of TTSs (Supplementary Table S2). Besides, the same triplexes verified in (12) were also predicted (Supplementary Figure S3).
Figure 2.

TDF detects known and novel DNA binding domains of Fendrr and MEG3. (A, B) The coverage of TFOs (y-axis) within Fendrr and MEG3 sequences (x-axis). Regions highlighted in red/grey indicate significant DBDs. (C) DBD-Capture-Seq signals and peaks for Domain I (blue), Domain II (green), and control (red) as well as ChOP-Seq peaks in the validated triplex-forming regions (orange). (D) A Venn diagram showing the overlap between DBD-Capture-Seq (MEG3 Domains I and II) and MEG3 ChOP-Seq. De novo motifs detected in the top 500 DBD-Capture-Seq regions are also presented. (E) TDF analysis reveals a high propensity (higher z-score) of Domain I RNA to form triple helices in Domain I DBD-Capture-Seq peaks in comparison with Domain II RNA sequence and vice versa. (F, G) DBD logos indicating the nucleotides from the MEG3 domain sequence, which are predicted to form triple helices in Capture-Seq peaks of the respective domain. Higher nucleotides indicates higher number of triple helices (TTSs).
LncRNA HOTAIR was identified to impact the differentiation of mesenchymal stem cells (16). HOTAIR was shown to form triple helices on the promoter of PCDH7 to repress its expression. In order to revalidate these results with independent data, we applied TDF analysis on the genes up-regulated after knockdown of HOTAIR in primary foot fibroblasts (28) and the DNA regions predicted to interact with HOTAIR in MDA-MB-231 cells by GRID-Seq (7). TDF identifies five enriched DBDs in fibroblasts and 6 DBDs in MDA-MB-231 cells, of which four of them coincide with the ones detected with HOTAIR ChIRP-Seq data (16) (Supplementary Figure S4 and Supplementary Table S3).
Moreover, TDF ranks PCDH7 as 6th target by coverage (out of 984) and predicts the same triple helices verified in (16) in foot fibroblasts (Supplementary Figure S3 and Supplementary Table S4). This indicates similar DBDs are also used by HOTAIR in distinct cellular contexts. Taken together, results suggest that TDF can detect known DBDs in RNAs and their target genes.
TDF detects a new DBD in MEG3
A recent study suggests that lncRNA MEG3 interacts with enhancers near TGFBR1, SMAD2 and TGFB2 via triplex formation (5). We therefore wanted to predict the triplex-forming potential of MEG3 more globally and to explore novel DBDs forming triple helices with DNA regions identified by 533 MEG3 ChOP-Seq peaks close to MEG3 dysregulated genes. As shown in Figure 2B, TDF detected three significant DBDs within MEG3 (Supplementary Table S5). The most significant one (Domain I, adjusted P-value 1.0e–5) corresponded to a previously validated domain (5). Additionally, TDF ranked regions near SMAD2 and TGFBR1 as the 14th and 122th (out of 533), respectively (Supplementary Figure S3), confirming the ability of TDF to identify reported triplexes.
To experimentally evaluate the capacity of TDF for identifying novel DBDs, we performed an RNA-based DNA capture assay (DBD-Capture-Seq; 31). In short, we incubated biotinylated RNA oligos corresponding to MEG3 Domain I (known) and Domain II (new) with sheared genomic DNA to allow for triplex formation. After binding to streptavidin-coated beads, RNA-associated DNA was eluted and subjected to deep sequencing. Control experiments were conducted in the absence of biotinylated RNA oligos. Differential peak calling detected 89,739 peaks for MEG3 Domain I and 73,546 peaks for Domain II, with 12,809 peaks being common to both domains (Figure 2D). DBD-Capture-Seq Domain I peaks coincided with DNA regions near TGFBR1, SMAD2, and TGFB2 among other genes that were shown to form triple helices with MEG3 (Figure 2C) (5). Of note, a repetitive GA motif is present in Domain I DNA targets (Figure 2D). This agrees with the formation of anti-parallel purine triple helices with the GA-rich Domain I RNA (Figure 2F). On the other hand, a G-rich motif was predominant in Domain II targets (Figure 2D); this finding is consistent with the formation of parallel pyrimidine triple helices with the C-rich Domain II RNA (Figure 2G).
