

Abstract
Gene maps, or annotations, enable us to navigate the functional landscape of our genome. They are a resource upon which virtually all studies depend, from single-gene to genome-wide scales and from basic molecular biology to medical genetics. Yet present-day annotations suffer from trade-offs between quality and size, with serious but often unappreciated consequences for downstream studies. This is particularly true for long non-coding RNAs (lncRNAs), which are poorly characterized compared to protein-coding genes. Long-read sequencing technologies promise to improve current annotations, paving the way towards a complete annotation of lncRNAs expressed throughout a human lifetime.
A fundamental goal of biology is to understand how the instructions to create and maintain an organism are encoded in its DNA sequence. From worm to man, the genomes of different species house remarkably similar numbers of protein-coding genes1, prompting the notion that many aspects of complex organisms arise from non-protein-coding regions. These non-coding regions comprise a rich diversity of regulatory and functional units, amongst the most numerous of which are loci encoding long non-coding RNAs (lncRNAs)2. Next-generation sequencing has identified tens of thousands of lncRNA loci, from single-celled eukaryotes to humans3. The sequences of lncRNAs are under purifying evolutionary selection4,5, and a substantial fraction yield clear phenotypic effects in both in vitro and in vivo loss of function studies6–10. Growing numbers of lncRNAs have been linked to human diseases11. However, their functionality remains contentious12, and the number of experimentally characterized or disease-associated lncRNAs lies in the hundreds, or ≤1% of identified loci13.
Closing this gulf between mapped and experimentally validated lncRNAs has prompted functional studies of growing scope. These studies have depended on the development of the fundamental resource of annotations, which describe the genomic locations, sequences and exon structure of lncRNA transcripts. As the basis of microarray designs, early lncRNA annotations enabled researchers to perform the first generation of functional genomics studies, implicating lncRNAs in processes as diverse as embryonic stem cell pluripotency14, reprogramming15, tumour suppression16, neuronal differentiation17 and cardiac differentiation18. More recently, large-scale functional screens based on the CRISPR–Cas system have been applied to hundreds or thousands of lncRNAs in a single experiment19.
Several different annotations exist for the human genome (TABLE 1), each with advantages and drawbacks that might not be immediately evident. They are based on two principal strategies of automated and manual annotation. Automated annotation typically employs transcriptome assembly approaches that are rapid and inexpensive but produce incomplete and inaccurate annotations. Manual annotation yields high-quality catalogues but at slow rates and requiring substantial long-term economic support. Both approaches suffer from a variety of deficiencies that are important for end users to understand.
Table 1 |.
lncRNA annotations
| name (version) | Reported size (gene loci) | Methodsa | comments | completeness | comprehensivenessb | exhaustivenessc |
|---|---|---|---|---|---|---|
| NONCODE (v5) | 96,308 | Integration of other databases | The most comprehensive resource | 8.9% | 67,276 | 2.3 |
| MiTranscriptome (v2) | 63,615 | Assembly from short reads | Mainly cancer samples | 4.4% | 45,088 | 4.4 |
| FANTOM CAT (v1) | 27,919 | Assembly, other annotations and CAGE evidence | Mapped 5′ ends using CAGE tags | 15.8% | 27,278 | 3.3 |
| RefSeq (GCF_000001405.37_GRCh38.p11) | 15,791 | Manual (based on cDNA) and automated annotation (based on RNA-seq data) | The oldest annotation | 11.0% | 14,889 | 1.9 |
| GENCODE (v27) | 15,778 | Manual annotation based on cDNA, ESTs and high-quality long-read data | Used by most consortia and integrated with Ensembl | 13.5% | 15,063 | 1.9 |
| BIGTranscriptome (v1) | 14,158 | Assembly, with CAGE and 3 P-seq evidence | Full-length transcripts | 27.7% | 12,632 | 2.1 |
| GENCODE+ | 13,434 | Union of GENCODE (v20) and CLS lncRNAs with anchor-merged CLS transcript models | Extension of GENCODE by CLS | 24.0% | 13,434 | 3.3 |
| CLS FL | 807 | lncRNAs from GENCODE+ with CAGE and poly(A) evidence | Full-length transcripts | 71.7% | 807 | 5.5 |
| Protein-codingd | 19,502 | GENCODE confident protein-coding transcripts | Not tagged mRNA_end_NF nor mRNA_start_NF in the original GENCODE v27 GTF file | 53.8% | 18,995 | 2.9 |
All numbers correct as of the end of 2017. MiTranscriptome, Functional Annotation of the Mammalian genome (FANTOM) cap analysis of gene expression (CAGE)-associated transcriptome (CAT) and BIGTranscriptome long non-coding RNA (lncRNA) catalogues were lifted over to the Genome Reference Consortium Human Build 38 (GRCh38) genome assembly. 3P-seq, poly(A)-position profiling by sequencing; CLS, capture long-read sequencing; EST, expressed sequence tag; RNA-seq, RNA sequencing.
