

Abstract
Vaccination represents one of the most effective means of preventing infectious disease. In order to maximize the utility of vaccines, highly potent formulations that are easy to administer and promote high patient compliance are desired. In the present work, a biomimetic self-propelling micromotor formulation is developed for use as an oral antivirulence vaccine. The propulsion is provided by a magnesium-based core, and a biomimetic cell membrane coating is used to detain and neutralize a toxic antigenic payload. The resulting motor toxoids leverage their propulsion properties in order to more effectively elicit mucosal immune responses. After demonstrating the successful fabrication of the motor toxoids, their uptake properties are shown in vitro. When delivered to mice via an oral route, it is then confirmed that the propulsion greatly improves retention and uptake of the antigenic material in the small intestine in vivo. Ultimately, this translates into markedly elevated generation of antibody titers against a model toxin. This work provides a proof-of-concept highlighting the benefits of active oral delivery for vaccine development, opening the door for a new set of applications in which biomimetic motor technology can provide significant benefits.
Keywords: oral vaccination, micromotor, biomimetic toxoid, cell membrane coating, mucosal vaccine
Graphical Abstract
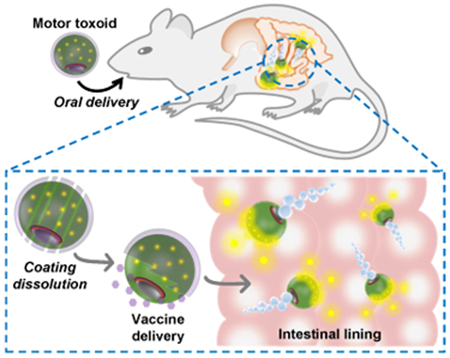
The discovery of vaccines for preventing infectious diseases has represented one of the most important advances in modern medicine,1 and widespread vaccination programs have had a significant impact on global health, likely saving millions of lives in the process.2–3 Vaccines work by leveraging the endogenous mechanisms of the immune system, training the human body to recognize and quickly generate sterilizing responses upon encountering a specific pathogen. In some cases, this strategy for disease management has been extremely successful, such as in the case of smallpox and polio. 4–5 Unfortunately, there are also other instances in which vaccination has not been nearly as effective, which can stem from issues with vaccine potency, specificity, or accessibility.6–8 Among the various types of vaccines, those delivered via the oral route are highly desirable. Their advantages include ease of administration, which greatly improves patient compliance, as well as the ability to generate a broader response by stimulating mucosal immunity.9–10 It is for this reason that there are a significant number of licensed oral vaccines currently being employed in the clinic. A major challenge, however, is developing formulations that possess sufficient potency to provide robust protection against a target of interest, and this is made difficult due to various spatial, physical, and tolerogenic barriers unique to gastrointestinal delivery.9–11 As such, significant efforts have been placed on addressing these hurdles in order to further expand the utility and applicability of oral vaccines.
Over the past several decades, particulate delivery systems have been widely explored for the encapsulation and delivery of antigenic and immunostimulatory payloads.12–13 Numerous types of systems have been reported, and many have shown the potential to greatly enhance vaccination potency when orally delivered.14–16 In addition to the inherent advantages afforded by particulate delivery, which include high loading capacity and sustained release characteristics, these platforms can further be improved with specific targeting mechanisms.17–18 Within the field of biomedical engineering, a recently growing topic of interest has been the development of active delivery systems, namely micro/nanomotors, that are capable of efficient propulsion, which can be used to improve cargo delivery and enhance tissue penetration.19–21 Nano/micromotors are synthetic vehicles, made from different materials and various shapes, capable of converting chemical fuels or external energies into rapid motion.22–29 These motors have been used for cargo transport, drug delivery, chemical sensing, and remediation applications, among others.22–29 Their dynamic behavior provides unique advantages that have motivated researchers to explore their use in vivo, where their ability to autonomously propel in biological environments can provide significant benefits.30–32 In particular, it has been shown that micromotors made of biodegradable magnesium (Mg) or zinc bodies can propel quickly in gastric and intestinal fluids, improve cargo delivery and retention in gastrointestinal tissue, and enhance therapeutic antibacterial efficacy.30–31, 33–34
Herein, we report on the development and application of a biomimetic micromotor toxoid platform for oral vaccination. To accomplish this, we take advantage of cell membrane coating technology in order to effectively immobilize and neutralize a model bacterial toxin onto the surface of the motor.35–37 This toxic detainment strategy has been shown previously to elicit potent systemic titers when vaccinating via the subcutaneous route, providing strong protective immunity against antibiotic-resistant bacterial infections.38–40 Delivery of toxins in their native form, as opposed to the harsh denaturation strategies employed for traditional toxoid synthesis, can greatly improve immunogenicity and antigenicity. This approach is also easy to generalize, as it works by leveraging the natural binding mechanisms between toxins and cell membranes.41 The present motor toxoid formulation is fabricated by a sequential coating process, where Mg-TiO2 core–shell micromotors are coated by a layer of toxin-inserted red blood cell (RBC) membrane to introduce the antigen material; this is followed by a layer of mucoadhesive chitosan and then a pH-responsive enteric polymer to promote intestinal localization (Figure 1a). Upon oral administration, the motor toxoids travel through the gastrointestinal tract and activate within the small intestine, where the active propulsion drives the payloads towards the intestinal wall, improving retention and stimulating mucosal immunity (Figure 1b). In this report, we demonstrate the successful fabrication of self-propelling motor toxoids, followed by physical and biological characterization of the formulation. The benefits of active propulsion for enhancing delivery and promoting mucosal immunity are investigated both in vitro and in vivo.
Figure 1.
