

Abstract
Background
Statins are the first-line therapy for lowering high lipid levels. Atorvastatin calcium (AtC) is the most commonly prescribed statin. It inhibits 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase which converts HMG-CoA into mevalonic acid, a cholesterol precursor.
Objective
To compound and evaluate the stability of AtC suspension (0.4% w/v) using commercial tablets or pure AtC powder as the source of the active pharmaceutical ingredient.
Method
Several AtC suspension formulations were produced using commercial AtC tablets or AtC pure powder as the source of the active ingredient. The most suitable one in terms of general organoleptic properties and dissolution was selected for stability studies. For this purpose, samples of final suspensions were stored at room temperature and in the refrigerator. Assay, pH, organoleptic properties and microbial contamination were evaluated according to the USP specifications. High performance liquid chromatography was used for the analysis and quantification of AtC in the studied samples.
Results
The obtained suspension (S4) had good organoleptic properties. It showed complete dissolution of AtC within 30 min. However, the suspension prepared from crushed tablet (St4) showed a better dissolution profile than that prepared from pure powder (Sp4). The prepared formula had unchanged pH, which remained around 9.9. St and Sp formulas were both free from microbial contamination. Both products showed good stability within at least the period of use of the 100 mL AtC bottles.
Conclusions
AtC extemporaneous suspension was successfully prepared using tablets as a source of AtC or pure AtC powder. However, St4 had a better dissolution profile than Sp4. This study provides a solution for patients with swallowing difficulties or feeding tubes who are unable to take medicines in solid oral dosage forms. Community pharmacists can prepare the suspension using AtC tablets as the source of the active ingredient.
Keywords: Atorvastatin calcium, Extemporaneous, Suspension, Stability, DRUG MANUFACTURING / PREPARATION / COMPOUNDING
Introduction
Statins are the first-line therapy for lowering high lipid levels.1 Atorvastatin calcium (AtC) chemically is (3R,5R)-7-(2-(4-fluorophenyl)-3-phenyl-4-(phenylcarbamoyl)-5-propan-2-ylpyrrol-l-yl)-3,5-dihydroxyheptanoic acid (figure 1).
Figure 1.

Chemical structure of atorvastatin calcium.
AtC is the most commonly prescribed statin.2 It inhibits 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase,3 the enzyme that converts HMG-CoA into mevalonic acid, a cholesterol precursor, thereby reducing endogenous cholesterol synthesis.4 5 This increases the expression of low density lipoprotein (LDL) receptor on hepatocytes resulting in a fall in serum LDL concentration to about 40%.6 Statins also increase high density lipoprotein (HDL) and cause a small reduction in the triglyceride level of about 10–20%, resulting in a reduction in coronary artery disease and death.6 7
AtC has several side effects including headache, lower back pain, stuffy or runny nose, constipation and abdominal pain. However, it is contraindicated in patients with unexplained persistent elevations of serum transaminases or in those with active liver disease; in addition, it is contraindicated in pregnancy and in breast feeding mothers.8
The prevalence of swallowing difficulties is a common problem among paediatric patients. This problem increases in elderly patients as a result of various causes including stroke, carcinoma and advanced Alzheimer's disease.9 One of the main factors that contribute to non-adherence is tablet swallowing.10 Children and adolescents often have difficulty with swallowing tablets and capsules.11 The National Health Service organisation Medicines Optimisation (Stoke on Trent) suggested a number of steps that pharmacists should follow in order to minimise this inconvenience (http://www.medicinesmanagementstoke.nhs.uk/). In fact, pharmacists can raise a series of questions in order to manage such cases, including whether there is a special formulation of the prescribed drug available and, if not, whether they can compound a suitable formulation of the drug.12
Recently, Rosemont Pharmaceuticals launched two licensed strengths of simvastatin oral suspension products. Both strengths were stable for 12 months in closed bottles and for 1 month in an open bottle.13 Unfortunately, this new formulation is not available on our market. In fact, statins are only available as solid oral dosage forms. The development of a stable statin suspension formulation for extemporaneous use would be useful for patients with swallowing problems.
