

ABSTRACT
Cell type specification during early nervous system development in Drosophila melanogaster requires precise regulation of gene expression in time and space. Resolving the programs driving neurogenesis has been a major challenge owing to the complexity and rapidity with which distinct cell populations arise. To resolve the cell type-specific gene expression dynamics in early nervous system development, we have sequenced the transcriptomes of purified neurogenic cell types across consecutive time points covering crucial events in neurogenesis. The resulting gene expression atlas comprises a detailed resource of global transcriptome dynamics that permits systematic analysis of how cells in the nervous system acquire distinct fates. We resolve known gene expression dynamics and uncover novel expression signatures for hundreds of genes among diverse neurogenic cell types, most of which remain unstudied. We also identified a set of conserved long noncoding RNAs (lncRNAs) that are regulated in a tissue-specific manner and exhibit spatiotemporal expression during neurogenesis with exquisite specificity. lncRNA expression is highly dynamic and demarcates specific subpopulations within neurogenic cell types. Our spatiotemporal transcriptome atlas provides a comprehensive resource for investigating the function of coding genes and noncoding RNAs during crucial stages of early neurogenesis.
KEY WORDS: Drosophila melanogaster, Embryogenesis, lncRNA, Spatiotemporal transcriptome, Neurogenesis
Summary: DIV-MARIS, an adapted technique for examining stage- and cell type-specific gene expression, reveals a complex network of mRNAs and lncRNAs expressed in specific cell types during early Drosophila embryonic nervous system development.
INTRODUCTION
Development of complex tissues from naïve primordia requires the precise spatiotemporal deployment of transcriptional programs as cells subdivide, specify and differentiate. Owing to the availability of tissue- and cell type-specific markers characteristic for neurogenic cell types in the fruit fly embryo (Heckscher et al., 2014), Drosophila neurogenesis is highly tractable and several crucial regulators of neurogenesis have been identified over the past several decades (Skeath and Thor, 2003; Beckervordersandforth et al., 2008; Broadus et al., 1995; Landgraf et al., 1997; Rickert et al., 2011; Wheeler et al., 2006; Doe, 2017; Heckscher et al., 2014; Skeath et al., 1994; Weiss et al., 1998; Wheeler et al., 2009). Among the earliest events in embryonic neurogenesis is the subdivision of the lateral neurogenic ectoderm into columnar domains along the dorsoventral axis (Von Ohlen and Doe, 2000; Cowden and Levine, 2003). This is followed by the formation of proneural clusters and consecutive phases of delamination, whereby neuroblasts cease contact with surrounding cells of the neuroectodermal columns and ingress into the embryo (Campos-Ortega, 1995). Embryonic neuroblasts – Drosophila neural stem cells – undergo a series of self-renewing asymmetric divisions that produce ganglion mother cells, which give rise to glia and neurons (Broadus et al., 1995; Sousa-Nunes et al., 2010; Homem and Knoblich, 2012; Heckscher et al., 2014). Importantly, each of the three neurogenic columns gives rise to molecularly and functionally distinct sets of neuroblasts (Doe, 1992), but the molecular mechanisms that link spatial origin to the ensuing distinct fates remain poorly understood. To date, a small set of marker genes specifically expressed in individual columnar domains and in emerging cell types has been identified, but it remains unclear how these cell populations differ with respect to the global gene expression programs that shape their identities.
Although expression dynamics of protein-coding transcripts have given important insights into the mechanisms that drive cellular differentiation, it should be noted that an emerging class of noncoding transcripts – the long noncoding RNAs (lncRNAs) – may well emerge as pivotal regulators of neurogenesis. In mammals, lncRNAs have been shown to be especially abundant in differentiated neuronal cells (Briggs et al., 2015), are expressed often with exquisite spatiotemporal specificity in the nervous system (Sauvageau et al., 2013; Goff et al., 2015), and some lncRNA species even exhibit neuronal subtype specificity (Molyneaux et al., 2015; Liu et al., 2016). Though the functional importance of some lncRNAs for development and cellular identity has been demonstrated in Drosophila (Wen et al., 2016), including in the nervous system (Li and Liu, 2015; Landskron et al., 2018), very little is known about the cell type-specific expression and function of lncRNAs over the course of early neurogenesis.
Large-scale efforts have characterized spatial gene expression in RNA in situ hybridization screens (Tomancak et al., 2002; Inagaki et al., 2005; Tomancak et al., 2007; Lécuyer et al., 2007; Wilk et al., 2016), but such efforts are qualitative rather than quantitative and largely exclude lncRNAs. In contrast, efforts to determine global transcriptome dynamics in the developing Drosophila embryo (Graveley et al., 2011; Brown et al., 2014; Young et al., 2012; Chen et al., 2016) may detect the expression of lncRNAs, but lack cell type resolution. As for most complex tissues, recapitulating early neurogenesis in cell culture is unfortunately not an option, because accurate specification and differentiation of cells depends on embryonic context, intricate interactions among cells within the neuroectoderm (Kunisch et al., 1994; Lai, 2004) and signaling gradients involving surrounding tissues (Bier and De Robertis, 2015; Rogers et al., 2017).
To overcome these limitations and to dissect stage- and cell type-specific transcriptomes in early neurogenesis, we adapted MARIS (Hrvatin et al., 2014) for use in developing Drosophila embryos. DIV-MARIS (Drosophila in vivo method for analyzing RNA following intracellular sorting) allows purification of chemically cross-linked cell types from staged developing embryos based on marker gene expression, followed by RNA extraction and next-generation sequencing. Here, we employ DIV-MARIS to determine the transcriptome dynamics in distinct neurogenic cell populations. We assess the gene expression programs of two principal neurogenic domains (the ventral and the intermediate columns) and of three differentiating cell types (neuroblasts, neurons and glia) at consecutive time points from primordial specification and subdivision to terminal differentiation.
DIV-MARIS reveals an extensive network of dynamic spatiotemporal gene expression during embryonic nervous system development. Our method reliably identifies known cell type-specific markers, but also reveals novel expression features. Furthermore, we uncover many genes – most of which have conserved homologs in human – that are expressed in distinct cell types throughout early neurogenesis and for which the functions remain to be elucidated. Hence, DIV-MARIS provides an accurate expression map of spatiotemporal transcriptional programs driving early nervous system development. Moreover, our analyses identified many lncRNAs expressed in cell type-specific patterns and for which no functional roles are yet known. Applying stringent criteria for selection, we characterize 13 neural cell type-enriched lncRNAs with varied temporal expression, abundance and subcellular localization. In situ visualization of lncRNA expression exposes an additional layer of specificity as neuroglial lncRNAs tend to be expressed highly, but only in extremely distinct subpopulations.
This study delivers a genome-wide, yet cell type-specific, view of gene expression during Drosophila neurogenesis from neurogenic columns to differentiated neurons and glia, provides insights into the expression properties of the coding and noncoding transcriptomes and will serve as a valuable tool for understanding how regulated coding and noncoding gene expression drives cell fate determination in early neurogenesis.
RESULTS
Isolation of neuroglial cell types with spatiotemporal resolution
Early Drosophila neurogenesis starts with the specification of the lateral neurogenic ectoderm at the onset of zygotic transcription. The neurogenic ectoderm is quickly subdivided into distinct neurogenic columns (Von Ohlen and Doe, 2000; Cowden and Levine, 2003), from which neuroblasts delaminate and undergo asymmetric division giving rise to ganglion mother cells (GMCs), followed by differentiation of GMCs into neurons and glia (Fig. 1A). To dissect the genome-wide transcriptional programs driving early neurogenesis, we purified specific cell populations comprising the neuroglial lineages using fluorescence-activated cell sorting (FACS) of chemically fixed cells. We isolated cells of the intermediate column (IC) and the ventral column (VC) using transgenic constructs by fusing IC- or VC-specific enhancers to reporter genes (Fig. S1A). Neuroblasts/GMCs, neurons and glia cells were purified using antibodies directed against the endogenous markers prospero (pros), embryonic lethal abnormal vision (elav) and reversed polarity (repo), respectively (Fig. 1D, Fig. S1B). Early neurogenesis is a rapidly unfolding process, with naïve primordia developing into differentiated cell types in a matter of hours (Fig. S2A). To assess the temporal dynamics of early neurogenesis, we collected these cell populations at developmental stages (bins) that encompass crucial events along the neurogenic lineages from early specification to terminal differentiation (Fig. 1B, Fig. S2A). Timed embryo collections were manually staged to confirm which neurogenic events were captured within the collection bins (Fig. S2B). The earliest collection bin (4-6 h after egg laying, AEL) primarily contains embryos immediately after specification and subdivision of the neurogenic ectoderm and encompasses the first rounds of neuroblast delamination. The second bin (6-8 h AEL) includes all waves of neuroblast delamination, proliferation and diversification, followed by early differentiation in the third bin (8-10 h AEL). A later collection towards the end of embryogenesis (18-22 h AEL) serves as a reference point for fully differentiated neurons and glia.
Fig. 1.

