

Abstract
Background
Metagenomic methods have been widely applied to study the relationship between gut microbiota and human health. To test whether metagenomic amplicon sequencing could be an effective method to diagnose and trace the pathogens of infantile infectious diarrhea, the fecal samples of 20 diarrheic and 13 healthy infants were collected. After 16S rDNA amplicon sequencing, diversity analyses were carried out. The relationship between the pathogens of the gut microbiota and geography of patients was analyzed.
Results
The diversity of the gut microbiota in diarrheic infants was significantly lower than that of the gut microbiota in healthy ones and that, the composition of gut microbiota in the diarrheic group was significantly different than that of the gut microbiota in the healthy group. The results also indicated that in some of the patients, the amounts of Escherichia coli were significantly increased in the diarrheic infants, which was in agreement with the result of the qPCR analysis. Using a geographical map, we found some patterns between pathogen source and geographical location. This is helpful for an early warning of the disease.
Conclusions
The method of using high-throughput DNA sequencing and a comprehensive and deep data analysis can be a new strategy to detect and trace pathogens in infantile infectious diarrhea.
Trial registration Diagnosing and tracing the pathogens of infantile infectious diarrhea by amplicon sequencing, ChiCTR-DDD-1701088, Registered 16 March 2017-Retrospectively registered, http://www.chictr.org.cn/showproj.aspx?proj=18477
Keywords: Infantile infectious diarrhea, Pathogens detection, Amplicon sequencing
Background
Worldwide, infantile infectious diarrheal diseases constitute a cardinal health problem in developing countries where both morbidity and mortality rates are very high [1]. Globally, 7.6 million children under the age of 5 died in 2010 [2]. Infectious diarrhea accounts for 10.5% of all deaths [3]. Infectious diarrhea is defined as ≥ 3 loose stools in the preceding 24-h period [4–7]. Infectious diarrheal disease is the result of infection by any of a number of different bacterial, fungal and parasitic pathogens. Many factors result in the high incidence of infectious diarrhea in young children [8–11]. Infectious diseases are caused by various pathogens. For an effective treatment, the clinical detection and identification of pathogens have become primary tasks. Traditionally, the detection methods include culture, microscopy and biochemical tests [12, 13]. Conventional examination protocols usually require substantial amounts of labor, time, and skill [8, 14, 16], thus forming an obstacle to a prompt diagnosis [15, 16]. Mixed infections are common, but they are difficult to interpret by traditional methods [17].
Currently, high-throughput sequencing analysis of the amplified 16S rDNA gene allows us to detect known and unknown pathogens [18]. Unlike traditional culture methods, this is a comprehensive, non-cultivable, non-targeted, quantitative detection strategy [19]. Some metagenomics’ studies of feces from a single or a few patients have shown the feasibility of detecting bacterial pathogens [20]. However, cohort studies of detecting pathogens by metagenomics are relatively lacking, and strategies for cohort metagenomics data analysis for diagnosis purposes haven’t been fully developed, which may impede the application of 16S rDNA gene sequencing in clinical bacterial diagnosis. Here, we show an example study of the amplicon-sequencing-based detection of pathogens in individuals from an infantile infectious diarrhea cohort. In this study, we analyzed the potential pathogen in the patients by comparing their gut microbial compositions with those of local, healthy infants.
Methods
Participants
Diarrheic infant patients, including seven females and thirteen males, were recruited from Tangshan Maternal and Child Health Care Hospital. Inclusion criteria were diarrheal episodes ≥ 3 times per 24 h and an admission to the hospital within 7 days of the onset of symptoms. All the infants enrolled in this study were diagnosed as bacterial enteritis, and viral enteritis samples have been excluded according to the clinic diagnose in the hospital. The healthy control group included seven female and six male infants with a similar age range to that of the patient group, and the control group participants were not related to the diarrheic infants. All parents signed parent consent forms.