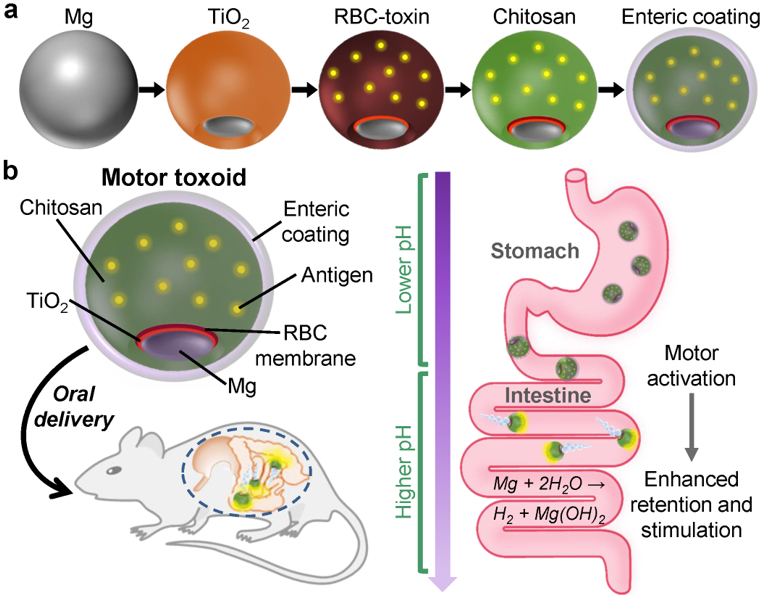
Schematic of micromotor toxoids for oral vaccination. (a) Motor toxoids are fabricated by a sequential process in which magnesium (Mg) microparticles are coated with an asymmetrical layer of TiO2, followed by toxin-inserted RBC membrane (RBC-toxin) as the antigenic material, mucoadhesive chitosan, and a pH-sensitive enteric coating. (b) When administered orally to mice, motor toxoids first enter the stomach, where the enteric coating protects the formulation from degradation in the low pH environment. Upon encountering the more neutral pH of the intestines, the enteric coating dissolves and the intestinal fluid activates the motors. The autonomous propulsion of the motors facilitates enhanced retention and penetration in the intestinal wall, enhancing immune stimulation against the antigen payload.
In order to develop the motorized vaccine formulation, Mg-TiO2 core–shell micromotors were fabricated by dispersing a layer of Mg microparticles onto a glass slide, followed by an asymmetrical coating of the microspheres with a thin TiO2 layer using atomic layer deposition (ALD). The ALD process leads to a uniform TiO2 coating over the Mg microparticles, while leaving a small opening at the sphere–glass contact point, as visualized by scanning electron microscopy (SEM) (Figure S1). This asymmetry is essential for achieving directional movement upon contact with gastrointestinal fluids.42 The TiO2 layer acts as a rigid shell scaffold that maintains the micromotor’s spherical shape and opening during the propulsion, leading to consistent and prolonged operation. After collection of the core–shell micromotors by soft mechanical scratching of the glass slide, an antigen payload was loaded onto the motors using a biomimetic cell membrane coating approach.38 It has previously been demonstrated that RBC membrane, supported by particulate substrates, enables the neutralization of pore-forming toxins, which are secreted by a wide range of pathogens and venomous animals. These detained virulence factors can then be safely administered in vivo to elicit potent immunity capable of preventing toxin-mediated damage.39–40 Staphylococcal α-toxin, one of the major hemolytic factors secreted by Staphylococcus aureus,43 was used as a model payload. After incubating the toxin with the RBC membrane to facilitate their complexation, the resulting toxin-inserted membrane was coated onto the bare micromotors by a sonication process. Per 1 mg of the motors, it was demonstrated that approximately 5 μg of protein content could be loaded; the majority could be attributed to material from the RBC membrane (Figure 2a). It should be noted that the main function of the RBC membrane in this case was to neutralize and load toxin onto the micromotor surface, which does not necessarily require the complete preservation of protein function or membrane sidedness. In order to confirm the presence of α-toxin on the formulation, western blotting analysis was conducted. Compared with motors coated only with RBC membrane, those coated with toxin-inserted membrane exhibited strong banding when probed with the appropriate primary immunostain (Figure 2b). Using a set of α-toxin standards, it was estimated that approximately 150 ng of antigen material was loaded per 1 mg of the motor toxoid formulation (Figure 2c).
Figure 2.
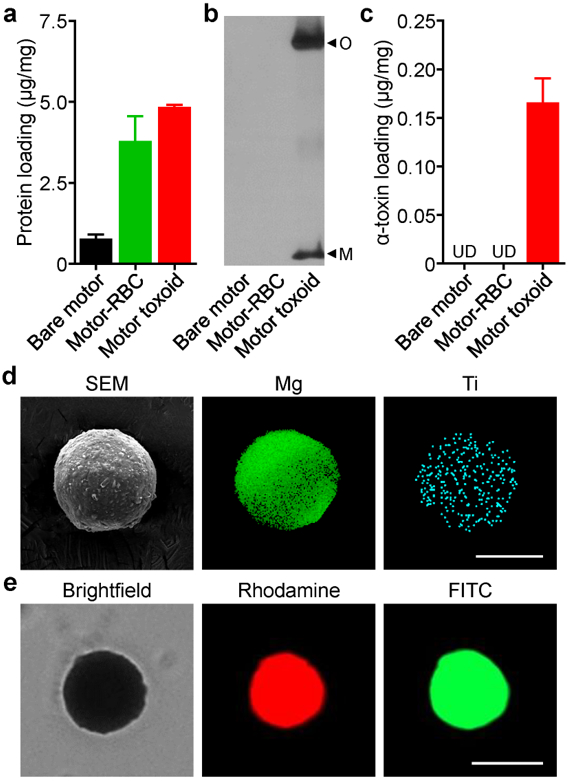
Motor toxoid synthesis and characterization. (a) Quantification of the total protein loaded onto bare Mg-TiO2 micromotors, RBC-coated motors (motor-RBC), and motor toxoids per 1 mg of motor (n = 3, mean + SD). (b) Representative western blots probing for α-toxin on bare motors, motor-RBC, and motor toxoids (O: oligomer band, M: monomer band). (c) Quantification of the amount of α-toxin loading onto bare motors, motor-RBC, and motor toxoids per 1 mg of motor (n = 3, mean + SD; UD: undetectable). (d) SEM visualization of motor toxoids; corresponding EDX spectroscopy confirmed the presence and distribution of Mg (green) and Ti (teal) in the particles (scale bar = 20 µm). (e) Brightfield and fluorescence microscopy visualization of motor toxoids fabricated with rhodamine-labeled (red) RBC membrane and FITC-labeled (green) chitosan (scale bar = 25 μm).