DIV-MARIS for the enrichment of staged neurogenic cell types. (A) Biological materials studied over the course of neurogenesis: intermediate column (IC); ventral column (VC); neuroblasts (NBs); neurons; glia. (B) Time windows (collection bins) of sorted materials. (C) Overview of the DIV-MARIS protocol. (D) Merge (right) of antibody (left) and RNA-FISH (middle) shows that the sorting strategy faithfully marks cell types of interest (NBs, Pros; neurons, Elav; glia, Repo); embryos at 6-8 h. (E) Expression of marker genes specific to the cell type of interest measured by qPCR in marker-enriched (M+), and marker-depleted (M−) populations, calculated relative to whole embryo (WE, dashed red line); embryos collected at 4-10 h AEL. Error bars represent s.e.m. Embryos are ∼500 µm in length, shown anterior leftwards and ventral downwards; stage 10/11.
To isolate cell type-specific RNA from specific neurogenic cell types, we adapted the MARIS protocol (Hrvatin et al., 2014), but had to introduce several modifications to temporally resolve cell types from complex and quickly developing Drosophila embryos in vivo. DIV-MARIS (outlined in Fig. 1C) is a flexible method for the isolation of high-quality RNA from specific fixed cell types within complex and rapidly developing embryos. Briefly, staged embryos are collected, dissociated into single-cell suspensions, and immediately cross-linked with formaldehyde. Neurogenic cell types were stained using antibodies, either against transgenic reporters (for the ventral and intermediate columns; Fig. S1A), or against endogenous markers (for neuroblasts/GMCs, neurons and glia; Fig. 1D, Fig. S2B). Positively marked and unmarked populations were purified by FACS (Fig. 1C). We used microscopy (e.g. Fig. S1C) and analytical cytometry (e.g. Fig. S3) to confirm that the sorting strategy reliably isolated marked cells of interest; samples generally had purities >95% and samples below 90% purity were discarded. Furthermore, we evaluated the enrichment of DIV-MARIS-sorted cell types by quantitative RT-PCR against several marker genes associated with the cell types of interest (i.e. pros and worniu in neuroblasts/GMCs, elav and Lim3 in neurons, repo and gcm in glia) as independent measures of cell-type enrichment (Fig. 1E). We confirmed specific enrichment of the expected markers in sorted cells compared with whole embryos, as well as their depletion in sorted marker-negative cells.
As DIV-MARIS robustly isolates neurogenic cell populations of interest, we extracted RNA from sorted populations at four developmental time points for whole-transcriptome sequencing. Principal component analysis demonstrated that variance between samples is primarily due to developmental time and cell type of origin (Fig. S4). The resulting cell type-specific gene expression atlas quantitatively assesses neurogenic transcription in five distinct neurogenic cell populations (enriched and depleted) across four developmental time points covering major neurogenic events (Fig. 1A,B).
Cell type-specific expression of protein-coding genes during neurogenesis
In addition to purity, we evaluated sorting specificity by assessing gene expression of the five cell type marker genes (ind, vnd, pros, elav and repo) across the sorted populations in terms of normalized counts (Table S1). In all cases, strong enrichment of marker gene expression levels in the marker-enriched compared with the depleted samples was observed, as expected (Fig. 2A, Fig. S5). For example, the high and near-exclusive enrichment of repo transcript in purified glia demonstrates sorting effectiveness of DIV-MARIS when using a highly specific and exclusive marker (Fig. 2A, Fig. S5E). Similarly, elav transcript levels were highly enriched in purified neurons compared with glia (Fig. 2A, Fig. S5D), whereas lower levels were detected in early neuroblasts and columnar material, which is in line with observations that the common neuronal marker elav is transiently expressed pan-neurogenically at the onset of differentiation (Berger et al., 2007). The columnar markers vnd and ind mark distinct columnar neurogenic territories that each give rise to neuroblasts, neurons and glia. Accordingly, although vnd and ind transcripts were largely exclusive to their respective neurogenic columns, each was detectable to some degree in neuroblasts, most likely because early neuroblasts stem from one of the respective neurogenic columns co-purified by FACS (Fig. 2A, Fig. S5A-C). Interestingly, normalized counts for vnd were higher than those for ind, which likely reflects the fact that the ventral column generates more neuroblasts in the first waves of delamination compared with the intermediate column (Doe, 1992).
Fig. 2.

Defining mRNA signatures of neuroglial cell types. (A) Normalized expression values for each marker gene used for FACS (ind, vnd, pros, elav and repo) across sorted samples. Error bars represent s.e.m. (B) Heat map of expression profiles of Drosophila nervous system genes. Row mean-centered expression values calculated by variance-stabilizing transformation (VST) of gene-level RNA-seq counts (scale=log2 ratio of row mean).
To validate cell type-specific gene expression, we examined genes with known neurogenic roles (Von Ohlen and Doe, 2000; Skeath and Thor, 2003; Sousa-Nunes et al., 2010; Crews, 2010; Sandler and Stathopoulos, 2016) and confirmed specificity of mRNA expression in cell types previously associated with gene function (Fig. 2B). exex, for example, is a homeodomain transcription factor required in motor neurons that project to ventral somatic muscles (Santiago et al., 2014) and we found it exclusively in young neurons (Fig. 2B). Whereas markers of neuroblast identity were not only enriched in neuroblasts, but also depleted in the differentiated cell types neuroblasts give rise to (neuronal and especially glial), neurogenic column marker expression was often maintained in neuroblasts, highlighting that neuroblasts retain columnar identity after delamination, as they adopt column-specific fates (Doe, 1992).
To systematically uncover protein-coding genes that demarcate columnar and cell-type identities in nervous system development, we looked for genes expressed in a similar pattern to known neurogenic genes by Pearson correlation (r>0.9). We uncovered 753 additional genes (summarized in Table S2) and though many have no known association with embryonic neurogenesis, in situ screens annotating expression using controlled anatomical imaging vocabulary (ImaGO; Hammonds et al., 2013; Tomancak et al., 2002, 2007) indicate that this gene set is indeed specifically expressed in components of the developing nervous system. For example, the most enriched ImaGO terms for this gene set include ‘ventral nerve cord primordium’, ‘brain primordium’ and ‘ventral nerve cord’ (Fig. S6A). GO analysis revealed the most enriched molecular function for this gene set to be ‘DNA binding’, and the most enriched biological processes were ‘chromosome organization’ and ‘nucleic acid metabolic process’ (Fig. S6B,C). Furthermore, protein domains enriched among the proteins specifically expressed in compartments of the developing nervous system included histone folds, chromatin interaction domains and sequence-specific DNA-interaction domains, such as zinc fingers and homeobox domains (Fig. S6D).
We were surprised that one-quarter of the genes deployed similarly to known neurogenic marker genes remain largely unstudied (199 ‘computed genes’) and though many of these candidates lack any described function, more than 62% can be directly mapped to human homologs and some have even been linked to nervous system function.
We focused on a subset (40) of these genes, which were predicted to be expressed in neuroglial cell types with clear spatiotemporal specificity (Fig. S7A). In concordance with DIV-MARIS predictions, RNA in situ hybridization data (Hammonds et al., 2013; Tomancak et al., 2002, 2007) confirmed that a selection of these candidate genes mark specific subsets of cells in the developing nervous system (Fig. S7B).
Thus, DIV-MARIS reliably captures and uncovers cell type-specific gene expression dynamics during embryogenesis. As many of the specifically expressed genes encode known and predicted transcription factors and signaling pathway components (Table S2), this cell type-specific expression map identifies new regulatory nodes that likely play central roles in the specification and differentiation of neuroglial cell types.
Specific expression and properties of long noncoding RNAs along the neuroglial lineage
To explore lncRNA expression during early neurogenesis, we first identified nervous system-specific lncRNAs by calculating enrichment of expression in marker-positive versus marker-depleted samples at each time point using DESeq2 (Love et al., 2014; log2FC>1.0, Padj<0.05). We found 325 such lncRNA candidates (Table S3) and evaluated them according to several criteria, including spatiotemporal regulation through neurogenesis, expression above an abundance threshold [transcript counts per million reads (TPM)>300] in at least one cell type, absence of sense overlap with a protein-coding gene, and transcript boundaries consistent with lncRNA annotations. Applying these stringent criteria, we selected 13 high-confidence lncRNA candidates that are strongly and specifically expressed in a variety of cell types of the Drosophila nervous system (Fig. 3, Fig. S8).
Fig. 3.

Neuroglial lncRNAs are highly regulated transcripts. (A) Row mean-centered expression values of lncRNAs in marker-enriched and -depleted samples (scale=log2 ratio of row mean, gene level VST). (B) PhyloCSF scores (ScorePerCodon) for the putative ORF with the highest coding potential within each transcript. Scale is from ∼1 s.d. above the mean score of coding regions (very high coding potential) down to ∼1 s.d. below the mean of noncoding regions (very low coding potential). (C) Row mean-centered expression profiles in 6-8 h and 18-22 h embryo nuclear and cytoplasmic fractions generated by Fractionation-Seq; values as in A. (D) Violin plot showing distribution of maximum TPM (maxTPM) values for all lncRNAs (red; n=325) and mRNAs (gray; n=3835) differentially expressed (log2FC>1.0, Padj<0.05) between any marker-positive and marker-negative cell type; lncRNAs presented in A are highlighted. (E) CR30009 genomic locus showing stranded RNA-seq data from sorted glia at 6-8 h AEL (negative strand; blue), overlay of smoothed PhyloCSF scores of individual codons in each of three frames (horizontal line is 0), and conservation among drosophilids (phastCons).