Fecal samples collection, DNA extraction and 16S rDNA sequencing
Fresh fecal samples of diarrheic and healthy infants were collected in sterile fecal collection tubes and frozen at − 80 °C. Microbial genomic DNA was extracted from each fecal sample using a previously described method [21]. The V3–V4 region of 16S rDNA from each sample was amplified by PCR using the bacterial universal primer pair 341F(CCTAYGGGRBGCASCAG)/806R(GGACTACNNGGGTATCTAA T). The library was constructed using a TruSeq® DNA PCR-Free Sample Preparation Kit (Illumina, San Diego, USA) and high throughput sequencing was conducted using a HiSeq 2500 platform according to the manufacturer’s instructions.
Bioinformatic analysis
Raw data were assigned to samples based on their unique barcode and truncated by cutting off the barcode and primer sequence. Paired-end sequencing reads were merged using FLASH to get the raw tags [22]. Then the raw tags were filtered by QIIME v1.7.0 [23] (http://qiime.sourceforge.net/) to remove low quality tags and by UCHIME Algorithm [24] to remove chimeric sequences. OTUs (operational taxonomic units) were picked from the remaining tags using Uparse v7.0.1001 [25] at a sequence similarity threshold of 97%, and the representative sequence of each OTU was annotated with RDP Classifier v2.2 [26] and Green Gene Database [27]. The OTU abundance data was normalized to 20,570, corresponding to the sample with the least sequences. All subsequent analyses were performed based on this normalized data.
For the diversity analysis, Bray–Curtis similarity coefficients were calculated based on OTUs data and plotted on a nonmetric multidimensional scaling (NMDS) graph to show the similarity among samples using the software PAST version 3.11 [28]. Analysis of similarities (ANOSIM) was performed with the distance measure set as Bray–Curtis. The Simpson index was calculated to indicate the α-diversity of each sample. The Bray–Curtis index was calculated to indicate the β-diversity for the healthy and diarrheic groups.
The OTU abundance data of healthy infants were used to form a healthy gut microbiota dataset of local infants. To find the key OTUs of the gut microbiota from diarrheal infants that could distinguish between the microbiota from the healthy infants and the microbiota from the infected infants, LEfSe analysis was conducted between the healthy gut microbiota dataset and OTU abundance from individual diarrheal patients. Due to the requirement of LEfSe that each class must contain more than one sample, the OTU data of individual patients were duplicated, which may have some minor effects LDA effect size values of OTUs. The Pathogenic Bacteria Database (http://www.globalrph.com/bacterial-strains.htm) was referred to annotating the pathogenic potential of bacteria. The cluster analysis of healthy and diarrheic infants based on the abundance of potential pathogens was conducted using PAST.
Analysis of the virulence gene of fecal microflora by real-time quantification PCR methodology
The culture conditions of E. coli O157 cells that were described by Leo Heijnen were chosen [29]. Briefly, E. coli O157 cells were grown in 5 ml m-TSB broth containing 0.02 g l−1 novobiocin for 8 h at 37 °C with agitation. The concentration of E. coli O157 colony forming units (CFU) in this suspension was determined by plating dilution series of the suspension on m-TSB (Trypticase Soy Broth Modified) agar plates (Beijing Foodsafety Biotechnology Co., China) and then counting colonies after 16 h of incubation at 42 °C. The concentration of cells in the suspension was also determined by epifluorescent microscopic counting after staining with acridine orange [30]. The CFU concentration was confirmed to be equivalent to the microscopically determined cell concentration resulting in a thoroughly quantified cell suspension. DNA was isolated and purified with a DNA extraction and purification kit according to the manufacturer’s protocol (Cat. A1120, Promega, USA).