After antigen loading, the micromotors were further coated with a thin layer of positively charged chitosan, which interacts directly with the negatively charged RBC membrane by electrostatic interaction, in order to promote adhesion to the mucosal layer of the intestinal wall.14–16 This was followed by a layer of enteric coating using Eudragit L100–55, a pH-responsive methacrylate-based polymer commonly used for protecting oral drugs from the harsh acidic conditions of the stomach.30 Previous studies have demonstrated the utility of enteric coatings for facilitating the delivery of micromotors to the intestine.31 After passing through the stomach, the increasingly neutral pH of the intestine results in the dissolution of the protective coating by deprotonating the polymer’s functional groups and raising its solubility. At the coating thickness obtained using a 6.5% (w/v) solution of Eudragit L100–55, activation occurs ~20 min after immersion in intestinal fluid, triggering motor propulsion and enabling spatial positioning. In order to visualize the spherical structure of the motor toxoids, SEM was employed (Figure 2d). Energy-dispersive X-ray (EDX) spectroscopy mapping analysis was concurrently used and confirmed the presence and distribution of both Mg from the microparticle core and Ti from the TiO2 shell. Further characterization of the cell membrane and chitosan layers was performed using 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) and fluorescein isothiocyanate-dextran to label each component, respectively (Figure 2e). Strong signal was observed for both dyes when the motors were visualized by fluorescence microscopy, confirming that each layer was successfully incorporated onto the formulation. In total, the data provided strong evidence that the proposed structure, with a micromotor core coated in antigen, followed by polymer coatings to facilitate intestinal delivery and retention, had been successfully fabricated.
The ability of the motor toxoid formulation to efficiently propel within the gastrointestinal tract was first tested in vitro using simulated gastric (pH ~1.3) and intestinal (pH ~6.5) fluids (illustrated in the tracking trajectories of Figure 3a). For the bare Mg-TiO2 micromotors, fast and prolonged autonomous propulsion was observed in both the gastric and intestinal fluids (Supporting Video S1). The motor toxoid formulation likewise exhibited strong propulsion in both types of media, which suggested that it could help to enhance antigen distribution and retention on the intestinal wall (Supporting Video S2). In contrast, no propulsion was observed in both media using control static microparticles based on complete coating of the Mg particle with TiO2, leaving no exposed surface (Supporting Video S3). Finally, the enteric-coated motor toxoid was evaluated (Supporting Video S4). Under low pH conditions, the coating prevented exposure of the Mg to media, completely inhibiting the propulsion characteristics of the motor toxoids. Once exposed to pH values above 5.5, the enteric coating material is designed to quickly dissolve, and it was observed that, in the simulated intestinal fluid, the propulsion characteristics of the enteric-coated motor toxoids were recovered. Quantitative analysis revealed that the bare motors achieved speeds greater than 200 µm/s (Figure 3b). In gastric fluid, the motor toxoids had nearly identical speeds compared with the bare motors, whereas the formulation exhibited a speed reduction of 24% in the intestinal fluid. This difference may be attributed to the fact that, in more neutral pH values, the integrity of the membrane and chitosan coatings is preserved, thus slightly inhibiting the fuel access of the motor core. For the motor toxoids with enteric coating, the propulsion speed matched well with that of uncoated motor toxoids when in intestinal fluid. Overall, the data indicated that it was possible to design an active delivery system that could be activated and propelled specifically in the intestines, which could help to greatly facilitate mucosal immunity.
Figure 3.
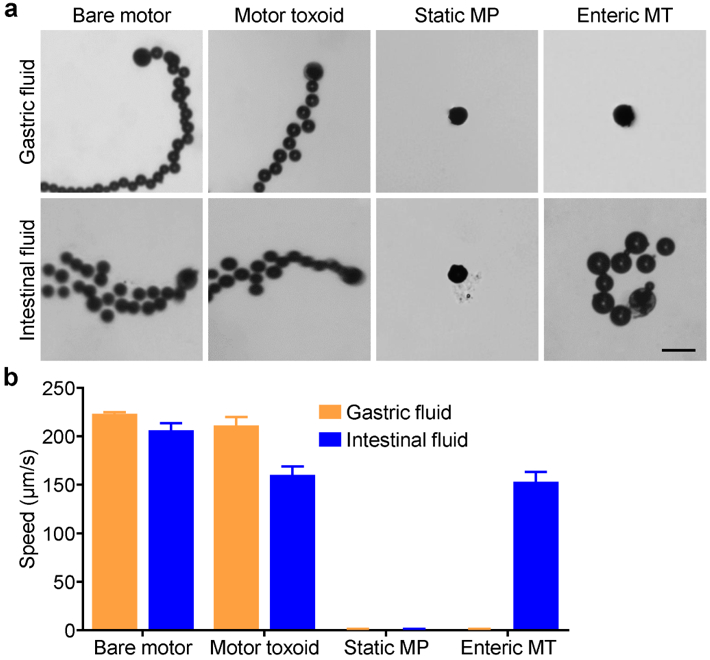
Propulsion characterization. (a) Representative tracking trajectories (captured from Supporting Videos S1, S2, S3, and S4) of bare Mg-TiO2 motors, motor toxoids (MT), static microparticles (MP), and enteric-coated MT in either simulated gastric fluid at pH ~1.3 or intestinal fluid at pH 6.5 (scale bar = 50 μm). (b) Quantification of the speed in each of the samples from (a) (n = 3, mean + SD).
Further in vitro characterization was performed using a murine dendritic cell line to assess the interaction of the motor toxoids with live cells. First, motor toxoids and static microparticles without enteric coatings were formulated using an RBC membrane coating labeled with a red fluorescent dye in order to facilitate tracking of the antigenic material. After incubating the samples with the cells for 6 h, fluorescence imaging was performed to assess the distribution of the membrane material that was released (Figure 4a). From the images, it was readily apparent that the cells incubated with motor toxoids had a significant amount of membrane fragment uptake. In comparison, the static microparticles exhibited much less uptake, despite being loaded with similar amounts of membrane and antigen material (Figure S2). The difference between the two groups was further confirmed by flow cytometry, where cells incubated with the motor toxoids had more than double the mean fluorescence intensity compared with those incubated with static microparticles (Figure 4b). Increased uptake of membrane material from the motor toxoid formulation suggests that active propulsion may help to promote cellular contact and interaction. Additionally, the effect of the formulations on cell viability was assessed after 3 days of incubation (Figure 4c). At up to 2 mg/mL of the particles, neither the motor toxoids nor the static microparticles had any effect on the percentage of live cells, indicating that they were safe for subsequent in vivo use.