This study was conducted in order to prepare an extemporaneous AtC suspension and to evaluate its stability using AtC commercial tablets or AtC pure powder as the source of the active pharmaceutical ingredient (API).
Methods
Materials
For the purpose of this study, Lipovast tablets were obtained as a gift from SAMA Pharmaceutical Manufacturing, Nablus, Palestine; Lipitor tablets were bought from a community pharmacy; AtC powder was obtained as a gift from Pharmacare Ltd; peppermint oil was obtained from Omega (Phyto Active); Xanthan gum was obtained from Omega from Balirom Ltd; and tri-sodium phosphate was supplied by Sun Pharma (China Shenzhen Ocean Industrial & Trading).
Instruments and chromatographic conditions
The high performance liquid chromatography (HPLC) system (Dionex, Thermo Scientific) consisted of an ultimate 3000 model pump (SN 8031808), autosampler (SN 8031972), column oven (SN 8031817), PDA detector (SN 8031310) and Chromeleon software V.6.8 (Dionex, Germany). Dissolution apparatus model (Labindia DS 8000), India, was supplied by PharmaAccess. Weights were measured using HR-202i analytical balance (AXID-Japan) and pH was identified using Thermoscientific pH meter.
Formulation of AtC suspension
Suspensions were extemporaneously prepared to a final concentration of 0.4 g/100 mL using 40 mg Lipovast tablets. Samples were taken for initial analysis and the remaining samples were stored at room temperature and in the refrigerator for analysis at 0, 7, 14, 21 and 30 days.
Several trials were carried out in order to achieve the most suitable formula that met the desired quality in terms of organoleptic properties, dissolution behaviour and stability, as shown in table 1.
Table 1.
Formulation of atorvastatin calcium (AtC) suspension from crushed tablets or pure AtC powder
| Quantities per 1000 mL (in g) |
||||
|---|---|---|---|---|
| Ingredients | S1 | S2 | S3 | S4 |
| AtC | 4 | 4 | 4 | 4 |
| Veegum | 3 | 3 | 2 | 0 |
| Xanthan gum | 1.5 | 1.5 | 0.5 | 0.5 |
| Peppermint oil | 0.5 | 0.5 | 0.5 | 0.5 |
| Aspartame | 8 | 8 | 8 | 0 |
| Saccharin sodium | 3 | 3 | 3 | 0 |
| Sucrose | 0 | 0 | 0 | 400 |
| Tri-sodium phosphate | 0 | 3 | 3 | 3 |
| Purified water up to | 1000 mL | 1000 mL | 1000 mL | 1000 mL |
Formulation procedure
Each formula was produced in triplicate according to the following general procedure.
Crush 10 tablets (Lipovast 40 mg) using a mortar and pestle.
Precisely calculate all required quantities of each component for the total volume to be prepared.
Weigh and/or take the volume of each ingredient precisely.
Crush and mill well the required atorvastatin tablets (in case of use of commercial tablets as source of AtC).
Add and mix well the remaining powder components using the geometric dilution method.
Add and mix well peppermint oil to the achieved mixture in step 4.
Add water and mix well until achieving a pourable mass.
Add water again up to the desired volume.
Shake well before filling suspension into amber glass bottles.
Place three bottles at room temperature and the remainder in the refrigerator.
For compounding AtC suspensions using AtC pure powder, the same excipients and procedures were used except that the drug used was pure AtC powder, as reported in table 1.
Quality control of suspension
The obtained suspension was tested for several properties including pH, organoleptic properties and dissolution behaviour and the formula that showed the best results was used for further investigation including stability assay and microbial contamination.