DBD-Capture-Seq peaks for Domains I and II overlapped with respectively 2,728 and 1,060 peaks of MEG3 ChOP-Seq peaks (5) (Figure 2D; P-value <1.0e−239 for Domain I and P-value <1.0e−51 for Domain II; intersection test (37)). Note that DBD-Capture-Seq is performed in vitro on naked DNA and therefore detects more potential DNA target sites of MEG3 than ChOP-Seq, which identifies in vivo MEG3 interactions with cross-linked chromatin. Moreover, ChOP-Seq and similar protocols (CHART-Seq, RAP-Seq or GRID-Seq) also include indirect RNA–DNA interactions (protein mediated), which explains why not all ChOP-Seq peaks overlaps with a DBD-Capture-Seq peaks. Altogether, both TDF and DBD-Capture-Seq confirms all three known triplex helices formed by MEG3.
To evaluate the power of TDF to rank DBDs from MEG3, we executed the TDF genomic region test using the top 5,000 regions identified by MEG3 DBD-Capture-Seq (Domain I, Domain II, and control peaks). This analysis ranked Domain I as the first and Domain II as the 10th for DNA sequences from Domain I DBD-Capture-Seq peaks (Supplementary Table S6). Similarly, Domain II was ranked first and Domain I fourth in the analysis of DNA sequences from Domain II DBD-Capture-Seq peaks (Figure 2E; Supplementary Table S7). No enriched DBD was detected in control peaks. The fact that Domain II is significant in forming triple helices in Domain I DBD-Capture-Seq peaks indicates that distinct DBDs target the same or very close genomic regions. This is supported by the overlap (18%) between Domain I and Domain II Capture-Seq peaks (Figure 2D). Altogether, these results confirmed the ability of TDF to detect novel DBDs and to rank them by their target DNA sequences.
Evaluation of triplex-forming potential of lncRNAs in cardiac differentiation
To investigate the capacity of TDF for ranking multiple lncRNAs by their ability to form triple helices, we evaluated lncRNAs expressed during cardiomyocyte development (Figure 3A). Human pluripotent stem cells (hPSCs) were differentiated into cardiomyocytes (38,39). Cells were harvested at Day 0 (undifferentiated hPSCs) and Day 4 (cardiac progenitors) and RNA sequencing was performed in triplicates. The identity of the cells where confirmed by the expression dynamics of pluripotency markers and cardiac progenitor markers as well as by the gene ontology enrichment analysis for the differentially expressed genes (Supplementary Figure S5).
Figure 3.

Characterisation of triple helices forming lncRNAs during cardiac differentiation. (A) The strategy for identification of lncRNAs forming triple helices during cardiac differentiation. (B) Distribution of statistics used to rank lncRNAs according to their triplex-forming potential. (C) The expression profile of GATA6-AS. (D) TDF showing the presence of two domains in GATA6-AS, which were predicted to form a triplex in promoters of the up-regulated genes. (E) A de novo identified G-rich motif in 332 Domain I DBD-Capture-Seq peaks (out of 500 top-ranked peaks). (F) DBD logo indicating the nucleotides from the GATA6-AS domain sequence, which are predicted to form triple helices in GATA6-AS Capture-Seq peaks. (G) TDF analysis showing high propensity (higher z-score) of Domain I RNA to form triple helices in corresponding Capture-Seq peaks but not in control peaks. (H) Area under the precision recall curve (blue) associating the overlap of GATA6-AS-Domain I-Capture-Seq peaks with the promoters of genes (±1 kb) as ranked by TDF.