The real-time PCR analysis of samples and E. coli O157 was performed with a quantification-PCR core kit (Toyobo) by an ABI Prism SDS 7500 instrument (Applied Biosystem). Standard curves of the 16S rDNA gene and the rfbE gene were established as described by Rinttila et al. [31]. The amount of specific bacteria was determined by the interpretation of the threshold cycle values to the standard curve, and the results were expressed as log10 CFU/g feces. The sequences of the primers that were used to detect the 16S rDNA gene of the total bacterial sample, total E. coli species and the specific rfbE gene of E. coli O157 are F(GTAAATATGTGGGAACATTTGG)/ R(GGCCTTTAAAATGTAAACAACGG) [32], F(ACCTGCGTTGCGTAAATA) /R(GGGCGGGAGAAGTTGATG) [33] and F(GTAAATATGTGGGAACATTTGG) /R(GGCCTTTAAAATGTAAACAACGG) [34] respectively. PCR conditions were 5 min at 95 °C, 40 cycles of 30 s at 95 °C, and 1 min at 60 °C. DNA amplification was monitored by measuring the accumulation of fluorescence resulting from the binding of SYBR green to double stranded DNA during the 60 °C incubation step. Finally, melting curve analysis was performed by heating the samples to 95 °C and then cooling them down to 55 °C, followed by stepwise temperature increasing of them using 0.1 °C steps, with a 10 s incubation at every step. The fluorescence was measured continuously throughout this analysis.
Statistics
The data in this study were analyzed with one-way analysis of variance (ANOVA) using SPSS v18.0 (SPSS Inc., Chicago, IL, USA). The differences were considered significant at P < 0.05. The resulting data were presented as the mean ± SD.
Results
Gut microbiota of local, healthy infants
The gut microbiota of humans differs across different ages and geographies [9]. To determine the potential pathogens that cause diarrhea in infants, the gut microbiota of local, healthy infants was first analyzed to get a healthy control profile. Gut microbiota data from seven female and six male infants ranging from 17- to 42-months-old were included in the healthy control profile of this study. The nonmetric multidimensional scaling (NMDS) plot of this healthy gut microbiota showed that there was not a significant cluster between different genders and that no trend with age was observed from the view of a comprehensive community structure (Fig. 1a). Firmicutes, Actinobacteria and Bacteroidetes were identified as the most abundant phyla in the gut microbiota of healthy infants, among which Firmicutes (66.5%) was predominant, followed by Actinobacteria (22.7%) and Bacteroidetes (7.6%). The most abundant OTU included OTU3 (Bifidobacterium, 12.8%), OTU6 (Roseburia, 9.4%), OTU7 (Faecalibacterium prausnitzii, 9.1%), OTU9 (Ruminococcus bromii, 5.8%), and OTU2 (Bifidobacterium, 4.2%). No known diarrhea-causing bacteria were found to be more abundant than 0.1% in the gut microbiota of healthy infants, and the abundance of opportunistic pathogens in healthy infant gut, such as E. coli, Enterococcus spp., Bacteroides fragilis, Acinetobacter was not more than 10% of the total bacteria.
Fig. 1.

NMDS plot of gut microbiota of healthy and diarrheic infants. a The gut microbiota of healthy infants showed no cluster by the factor of gender or age. b The gut microbiota of diarrheic infants showed disassociation with those of healthy infants. Blue circular: male healthy, blue triangle: female healthy, brown circular: male diarrheic; brown triangle: female diarrheic
Community structure and diversity analyses of gut microbiota in diarrheic infants
Fecal samples from twenty diarrheic infants were collected from a local children’s hospital. All of these diarrheic infants were no older than 42 months, and seven of them were females. The gut microbiota of diarrheic infants showed a disassociation with that of healthy infants in the NMDS plot (Fig. 1b), and the ANOSIM analysis implied a significant difference between the gut microbiota of the diarrheic infants and the gut microbiota of the healthy infants (P < 0.05). From the NMDS plot of the diarrheic gut microbiota, there was no relationship observed between community structure and age or gender of the infants (Fig. 1a). The distance between the diarrheic infant points in the plot were, on average, much larger than the distance between the healthy infant points (Fig. 1b) and this was confirmed by the result that the β-diversity of the microbiota from the diarrheic group was significantly larger than that of the microbiota from the healthy group (P < 0.05, Fig. 2a). However, the α-diversity of the gut microbiota from the diarrheic group was significantly lower than that of the gut microbiota from the healthy group (P < 0.05, Fig. 2b).