Assay of AtC in the suspension
Means and SDs were calculated for the samples analysed. For each study period a 5 mL sample was withdrawn from each of the bottles (3 bottles at room temperature and 7 bottles in the refrigerator) and analysed in triplicate. The percentage of the remaining concentration of AtC was estimated in order to assess the chemical stability of the suspension. The amount of AtC in the obtained suspension was assessed using the HPLC analytical technique according to the reported USP analytical method for AtC.14
This method was validated using validation parameters including specificity, linearity, precision, robustness and system suitability. The linearity and range of the method were evaluated using a series of standards at different concentrations of AtC (100±20%). Each concentration was analysed in triplicate; a linear regression analysis was performed. In order to evaluate the precision of the method, six samples of AtC suspension were tested. The acceptance criteria for the analysed data were %RSD of not more than 2.0% and all accuracy results had to be within 10.0% of the actual amount of AtC. The ruggedness of the analytical method was assessed by running samples on each of the two days by different analysts using different instruments. The results of the two days had to have %RSD of not more than 5.0% and all accuracy results had to be within ±10.0% of the actual amount. The results between days 1 and 2 had to have %RSD of not more than 10.0%. Accuracy was also examined in order to determine if any of the used excipients interfered with the peak of AtC. The analytical method was considered accurate if the percentage accuracy was within ±2.0% of the actual amount (98–102%). The accuracy was established using three concentrations (80%, 100% and 120%) and three replicates of each concentration. The percentage recovery and %RSD were calculated for each of the replicate samples. System suitability parameters including column efficiency and peak symmetry/tailing were checked. The system was considered to be adequate if the tailing factor was not more than 5 and the theoretical plates were not less than 10 000.
A specificity test was performed by adding a known amount of placebo with solvent, then analysing the resulting solution under the same conditions as those for the assay samples and comparing with a 100% standard solution.
The HPLC experimental conditions were optimised on octadecylsilyl silica gel (C18), 25 cm×4.6 column (Luna phenomenex, USA), the injection volume was 20 µL and the detection wavelength was 246 nm.
The gradient mobile phase consisted of:
Mobile phase A: mixture of acetonitrite, methanol and buffer (45:45:10) in which buffer solution was prepared by adjusting a solution of 0.02 M potassium dihydrogen phosphate (Merck, Germany) in water to pH 6.9 with potassium hydroxide.
Mobile phase B: buffer solution with a flow rate of 1 mL/min and methanol (JT-Baker) was used as a solvent to prepare the sample and standard solution.
Peak quantification was obtained by comparing the sample and standard peak area ratios as a function of concentration.
Dissolution study
An in vitro dissolution study was carried out using a dissolution apparatus USP (type II) at a paddle speed of 75 rpm.15 The dissolution medium was 900 mL phosphate buffer at pH 6.8. The buffer was prepared by dissolving 32.86 g dihydrogen potassium phosphate in water, adjusting the pH to 6.8 using 0.05 M sodium hydroxide and then diluting to 7000 mL using purified water. The dissolution profile was performed under sink conditions using the de-aerated media. The dissolution apparatus was kept at 37°C for the entire period of the study. Five mL of suspension was placed in each paddle and six paddles were used for a total of six reads for each time point. Samples (10 mL) were taken periodically at 15, 30 and 45 min with replacement of the same volume of medium buffer. The mean for each six reads was recorded in order to design the dissolution profiles using Excel program 2013.
Similarity and non-similarity factors (f2 and f1 respectively) were calculated according to equations 1 and 2, respectively.
 |
1 |
 |
2 |
where Rt and Tt are the percentages of drug dissolved at each time point for the reference listed product and test products, respectively. The f2 factor measures the similarity between two profiles and f1 measures the difference between two profiles. An f1 value <15 indicates significant non-similarity and an f2 value >50 indicates significant similarity.13 16–18
Results
The USP analytical method for AtC was adopted and the validation parameters were found to be within the validation criteria. The method was found to be specific since there was no interference between the AtC peak and the peaks of any other excipients. It also showed linearity in the range between 5 and 20 µg/mL with a correlation coefficient higher than 0.99. Injection precision and tailing factor for the standard solution were within the accepted limits (less than 2% and higher than 2%, respectively). Accuracy was conducted on three concentration levels (100±20%) and the mean±SD values were 99.8±0.244%, 101.0±0.165% and 100.3±0.596%, respectively.