Figure 4.
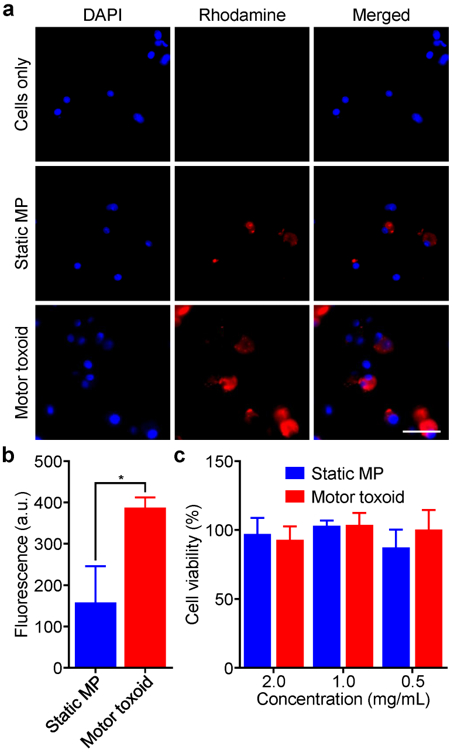
In vitro uptake and safety. (a) JAWSII dendritic cells were incubated for 6 h with either static microparticles (MP) or motor toxoids fabricated using rhodamine-labeled membrane (red). After washing the cells and staining the nuclei with DAPI (blue), membrane uptake was visualized by fluorescence microscopy (scale bar = 50 μm). (b) Rhodamine-labeled static MP or motor toxoids were incubated with JAWSII cells for 24 h and the fluorescence of the cells was quantified by flow cytometry (n = 3, mean + SD). *p < 0.05; Student’s t-test. (c) Static MP or motor toxoids were incubated with JAWSII cells at varying concentrations for 72 h, after which the cell viability was quantified (n = 3, mean + SD).
Following the fabrication, characterization, and in vitro testing of the motor toxoids, their use was further investigated under an in vivo setting. To facilitate imaging in biological tissue, a version of the particles labeled with a far-red fluorescent dye was developed (Figure S3). Motor toxoids and static microparticles with enteric coatings were administered into fasted mice by oral gavage in order to study their distribution and retention. After 6 h, the mice were euthanized, and their gastrointestinal tracts were isolated for ex vivo imaging (Figure 5a and Figure S4). Whereas fluorescent signal for the static formulation was found mainly in the stomach, the motor toxoid formulation was highly present within the intestinal region. Quantitative analysis of the total fluorescence within the gastrointestinal tract confirmed that the motor toxoids exhibited significantly more retention compared to the static microparticles (Figure 5b). This observed distribution was in line with the design of the enteric-coated motor toxoids, which were expected to be activated in the higher pH environment of the intestines. Note that the micromotors will travel down the intestine over time,31 leading to eventual passage through the gastrointestinal tract. In order to further analyze this effect, histological sections were taken from the small intestine and the cell nuclei were stained with a blue fluorescent dye for visualization (Figure 5c). While negligible signal was observed in the sections for the static microparticle group, fluorescent remnants of the particles were highly visible on the sections from mice receiving the motor toxoids. Not only was signal present on the apical side of the epithelial cells, a high amount of fluorescence was also observed within the individual villus structures. The data provided promising evidence that the active propulsion of the motor toxoids played an important role for improving payload retention on the intestinal wall and within the villi.
Figure 5.
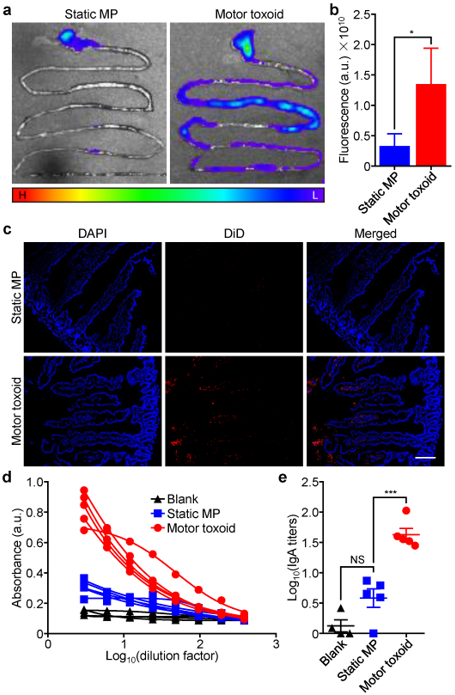
In vivo delivery and antibody titer generation. (a) Representative images of the gastrointestinal tract of male CD-1 mice 6 h after administration of DiD-labeled static microparticles (MP) or motor toxoids by oral gavage (H: high fluorescence, L: low fluorescence). (b) Quantification of the fluorescence from (a) (n = 3, mean + SD). (c) Histological sections from the intestine 6 h after administration of DiD-labeled (red) static MP or motor toxoids; DAPI (blue) was used to label cell nuclei (scale bar = 100 μm). (d) Absorbance data from an ELISA assay probing for IgA antibody production against staphylococcal α-toxin in the feces of mice one week after the administration of blank solution, static MP, or motor toxoids (n = 4 or 5; four-parameter dose-response curve). (e) IgA titers against α-toxin as calculated using the data from (d) (n = 4 to 5, geometric mean ± SEM). *p < 0.05, ***p < 0.001, NS: not significant; one-way ANOVA.
Finally, a study was performed to assess the effect of the enhanced retention of the motor toxoids on their ability to induce immune responses against α-toxin, the model antigen of interest. Mice were orally administered with either the static microparticles or the motor toxoids, and their feces were collected after a period of 1 week to assess IgA antibody titers using an enzyme-linked immunosorbent assay. When looking at absorbance values, it was readily apparent that mice orally vaccinated with the motor toxoids produced significantly more IgA antibodies against α-toxin compared with the static microparticle group, which only demonstrated a modest amount of signal near the level of mice administered with blank solution (Figure 5d). When plotted as titer data, it could be seen that the propulsion properties afforded by the motor toxoid formulation enhanced the anti-toxin IgA titer production by approximately one order of magnitude, whereas the static formulation did not significantly elicit titers compared to the blank solution (Figure 5e). In terms of generating mucosal immunity, the results of this study indicate that there is a significant advantage in using micromotor technology to facilitate the active delivery and retention of antigenic payloads.