Fig. 2.

The box plots of a α-diversity (Simpson index) and b β-diversity (Bray–Curtis index) of gut microbiota of healthy and diarrheic infants. The circles represent the outliers and the asterisks represent the extremes
Pathogen determination of individual diarrheic infants
According to the obvious increased β-diversity of gut microbiota in diarrheic infants, the diarrhea in the infants in our study was most likely to have a heterogenous etiology. Thus, instead of analyzing the common pathogens in the diarrheic infant group, the potential pathogens were determined by analyzing each individual diarrheic infant. We designed the following two-step procedures to pick up the potential pathogens from individual diarrheic infants. First, the OTU were filtered by LEfSe analysis, and only the OTUs that were more present in at least one of the diarrheic individuals than in the healthy controls and that had value of LDA > 3 were picked out. This step was actually used to find biomarker OTUs in each diarrheic individual. Second, from the filtered OTUs of the first step, two kinds of OTUs were focused on: the OTUs that can be annotated to a known pathogen or opportunistic pathogen and the OTUs that were less than 0.5% abundant in each healthy individual and more than 2.5% abundant in at least one of the diarrheic individuals. The results of this analysis were shown in a heatmap (Fig. 3).
Fig. 3.

Heatmap of significantly different bacterial genera among healthy and diarrheic infants
Our results showed that the diarrhea of PE1 was caused by the known pathogen Clostridium neonatale [35] which was not found in any of the healthy individuals and was found at an abundance of less than 0.5% in any of the other diarrheic individuals but was presented in PE1 at an abundance of 21.8%. The gut microbiota of PE10 was infected by another clostridium pathogen, Clostridium perfringens [36], with an abundance of 16.0%, followed by two Bacteroides species, Bacteroides ovatus (14.4%) and Bacteroides fragilis (10.6%). PE4 and PE15 showed similar pathogen combinations including Acinetobacter guillouiae (5.4% and 5.6%), Pseudomonas stutzeri (12.2% and 10.3%), and Enterococcus spp., with the former two bacteria in distinctively high abundances compared with the gut microbiota of the healthy infants and other diarrheic infants. PE15 presented with higher abundances of Rothia mucilaginosa (15.4%) and Streptococcus peroris (15.8%), while PE4 did not. Yersinia enterocolitica was detected in PE4 (0.54%), while in another subject, this percentage was less than 0.1%. Escherichia coli dominated the gut microbiota of PE6 (76.0%) and PE14 (57.1%), and except for PE12, PE16 and PE 17, E. coli showed significant difference in all of the diarrheic infants. Enterococcus spp. dominated the gut microbiota of PE1 (55.0%), PE3 (53.1%), PE12 (95.2%), PE13 (73.2%), PE16 (91.2%), PE17 (96.3%), PE18 (92.0%), PE19 (67.3%), and PE20 (57.0%). Dialister pneumosintes (2.8%) in the gut microbiota of PE2, Citrobacter spp. (3.6%) in the gut microbiota of PE5, Megasphaera elsdenii (8.1%) and S. peroris (8.6%) in the gut microbiota of PE11 were significantly higher than those in healthy control group. However, no obvious changes in the abundances of potential bacterial pathogens were found for PE7, PE8, and PE9.
Relationship of gut microbiota pathogen and geography of patients
From the cluster analysis of healthy and diarrheic infants based on the abundance of potential pathogens, PE3 and PE19 showed clusters with distinctive characteristics in the abundance of Enterococcus faecium and E. coli. Interestingly, from the geographical map of patients (Fig. 4), the habitations of PE3 and PE19 showed a close geographical distance. Additionally, PE5 and PE7 formed a cluster (Fig. 3), and these two patients live in the same community (Fig. 4). This relationship indicated that there might be some common source of the pathogen around these geographical points.
Fig. 4.