Four suspension formulations were prepared using crushed tablets and pure AtC powder as the source of API. These formulations were tested for several properties including pH, assay, organoleptic properties, microbial contamination and dissolution behaviour (table 2). Among the obtained suspensions, both S3 and S4 showed the best organoleptic properties such as colour, odour, pourability and taste, so they were used for further investigations including dissolution behaviour in order to select the one with the best AtC release. As shown in table 2, S4 showed the best dissolution of AtC within 30 min. Accordingly, this formula was selected to produce an AtC suspension from crushed tablets (St4) and from pure AtC powder (Sp4).
Table 2.
Quality control tests for suspensions from crushed tablets
| Test | S1 | S2 | S3 | S4 |
|---|---|---|---|---|
| pH | 6.5–7 | 9.5–10 | 7.5–8.5 | 8–9.5 |
| Dissolution after 30 min | 53% | 71% | 73% | 99.4% |
However, the dissolution profile of St4 had a higher f2 and lower f1 than Sp4, as shown in table 3 and figure 2. This suggests that using commercial AtC tablets would be much better in terms of predicted bioavailability.
Table 3.
Similarity and non-similarity factors for suspensions
| Suspension | Commercial tablets | f2 | f1 |
|---|---|---|---|
| St4 | Lipovast | 68.5 | 0.4 |
| Lipitor | 65.66 | 2.15 | |
| Sp4 | Lipovast | 56 | 13 |
| Lipitor | 52 | 9.6 |
Figure 2.
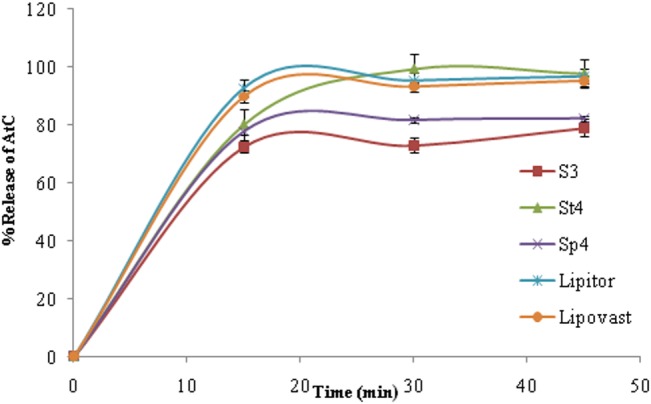
Dissolution profile of atorvastatin calcium (AtC) from St4 and Sp4 (20 mg/5 mL) versus Lipovast and Lipitor tablets.
Once the dissolution profile was optimised, St4 was stored at two storage conditions (room temperature and in the refrigerator) in order to assess its stability. The suspension was chemically stable throughout the 30-day study period in all suspensions. In fact, when the starting amount of AtC is 100%, the mean percentage of remaining AtC was estimated to be around 97.4±0.18% at room temperature and 98.4±0.2% in the refrigerator, and the pH remained unchanged at around 9.5. St4 and Sp4 were also tested for microbial contamination and no microbial contamination was found, as shown in table 4.
Table 4.