To assess spatiotemporal expression of these lncRNAs, we calculated their relative abundance among all cell types and collection bins. The lncRNAs were depleted in marker-negative non-neurogenic cells and exhibited dynamic spatiotemporal enrichment in specific marker-positive cell types (Fig. 3A). Strikingly, although we found very few lncRNAs with distinct expression in the earlier and more naïve intermediate and ventral columns, specific lncRNA deployment could be readily observed in more mature and differentiated cell types, such as neuroblasts, neurons and/or glia, indicating that lncRNA expression is a hallmark of differentiated cells more so than of primordia.
To confirm that these transcripts are bona fide lncRNAs, we evaluated the coding potential of each by phylogenetic codon substitution frequency (PhyloCSF) (Lin et al., 2011). Each lncRNA locus exhibited a total PhyloCSF score below zero across all frames, consistent with a complete lack of coding potential (Fig. 3B). Given that some lncRNAs have been shown to exhibit variable subcellular localization with localized functions (Chen, 2016), we assessed the general subcellular expression of these transcripts by Fractionation-Seq. Briefly, we generated a subcellular reference transcriptome of the cytoplasmic and nuclear compartments of 6-8 h and 18-22 h embryos and examined abundance of each of these lncRNA transcripts between these fractions. Intriguingly, the 13 lncRNAs exhibited distinct subcellular localization patterns with varying degrees of nuclear/cytoplasmic restriction (Fig. 3C), ranging from almost exclusively cytoplasmic (e.g. CR30009 and cherub) to almost exclusively nuclear (e.g. CR45312) detection, including instances in which location appeared to be temporally regulated (e.g. CR44978).
To assess lncRNA abundance relative to other transcripts (noncoding and protein-coding) in the neurogenic cell types, we normalized read counts for each transcript in each sample (TPM, Table S4). The maximum expression score for lncRNAs across cell types (maxTPM) showed that although expression varies among lncRNAs, they are generally not expressed at low levels; rather, lncRNA expression was well within the range of what may be expected for protein-coding genes significantly regulated during neurogenesis (Fig. 3D). That these lncRNAs are bona fide regulated transcripts is further supported by specific splicing, which was observed for several of the neurogenic lncRNAs (Fig. 3E, Fig. S8). Thus, these lncRNAs are unlikely to be merely by-products of spurious transcription; rather, they are subject to regulated expression, RNA processing, and controlled export, which supports a potential role in neurogenesis.
One intriguing example of a lncRNA demonstrating specific expression over the course of early neurogenesis is CR30009. This lncRNA showed increased expression in the early intermediate column and in neuroblasts, but was most highly enriched in glial cells during all assayed time windows (Fig. 3A). Furthermore, CR30009 is spliced and primarily exported to the cytoplasm (Fig. 3C,E), features indicative of specific co- and post-transcriptional regulation. However, CR30009 has the lowest coding potential out of all tested lncRNAs: its PhyloCSF score per codon (−42.647) was more than three standard deviations below the mean for noncoding regions in Drosophila (−18.7±7.2, Fig. 3B). Furthermore, CR30009 is one of the most highly abundant transcripts in glia – noncoding or protein-coding (log2 maxTPM=15.75, Fig. 3D, Table S3) – which underscores the potential functional importance of CR30009 in gliogenesis. Notably, this lncRNA appears to exist predominantly as an unannotated short isoform and exhibits regions of high noncoding sequence conservation among drosophilids within the first exon and at the 3′ end of the transcript (Fig. 3E).
A second example, CR43283 (also known as cherub), exhibited dynamic temporal regulation. Expression of cherub was strongly enriched in the earliest neuroblasts at 4-6 h, but enrichment quickly waned in later neuroblasts (6-8 h); however, over time cherub became specifically expressed being strongly enriched in differentiated neurons and glia by the end of neurogenesis at 18-22 h AEL (Fig. 3A). We note that enriched expression of the lncRNA in Elav- and Repo-negative samples may be caused by cherub-positive glia in the neuron-depleted fraction and cherub-positive neuroblasts/neurons in the glia-depleted fraction. cherub was also specifically localized to the cytoplasm throughout embryogenesis and is clearly spliced, but harbors no coding potential (Fig. 3B,C, Fig. S8D).
CR32730 was first detected in 4-6 h neuroblasts and was moderately enriched at 8-10 h in the neuronal, but not in the glial, population (Fig. 3A). CR32730 is transcribed antisense to the intron of CG9650 (Fig. S8C), a putative neurogenic transcription factor that has been implicated in CNS development (McGovern et al., 2003). However, CR32730 appears to be transcribed independently of CG9650, which was expressed at low levels in early neuroblasts according to DIV-MARIS (Fig. S8C), suggesting that their roles could be independent. Fractionation-Seq predicts that CR32730 is moderately enriched in the nuclear fraction in early and late embryos (Fig. 3C).
Expression of another lncRNA, CR46003, was first detected in the ventral column and was most highly enriched in early neuroblasts, but expression persisted in neuroblasts and early neurons (Fig. 3A). CR46003 was one of the most abundant lncRNAs in our dataset and did not exhibit clear subcellular enrichment in either early or late embryos (Fig. 3C,D). Intriguingly, the transcription start site of CR46003 is antisense to CR46004, which contains a miRNA implicated in behavior (Picao-Osorio et al., 2017) (Fig. S8B).
CR44024 expression was first enriched in early neuroblasts and persisted through neuronal differentiation, and is predicted to be excluded from the intermediate and ventral columns and glia (Fig. 3A). This lncRNA is not predicted to exhibit distinct subcellular localization in early (6-8 h) embryos, but was moderately enriched in the cytoplasm at the end of embryogenesis (18-22 h, Fig. 3C). CR44024 was also one of the highly expressed lncRNAs in our dataset with expression on a par with protein-coding genes (Fig. 3D). The transcript is intergenic, and appears to be spliced, although not in accordance with its annotated transcript model (Fig. S8E).
In summary, DIV-MARIS predicted spatiotemporal expression of a number of lncRNAs during neurogenesis. Through the application of stringent criteria, we refined this list to a high-confidence selection of noncoding transcripts with diverse predicted expression patterns and properties. To confirm these predictions for several lncRNA candidates, we first visualized their expression in the context of a whole developing embryo.
Neurogenic lncRNAs mark specific neuroglial subsets
To visualize lncRNA expression, we performed multiplex RNA fluorescent in situ hybridization (RNA-FISH; Kosman et al., 2004) against the five examples discussed above (CR30009, cherub, CR46003, CR32730 and CR44024) together with neurogenic marker genes. Remarkably, RNA-FISH revealed exquisite spatiotemporal specificity of lncRNA expression for each of the lncRNAs tested.
CR30009 – predicted by DIV-MARIS to be highly enriched in glia – was indeed co-expressed with repo as expected in clusters of glial cells as early as stage 9/10 (Fig. 4A,B, Fig. S9). CR30009 remained co-expressed with most repo-expressing cells through stage 13/14 (Fig. 4C,D). However, timing of CR30009 expression suggests it to be independent of repo, indicating that this lncRNA constitutes an earlier marker of the glial lineage than currently known. Although most repo-positive cells also expressed CR30009 in stage 9-12 embryos, the lncRNA was largely expressed in small puncta within other cells in the ventral nerve cord and brain that are likely to be neuroblasts (Fig. 4A,B, Fig. S9). Accordingly, DIV-MARIS predicts CR30009 expression in 4-6 h and 6-8 h pros-positive cells (Fig. 3A, Fig. S10). It is feasible, therefore, that the lncRNA CR30009 constitutes the earliest neuroblast marker of the glial lineage identified to date, which accumulates into larger, brighter foci during early phases of glial differentiation (Figs S9, S10).
Fig. 4.