Geography map of patients. Each red point presents the habitation of one patient
qPCR analysis of the virulence genes in the gut microbiota of diarrheic infants
To further analyze the presence of pathogenic E. coli in the gut microbiota of patients, the abundance of the virulent gene rfbE was detected by qPCR. Standard curves of the 16S rDNA and rfbE genes displayed a good linear relationship between Ct and the starting amount of DNA (R2 > 0.996), with an amplification efficiency ranging from 97 to 110%. From the results of qPCR, PE3, PE6, PE7, PE 11, PE14 showed high abundance of total E. coli species, while the rfbE gene can be detected in only PE 10, PE15 and PE19, and rfbE, as a specific gene of E. coli O157, can reflect the pathogenicity of E. coli. The result revealed that the E. coli in PE 10, PE15 and PE19 include both O157 and non-O157 serotype (Fig. 5).
Fig. 5.
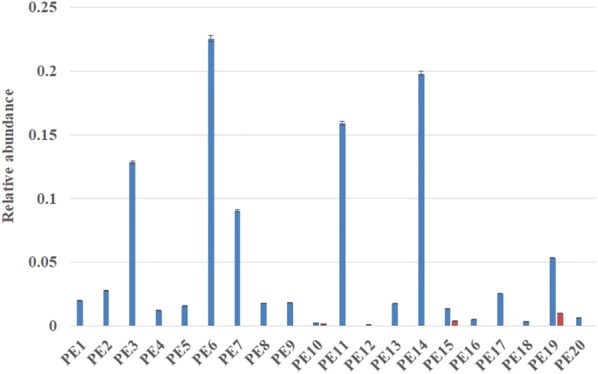
The relative abundance of E. coli species (blue bar) and E. coli O157 (red bar)
Discussion
16S rDNA sequencing based metagenomics can accurately detect the changes of gut microbiota composition. Based on the results of high-throughput sequencing, we can further tap into the relative information of the gut microbiota. By means of sequencing and in-depth data analysis described in this study and in addition to finding a causative pathogenic microbe, we analyzed the possible relationship between the microbial population’s diversity and living environment. Our main goal was to provide a new strategy for bacterial pathogen diagnosis and etiological analysis.
A variety of factors, such as environment, geographic district, dietary habits and cultural traditions are closely connected with the composition of gut microbiota [9, 31]. The features of gut microbiomes are unique to different locations. For accurate results, when conducting research involved in determining the diversity of gut microbiota, the above aspects should be considered. Therefore, for the controls in our study, we selected healthy infants from the same district as the infants with infectious diarrhea so that we could ensure a more meaningful result. This study revealed that Firmicutes accounted for the largest proportion of bacteria, which is similar to the findings of previous studies [37].
A pathogen infection would change the microbial community’s configuration. NMDS analysis of Bray–Curtis similarity coefficients can clearly show these changes in microbial community structure [38]. Using ANOSIM analysis, the result of our study revealed the significant difference in microbial community composition between healthy infants and infectious diarrhea infants. The obvious distinction was the result of enteropathogen invasion into the infectious diarrheic infants [11, 17]. In the diarrheic infant group, the points of the NMDS plot presented a more dispersed distribution. Therefore, we can predict that the increased diversity is due to a different host being infected by different enteropathogens.
The above analytical methods have been presented to discover changes in intestinal microbial profile. However, for the diagnosis of the disease, it is essential to find pathogenic bacteria and explore the etiology of symptoms. Thus, we used then LEfSe method. LEfSe is a computational approach to detail biomarker class comparisons, that helps to understand microbial communities and guide biologists or doctors in detecting novel metagenomic biomarkers [17]. The method can support high-dimensional class comparisons with a particular focus on metagenomic analysis [39]. LEfSe, as a strict tool, was utilized to identify dominant OTUs. From the result of the heatmap, the main pathogenic microbe of every sample was clear, which is something that is usually difficult to achieve by conventional examination protocols.
The geographic information system (GIS) can transfer spatial data into the geographic coordinate system or projected coordinate system, and ultimately lead to related data visualization [40]. In disease surveillance, through the analysis of time and the spatial distribution of the disease, we can find the disease propagation law. GIS has been widely applied in many infectious diseases [40, 41]. We labeled the home addresses of the participants on the geographic map. According to the spatial distribution of children, we tried to find the epidemic trend of the disease of infectious diarrhea and provide an early warning of the disease. Therefore, the strategy demonstrates its unique and significant superiority with which other examination procedures cannot compared.