Then, we identified all differentially expressed genes during differentiation. Of the 2,101 up-regulated genes, 75 are annotated as noncoding in GENCODE (35). Next, we carried out the TDF promoter test to evaluate the triplex formation of these 75 lncRNAs in the promoters of either up-regulated or down-regulated genes. This analysis revealed that 38 of these lncRNAs have at least one enriched DBD in either up ( for 37 lncRNAs) or down (for 18 lncRNAs) regulated genes (adjusted P-value <0.05; Supplementary Table S8). Next, we ranked the combination of lncRNAs and target genes (up or down) using the following criteria: the number of predicted TTSs, the number of DBDs per kilobase, the fold change in their expression during cardiomyocyte differentiation, and the sum of the above ranks (Figure 3B). The top-ranked lncRNA was GATA6-AS on up-regulated genes. Besides, LINC00261 (40,41), which plays a role in mesendodermal differentiation by regulating the expression of transcription factor FOXA2, ranked 6th.
GATA6-AS forms triple helices and affects cardiac mesoderm differentiation
For functional characterization of the top-ranked lncRNA GATA6-AS, we confirmed its expression with rapid amplification of cDNA ends (RACE), which indicated a larger GATA6-AS transcript than the one in GENCODE (Figure 3C and Figure S5). Moreover, RNA fractionation experiments confirms that GATA6-AS is prominently localized in the nucleus (Supplementary Figure S6). TDF analysis of GATA6-AS regarding the promoters of genes up-regulated during cardiac differentiation identified two enriched DBDs (regions 80–112 and 778–811) (Figure 3D; Supplementary Table S9). Furthermore, TDF ranked several important transcription factors as targets of GATA6-AS: MEIS1 (3rd), ID2 (4th), BMI (9th), GATA6 (12th), FOXP1 (15th), and HCN4 (79th) (Supplementary Table S10).
To validate the triplex-forming potential of GATA6-AS, we conducted a DBD-Capture experiment with an RNA oligo corresponding to Domain I of GATA6-AS (positions 80–112) and sequenced the associated DNA. Differential peak calling identified 104,786 peaks for GATA6-AS Domain I and 36,330 control peaks in experiments without RNA oligo. Captured regions were enriched with a G-rich motif (Figure 3E), in agreement with the formation of a pyrimidine parallel motif with the C-rich Domain I sequence (Figure 3F). Moreover, the TDF genomic region test revealed a high triplex-forming potential of Domain I toward the respective captured DNA regions, whereas no significant potential was found for control peaks (Figure 3G).
It is noteworthy that we found an association between the ranking of promoters proposed by TDF and GATA6-AS-Capture-Seq peaks. The TDF ranking obtained a precision higher than 80% among the top 15 genes (Figure 3H). Indeed, GATA6-AS-Capture-Seq also finds peaks in the promoter of top ranked mesoderm genes (Figure 4A). To confirm these interactions in vivo, we assessed the association of GATA6-AS with these regions in a GATA6-AS ChOP experiment. PCR-based analysis uncovered an association with predicted MEIS1, ID2, BMI1, GATA6, FOXP1 and HCN4 but not with E2F2 or MNS1 (Figure 4B).
Figure 4.

Functional characterization of GATA6-AS targets: (A) GATA6-AS DBD-Capture-Seq peaks are localized within the promoter of genes predicted to be targets of GATA6-AS by TDF. As examples of non-target regions, promoters of genes not up-regulated during cardiac differentiation are shown. (B) ChOP-PCR showing the in vivo association of GATA6-AS with the target genes identified by GATA6-AS DBD-Capture-Seq. (C) Quantitative reverse-transcription PCR (RT-qPCR) analysis of GATA6-AS and the predicted targets (GATA6 and MEIS1) after an ASO-based knockdown of GATA6-AS. (D) RT-qPCR analysis of mesodermal and cardiac mesodermal genes after the ASO-based knockdown of GATA6-AS. Error bars represent standard deviations for n = 3. The P-values were generated by a two-tailed t-test.