In conclusion, we have successfully fabricated an active oral vaccine formulation that leverages the unique benefits of micromotor technology and biomimetic membrane coatings to facilitate the enhanced generation of mucosal immunity. In contrast to particulate delivery systems that rely on passive transport, the autonomous propulsion characteristics afforded by the micromotor core of the presently reported toxoid formulation offer an active mechanism for improving the retention and uptake of antigenic material within the intestinal tract. Combined with both the mucoadhesive and enteric coatings that further improve intestinal localization, the effectiveness of the formulation may be explained by its ability to simulate invasive infection. Using the cell membrane-based toxin detainment strategy, a wide range of toxic antigenic payloads can be neutralized and safely delivered by modulating the source of the membrane material.44–48 In the future, it may be possible to explore the development of self-propelling motor toxoids that incorporate immunological adjuvants or take advantage of different mucosal vaccination routes to further boost efficacy. The potential for generating systemic immunity should also be studied, and this would expand the applications of this platform far beyond infectious diseases. Ultimately, continued research along these lines may lead to a paradigm shift from passive to active delivery mechanisms in vaccine development, leading to formulations that are easy to administer, potent, and broadly applicable.
Materials and Methods
Fabrication of Motor Toxoids.
The Mg-based core–shell micromotors were prepared using commercially available Mg microparticles (TangShan WeiHao Magnesium Powder Co.) with an average size of 20 ± 5 μm as the core. The Mg microparticles were initially washed with acetone to eliminate the presence of impurities. After being dried under nitrogen gas, the Mg microparticles were dispersed onto glass slides (~2 mg per glass slide), followed by atomic layer deposition (ALD) of TiO2 at 100 °C for 3000 cycles using a Beneq TFS 200 system. The ALD process utilizes gas phase reactants, leading to uniform coatings over the Mg microparticles while leaving a small opening at the contact point of the particle to the glass slide, which is essential for micromotor propulsion. Finally, the micromotors were collected by lightly scratching them off the glass slide.
For the antigen loading, CD-1 mouse red blood cells (BioreclamationIVT) were treated by a hypotonic lysis procedure to obtain membrane ghosts,49 which were then incubated with staphylococcal α-toxin (Sigma-Aldrich) at a ratio of 1 mg of RBC membrane to 100 μg of toxin. For coating, 2 mg of Mg-TiO2 micromotors were coated with 100 μL of the toxin-bound RBC membrane at 1 mg/mL by 15 min of sonication at room temperature in a Fisher Scientific CPXH 2800 ultrasonic bath. For the mucoadhesive coating, the Mg-based micromotors were incubated in 0.05% (w/v) chitosan (Sigma-Aldrich) and 0.01% (w/v) sodium dodecyl sulfate (Sigma-Aldrich) prepared in 3 mM acetic acid (Sigma-Aldrich) under a gentle stirring process for 30 min.
The commercial enteric polymer Eudragit L100–55 (Evonik Industries) was chosen to coat the Mg-TiO2 micromotors to prevent the Mg core from reacting in gastric fluid. First, a batch of motor toxoids was added in 100 μL of a 6.5% (w/v) Eudragit L100–55 solution prepared in pure isopropyl alcohol. The micromotor suspension was dispersed into a paraffin oil (Sigma-Aldrich) and Span 85 (Sigma-Aldrich) matrix (100:1 ratio) following a solvent evaporation process. The micromotors were then solidified with hexanes followed by a subsequent drying process. Finally, a period of soft annealing at 70 °C for 2 h ensured the complete sealing of the enteric-coated motor toxoids. The static control microparticles were fabricated following a similar process. Mg-TiO2 microparticles were fully covered with TiO2 by a double ALD process, and the resulting particles were coated with toxin-loaded membrane, chitosan, and Eudragit L100–55 as described above.
For characterization purposes, various fluorescent-labeled motor toxoids were also fabricated. To confirm membrane coating and evaluate antigen delivery in vitro, (1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (DMPE-rhodamine, ex/em = 560/583 nm, Avanti Polar Lipids) was used to label RBC membrane by resuspending 10 μg of the dried dye in 1 mL of 1 mg/mL membrane solution and incubating at 70 °C for 30 min. To confirm the mucoadhesive coating, fluorescein isothiocyanate-dextran (FITC-dextran, ex/em = 492/520 nm, Sigma-Aldrich) was used to label the chitosan by including ~4% (v/v) of FITC-dextran at 1 μg/mL with the coating solution. Finally, for the in vivo distribution and retention studies, far-red dye-labeled motor toxoids were fabricated by first coating the Mg-TiO2 micromotors with a 0.05% (w/v) chitosan aqueous solution containing 4% (v/v) of a 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine (DiD, ex/em = 644/665 nm, Life Technologies) solution prepared at 10 μg/mL in dimethyl sulfoxide.
Motor Toxoid Characterization.
SEM images were obtained with a Phillips XL30 ESEM instrument, using an acceleration voltage of 10 kV. EDX mapping analysis for Mg and Ti was performed using an Oxford EDX detector attached to the SEM instrument and operated by INCA software. Brightfield and fluorescent images of the motor toxoids were captured using an Invitrogen EVOS FL microscope coupled with a 40× microscope objective using the GFP, RFP, and Cy5 fluorescence filters. The autonomous propulsion of the various samples was tested in vitro using simulated gastric fluid without enzyme (pH ~1.3, Sigma-Aldrich) and simulated intestinal fluid (pH 6.5, Sigma-Aldrich) supplemented with 0.2% Triton X-100 (Fisher Scientific) as a surfactant. A Nikon Eclipse Ti-S/L100 inverted optical microscope with a 20× objective, along with a Hamamatsu C11440 digital camera and NIS Elements AR 3.2 software, was used to capture the videos. The speed of the micromotors was tracked using an NIS Elements tracking module.