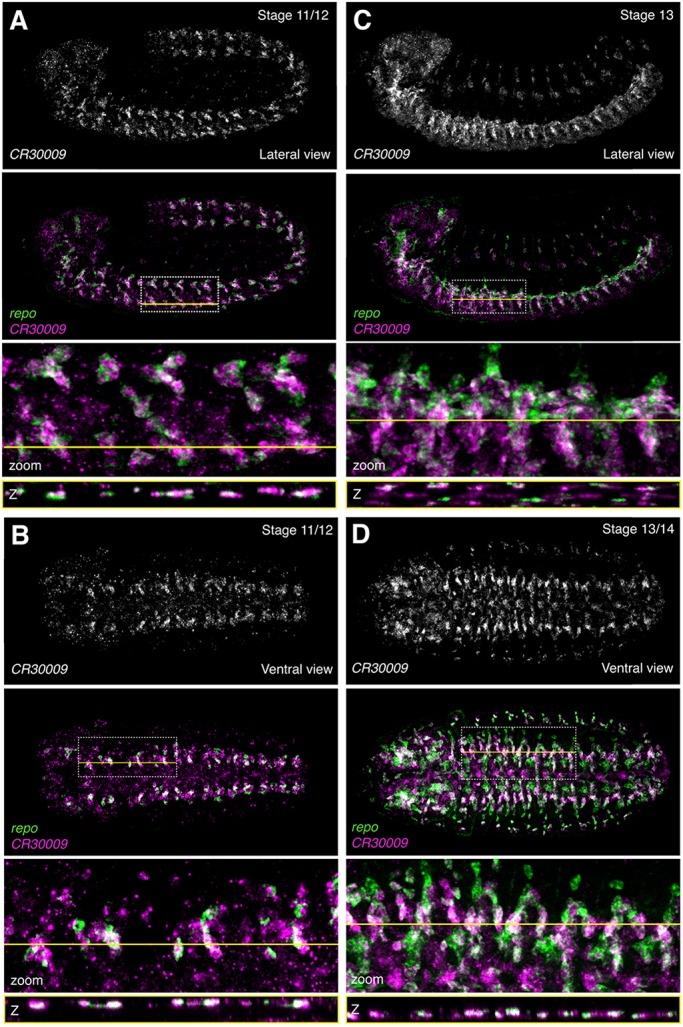
The lncRNA CR30009 is expressed in glial subsets. RNA-FISH against CR30009 and the glial marker repo. (A) Lateral view, stage 11/12. (B) Ventral view, stage 11/12. (C) Lateral view, stage 13. (D) Ventral view; stage 13/14. Top: CR30009 alone; below: merge of CR30009 (magenta) with repo (green). Second from bottom: enlargement of the region of interest (ROI) indicated by the dotted white box. Bottom: Slice through z-stack at the level indicated by the yellow line. Embryos are ∼500 µm along the long axis, oriented anterior leftwards and ventral downwards in lateral views.
RNA-FISH against the lncRNA cherub revealed strong spatiotemporal regulation of cherub broadly in accordance with DIV-MARIS, which predicted cherub to be strongly and specifically enriched in early (4-6 h) neuroblasts, and late (18-22 h) neurons and glia (Fig. 3A). We observed clear induction of cherub expression within six small clusters of cells in the ventral nerve cord during stage 12, each of which also expressed pros (Fig. 5A) and to a lesser degree, elav (Fig. 5B); both of these observations are in line with cherub constituting a neuroblast marker. During stage 13, cherub was seen in several additional clusters in the brain (Fig. 5B, Fig. S11A,B). By stage 14-15, cherub was very strongly expressed in multiple defined pros neuroblast clusters, but appeared to be excluded from mature neurons and glia (Fig. 5C,D, Figs S11C,D, S12), and remained strongly expressed through the remainder of embryogenesis (stage 16/17, Fig. S12B), in line with DIV-MARIS predictions (Fig. 3A).
Fig. 5.
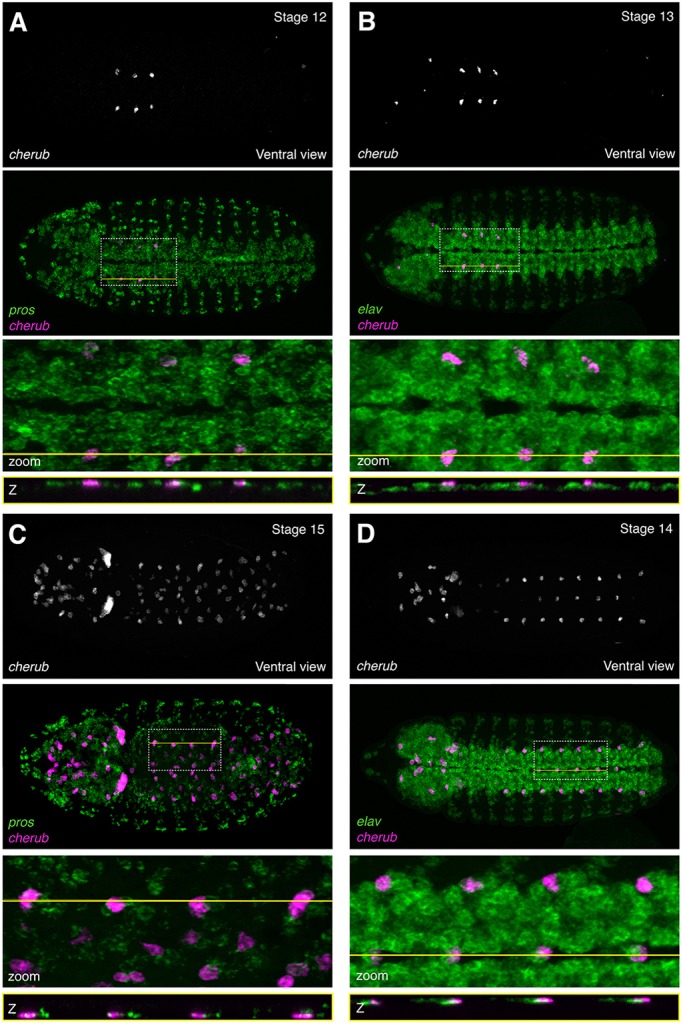
The lncRNA cherub is expressed with strict spatiotemporal specificity primarily in a subset of neuroblasts. RNA-FISH against cherub, the neuroblast marker pros and the neuronal marker elav. Ventral views. (A) cherub with pros; stage 12. (B) cherub with elav; stage 13. (C) cherub with pros; stage 15. (D) cherub with elav; stage 14. Top: cherub alone. Second from top: cherub (magenta) overlaid with marker (green). Second from bottom: enlargement of the region of interest (ROI) indicated by the dotted white box. Bottom: Slice through z-stack at the level indicated by the yellow line. Embryos are ∼500 µm along the long-axis, oriented anterior leftwards.
DIV-MARIS predicts similar spatiotemporal expression of CR46003 and CR32730 in neuroblasts and neurons (Fig. 3A). Indeed, RNA-FISH revealed very similar patterns of expression of the two lncRNAs. CR46003 exhibited the earliest expression of all lncRNAs tested here and is was in a small cell cluster already at stage 5-6 (Fig. S13). By stage 9-10, punctate expression of CR46003 appeared in defined pros-expressing clusters along the embryonic ventral midline (Fig. 6A), in agreement with the DIV-MARIS-predicted enrichment in cells of the ventral column and neuroblasts at 4-6 and 6-8 h AEL (Fig. 3A). CR46003 expression expanded to a greater number of cells within and beyond the ventral nerve cord and brain from stage 11-13, many of which also expressed pros (Fig. 6B, Fig. S14A-C) and some expressed elav as well (Fig. S15). As predicted by DIV-MARIS, RNA-FISH demonstrated that CR32730 follows a very similar pattern of expression to CR46003 from stage 9-10 to stage 13 (Fig. 6, Fig. S14).
Fig. 6.
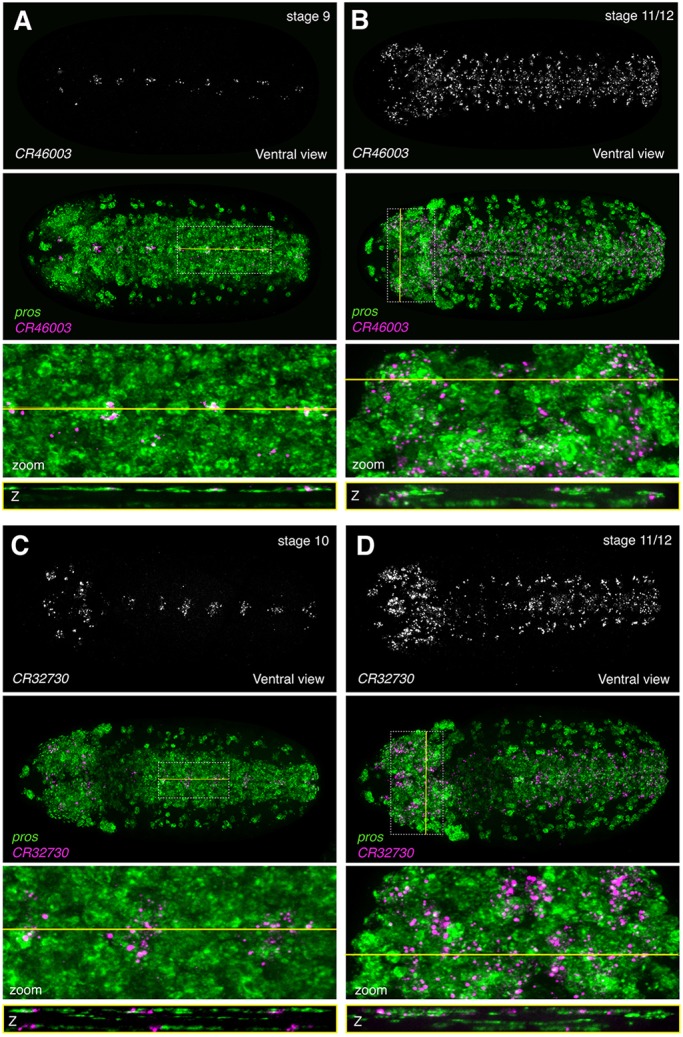
The lncRNAs CR46003 and CR32730 are expressed with similar spatiotemporal specificity in a subset of neuroblasts. RNA-FISH against CR46003 and CR32730 together with the neuroblast marker pros. Ventral views. (A) CR46003; stage 9. (B) CR46003; stage 11/12. (C) CR32730; stage 10. (D) CR32730; stage 11/12. Top: lncRNA alone. Second from top: lncRNA (magenta) overlaid with pros (green). Second from bottom: enlargement of the region of interest (ROI) indicated by the dotted white box. Bottom: Slice through z-stack at the level indicated by the yellow line. Embryos are ∼500 µm along the long axis, oriented anterior leftwards.