Finally, to test the functional relevance of GATA6-AS, we employed antisense oligos (ASOs) to deplete the GATA6-AS transcript after mesoderm induction in human embryonic stem cells (hESCs). Targeted depletion of GATA6-AS led to a decrease in GATA6-AS levels upon differentiation into cardiac mesoderm as compared to the control ASO condition (Figure 4C). Moreover, depletion of GATA6-AS down-regulated the sense transcript of GATA6 and up-regulated transcripts of mesodermal markers MEIS1 and CDX2 (Figure 4C and D). As expression and function of GATA6 are essential for differentiation of mesodermal cells into the cardiac mesoderm, our data suggest that downregulation of GATA6 owing the depletion of GATA6-AS in mesodermal cells may impact on differentiation into the cardiac mesoderm. These results reveal a novel regulatory role of GATA6-AS, which interacts with the promoters of GATA6 and other mesoderm genes via triple helices to regulate differentiation of mesodermal cells into the cardiac mesoderm.
Comparative analysis of TRIPLEXATOR, TRIPLEXES and TDF
First, we evaluate the time performance of TRIPLEXES and the two algorithms of TRIPLEXATOR (brute force and q-gram) on detection of triple helices in the sequences of 75 lncRNAs from our case study on cardiac differentiation (total combined length 283,501 nucleotides) and DNA from chromosome 22 (846,976 bp). The other triplex prediction tool, LongTarget (21), cannot be evaluated since it does not support multiple RNA sequences. Moreover, recent benchmarking work indicates its poor performance contrasted to TRIPLEXATOR (22). Time performance analyses show that TRIPLEXES is faster than TRIPLEXATOR algorithms in most parameterizations (Supplementary Figure S7). For high mismatch rates (20%), TRIPLEXES is 1.86-fold faster than TRIPLEXATOR. The use of high mismatch rates is important because they were adopted during the detection of triple helices for lncRNAs HOTAIR (16) and MEG3 (5). As expected, both TRIPLEXES and TRIPLEXATOR return the same triple helices under same input parameters.
Next, we evaluate the predictive performance of TDF and TRIPLEXATOR in finding functionally relevant triple helices. For this, we evaluate how well TDF and TRIPLEXATOR ranked DNA target regions, which overlap with DBD-Capture-Seq peaks in MEG3 or GATA6-AS. TRIPLEXATOR predictions are ranked by its criteria ‘length-adjusted triplex potential (Total Rel)’, which consider the number of TFO-TTS pairs normalized by RNA/DNA sequence size. As indicated in the Precision-Recall curves (Figure 5) and Receiver operating characteristic (ROC) curves (Figure S8), TDF has higher AUPR and ROC values than TRIPLEXATOR for both GATA6-AS and MEG3. Moreover, TDF indicates higher rankings of known MEG3 and GATA6-AS DNA targets than TRIPLEXATOR. This reinforces the unique ability of TDF to rank DNA target regions.
Figure 5.

Benchmarking of TDF and TRIPLEXATOR. (A,B) Precision recall curves based on ranking of DNA target regions from TDF and TRIPLEXATOR for MEG3 and GATA6-AS. TDF has an area under the curve of 0.54 for GATA6-AS and 0.04 for MEG3, while TRIPLEXATOR has AUPR of 0.46 for GATA6-AS and 0.03 for MEG3. Background precision corresponds to the proportion of possible DNA target regions (promoters for GATA6-AS and whole genome for MEG3), which overlap with a DBD-Capture-Seq peak of the corresponding lncRNA.