Assembly in the Methods column refers to transcriptome assembly using short reads from RNA-seq.
Comprehensiveness is the total number of gene loci boundaries defined using buildLoci. To compare gene sets in a consistent way, the assembly patches were excluded, and the gene loci boundaries were redefined using buildLoci, which explains discrepancies between gene numbers presented here and those reported in original publications.
Exhaustiveness is the average number of isoforms per gene locus. Figures for completeness, comprehensiveness and exhaustiveness as presented in FIG. 3 are shown here.
A set of protein-coding transcripts was used as a reference.
Recent technical developments promise to revolutionize annotation methods. Third-generation sequencing technologies are capable of reading entire RNA or cDNA molecules. Combined with methods to capture desired transcripts, third-generation sequencing promises to extend and improve existing lncRNA annotations rapidly and cost-effectively. These advances make it feasible to envisage the eventual complete annotation of the genome, whereby the entirety of biologically relevant genes, transcripts and exons is catalogued in all cell types throughout the human lifespan. A key subsidiary aim will be to define what threshold constitutes biological relevance and hence whether expression (or other) thresholds should be used for inclusion in final annotations20.
This Review has two main objectives. The first is to provide an overview of the current state of lncRNA annotations: how they are created, how good they are, best practice in their use, and the development of quantitative standards by which they might be evaluated and compared. The second is to discuss how emerging technologies will have an impact on these annotations and may alter our understanding of what constitutes the human lncRNA transcriptome. Although we focus mainly on human studies, the following discussions are of relevance to other model and non-model organisms. Of note, the lncRNAs discussed here are almost exclusively those of the olyadenylated (polyA+) fraction, owing to the fact that most transcriptomic surveys have been performed on conventional, oligo-dT-primed cDNA. The universe of polyA– lncRNAs remains largely unexplored and may hold many functional molecules21.
lncRNA annotations: a research foundation
Structure of lncRNA annotations and biotypes.
Annotations, whether of protein-coding or lncRNA-encoding genes, are hierarchical: they are composed of gene loci, each of which is composed of one or more partially overlapping transcripts, themselves composed of one or more exons (FIG. 1a). In the absence of a clear understanding of their sequence–structure–function relationship, lncRNAs have tended to be classified by their genomic organization, in other words, the relationship of their encoding locus to the nearest protein-coding gene (FIG. 1b). In the context of genome annotation, this can be used as a biotype label. The principal dichotomy of genomic organization is genic versus intergenic, or lncRNAs that overlap or do not overlap a protein-coding gene, respectively. The latter are also referred to as long intergenic non-coding RNAs (lincRNAs). Genic lncRNAs may be subdivided by the precise nature of their overlap with the protein-coding gene, and there is some evidence for distinct functions and features between these classes22. By numbers, lncRNAs tend to be approximately equally divided into genic and intergenic classes.
Fig. 1 |. Basic concepts of lncRNA annotations.
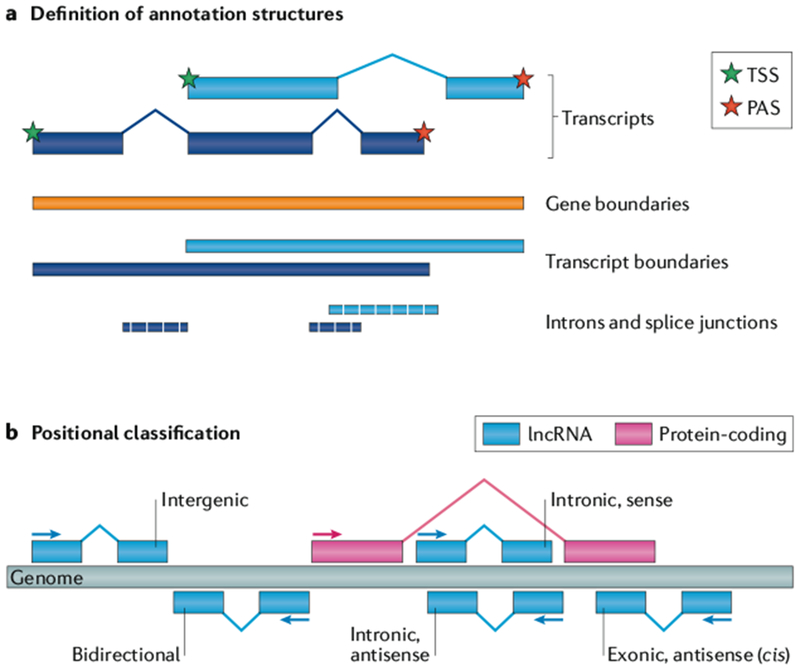
a | The principal structures of a long non-coding RNA (lncRNA) to be annotated. Annotations are hierarchical: they are composed of gene loci, each of which is composed of one or more partially overlapping transcripts, themselves composed of one or more exons (blue rectangles). b | Positional classification of lncRNAs with respect to the nearest protein-coding gene. Genic lncRNAs overlap a protein-coding gene locus, whereas intergenic lncRNAs, also known as long intergenic non-coding RNAs (lincRNAs), do not. Transcripts that overlap a protein-coding gene on the opposite strand are identified as antisense. PAS, polyadenylation site; TSS, transcription start site.