Although we were not able to detect the transient CR44024 expression in early (stage 9-10) pros-positive neuroblasts as predicted by DIV-MARIS, we did observe that this lncRNA exhibits highly dynamic temporal regulation. At stage 12, CR44024 was induced within small elav-positive clusters flanking the midline (Fig. S16). Starting at stage 13, CR44024 was expressed much more broadly, yet was still restricted to subsets of elav- and pros-expressing cells within the ventral nerve cord and central brain (Fig. 7).
Fig. 7.
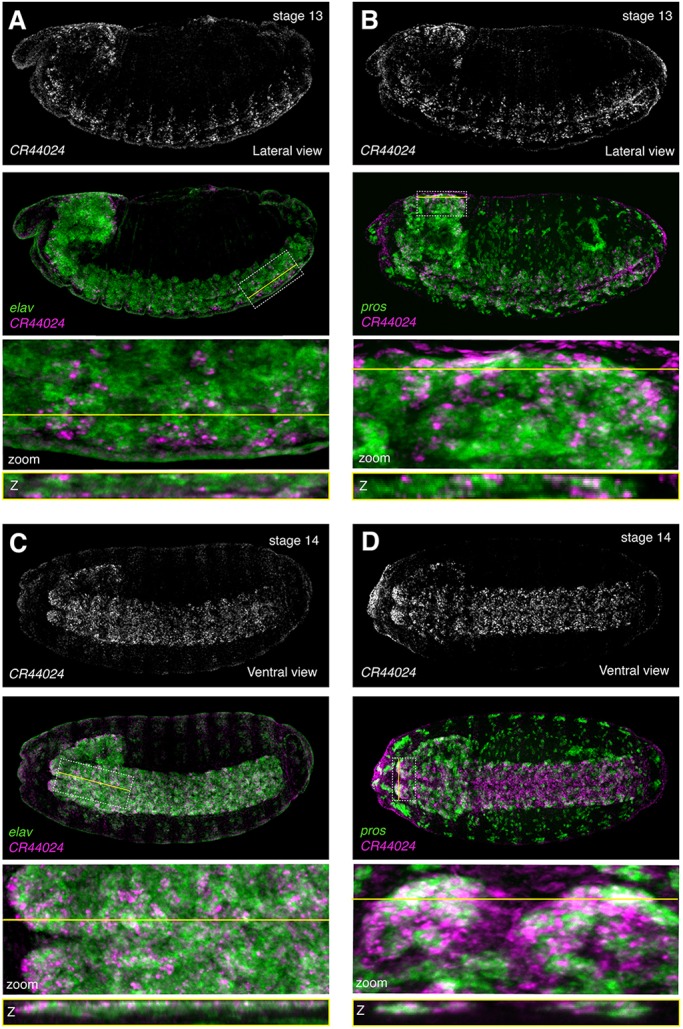
The lncRNA CR44024 is expressed later in embryogenesis in neuronal subsets. RNA-FISH against CR44024, the neuroblast marker pros and the neuronal marker elav. (A) CR44024 and elav; lateral view; stage 13. (B) CR44024 and pros; lateral view; stage 13. (C) CR44024 and elav; ventral view; stage 14. (D) CR44024 and pros; ventral view; stage 14. Top: CR44024 alone. Second from top: CR44024 (magenta) overlaid with marker (green). Second from bottom: enlargement of the region of interest (ROI) indicated by the dotted white box. Bottom: Slice through z-stack at the level indicated by the yellow line. Embryos are ∼500 µm along the long axis, oriented anterior leftwards and ventral downwards in lateral views.
Lastly, we assessed the subcellular localization of individual lncRNAs. For example, Fractionation-Seq (Fig. 3C) predicted CR30009 and cherub to be predominantly cytoplasmic. This is supported for both lncRNAs by high-resolution confocal microscopy, as both transcripts were primarily detected in the cytoplasm (Fig. S17A,B). CR46003 and CR32730 both showed a slight bias for nuclear localization by Fractionation-Seq, which was confirmed by microscopy as both lncRNAs were clearly stained within the nucleus, though it should be noted that subnuclear puncta were observed (Fig. S17C,D). Similarly, CR44024 appeared to be restricted primarily to the nucleus in the ventral nerve cord at stage 14 (Fig. S17E), matching the prediction.
The identification of such complex, yet specific, expression patterns highlights the importance of tissue- and cell type-specific expression analysis. Whole embryo studies, for example, not only lack spatial resolution, but expression signatures – even of highly expressed genes – may be lost if their expression is specific to a small-enough subset of cells. Here, we provide a map for the cell type-specific expression of coding, as well as noncoding, RNAs over the course of embryonic neurogenesis in the developing Drosophila embryo. Although hundreds of coding and dozens of lncRNAs are deployed with specific spatial and temporal dynamics, it should be noted that direct imaging of expression within a spatial context can reveal nuances of expression that is beyond the resolution of many cell type-specific genomic approaches.
DISCUSSION
Complex tissues are defined by the intricate interplay of individual cell types that differ in their gene expression programs. Tissue culture has long been an important tool for the genome-wide investigation of cellular responses as it avoids much of the heterogeneity inherent to living tissues. Unfortunately, it is often precisely this heterogeneity and the dynamic contacts between cells and tissues that shape cellular identities and transcriptomic responses. Hence, to determine the gene regulatory programs that drive complex organismal development, it is crucial to (1) preserve the cellular interactions in vivo, (2) acquire genome-wide transcriptomic data with spatial and/or cell-type resolution, and (3) assure temporal resolution.
DIV-MARIS to investigate global cell type-specific gene expression dynamics
To investigate the transcriptome dynamics over the course of neurogenesis from primordial to neuronal and glial identities, we developed a method of isolating specific cell types from Drosophila embryos with resolution in time and space. DIV-MARIS is widely applicable and can be employed for spatiotemporal transcriptional profiling of basically any cell type of interest in the Drosophila embryo and other complex tissues, as long as markers allowing for sorting a cell type of interest are available (i.e. appropriate antibodies or transgenic markers e.g. enhancer-reporter constructs). DIV-MARIS employs chemical cross-linking of the cellular material, thus ensuring that the developmental status quo is preserved, and elaborate sorting strategies based on multiple markers, which could be devised to fine-tune the sub-population selection one wishes to purify (Molyneaux et al., 2015).
Here, we purified fixed cells based on markers of specific neurogenic cell populations in the early Drosophila embryo. DIV-MARIS faithfully resolved known expression patterns of neurogenic protein-coding genes, but also identified cell type-specific expression of additional genes with yet unknown neurogenic functions; neuroglial expression was confirmed by in situ hybridization for a few dozen mRNAs, but hundreds more are predicted to exhibit spatiotemporal expression over the course of early neurogenesis. This compendium lays the groundwork for a comprehensive understanding of the mechanisms driving early neurogenesis and, given that many of the spatiotemporally expressed genes encode regulatory factors such as transcription factors and signaling molecules, careful examination of their neurogenic roles will be required.
Identification of spatiotemporal lncRNA expression
This study has identified many cell type-specific lncRNAs with potential neurogenic function. We emphasize that this is not yet an exhaustive list of lncRNAs expressed in the nervous system, as our filtering criteria were conservative. Instead, we focused on a high-confidence set of 13 lncRNAs with a variety of expression and transcript characteristics. Given that these noncoding transcripts are (1) temporally expressed in specialized cell types and subtypes of the nervous system, (2) moderately-to-highly abundant and (3) often exhibit hallmarks of RNA processing (such as splicing and nuclear-cytoplasmic shuttling), these lncRNAs appear to be subject to regulated expression rather than being by-products of spurious transcription.
Notably, we did not identify any lncRNAs with expression restricted to the early neuroectodermal columns. According to DIV-MARIS, there is some enrichment of CR30009 in the intermediate column and CR46003 in the ventral column (Fig. 3A), but as the respective territorial markers of the ventral and intermediate columns are still detectable in the neuroblast progenitors, this expression may be specific to neuroblasts, in which higher enrichment is observed. High and specific lncRNA expression appears to be a feature of differentiating and differentiated cell types of the nervous system, rather than of primordial territories.