DISCUSSION
LncRNAs have the unique capacity for interaction with multiple proteins, DNA, and RNA simultaneously (1,2). While the ability of RNAs to form triplexes with DNA sequences have been studied extensively in vitro (42), psoralen-labeled oligo- and triplex-specific antibody-based assays indicate the occurrence of triplex structures in vivo as well (5,11,13). As many lncRNAs, such as HOTAIR, MEG3 and PARTICLE, have been suggested to target genomic loci in trans via triplex-formation (5,14,16), prediction of domains that participate in such interactions is crucial to understand lncRNA function. Here, we describe TDF, the first method to detect DBDs in RNAs by statistical analysis of multiple RNA sequences and target DNA sequences. We also present TRIPLEXES, which is a novel algorithm for prediction of triple helices among RNA–DNA pairs of sequences. Our results show that TRIPLEXES is faster than TRIPLEXATOR in the evaluation of long RNA–DNA sequences when high mismatch rates are used, which are crucial in the search for triple helices (Supplementary Figure S7). Moreover, TDF was more accurate in ranking DNA regions supported by DBD-Capture-Seq peaks than TRIPLEXATOR.
We show that TDF can recapitulate previously described DBDs and DNA targets of known lncRNAs Fendrr, HOTAIR and MEG3 by analysis of target regions from genome-wide data, i.e. ChOP-Seq for the RNA-interacting chromatin regions or expression data for the genes showing misregulation after a knockdown of respective triplex-forming RNA. Additionally, we identified a novel DBD in MEG3 by TDF analysis and validated its ability to form a triplex in a genome-wide DNA capture experiment. Sequencing of the captured DNA indicated that the two triplex-forming domains of MEG3 target distinct genomic regions and have distinct sequence specificity (GA repeats for Domain I and G-rich sequences for Domain II). Notably, modularity, i.e. the ability to have several interactive domains and to use them in a context-specific manner, is an important characteristic of lncRNAs (1,2). To our knowledge, this is the first study showing that a single lncRNA may utilize two DBDs to target distinct genomic loci in a sequence-specific manner.
Using TDF and lncRNAs up-regulated during cardiac differentiation, we obtained an unbiased ranking of lncRNAs by their triplex-forming potential. We next studied the lncRNA with the highest triplex-forming potential—GATA6-AS—and validated its triplex formation in a genome-wide DNA capture assay. Our finding that Domain I of GATA6-AS can form triple helices in the promoter of genes up-regulated in cardiac differentiation, e.g., GATA6 and MEIS1, points to the importance of this lncRNA and triplex formation for this process. In accord with this result, our ChOP-qPCR and GATA6-AS knockdown experiments confirmed that GATA6-AS binds to promoter regions of these target genes in vivo and controls the expression of GATA6 and of early mesodermal genes. Notably, in a recent study, GATA6-AS was shown to be associated with angiogenesis via negative regulation of repressive chromatin remodeler LOXL2 (43). It is conceivable that tethering of GATA6-AS via triple helices protects promoters of target genes from LOXL2 repression.
Application of TDF to functional data on a specific lncRNA, e.g., chromatin association or knockdown studies of MEG3 and Fendrr, helped delineate known and novel DBDs of these lncRNAs. Our analysis of cardiac differentiation indicates that TDF can also be used to identify de novo triplex-forming lncRNAs by evaluating standard RNA-Seq data. Novel protocols as GRID-Seq (7), ChAR-Seq (8), and SPRITE-Seq (9) are great resources for genome-wide RNA–DNA interactions in cells. Note however that these protocols cannot discern between direct RNA–DNA interactions as triple helices from protein mediated interactions. TDF and DBD-Capture-Seq are unique tools for detection and characterization of triple-helix forming RNAs predicted by these protocols. This brings us a step closer to dissect mechanisms and function of DNA-binding lncRNAs.
DATA AVAILABILITY
DBD-Capture-Seq of control RNAs, MEG3 Domain I, MEG3 Domain II, GATA6-AS Domain I as well as Total RNA-Seq data for Day 0 and Day 4 of cardiac differentiation are deposited in GEO with accession numbers GSE119638 and GSE115575.