Microbial limit tests after 30 days of storage at room temperature
| Microbiology | Results | Limits |
|---|---|---|
| Escherichia coli bacteria | Absent | Absent |
| Yeast and mould | Not more than 10 cfu/mL | Not more than 10 cfu/mL |
| Total aerobic count | Not more than 10 cfu/mL | Not more than 100 cfu/mL |
Discussion
Patients with swallowing difficulties or feeding tubes are unable to take medicines in solid oral dosage forms. Special care is therefore necessary in order to provide their medicine.18 Many research projects have been conducted in order to compound extemporaneous liquid dosage forms for drugs that are available in the market only as solid dosage forms.12 19–22
To our knowledge, statins are not available in any dosage form that could be suitable for patients with swallowing problems, except for simvastatin. AtC tablets are available only as conventional tablets. This study aimed to develop and evaluate the quality and stability of an extemporaneous AtC suspension using commercially available AtC tablets and pure AtC powder as the source of API. Among the obtained formulas, S3 and S4 showed the best organoleptic properties and were subjected to dissolution testing. The dissolution test was carried out because it could be used as in vitro tool to predict bioavailability. St4 (crushed AtC tablets) showed complete AtC release within 30 min, while the dissolution profile of Sp4 (pure AtC powder) showed approximately 80% AtC release after 30 min. This difference may be explained by the fact that AtC tablets contain other excipients such as Tween 80 which is a wetting agent used to increase the dissolution of water insoluble drugs. The bioavailability of the AtC suspension in this study was not evaluated; however, the absorption and therapeutic efficacy of a drug in a suspension compounded from crushed tablets is unlikely to differ from that of the original dosage form used in its compounding. In fact, to the best of our knowledge, bioavailability and pharmacokinetic studies are not conducted for most extemporaneously prepared medications since conventional tablets or capsules containing the active ingredients are used as the source of the drug. If conventional tablets or capsules with immediate release profiles are used to prepare a suspension, it is assumed that the drug absorption and hence its bioavailability and pharmacokinetics will not be compromised in patients.
Stability was defined as retention of more than 90% of the initial concentration of the API. St4 was selected for further stability studies since it showed the best similarity and non-similarity compared with the tablet brand used in the formulation.
In our study, at least 97% of the initial concentration of AtC remained throughout the 30-day study period in the suspensions regardless of the storage conditions. There was no detectable change in colour, odour or taste, and no visible microbial growth was observed in any sample. The preparation was a well-distributed suspension after gentle shaking. It was palatable with a slight aftertaste. Regarding the pH, AtC is an acid sensitive drug and it needs an alkaline pH for better chemical stabilisation. No appreciable change in mean pH occurred in any of the tested samples, regardless of whether they were stored at room temperature or in the refrigerator. Pharmaceutical manufacturers should benefit from this information as they can prepare such a formula upon prescription using pure powder or their commercial AtC tablets since an AtC suspension dosage form is not commercially available. This could be very useful for a wide range of patients who need statins in a suitable dosage form.
Conclusion
An extemporaneous suspension of AtC was successfully prepared using tablets or pure AtC powder as the source of active ingredient. However, St4 had a better dissolution profile than Sp4. Community pharmacists can extemporaneously prepare the suspension using commercial tablets as the source of active ingredient. Pharmaceutical manufacturers should provide their package inserts with scientific information and directions on how to compound their commercial immediate release tablets into a suspension as well as information on the expiry date of the obtained suspensions.
Key messages.
What is already known on this subject
Statins are available as solid dosage forms and only one drug of this class, Simvastatin was approved in 2014 as a suspension in the UK.
Liquid dosage forms are very important for patients with swallowing difficulties.
Statins are usually unstable in acidic conditions and it is not easy to prepare a stable water suspension.
What this study adds
This will help many hospitals and community pharmacies in providing Atorvastatin suspension for paediatric and elderly patients with swallowing difficulties worldwide.
This suspension is very helpful when dose adjustments are required for patients with impaired urinary or metabolic function.
Acknowledgments
The authors wish to express their sincere gratitude to Sama Pharm Co and all who directly or indirectly have contributed in any form to this work.