To quantify the protein content coated onto the Mg-TiO2 micromotors, 2% (w/v) sodium dodecyl sulfate was used to extract the protein from 2 mg of motors. After heating at 37 °C for 30 min and 2 min of sonication in a Fisher FS30D bath sonicator, the sample was spun down at 10,000 g for 5 min, and the supernatant was collected for protein concentration determination using a Pierce BCA protein assay kit (Thermo Scientific). Western blotting was carried out to quantitatively determine the amount of α-toxin. Samples were prepared at a final particle concentration of 6.7 mg/mL using lithium dodecyl sulfate sample loading buffer (Invitrogen), followed by heating at 70 °C for 30 min and sonication for 2 min. Purified α-toxin at various concentrations were used as standards to determine the toxin amount on the motor toxoids. Then, 20 μL of each sample was run on Bolt 4–12% Bis-Tris Plus minigels (Invitrogen) in MOPS running buffer (Invitrogen). After transferring onto nitrocellulose membrane (Thermo Scientific), a polyclonal rabbit anti-staphylococcal α-toxin (Sigma Aldrich) was used as the primary immunostain, followed by an HRP-conjugated anti-rabbit IgG (Biolegend) as the secondary. Blots were developed with western blotting substrate (Thermo Scientific) using an ImageWorks Mini-Medical/90 Developer. Band intensities were measured using Adobe Photoshop.
In Vitro Cellular Uptake and Safety.
The JAWSII murine dendritic cell line (CRL-11904, American Type Culture Collection) was cultured and maintained in growth media consisting of 500 mL Iscove’s Modification of DMEM supplemented with 50 mL USDA-certified fetal bovine serum (Omega Scientific), 55 μM β-mercaptoethanol (Gibco), 2 mM L-glutamine (Gibco), 100 U/mL penicillin-streptomycin (Gibco), and 10 ng/mL granulocyte/macrophage-colony stimulating factor (Biolegend). For the cellular uptake study, 1 × 104 JAWSII cells were seeded into 4-well chamber slides and incubated with rhodamine-labeled motors toxoids or static microparticles for 6 h at a concentration of 1 mg/mL. Cells were then washed three times with phosphate buffered saline (PBS), fixed with 10% phosphate buffered formalin (Fisher Chemical) for 15 min, then washed again three times with PBS and mounted in Vectashield mounting media with DAPI (Vector Laboratories). Samples were imaged under a Keyence BZ-9000 fluorescence microscope with a 60× oil objective using the DAPI and TRITC filters. For flow cytometry, 1 × 105 JAWSII cells were seeded into 6-well tissue culture plates and then incubated with rhodamine-labeled samples at 1 mg/mL for 24 h. Cells were then washed three times with PBS, detached and resuspended in 250 μL of PBS containing 0.015 μM Calcein Violet-AM (Biolegend) for live cell staining. Data was collected using a BD Biosciences FACSCanto-II flow cytometer and analysis was performed using Flowjo software. The mean fluorescence intensity of the live cell population was reported. To assess cytotoxicity, 6 × 103 JAWSII cells were plated into 96-well plates overnight and then incubated with 0.5 mg/mL, 1 mg/mL, or 2 mg/mL motor toxoids or static microparticles. After 72 h of incubation, cell viability was assayed using an MTS reagent (Promega) following the manufacturer’s instructions. Untreated cells were used as the 100% viability control.
In Vivo Distribution and Titer Studies.
All animal experiments followed protocols that were reviewed, approved, and performed under the regulatory supervision of the University of California San Diego’s Institutional Animal Care and Use Committee (IACUC). To perform the in vivo distribution and retention study, male CD-1 mice (Envigo) were fed with alfalfa-free food (LabDiet) for one week prior to the experiment. Mice were administered with 200 μL of a 10 mg/mL suspension of DiD-labeled enteric-coated motor toxoids or static microparticles by oral gavage after a period of overnight fasting. The mice were euthanized, and their gastrointestinal tracts were collected 6 h after administration. The samples were rinsed with PBS and the fluorescent signal was imaged and quantified using a Xenogen IVIS 200 imaging system under the Cy5.5 filter. For histological analysis, the small intestines were fixed in 10% formalin for 24 h. They were then placed in 15% (w/v) sucrose in PBS until the tissue became submerged and then in 30% (w/v) sucrose in PBS until tissue became submerged again. The tissues were embedded in Tissue-Tek OCT compound (Sakura Finetek) for cryosectioning. The sections were mounted in Vectashield mounting media with DAPI and imaged under a Keyence BZ-9000 fluorescence microscope with a 20× using the DAPI and Cy5 filters.
For the antibody titer study, male CD-1 mice (Envigo) were administered with 200 μL of a 10 mg/mL suspension of enteric-coated motor toxoids or static microparticles by oral gavage after a period of overnight fasting. One week later, feces samples were collected and resuspended to 200 mg/mL in PBS containing a protease inhibitor cocktail (Sigma-Aldrich). After centrifugation at 15,000 g for 5 min, the supernatant was collected to assay for α-toxin IgA antibody titers by an enzyme-linked immunosorbent assay (ELISA). A 96-well plate was first coated overnight with 5 μg/mL α-toxin using an ELISA coating buffer (Biolegend). The wells were then blocked with 1% (w/v) bovine serum albumin (Sigma-Aldrich) in PBS containing 0.05% Tween 20 (National Scientific) for 1 h before adding serially diluted feces samples as the primary antibody. HRP-conjugated goat anti-mouse IgA (SouthernBiotech) was then used as the secondary. The plate was developed with TMB-ELISA substrate (Biolegend) and absorbance was measured at 450 nm with a Tecan Infinite M200 multiplate reader.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Defense Threat Reduction Agency Joint Science and Technology Office for Chemical and Biological Defense (Grant Numbers HDTRA1–18-1–0014 and HDTRA1–13-1–0002). M.B.-G. acknowledges a postdoctoral fellowship from Consejo Nacional de Ciencia y Tecnología (CONACyT). E.K. acknowledges support from the Charles Lee Powell Foundation. J.Z. acknowledges support from a National Institutes of Health 5T32CA153915 training grant from the National Cancer Institute.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
The authors declare no competing financial interest.