Source code, tutorial and examples of the use of TRIPLEXES and TDF are found at www.regulatory-genomics.org/TDF.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Elmar Weinhold (RWTH Aachen) for discussions and the IZKF Genomics Core Facility (RWTH Aachen Medical School) for sequencing experiments.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
This work was supported by the Start Grant and the Interdisciplinary Center for Clinical Research RWTH Aachen Medical Faculty (IZKF Aachen) to IC; by the Excellence Initiative of the German federal and state governments to IC; and computing resources by ITC RWTH Aachen University to IC; by the NRW Stem Cell Network Independent Group Leader Grant [3681000801 and 2681101801 to LK]; by the Else Kroener-Fresenius Foundation (EKFS) to LK; by the Deutsche Forschungsgemeinschaft (DFG) to LK; by the KAW, Swedish Foundation for Strategic Research, Swedish Cancer Research foundation, Swedish Research Council, Barncancerfonden and LUA/ALF to C.K.. Funding for open access charge: institutional funding.
Conflict of interest statement. None declared.
REFERENCES
- 1. Guttman M., Rinn J.L.. Modular regulatory principles of large non-coding RNAs. Nature. 2012; 482:339–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Johnson R., Guigó R.. The RIDL hypothesis: transposable elements as functional domains of long noncoding RNAs. RNA. 2014; 20:959–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chu C., Qu K., Zhong F., Artandi S., Chang H., Zhong L.F., Artandi E.S., Chang Y.H.. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol. Cell. 2011; 44:667–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. West J.a., Davis C.P., Sunwoo H., Simon D.M., Sadreyev R.I., Wang P.I., Tolstorukov M.Y., Kingston R.E.. The long noncoding RNAs NEAT1 and MALAT1 bind active chromatin sites. Mol. Cell. 2014; 55:791–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mondal T., Subhash S., Vaid R., Enroth S., Uday S., Reinius B., Mitra S., Mohammed A., James A.R., Hoberg E. et al.. MEG3 long noncoding RNA regulates the TGF-β pathway genes through formation of RNA-DNA triplex structures. Nat. Commun. 2015; 6:7743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Engreitz J.M., Sirokman K., McDonel P., Shishkin A.A., Surka C., Russell P., Grossman S.R., Chow A.Y., Guttman M., Lander E.S.. RNA-RNA interactions enable specific targeting of noncoding RNAs to nascent pre-mRNAs and chromatin sites. Cell. 2014; 159:188–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li X., Zhou B., Chen L., Gou L.T., Li H., Fu X.D.. GRID-seq reveals the global RNA-chromatin interactome. Nat. Biotechnol. 2017; 35:940–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bell J.C., Jukam D., Teran N.A., Risca V.I., Smith O.K., Johnson W.L., Skotheim J.M., Greenleaf W.J., Straight A.F.. Chromatin-associated RNA sequencing (ChAR-seq) maps genome-wide RNA-to-DNA contacts. eLife. 2018; 7:e27024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Quinodoz S.A., Ollikainen N., Tabak B., Palla A., Schmidt J.M., Detmar E., Lai M., Shishkin A., Bhat P., Takei Y. et al.. Higher-order inter-chromosomal hubs shape 3D genome organization in the nucleus. Cell. 2018; 174:744–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Felsenfeld G., Davies D.R., Rich A.. Formation of a three-stranded polynucleotide molecule. J. Am. Chem. Soc. 1957; 79:2023–2024. [Google Scholar]