The method of high-throughput sequencing has the characteristics of a comprehensive assessment. It ensures that we can select out all pathogens without leaving any out. For the precise quantitative analysis of some pathogens, the qPCR analysis has unique advantages. In this study, we verified pathogens by further qPCR analysis.
Conclusions
High throughput amplicon sequencing of fecal samples could be used to determine the pathogen of individual diarrheic infants, and the microbiota pathogen pattern from sequencing data could be combined with geography map to help to realize the process of diarrhea outbreak. These results indicated that high-throughput DNA sequencing combined with comprehensive and deep data analysis can used as a new strategy to detect and trace pathogens in infantile infectious diarrhea.
Authors’ contributions
HL conceptualized and designed the study, collected the samples, drafted the initial manuscript, reviewed and revised the manuscript. MG carried out the initial bioinformatic analysis, reviewed and revised the manuscript. YJ and JZ performed the DNA extraction. YC and QQ collected the fecal sample. XH and KH designed the study, carried out the bioinformatic analysis. WX conceptualized and designed the study and reviewed and revised the manuscript. All authors read and approved the final manuscript.
Acknowledgements
Thank you to Dr. Liu Junying from Tangshan Worker’s Hospital and Dr. Han Bing from Tangshan Maternal and Child Care Health Hospital to provide samples.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Written informed consent and questionnaires were obtained from the infants’ parents and investigations were conducted according to the principles of the Declaration of Helsinki. The consent procedure and experimental design were approved by the Ethics Committee of North China University of Science and Technology.
Funding
Funded by the National Natural Science Foundation of P.R. (Grant/Award Number: 31501586); Provincial Science and Technology Research Projects of Hebei (Grant/Award Number: ZD2015122); Outstanding Youth Foundation of North China University of Science and Technology (Grant/Award Number: JQ201716); Cultivating Fund of North China University of Science and Technology (Grant/Award Number: GP201524); Key Laboratory of Assessment of Genetically Modified Organism (Food Safety), Ministry of Agriculture, P.R. China.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- NMDS
nonmetric multidimensional scaling
- qPCR
quantitative polymerase chain reaction
- OTU
operational taxonomic unit
- ANOSIM
analysis of similarities
- Ct
cycle threshold
- GIS
geographic information system
- CFU
colony forming units
- ANOVA
analysis of variance
References
- 1.Myron M, Levine MD. Vaccines and milk immunoglobulin concentrates for prevention of infectious diarrhea. Pediatrics. 1991;118(4):S129–S136. doi: 10.1016/S0022-3476(05)81440-4. [DOI] [PubMed] [Google Scholar]
- 2.Liu L, Johnson HL, Cousens S, et al. Global, regional, and national causes of child mortality: an updated systematic analysis for 2010 with time trends since 2000. Lancet. 2012;379(9832):2151–2161. doi: 10.1016/S0140-6736(12)60560-1. [DOI] [PubMed] [Google Scholar]
- 3.Christie GT, Theresa JO. The role of maternal breast milk in preventing infantile diarrhea in the developing world. Curr Trop Med Rep. 2014;1(2):97–105. doi: 10.1007/s40475-014-0015-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.WHO Persistent diarrhoea in children in developing countries: memorandum from a WHO meeting. Bull World Health Organ. 1988;66(6):709–717. [PMC free article] [PubMed] [Google Scholar]
- 5.Guerrant RL, Oriá RB, Moore SR, et al. Malnutrition as an enteric infectious disease with long-term effects on child development. Nutr Rev. 2008;66(9):487–505. doi: 10.1111/j.1753-4887.2008.00082.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Petri WA, Jr, Miller M, Binder HJ, et al. Enteric infections, diarrhea, and their impact on function and development. J Clin Invest. 2008;118(4):1277–1290. doi: 10.1172/JC134005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walker SP, Wachs TD, Grantham-McGregor S, et al. Inequality in early childhood: risk and protective factors for early child development. Lancet. 2011;378(9799):1325–1338. doi: 10.1106/S0140-6736(11)-2. [DOI] [PubMed] [Google Scholar]