Multiplex RNA-FISH shows that lncRNAs often exhibit a high degree of cell-type specificity. Though co-expression was generally detected with cell type-specific markers, as predicted by DIV-MARIS, we could observe much more nuanced spatiotemporal lncRNA regulation than we could have predicted – the noncoding transcripts investigated here tended to be expressed in highly specific subsets of neurogenic cell types (Figs 4–7). It is therefore feasible that these lncRNAs perform highly specialized functions in subsets of cells contributing to discrete regions of the nervous system.
For example, CR46003 and CR32730 are the first lncRNAs that appear to specifically mark midline and midline-proximal structures (Fig. 6, Fig. S14). Given the midline's highly specialized role as a signaling and organizing center (Wheeler et al., 2006; Crews, 2010; Zhou et al., 1995), it is intriguing to speculate that such lncRNAs may help shape the midline fates. Although lncRNAs were enriched in a variety of neurogenic populations, CR30009 was consistently and highly enriched in repo-positive glia and to some degree in pros-expressing neuroblasts (Fig. 4, Figs S9, S10). It is feasible that CR30009 may play a role in the priming of glial fates from the earliest stages of differentiation, possibly mediating the transition from neuroblasts and GMCs to specifically the glial fate. As most glia in the Drosophila embryonic CNS originate from the lateral column, it will be of interest to determine whether CR30009 expression and function are limited to glia of the lateral neurogenic ectoderm, or present in ventral column-derived glia as well.
Are these lncRNAs functional? cherub serves as a nice example arguing that several of them likely are. The lncRNA cherub was recently identified as a highly upregulated transcript in neuroblast-derived tumors in larvae (Landskron et al., 2018). In larvae, cherub is asymmetrically inherited by the self-renewing neuroblast to allow fate progression of the sibling cell and cherub’s specific predicted enrichment in embryonic neuroblasts (Fig. 3A) indicates that this lncRNA could exhibit a similar function in the early embryo. However, the precise temporal regulation of cherub was surprising, as RNA-FISH identified its presence not in early, but in differentiating and fully differentiated neurons and glia by the end of embryogenesis (Fig. 5, Figs S11, S12).
Intricate spatiotemporal expression regulation is a hallmark of many lncRNAs (Wilk et al., 2016; Karaiskos et al., 2017; Landskron et al., 2018). Various lncRNAs have been demonstrated to play diverse biological roles – nuclear and cytoplasmic – from integral parts of riboprotein complexes, to regulating dosage compensation, to affecting genome topology. lncRNA complexity has been reported to be especially pronounced in the nervous system (Briggs et al., 2015; Molyneaux et al., 2015) and even early stages of embryonic neurogliogenesis appear to be no exception. However, the challenge clearly remains to unravel the neurogenic roles of these putative noncoding regulators, and the molecular mechanisms by which they act. This study represents a valuable resource for understanding transcriptome complexity in the emerging nervous system and it lays the basis for further studies into the mechanisms by which noncoding genes, but also hundreds of specifically deployed coding genes, shape nervous system development.
MATERIALS AND METHODS
Fly lines
For details of fly strains and husbandry, see supplementary Materials and Methods.
FACS purification and RNA isolation using DIV-MARIS
Briefly, embryos were dissociated into single-cell suspensions, and cells were fixed in 4% formaldehyde. Fixed cell suspensions were immunostained under RNase-free conditions and FACS-purified using a FACS-AriaII cell sorter (BD Biosciences). Marker-enriched and -depleted cell populations were collected in biological duplicates. FACS-purified cells were subject to cross-link reversal and proteinase K digestion prior to RNA isolation. Additional experimental details for DIV-MARIS are provided in the supplementary Materials and Methods; primary and secondary antibodies used in this study are listed in Table S6.
Nuclear-cytoplasmic fractionation
Cytoplasmic and nuclear extracts were isolated from whole Drosophila embryos by detergent-based hypotonic lysis for RNA isolation. Additional experimental details are provided in the supplementary Materials and Methods.
Quantitative RT-PCR (qPCR)
qPCR was performed using standard SYBR Green, with the Bio-Rad CFX96 Touch Real-Time PCR Detection System. Additional information is available in the supplementary Materials and Methods; qPCR primer sequences are listed in Table S7.
Library preparation and RNA-sequencing
All RNA-seq libraries were constructed using the NuGEN Ovation Drosophila RNA-Seq System with 10-100 ng total RNA input. Library concentration was quantified using the Qubit dsDNA HS Assay (Thermo Fisher Scientific, Q32854) and quality was determined on a BioAnalyzer using Agilent High Sensitivity DNA Kits (Agilent, 5067-4626). All libraries were sequenced on the Illumina HiSeq4000 at a mean depth of 62.5 million 75 bp paired-end reads per sample. RNA-seq datasets generated for this study are detailed in Tables S10 and S11.
Bioinformatic analysis of RNA-seq data
Sequencing files were demultiplexed using bcl2fastq (v2.19, Illumina), and quality determined using FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). A genomic reference index for Drosophila melanogaster was constructed with RSEM using the most recent genome build (BDGP release 6) and transcriptome annotation (Release 6.15) obtained from Flybase (www.flybase.org). Annotations used for lncRNAs have been described by Young et al. (2012). Paired-end reads were pseudo-aligned to the RSEM reference index using Salmon (Release 0.8.1) using the following parameters: $ salmon quant --libType ISF –seqBias –gcBias –posBias -p 8 --numBootstraps 100.
Gene-level counts were prepared for differential expression analysis with tximport as part of the Bioconductor package (Release 3.5). Feature length-scaled TPM values were calculated with tximport using the following command: >tximport(files, type=“salmon”, countsFromAbundance=“lengthScaledTPM”, tx2gene=tx2gene).
Given the cell-type heterogeneity between samples in this dataset, we used normalized counts instead of TPM or FPKM for more accurate inter-sample comparisons of gene abundance. We normalized gene-level counts via variance stabilizing transformation (Table S1). Variance-stabilized transformed counts, principal component analysis, and differential expression were calculated using DESeq2 (Love et al., 2014) as part of the Bioconductor package (Release 3.5), using default parameters.
PhyloCSF
PhyloCSF uses substitutions and codon frequencies in a genome alignment of 23 drosophilid species to distinguish the evolutionary signature of selection for protein-coding function (Lin et al., 2011). For each transcript, PhyloCSF generates a score for the putative open reading frame (ORF) with highest coding potential; transcripts with positive scores are more likely to be protein coding. The candidate ORFs, their PhyloCSF scores, and other related information are included in Table S5.
Briefly, local alignments used for PhyloCSF were extracted from the 23-drosophilid subset of the 27-way MULTIZ insect whole-genome alignments (Blanchette et al., 2004), downloaded from UCSC: http://hgdownload.soe.ucsc.edu/goldenPath/dm6/multiz27way/ (Tyner et al., 2017). PhyloCSF scores were computed using the 23flies parameters with the options ‘-f3 --orf=ATGStop --allScores --bls’, which computes the score of every ORF within the transcript that begins with ATG, is followed by a stop codon, and is at least the default length of 25 codons. Because CR44272 has no putative ORFs that long, we used ‘--minCodons=19’ to lower the threshold for that gene to the length of its longest putative ORF. We then selected the ORF in each transcript having the highest PhyloCSF score. The reported ‘ScorePerCodon’ is the PhyloCSF score divided by the number of codons in the putative ORF. To identify potential cases in which one of the transcripts under consideration contains part of a coding ORF but the complete ORF is in an unidentified overlapping transcript, we also ran PhyloCSF using the --orf=StopStop3 option, with --minCodons=10, which looks for ORF fragments ending in a stop codon. However, that did not identify any plausible partial coding ORFs. The PhyloCSF track images in Fig. 3 and Fig. S6 are overlays of the ‘Smoothed PhyloCSF’ tracks in all three frames on the appropriate strand, from the PhyloCSF track hub in the UCSC genome browser, documented at: https://data.broadinstitute.org/compbio1/PhyloCSFtracks/trackHub/hub.DOC.html.
Generation of coverage plots
The strand-specific and paired-end RNA-seq reads were mapped to the Drosophila melanogaster reference genome dm6 with the splicing-aware mapper STAR v2.5.3a (Dobin et al., 2013) using default parameters and a Drosophila-specific adjustment for maximum intron length and mate distance of 50 kb. The resulting BAM files were filtered to include only uniquely mapping read pairs and then converted into strand-specific genome coverage tracks in BigWig format for visualization in the UCSC genome browser (Kent et al., 2010; Raney et al., 2014) using the stranded-coverage (https://github.com/pmenzel/stranded-coverage) and wigToBigWig from the UCSC genome browser tools.
Immunohistochemistry and FISH
Immunohistochemistry and RNA-FISH were performed as previously described (Kosman et al., 2004; Karaiskos et al., 2017). Primary and secondary antibodies used in this study are listed in Table S6. The procedure for probe synthesis is detailed in supplementary Materials and Methods, and RNA probes are listed in Tables S8 and S9.