REFERENCES
- (1).Plotkin S, History of Vaccination. P. Natl. Acad. Sci. USA 2014, 111, 12283–12287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Orenstein WA; Ahmed R, Simply Put: Vaccination Saves Lives. P. Natl. Acad. Sci. USA 2017, 114, 4031–4033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Bloom DE, The Value of Vaccination. Adv. Exp. Med. Biol 2011, 697, 1–8. [DOI] [PubMed] [Google Scholar]
- (4).Parrino J; Graham BS, Smallpox Vaccines: Past, Present, and Future. J. Allergy Clin. Immunol 2006, 118, 1320–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Blume S; Geesink I, A Brief History of Polio Vaccines. Science 2000, 288, 1593–1594. [DOI] [PubMed] [Google Scholar]
- (6).Oyston P; Robinson K, The Current Challenges for Vaccine Development. J. Med. Microbiol 2012, 61, 889–894. [DOI] [PubMed] [Google Scholar]
- (7).Kaufmann SHE; McElrath MJ; Lewis DJM; Del Giudice G, Challenges and Responses in Human Vaccine Development. Curr. Opin. Immunol 2014, 28, 18–26. [DOI] [PubMed] [Google Scholar]
- (8).Maslow JN, The Cost and Challenge of Vaccine Development for Emerging and Emergent Infectious Diseases. Lancet Glob. Health 2018, 6, E1266–E1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Shalaby WSW, Development of Oral Vaccines to Stimulate Mucosal and Systemic Immunity - Barriers and Novel Strategies. Clin. Immunol. Immunopathol 1995, 74, 127–134. [DOI] [PubMed] [Google Scholar]
- (10).Ramirez JEV; Sharpe LA; Peppas NA, Current State and Challenges in Developing Oral Vaccines. Adv. Drug Deliv. Rev 2017, 114, 116–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Neutra MR; Kozlowski PA, Mucosal Vaccines: The Promise and the Challenge. Nat. Rev. Immunol 2006, 6, 148–158. [DOI] [PubMed] [Google Scholar]
- (12).Fang RH; Kroll AV; Zhang LF, Nanoparticle-Based Manipulation of Antigen-Presenting Cells for Cancer Immunotherapy. Small 2015, 11, 5483–5496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Fang RNH; Zhang LF, Nanoparticle-Based Modulation of the Immune System. Annu. Rev. Chem. Biomol. Eng 2016, 7, 305–326. [DOI] [PubMed] [Google Scholar]
- (14).des Rieux A; Fievez V; Garinot M; Schneider YJ; Preat V, Nanoparticles as Potential Oral Delivery Systems of Proteins and Vaccines: A Mechanistic Approach. J. Control. Release 2006, 116, 1–27. [DOI] [PubMed] [Google Scholar]
- (15).Agnihotri SA; Mallikarjuna NN; Aminabhavi TM, Recent Advances on Chitosan-Based Micro- and Nanoparticles in Drug Delivery. J. Control. Release 2004, 100, 5–28. [DOI] [PubMed] [Google Scholar]
- (16).Ponchel G; Irache JM, Specific and Non-Specific Bioadhesive Particulate Systems for Oral Delivery to the Gastrointestinal Tract. Adv. Drug Deliv. Rev 1998, 34, 191–219. [DOI] [PubMed] [Google Scholar]
- (17).Dehaini D; Fang RH; Zhang L, Biomimetic Strategies for Targeted Nanoparticle Delivery. Bioeng. Transl. Med 2016, 1, 30–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Brannon-Peppas L; Blanchette JO, Nanoparticle and Targeted Systems for Cancer Therapy. Adv. Drug Deliv. Rev 2004, 56, 1649–1659. [DOI] [PubMed] [Google Scholar]
- (19).Wang J, Nanomachines: Fundamentals and Applications. Wiley-VCH: Weinheim, 2013. [Google Scholar]
- (20).Burdick J; Laocharoensuk R; Wheat PM; Posner JD; Wang J, Synthetic Nanomotors in Microchannel Networks: Directional Microchip Motion and Controlled Manipulation of Cargo. J. Am. Chem. Soc 2008, 130, 8164–8165. [DOI] [PubMed] [Google Scholar]
- (21).Campuzano S; Esteban-Fernandez de Avila B; Yanez-Sedeno P; Pingarron JM; Wang J, Nano/Microvehicles for Efficient Delivery and (Bio) Sensing at the Cellular Level. Chem. Sci 2017, 8, 6750–6763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Chen CR; Karshalev E; Guan JG; Wang J, Magnesium-Based Micromotors: Water-Powered Propulsion, Multifunctionality, and Biomedical and Environmental Applications. Small 2018, 14, 1704252. [DOI] [PubMed] [Google Scholar]
- (23).Gao W; Peng XM; Pei A; Kane CR; Tam R; Hennessy C; Wang J, Bioinspired Helical Microswimmers Based on Vascular Plants. Nano Lett. 2014, 14, 305–310. [DOI] [PubMed] [Google Scholar]
- (24).Li JX; Esteban-Fernandez de Avila B; Gao W; Zhang LF; Wang J, Micro/Nanorobots for Biomedicine: Delivery, Surgery, Sensing, and Detoxification. Sci. Robot 2017, 2, eaam6431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Li JX; Sattayasamitsathit S; Dong RF; Gao W; Tam R; Feng XM; Ai S; Wang J, Template Electrosynthesis of Tailored-Made Helical Nanoswimmers. Nanoscale 2014, 6, 9415–9420. [DOI] [PubMed] [Google Scholar]
- (26).Pourrahimi AM; Pumera M, Multifunctional and Self-Propelled Spherical Janus Nano/Micromotors: Recent Advances. Nanoscale 2018, 10, 16398–16415. [DOI] [PubMed] [Google Scholar]
- (27).Wang H; Pumera M, Emerging Materials for the Fabrication of Micro/Nanomotors. Nanoscale 2017, 9, 2109–2116. [DOI] [PubMed] [Google Scholar]
- (28).Sanchez S; Soler L; Katuri J, Chemically Powered Micro- and Nanomotors. Angew. Chem. Int. Ed 2015, 54, 1414–1444. [DOI] [PubMed] [Google Scholar]
- (29).Peyer KE; Zhang L; Nelson BJ, Bio-Inspired Magnetic Swimming Microrobots for Biomedical Applications. Nanoscale 2013, 5, 1259–1272. [DOI] [PubMed] [Google Scholar]