Footnotes
Competing interests: None declared.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Tsiara S, Elisaf M, Mikhailidis DP. Early vascular benefits of statin therapy. Curr Med Res Opin 2003;19:540–56. 10.1185/030079903125002225 [DOI] [PubMed] [Google Scholar]
- 2.Corsini A, Bellosta S, Baetta R, et al. New insights into the pharmacodynamic and pharmacokinetic properties of statins. Pharmacol Ther 1999;84:413–28. 10.1016/S0163-7258(99)00045-5 [DOI] [PubMed] [Google Scholar]
- 3.Auer J, Berent R, Weber T, et al. Clinical significance of pleiotropic effects of statins: lipid reduction and beyond. Curr Med Chem 2002;9:1831–50. 10.2174/0929867023369024 [DOI] [PubMed] [Google Scholar]
- 4.Choi JW, Jung SE. Lovastatin-induced proliferation inhibition and apoptosis in c6 glial cells. J Pharmacol Exp Ther 1999;289:572–9. [PubMed] [Google Scholar]
- 5.Stancu C, Sima A. Statins: mechanism of action and effects. J Cell Mol Med 2001;5:378–87. 10.1111/j.1582-4934.2001.tb00172.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lippincott-Schwartz J. Profile of Jennifer Lippincott-Schwartz. Proc Natl Acad Sci USA 2009;106:10881–3. 10.1073/pnas.0905805106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dimmeler S, Aicher A, Vasa M, et al. HMG-CoA reductase inhibitors (statins) increase endothelial progenitor cells via the PI 3-kinase/Akt pathway. J Clin Invest 2001;108:391–7. 10.1172/JCI13152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.AbdulRazzaq HA, Abd Aziz N, Sulaiman SAS, et al. Development tool for self-reporting of adverse drug reactions of statin. Int J Collab Res Intern Med Public Health 2011;3:847. [Google Scholar]
- 9.Morris H. Dysphagia in the elderly—a management challenge for nurses. Br J Nurs 2006;15:558–62. 10.12968/bjon.2006.15.10.21132 [DOI] [PubMed] [Google Scholar]
- 10.Vlasnik JJ, Aliotta SL, DeLor B. Medication adherence: factors influencing compliance with prescribed medication plans. Case Manager 2005;16:47–51. 10.1016/j.casemgr.2005.01.009 [DOI] [PubMed] [Google Scholar]
- 11.Hansen DL, Tulinius D, Hansen EH. Adolescents’ struggles with swallowing tablets: barriers, strategies and learning. Pharm World Sci 2008;30:65–9. 10.1007/s11096-007-9142-y [DOI] [PubMed] [Google Scholar]
- 12.Zaid AN, Assali M, Qaddomi A, et al. Preparation and stability evaluation of extemporaneous oral suspension of valsartan using commercially available tablets. Int J Pharm Compd 2014;18:169–74. [PubMed] [Google Scholar]
- 13.Emami J. In vitro–in vivo correlation: from theory to applications. J Pharm Pharm Sci 2006;9:169–89. [PubMed] [Google Scholar]
- 14.United States Pharmacopeial Convention. Rockville, MD, 2005. [Google Scholar]
- 15.Unites States Food and Drug Administration. Dissolution methods. http://www.accessdata.fda.gov/scripts/cder/dissolution/dsp_getallData.cfm
- 16.Gupta A, Gaud RS, Ganga S. Development of discriminating dissolution method for an insoluble drug: nisoldipine. Int J PharmTech Res 2010;2:931–9. [Google Scholar]
- 17.Fortunato D. Dissolution method development for immediate release solid oral dosage forms. Dissolut Technol 2005;3:12–14. [Google Scholar]
- 18.Gupta E, Barends DM, Yamashita E, et al. Review of global regulations concerning biowaivers for immediate release solid oral dosage forms. Eur J Pharm Sci 2006;29:315–24. 10.1016/j.ejps.2006.05.001 [DOI] [PubMed] [Google Scholar]
- 19.Garner SS, Wiest DB, Reynolds ER Jr. Stability of atenolol in an extemporaneously compounded oral liquid. Am J Hosp Pharm 1994;51:508–11. [PubMed] [Google Scholar]
- 20.Mahmoud IM, Ibrahim YM, Shakir MA. Extemporaneous preparations of pediatric oral formulations: stability studies conducted in spironolactone suspensions, powders and capsules in Saudi hospital pharmacies. Global Trends Pharm Sci 2014;5:1595–602. [Google Scholar]
- 21.Nahata MC, Morosco RS, Brady MT. Extemporaneous sildenafil citrate oral suspensions for the treatment of pulmonary hypertension in children. Am J Health Syst Pharm 2006;63:254–7. 10.2146/ajhp050208 [DOI] [PubMed] [Google Scholar]
- 22. http://www.roche.com/tamiflu_pil_eu.pdf.