Microscopy
Confocal stacks were imaged at BIMSB/MDC using a Leica SP8 equipped with 405 nm laser diode, white light laser, and hybrid detectors, with a 20× glycerol objective. For each field of view, 65-85 slices were acquired using ∼AU=1 pinholes and taking care not to saturate signal. Appropriate slices were maximum intensity projected. Imaging work at the BioFrontiers Institute's Advanced Light Microscopy Core was carried out on either a Nikon A1R laser scanning confocal microscope (NIST-CU Cooperative Agreement 70NANB15H226) or on a Nikon Ti-E spinning disc confocal microscope (BioFrontiers Institute, Howard Hughes Medical Institute).
Supplementary Material
Acknowledgements
We are grateful to John L. Rinn for insightful comments and extensive discussions, to Petar Glažar and Panagiotis Papavasileiou for considerable RNA-seq troubleshooting and preliminary data analysis, Andrew Woehler and Joe Dragavon for assistance with confocal imaging, David Schechner for providing the cell fractionation protocol, Sara Ugowski and Claudia Kipar for maintaining the fly facility at the BIMSB/MDC, and Sabrina Krüger and Agnieszka Klawiter for help generating transgenic fly lines. Moreover, we are grateful to all members of the Zinzen Laboratory for in-depth discussions and technical assistance on experiments presented in this study. We also acknowledge the BIMSB Genomics Platform and the Systems Biology Imaging Platform at the MDC.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: A.L.M., R.P.Z.; Methodology: A.L.M., P.W., I.J., R.A.I., M.K., R.P.Z.; Validation: A.L.M., S.W., R.L.P.A., R.P.Z.; Formal analysis: A.L.M., I.J., P.M., C.J.S., M.K., R.P.Z.; Investigation: A.L.M., S.W., R.P.Z.; Data curation: A.L.M., R.A.I., I.M.M., R.P.Z.; Writing - original draft: A.L.M., R.P.Z.; Writing - review & editing: A.L.M., P.W., S.W., I.J., P.M., C.J.S., R.L.P.A., R.A.I., I.M.M., M.K., R.P.Z.; Visualization: A.L.M., P.W., S.W., R.L.P.A., R.P.Z.; Supervision: R.P.Z.; Project administration: R.P.Z.; Funding acquisition: R.P.Z.
Funding
A.L.M. and P.W. were supported by a generous grant from the Deutsche Forschungsgemeinschaft (DFG; Priority Program SPP1738). I.J. was supported by the National Institutes of Health [HG004037] and GENCODE Wellcome Trust [U41 HG007234]. Deposited in PMC for immediate release.
Data availability
RNA-seq data have been deposited in NCBI Gene Expression Omnibus under accession number GSE106095.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.175265.supplemental
References
- Beckervordersandforth R. M., Rickert C., Altenhein B. and Technau G. M. (2008). Subtypes of glial cells in the Drosophila embryonic ventral nerve cord as related to lineage and gene expression. Mech. Dev. 125, 542-557. 10.1016/j.mod.2007.12.004 [DOI] [PubMed] [Google Scholar]
- Berger C., Renner S., Lüer K. and Technau G. M. (2007). The commonly used marker ELAV is transiently expressed in neuroblasts and glial cells in the Drosophila embryonic CNS. Dev. Dyn. 236, 3562-3568. 10.1002/dvdy.21372 [DOI] [PubMed] [Google Scholar]
- Bier E. and De Robertis E. M. (2015). Embryo development. BMP gradients: a paradigm for morphogen-mediated developmental patterning. Science 348, aaa5838-aaa5838 10.1126/science.aaa5838 [DOI] [PubMed] [Google Scholar]
- Blanchette M., Kent W. J., Riemer C., Elnitski L., Smit A. F., Roskin K. M., Baertsch R., Rosenbloom K. and Clawson H. (2004). Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res. 14, 708-715. 10.1101/gr.1933104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs J. A., Wolvetang E. J., Mattick J. S., Rinn J. L. and Barry G. (2015). Mechanisms of long non-coding RNAs in mammalian nervous system development, plasticity, disease, and evolution. Neuron 88, 861-877. 10.1016/j.neuron.2015.09.045 [DOI] [PubMed] [Google Scholar]
- Broadus J., Skeath J. B., Spana E. P., Bossing T., Technau G. and Doe C. Q. (1995). New neuroblast markers and the origin of the aCC/pCC neurons in the Drosophila central nervous system. Mech. Dev. 53, 393-402. 10.1016/0925-4773(95)00454-8 [DOI] [PubMed] [Google Scholar]
- Brown J. B., Boley N., Eisman R., May G. E., Stoiber M. H., Duff M. O., Booth B. W., Wen J., Park S., Suzuki A. M. et al. (2014). Diversity and dynamics of the Drosophila transcriptome. Nature 512, 393-399. 10.1038/nature12962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos-Ortega J. A. (1995). Genetic mechanisms of early neurogenesis in Drosophila melanogaster. Mol. Neurobiol. 10, 75-89. 10.1007/BF02740668 [DOI] [PubMed] [Google Scholar]
- Chen B., Zhang Y., Zhang X., Jia S., Chen S. and Kang L. (2016). Genome-wide identification and developmental expression profiling of long noncoding RNAs during Drosophila metamorphosis. Sci. Rep. 6, 23330 10.1038/srep23330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L.-L. (2016). Linking long noncoding RNA localization and function. Trends Biochem. Sci. 41, 761-772. 10.1016/j.tibs.2016.07.003 [DOI] [PubMed] [Google Scholar]
- Cowden J. and Levine M. (2003). Ventral dominance governs sequential patterns of gene expression across the dorsal-ventral axis of the neuroectoderm in the Drosophila embryo. Dev. Biol. 262, 335-349. 10.1016/S0012-1606(03)00395-6 [DOI] [PubMed] [Google Scholar]
- Crews S. T. (2010). Axon-glial interactions at the Drosophila CNS midline. Cell Adh. Migr. 4, 1-5. 10.4161/cam.4.1.10208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M. and Gingeras T. R. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15-21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doe C. Q. (1992). Molecular markers for identified neuroblasts and ganglion mother cells in the Drosophila central nervous system. Development 116, 855-863. [DOI] [PubMed] [Google Scholar]
- Doe C. Q. (2017). Temporal patterning in the Drosophila CNS. Annu. Rev. Cell Dev. Biol. 33, 219-240. 10.1146/annurev-cellbio-111315-125210 [DOI] [PubMed] [Google Scholar]
- Goff L. A., Groff A. F., Sauvageau M., Trayes-Gibson Z., Sanchez-Gomez D. B., Morse M., Martin R. D., Elcavage L. E., Liapis S. C., Gonzalez-Celeiro M. et al. (2015). Spatiotemporal expression and transcriptional perturbations by long noncoding RNAs in the mouse brain. Proc. Natl Acad. Sci. USA 112, 6855-6862. 10.1073/pnas.1411263112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graveley B. R., Brooks A. N., Carlson J. W., Duff M. O., Landolin J. M., Yang L., Artieri C. G., Van Baren M. J., Boley N., Booth B. W. et al. (2011). The developmental transcriptome of Drosophila melanogaster. Nature 471, 473-479. 10.1038/nature09715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammonds A. S., Bristow C. A., Fisher W. W., Weiszmann R., Wu S., Hartenstein V., Kellis M., Yu B., Frise E. and Celniker S. E. (2013). Spatial expression of transcription factors in Drosophila embryonic organ development. Genome Biol. 14, R140 10.1186/gb-2013-14-12-r140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckscher E. S., Long F., Layden M. J., Chuang C.-H., Manning L., Richart J., Pearson J. C., Crews S. T., Peng H., Myers E. et al. (2014). Atlas-builder software and the eNeuro atlas: resources for developmental biology and neuroscience. Development 141, 2524-2532. 10.1242/dev.108720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homem C. C. F. and Knoblich J. A. (2012). Drosophila neuroblasts: a model for stem cell biology. Development 139, 4297-4310. 10.1242/dev.080515 [DOI] [PubMed] [Google Scholar]
- Hrvatin S., Deng F., O'Donnell C. W., Gifford D. K. and Melton D. A. (2014). Maris: method for Analyzing RNA following intracellular sorting K. Aalto-Setala, ed. PLoS ONE 9, e89459–6 10.1371/journal.pone.0089459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki S., Numata K., Kondo T., Tomita M., Yasuda K., Kanai A. and Kageyama Y. (2005). Identification and expression analysis of putative mRNA-like non-coding RNA in Drosophila. Genes Cells 10, 1163-1173. 10.1111/j.1365-2443.2005.00910.x [DOI] [PubMed] [Google Scholar]
- Karaiskos N., Wahle P., Alles J., Boltengagen A., Ayoub S., Kipar C., Kocks C., Rajewsky N. and Zinzen R. P. (2017). The Drosophila embryo at single-cell transcriptome resolution. Science 358, 194-199. 10.1126/science.aan3235 [DOI] [PubMed] [Google Scholar]
- Kent W. J., Zweig A. S., Barber G., Hinrichs A. S. and Karolchik D. (2010). BigWig and BigBed: enabling browsing of large distributed datasets. Bioinformatics 26, 2204-2207. 10.1093/bioinformatics/btq351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosman D., Mizutani C. M., Lemons D., Cox W. G., McGinnis W. and Bier E. (2004). Multiplex detection of RNA expression in Drosophila embryos. Science 305, 846-846 10.1126/science.1099247 [DOI] [PubMed] [Google Scholar]