- 11. Schmitz K.M., Mayer C., Postepska A., Grummt I.. Interaction of noncoding RNA with the rDNA promoter mediates recruitment of DNMT3b and silencing of rRNA genes. Genes Dev. 2010; 24:2264–2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Grote P., Wittler L., Hendrix D., Koch F., Währisch S., Beisaw A., Macura K., Bläss G., Kellis M., Werber M. et al.. The tissue-specific lncRNA Fendrr is an essential regulator of heart and body wall development in the mouse. Dev. Cell. 2013; 24:206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Postepska-Igielska A., Giwojna A., Gasri-Plotnitsky L., Schmitt N., Dold A., Ginsberg D., Grummt I.. LncRNA Khps1 regulates expression of the Proto-oncogene SPHK1 via triplex-mediated changes in chromatin structure. Mol. Cell. 2015; 60:626–636. [DOI] [PubMed] [Google Scholar]
- 14. O’Leary V.V., Ovsepian S.V., Carrascosa L.G., Buske F.A., Radulovic V., Niyazi M., Moertl S., Trau M., Atkinson M.J., Anastasov N.. PARTICLE, a triplex-forming long ncRNA, regulates locus-specific methylation in response to low-dose irradiation. Cell Rep. 2015; 11:474–485. [DOI] [PubMed] [Google Scholar]
- 15. O’Leary V.B., Smida J., Buske F.A., Carrascosa L.G., Azimzadeh O., Maugg D., Hain S., Tapio S., Heidenreich W., Kerr J. et al.. PARTICLE triplexes cluster in the tumor suppressor WWOX and may extend throughout the human genome. Scientific Rep. 2017; 7:7163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kalwa M., Hänzelmann S., Otto S., Kuo C.-C., Franzen J., Joussen S., Fernandez-Rebollo E., Rath B., Koch C., Hofmann A. et al.. The lncRNA HOTAIR impacts on mesenchymal stem cells via triple helix formation. Nucleic Acids Res. 2016; 44:10631–10643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhao Z., Sentürk N., Song C., Grummt I.. lncRNA PAPAS tethered to the rDNA enhancer recruits hypophosphorylated CHD4/NuRD to repress rRNA synthesis at elevated temperatures. Genes Dev. 2018; 32:836–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Goñi J.R., de la Cruz X., Orozco M.. Triplex-forming oligonucleotide target sequences in the human genome. Nucleic Acids Res. 2004; 32:354–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gaddis S.S., Wu Q., Thames H.D., DiGiovanni J., Walborg E.F., MacLeod M.C., Vasquez K.M.. A web-based search engine for triplex-forming oligonucleotide target sequences. Oligonucleotides. 2006; 16:196–201. [DOI] [PubMed] [Google Scholar]
- 20. Buske F.A., Bauer D.C., Mattick J.S., Bailey T.L.. Triplexator: detecting nucleic acid triple helices in genomic and transcriptomic data. Genome Res. 2012; 22:1372–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. He S., Zhang H., Liu H., Zhu H.. LongTarget: a tool to predict lncRNA DNA-binding motifs and binding sites via Hoogsteen base-pairing analysis. Bioinformatics. 2014; 31:178–186. [DOI] [PubMed] [Google Scholar]
- 22. Antonov I.V., Mazurov E., Borodovsky M., Medvedeva Y.A.. Prediction of lncRNAs and their interactions with nucleic acids: benchmarking bioinformatics tools. Brief. Bioinformatics. 2018; 2018:1–14. [DOI] [PubMed] [Google Scholar]
- 23. Guttman M., Donaghey J., Carey W.B., Garber M., Grenier K.J., Munson G., Young G., Lucas B.A., Ach R., Bruhn L. et al.. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature. 2011; 477:295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pauli A., Valen E., Lin F.M., Garber M., Vastenhouw L.N., Levin Z.J., Fan L., Sandelin A., Rinn L.J., Schier A.F.. Systematic identification of long noncoding RNAs expressed during zebrafish embryogenesis. Genome Res. 2012; 22:577–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dieterich C., Stadler P.F.. Computational biology of RNA interactions. Wiley Interdiscipl. Rev. RNA. 2012; 4:107–120. [DOI] [PubMed] [Google Scholar]
- 26. Myers G. Farach-Colton M. A fast bit-vector algorithm for approximate string matching based on dynamic programming. Journal of the ACM. 1998; 46:Berlin Heidelberg: Springer; 395–415. [Google Scholar]