- (30).Gao W; Dong RF; Thamphiwatana S; Li JX; Gao WW; Zhang LF; Wang J, Artificial Micromotors in the Mouse’s Stomach: A Step toward In Vivo Use of Synthetic Motors. ACS Nano 2015, 9, 117–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Li JX; Thamphiwatana S; Liu WJ; Esteban-Fernandez de Avila B; Angsantikul P; Sandraz E; Wang JX; Xu TL; Soto F; Ramez V; Wang XL; Gao WW; Zhang LF; Wang J, Enteric Micromotor Can Selectively Position and Spontaneously Propel in the Gastrointestinal Tract. ACS Nano 2016, 10, 9536–9542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Esteban-Fernandez de Avila B; Angsantikul P; Li JX; Gao W; Zhang LF; Wang J, Micromotors Go In Vivo: From Test Tubes to Live Animals. Adv. Funct. Mater 2018, 28, 1705640. [Google Scholar]
- (33).Esteban-Fernandez de Avila B; Angsantikul P; Li JX; Lopez-Ramirez MA; Ramirez-Herrera DE; Thamphiwatana S; Chen CR; Delezuk J; Samakapiruk R; Ramez V; Zhang LF; Wang J, Micromotor-Enabled Active Drug Delivery for In Vivo Treatment of Stomach Infection. Nat. Commun 2017, 8, 272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Li JX; Angsantikul P; Liu WJ; Esteban-Fernandez de Avila B; Thamphiwatana S; Xu ML; Sandraz E; Wang XL; Delezuk J; Gao WW; Zhang LF; Wang J, Micromotors Spontaneously Neutralize Gastric Acid for pH-Responsive Payload Release. Angew. Chem. Int. Ed 2017, 56, 2156–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Fang RH; Kroll AV; Gao WW; Zhang LF, Cell Membrane Coating Nanotechnology. Adv. Mater 2018, 30, 1706759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Kroll AV; Fang RH; Zhang LF, Biointerfacing and Applications of Cell Membrane-Coated Nanoparticles. Bioconjug. Chem 2017, 28, 23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Hu CMJ; Fang RH; Copp J; Luk BT; Zhang LF, A Biomimetic Nanosponge that Absorbs Pore-Forming Toxins. Nat. Nanotechnol 2013, 8, 336–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Hu CMJ; Fang RH; Luk BT; Zhang LF, Nanoparticle-Detained Toxins for Safe and Effective Vaccination. Nat. Nanotechnol 2013, 8, 933–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Wang F; Fang RH; Luk BT; Hu CJ; Thamphiwatana S; Dehaini D; Angsantikul P; Kroll AV; Pang Z; Gao W; Lu W; Zhang L, Nanoparticle-Based Antivirulence Vaccine for the Management of Methicillin-Resistant Staphylococcus aureus Skin Infection. Adv. Funct. Mater 2016, 26, 1628–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Wei X; Gao J; Wang F; Ying M; Angsantikul P; Kroll AV; Zhou J; Gao W; Lu W; Fang RH; Zhang L, In Situ Capture of Bacterial Toxins for Antivirulence Vaccination. Adv. Mater 2017, 29, 1701644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Fang RH; Luk BT; Hu CM; Zhang L, Engineered Nanoparticles Mimicking Cell Membranes for Toxin Neutralization. Adv. Drug Deliv. Rev 2015, 90, 69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Karshalev E; Esteban-Fernandez de Avila B; Beltran-Gastelum M; Angsantikul P; Tang SS; Mundaca-Uribe R; Zhang FY; Zhao J; Zhang LF; Wang J, Micromotor Pills as a Dynamic Oral Delivery Platform. ACS Nano 2018, 12, 8397–8405. [DOI] [PubMed] [Google Scholar]
- (43).Kennedy AD; Wardenburg JB; Gardner DJ; Long D; Whitney AR; Braughton KR; Schneewind O; DeLeo FR, Targeting of Alpha-Hemolysin by Active or Passive Immunization Decreases Severity of USA300 Skin Infection in a Mouse Model. J. Infect. Dis 2010, 202, 1050–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Gao WW; Fang RH; Thamphiwatana S; Luk BT; Li JM; Angsantikul P; Zhang QZ; Hu CMJ; Zhang LF, Modulating Antibacterial Immunity via Bacterial Membrane-Coated Nanoparticles. Nano Lett. 2015, 15, 1403–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Thamphiwatana S; Angsantikul P; Escajadillo T; Zhang QZ; Olson J; Luk BT; Zhang S; Fang RH; Gao WW; Nizet V; Zhang LF, Macrophage-Like Nanoparticles Concurrently Absorbing Endotoxins and Proinflammatory Cytokines for Sepsis Management. P. Natl. Acad. Sci. USA 2017, 114, 11488–11493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Dehaini D; Wei XL; Fang RH; Masson S; Angsantikul P; Luk BT; Zhang Y; Ying M; Jiang Y; Kroll AV; Gao WW; Zhang LF, Erythrocyte-Platelet Hybrid Membrane Coating for Enhanced Nanoparticle Functionalization. Adv. Mater 2017, 29, 1606209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Hu CMJ; Fang RH; Wang KC; Luk BT; Thamphiwatana S; Dehaini D; Nguyen P; Angsantikul P; Wen CH; Kroll AV; Carpenter C; Ramesh M; Qu V; Patel SH; Zhu J; Shi W; Hofman FM; Chen TC; Gao WW; Zhang K, et al. , Nanoparticle Biointerfacing by Platelet Membrane Cloaking. Nature 2015, 526, 118–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Fang RH; Hu CMJ; Luk BT; Gao WW; Copp JA; Tai YY; O’Connor DE; Zhang LF, Cancer Cell Membrane-Coated Nanoparticles for Anticancer Vaccination and Drug Delivery. Nano Lett. 2014, 14, 2181–2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Hu CMJ; Zhang L; Aryal S; Cheung C; Fang RH; Zhang LF, Erythrocyte Membrane-Camouflaged Polymeric Nanoparticles as a Biomimetic Delivery Platform. P. Natl. Acad. Sci. USA 2011, 108, 10980–10985. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.