- 8.Sean RM, Noelial LL, Alberto MS, et al. Prolonged episodes of acute reduce growth and increase risk of persistent diarrhea in children. Gastroenterology. 2010;139:1156–1164. doi: 10.1053/j.gastro.2010.05.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yatsunenko T, Rey FE, Manary MJ, et al. Human gut microbiome viewed across age and geography. Nature. 2012;486(7402):222–227. doi: 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Becker-Dreps S. Changes in childhood diarrhea incidence in nicaragua following 3 years of universal infant rotavirus immunization. Pediatr Infect Dis J. 2011;3(30):243–247. doi: 10.1097/INF.0b013e3181f87ffe. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moore S, LimaI NL, Soares AM, et al. Prolonged episodes of acute diarrhea reduce growth and increase risk of persistent diarrhea in children. Gastroenterology. 2010;139(4):1156–1164. doi: 10.1053/j.gastro.2010.05.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tannock GW, Munro K, Harmsen HJM, et al. Analysis of the fecal microflora of human subjects consuming a probiotic containing Lactobacillus rhamnosus DR20. Appl Environ Microbiol. 2000;66(6):2578–2588. doi: 10.1128/AEM.66.6.2578-2588.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson HL, Liu L, Fischer-Walker C, et al. Estimating the distribution of causes of death among children age 1–59 months in high mortality countries with incomplete death certification. Int J Epidemiol. 2010;39(4):1103–1114. doi: 10.1093/ije/dyq074. [DOI] [PubMed] [Google Scholar]
- 14.Walker CLF, Friberg IK, Binkin N, et al. Scaling up diarrhea prevention and treatment interventions: a Lives Saved Tool analysis. PLoS Med. 2011;8:e1000428. doi: 10.1371/journalpmed.1000428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schlaberg R, Simmon KE, Fisher MA. A systematic approach for discovering novel, clinically relevant bacteria. Emerg Infect Dis. 2012;18(3):422–430. doi: 10.3201/eid1803.111481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stephen JS, Dhruba JS, Christopher R, et al. Rapid 16S rRNA next-generation sequencing of polymicrobial clinical samples for diagnosis of complex bacterial infections. PLoS ONE. 2013;8(5):e65226. doi: 10.1371/journal.pone.0065226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taniuchi M, Sobuz SU, Begum S, et al. Etiology of diarrhea in bangladeshi infants in the first year of life analyzed using molecular methods. J Infect Dis. 2013;208(11):1794–1802. doi: 10.1093/infdis/jit507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shota N, Norihiro M, Ionut MM, et al. Metagenomic diagnosis of bacterial infections. Emerg Infect Dis. 2008;14(11):1784–1786. doi: 10.3201/eid1411.080589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.von Bubnoff A. Next-generation sequencing: the race is on. Cell. 2008;132:721–723. doi: 10.1016/j.cell.2008.02.028. [DOI] [PubMed] [Google Scholar]
- 20.Nakamura S, Maeda N, Miron IM, et al. Metagenomic diagnosis of bacterial infections. Emerg Infect Dis. 2008;14:1784–1786. doi: 10.3201/eid1411.080589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo M, Huang K, Chen S, et al. Combination of metagenomics and culture-based methods to study the interaction between ochratoxin a and gut microbiota. Toxicol Sci. 2014;141(1):314–323. doi: 10.1093/toxsci/kfu128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Magoč T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27(21):2957–2963. doi: 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Caporaso JG, Kuczynski J, Stombaugh J, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7(5):335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Edgar RC, Haas BJ, Clemente JC, et al. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27(16):2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods. 2013;10(10):996–998. doi: 10.1038/nmeth.2604. [DOI] [PubMed] [Google Scholar]