- 27. Sui Ho S.J. Fulton D.L., Arenillas D.J., Kwon A.T., Wasserman W.W.. OPOSSUM: integrated tools for analysis of regulatory motif over-representation. Nucleic Acids Res. 2007; 35:W245–W252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Khalil A.M., Guttman M., Huarte M., Garber M., Raj A., Rivea Morales D. Thomas K., Presser A., Bernstein B.E., van Oudenaarden A. et al.. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:11667–11672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Benjamini Y., Hochberg Y.. Controlling the false discovery Rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. B (Methodological). 1995; 57:289–300. [Google Scholar]
- 30. Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W. et al.. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008; 9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sentürk Cetin N., Kuo C.C., Ribarska T., Li R., Costa I.G., Grummt I.. Isolation and genome-wide characterization of cellular DNA:RNA triplex structures. Nucleic Acids Res. 2019; doi: 10.1093/nar/gky1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li H., Durbin R.. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics. 2010; 26:589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Allhoff M., Pires J.F., Zenke M., Costa I.G., Allhoff M., Ser K., Costa G.. Differential peak calling of ChIP-seq signals with replicates with THOR. Nucleic Acids Res. 2016; 44:e153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Harrow J., Frankish A., Gonzalez J.M., Tapanari E. et al.. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 2012; 22:1760–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lorenz R., Bernhart S.H., Höner Zu Siederdissen C., Tafer H., Flamm C., Stadler P.F., Hofacker I.L.. ViennaRNA Package 2.0. Algorithms Mol. Biol. : AMB. 2011; 6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. De S., Pedersen S.B., Kechris K.. The dilemma of choosing the ideal permutation strategy while estimating statistical significance of genome-wide enrichment. Brief. Bioinformatics. 2013; 15:919–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rao J., Pfeiffer M.J., Frank S., Adachi K., Piccini I., Quaranta R., Araúzo-Bravo M., Schwarz J., Schade D., Leidel S. et al.. Stepwise clearance of repressive roadblocks drives cardiac induction in human ESCs. Cell Stem Cell. 2016; 18:341–353. [DOI] [PubMed] [Google Scholar]
- 39. Frank S., Ahuja G., Bartsch D., Russ N., Yao W., Kuo J.C., Derks J.P., Akhade V.S., Kargapolova Y., Georgomanolis T. et al.. yylncT Defines a Class of Divergently Transcribed lncRNAs and Safeguards the T-mediated Mesodermal Commitment of Human PSCs. Cell Stem Cell. 2018; doi:10.1016/j.stem.2018.11.005. [DOI] [PubMed] [Google Scholar]
- 40. Kurian L., Aguirre A., Sancho-Martinez I., Benner C., Hishida T., Nguyen T.B., Reddy P., Nivet E., Krause M.N., Nelles D.A. et al.. Identification of novel long noncoding RNAs underlying vertebrate cardiovascular development. Circulation. 2015; 131:1278–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jiang W., Liu Y., Liu R., Zhang K., Zhang Y.. The lncRNA DEANR1 facilitates human endoderm differentiation by activating FOXA2 expression. Cell Rep. 2015; 11:137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li Y., Syed J., Sugiyama H.. RNA-DNA triplex formation by long noncoding RNAs. Cell Chem. Biol. 2016; 23:1325–1333. [DOI] [PubMed] [Google Scholar]
- 43. Neumann P., Jaé N., Knau A., Glaser S.F., Fouani Y., Rossbach O., Krüger M., John D., Bindereif A., Grote P. et al.. The lncRNA GATA6-AS epigenetically regulates endothelial gene expression via interaction with LOXL2. Nat.Commun. 2018; 9:237. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
DBD-Capture-Seq of control RNAs, MEG3 Domain I, MEG3 Domain II, GATA6-AS Domain I as well as Total RNA-Seq data for Day 0 and Day 4 of cardiac differentiation are deposited in GEO with accession numbers GSE119638 and GSE115575.
Source code, tutorial and examples of the use of TRIPLEXES and TDF are found at www.regulatory-genomics.org/TDF.