- Kunisch M., Haenlin M. and Campos-Ortega J. A. (1994). Lateral inhibition mediated by the Drosophila neurogenic gene delta is enhanced by proneural proteins. Proc. Natl Acad. Sci. USA 91, 10139-10143. 10.1073/pnas.91.21.10139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai E. C. (2004). Notch signaling: control of cell communication and cell fate. Development 131, 965-973. 10.1242/dev.01074 [DOI] [PubMed] [Google Scholar]
- Landgraf M., Bossing T., Technau G. M. and Bate M. (1997). The origin, location, and projections of the embryonic abdominal motorneurons of Drosophila. J. Neurosci. 17, 9642-9655. 10.1523/JNEUROSCI.17-24-09642.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landskron L., Steinmann V., Bonnay F., Burkard T. R., Steinmann J., Reichardt I., Harzer H., Laurenson A.-S., Reichert H. and Knoblich J. A. (2018). The asymmetrically segregating lncRNA cherub is required for transforming stem cells into malignant cells. eLife 7, R106 10.7554/eLife.31347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lécuyer E., Yoshida H., Parthasarathy N., Alm C., Babak T., Cerovina T., Hughes T. R., Tomancak P. and Krause H. M. (2007). Global analysis of mRNA localization reveals a prominent role in organizing cellular architecture and function. Cell 131, 174-187. 10.1016/j.cell.2007.08.003 [DOI] [PubMed] [Google Scholar]
- Li M. and Liu L. (2015). Neural functions of long noncoding RNAs in Drosophila. J. Comp. Physiol. A Neuroethol. Sens. Neural Behav. Physiol. 201, 921-926. 10.1007/s00359-014-0937-8 [DOI] [PubMed] [Google Scholar]
- Lin M. F., Jungreis I. and Kellis M. (2011). PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics 27, i275-i282. 10.1093/bioinformatics/btr209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S. J., Nowakowski T. J., Pollen A. A., Lui J. H., Horlbeck M. A., Attenello F. J., He D., Weissman J. S., Kriegstein A. R., Diaz A. A. et al. (2016). Single-cell analysis of long non-coding RNAs in the developing human neocortex. Genome Biol. 17, 67 10.1186/s13059-016-0932-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M. I., Huber W. and Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcgovern V. L., Pacak C. A., Sewell S. T., Turski M. L. and Seeger M. A. (2003). A targeted gain of function screen in the embryonic CNS of Drosophila. Mech. Dev. 120, 1193-1207. 10.1016/S0925-4773(03)00159-X [DOI] [PubMed] [Google Scholar]
- Molyneaux B. J., Goff L. A., Brettler A. C., Chen H.-H., Brown J. R., Hrvatin S., Rinn J. L. and Arlotta P. (2015). DeCoN: genome-wide analysis of in vivo transcriptional dynamics during pyramidal neuron fate selection in neocortex. Neuron 85, 275-288. 10.1016/j.neuron.2014.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picao-Osorio J., Lago-Baldaia I., Patraquim P. and Alonso C. R. (2017). Pervasive behavioral effects of microRNA regulation in Drosophila. Genetics 206, 1535-1548. 10.1534/genetics.116.195776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raney B. J., Dreszer T. R., Barber G. P., Clawson H., Fujita P. A., Wang T., Nguyen N., Paten B., Zweig A. S., Karolchik D. et al. (2014). Track data hubs enable visualization of user-defined genome-wide annotations on the UCSC Genome Browser. Bioinformatics 30, 1003-1005. 10.1093/bioinformatics/btt637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickert C., Kunz T., Harris K.-L., Whitington P. M. and Technau G. M. (2011). Morphological characterization of the entire interneuron population reveals principles of neuromere organization in the ventral nerve cord of Drosophila. J. Neurosci. 31, 15870-15883. 10.1523/JNEUROSCI.4009-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers W. A., Goyal Y., Yamaya K., Shvartsman S. Y. and Levine M. S. (2017). Uncoupling neurogenic gene networks in the Drosophila embryo. Genes Dev. 31, 634-638. 10.1101/gad.297150.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandler J. E. and Stathopoulos A. (2016). Stepwise progression of embryonic patterning. Trends Genet. 32, 432-443. 10.1016/j.tig.2016.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago C., Labrador J.-P. and Bashaw G. J. (2014). The homeodomain transcription factor Hb9 controls axon guidance in Drosophila through the regulation of Robo receptors. Cell Rep. 7, 153-165. 10.1016/j.celrep.2014.02.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauvageau M., Goff L. A., Lodato S., Bonev B., Groff A. F., Gerhardinger C., Sanchez-Gomez D. B., Hacisuleyman E., Li E., Spence M. et al. (2013). Multiple knockout mouse models reveal lincRNAs are required for life and brain development. eLife 2, e01749 10.7554/eLife.01749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skeath J. B. and Thor S. (2003). Genetic control of Drosophila nerve cord development. Curr. Opin. Neurobiol. 13, 8-15. 10.1016/S0959-4388(03)00007-2 [DOI] [PubMed] [Google Scholar]
- Skeath J. B., Panganiban G. F. and Carroll S. B. (1994). The ventral nervous system defective gene controls proneural gene expression at two distinct steps during neuroblast formation in Drosophila. Development 120, 1517-1524. [DOI] [PubMed] [Google Scholar]
- Sousa-Nunes R., Cheng L. Y. and Gould A. P. (2010). Regulating neural proliferation in the Drosophila CNS. Curr. Opin. Neurobiol. 20, 50-57. 10.1016/j.conb.2009.12.005 [DOI] [PubMed] [Google Scholar]
- Tomancak P., Beaton A., Weiszmann R., Kwan E., Shu S. Q., Lewis S. E., Richards S., Ashburner M., Hartenstein V., Celniker S. E. et al. (2002). Systematic determination of patterns of gene expression during Drosophila embryogenesis. Genome Biol. 3, RESEARCH0088 10.1186/gb-2002-3-12-research0088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomancak P., Berman B. P., Beaton A., Weiszmann R., Kwan E., Hartenstein V., Celniker S. E. and Rubin G. M. (2007). Global analysis of patterns of gene expression during Drosophila embryogenesis. Genome Biol. 8, R145 10.1186/gb-2007-8-7-r145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyner C., Barber G. P., Casper J., Clawson H., Diekhans M., Eisenhart C., Fischer C. M., Gibson D., Gonzalez J. N., Guruvadoo L. et al. (2017). The UCSC genome browser database: 2017 update. Nucleic Acids Res. 45, D626-D634. 10.1093/nar/gkw1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Ohlen T. and Doe C. Q. (2000). Convergence of dorsal, dpp, and egfr signaling pathways subdivides the drosophila neuroectoderm into three dorsal-ventral columns. Dev. Biol. 224, 362-372. 10.1006/dbio.2000.9789 [DOI] [PubMed] [Google Scholar]
- Weiss J. B., Von Ohlen T., Mellerick D. M., Dressler G., Doe C. Q. and Scott M. P. (1998). Dorsoventral patterning in the Drosophila central nervous system: the intermediate neuroblasts defective homeobox gene specifies intermediate column identity. Genes Dev. 12, 3591-3602. 10.1101/gad.12.22.3591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen K., Yang L., Xiong T., Di C., Ma D., Wu M., Xue Z., Zhang X., Long L., Zhang W. et al. (2016). Critical roles of long noncoding RNAs in Drosophila spermatogenesis. Genome Res. 26, 1233-1244. 10.1101/gr.199547.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler S. R., Kearney J. B., Guardiola A. R. and Crews S. T. (2006). Single-cell mapping of neural and glial gene expression in the developing Drosophila CNS midline cells. Dev. Biol. 294, 509-524. 10.1016/j.ydbio.2006.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler S. R., Stagg S. B. and Crews S. T. (2009). MidExDB: a database of Drosophila CNS midline cell gene expression. BMC Dev. Biol. 9, 56 10.1186/1471-213X-9-56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilk R., Hu J., Blotsky D. and Krause H. M. (2016). Diverse and pervasive subcellular distributions for both coding and long noncoding RNAs. Genes Dev. 30, 594-609. 10.1101/gad.276931.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young R. S., Marques A. C., Tibbit C., Haerty W., Bassett A. R., Liu J.-L. and Ponting C. P. (2012). Identification and Properties of 1,119 Candidate LincRNA Loci in the Drosophila melanogaster Genome. Genome Biol. Evol. 4, 427-442. 10.1093/gbe/evs020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L., Hashimi H., Schwartz L. M. and Nambu J. R. (1995). Programmed cell death in the Drosophila central nervous system midline. Curr. Biol. 5, 784-790. 10.1016/S0960-9822(95)00155-2 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.