- 26.Wang Q, Garrity GM, Tiedje JM, et al. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73(16):5261–5267. doi: 10.1128/AEM.0062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DeSantis TZ, Hugenholtz P, Larsen N, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72(7):5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hammer Ø, Harper D, Ryan P. Past: paleontological statistics software package for education and data analysis. Paleontol Electron. 2001;4(1):1–9. [Google Scholar]
- 29.Heijnen L, Medema G. Quantitative detection of E. coli, E. coli O157 and other shiga toxin producing E. coli in water samples using a culture method combined with real-time PCR. J Water Health. 2006;4:487–498. doi: 10.2166/wh2006.026. [DOI] [PubMed] [Google Scholar]
- 30.Hobbie JE, Daley RJ, Jasper S. Use of nuclepore filters for counting bacteria by fluorescence microscopy. Appl Environ Microbiol. 1977;33(5):1225–1228. doi: 10.1128/aem.33.5.1225-1228.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rinttila T, Kassinen A, Malinen E, et al. Development of an extensive set of 16S rDNA-targeted primers for quantification of pathogenic and indigenous bacteria in faecal samples by real-time PCR. J Appl Microbiol. 2004;97(6):1166–1177. doi: 10.1111/j/1365-2672.2004.02409.x. [DOI] [PubMed] [Google Scholar]
- 32.Fortin NY, Mulchandani A, Chen W. Use of real-time polymerase chain reaction and molecular beacons for the detection of Escherichia coli O157:H7. Anal Biochem. 2001;289(2):281–288. doi: 10.1006/abio.2000.4935. [DOI] [PubMed] [Google Scholar]
- 33.Mcdaniels AE, Rice EW, Reyes AL. Confirmational identification of Escherichia coli, a comparison of genotypic and phenotypic assays for glutamate decarboxylase and beta-d-glucuronidase. Appl Environ Microbiol. 1998;64(10):4113. doi: 10.1128/aem.64.10.4113-4113.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nadkarni MA, Martin FE, Jacques NA, Hunter N. Determination of bacterial load by real-time PCR using a broad-range (universal) probe and primers set. Microbiology. 2002;148(1):257–266. doi: 10.1099/00221287-148-1-257. [DOI] [PubMed] [Google Scholar]
- 35.Alfa MJ, Robson D, Davi M, et al. An outbreak of necrotizing enterocolitis associated with a novel clostridium species in a neonatal intensive care unit. Clin Infect Dis. 2002;35(Supplement 1):S101–S105. doi: 10.1086/341929. [DOI] [PubMed] [Google Scholar]
- 36.Asha NJ, Tompkins D, Wilcox MH. Comparative analysis of prevalence, risk factors, and molecular epidemiology of antibiotic-associated diarrhea due to Clostridium difficile, Clostridium perfringens, and Staphylococcus aureus. J Clin Microbiol. 2006;44(8):2785–2791. doi: 10.1128/JCM.0165-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Backhed F, Roswall J, Peng YQ, et al. Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microbe. 2015;17(5):690–703. doi: 10.1016/j.chom.2015.04.004. [DOI] [PubMed] [Google Scholar]
- 38.Looft T, Johnson TA, Allen HK, et al. In-feed antibiotic effects on the swine intestinal microbiome. PNAS. 2012;109(5):1691–1696. doi: 10.2307/41477160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang CH, Li SF, Yang L, et al. Structural modulation of gut microbiota in life-long calorie-restricted mice. Nat Commun. 2013;4(2163):1–35. doi: 10.1038/ncomms3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fradelos EC, Papathanasiou IV, Mitsi D, et al. Health based geographic information systems (GIS) and their applications. Acta Inform Med. 2014;22(6):402–405. doi: 10.5455/aim.2014.22.402-405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ezatti M, Utzinger J, Cairncross S, et al. Environmental risks in the developing world: exposure indicators for evaluating interventions, programmes and policies. J Epidemiol Commun Health. 2005;59(1):15–22. doi: 10.1136/jech.2003.019471. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.