Why are lncRNAs difficult to annotate?.
lncRNA annotations lag considerably behind those of protein-coding genes, for reasons that go beyond their more recent discovery. There are at least three factors that make lncRNA annotation challenging. First, lncRNAs are relatively lowly expressed, meaning that their transcripts will be weakly sampled in any unbiased transcriptomic data, including expressed sequence tags (ESTs), RNA sequencing (RNA-seq) and cap analysis of gene expression (CAGE) data2,23. Second, our understanding of the lncRNA sequence–function relationship is poor (BOX 1). Thus, in contrast to the information-rich, readily idenifiable open reading frame (ORF) of protein-coding genes, sequence features or functional elements can-not presently be used to identify novel lncRNAs. Third, lncRNAs tend to be weakly conserved during evolution24,25, making it challenging to identify their orthologues or paralogues by sequence similarity. Consequently, lncRNA annotation relies almost entirely on physical transcriptomic evidence.
Box 1 |. Beyond gene annotation: mapping functions and domains
In tandem with complete gene annotations, an additional objective is to predict and label molecular, biological and disease functions of long non-coding RNAs (lncRNAs). This aim is held back by our poor understanding of the sequence–function relationship of lncRNAs, in contrast to protein-coding genes whose functions can usually be predicted from primary sequence alone52. Here, we discuss a selection of promising methods to predict the functions and functional domains of lncRNAs. It will be interesting in the future to see such information integrated with annotation databases, LNCipedia and LncRNAWiki being the only resources thus far to do this61,62.
Gene-level functional annotation
Strategies to predict lncRNA functions have traditionally involved reassigning functional labels from protein-coding mRNAs to lncRNAs based on expression patterns. Tissue profiles of lncRNAs and mRNAs are determined from RNA sequencing (RNA-seq) or microarray data and then used to create mixed gene clusters by correlation. Significantly enriched functional labels attached to mRNAs in each cluster, such as Gene Ontology, Kyoto encyclopedia of Genes and Genomes (KEGG) or disease association terms103–105, are assigned to any lncRNAs in the same cluster. This widely used approach is often referred to as guilt by association23. However, it assumes that expression patterns hold information on molecular functionality. Algorithms of growing sophistication, often integrating additional data, are being applied to this problem106–108. A lack of gold standard data means that it is difficult to assess the power of such techniques, although new databases may help resolve this11,109,110.
The expression of lncRNAs within the cell, or subcellular localization, may hold more useful clues for molecular functions. RNA-seq-based maps of lncRNA levels in compartments of the cell, including nucleus, cytoplasm and other organelles, can be used to create maps of localization111–114. These data are then used to classify lncRNAs by their localization according to defined cut-offs114. Although this approach does not make specific functional predictions, it can provide broad pointers; for example, nuclear-enriched transcripts may regulate transcription, while cytoplasmic transcripts are more likely to play post-transcriptional roles. Localization data may also be used to search for domains or motifs that promote lncRNA trafficking to specific cellular sites115–117.
Mapping lncRNA functional elements
The prevailing view is that lncRNAs, similar to proteins, are modular and composed of separable ‘functional elements’ (REFS117–119). Convincing evidence is available for a limited number of cases117,118,120–124, but the global annotation of elements would be a powerful basis for predicting lncRNA functions.
Elements can be predicted by a variety of methods. Evolutionary conservation of RNA structures is a statistically rigorous way of finding putative functional elements88. Protein-binding data are useful in identifying molecular interactors and their binding sites, although they have the drawback that sensitivity depends on expression, which is usually low for lncRNAs125. Maps of inferred or experimentally identified microRNA sites may point to post-transcriptional regulatory roles, such as for competitive endogenous RNAs (ceRNAs)125,126. lncRNAs may interact with genomic DNA through the formation of triplex structures that can be predicted bioinformatically127. Other studies have attempted to map functional sites through transposable elements98,115,116,128.
The importance of accurate annotations.
The fundamental nature of lncRNA annotations means that uncertainties or inaccuracies can have a profound impact on downstream projects. For example, during studies on the developing bat wing, researchers used microarrays to identify what seemed to be an intergenic lncRNA upstream of the gene encoding the developmental factor Meis2 (REF.26). However, careful analysis revealed that the cDNA sequence upon which the annotation had been based was most likely an internally primed fragment of the Meis2 5′ untranslated region (UTR)26. Similarly, an annotated lncRNA whose orthologue was knocked out in mouse, Kantr, was identified through an analysis of full-length transcript models from long-read sequencing to be a protein-coding transcript with an ORF in a previously unannotated exon9,27. Finally, 1/2-sbsRNA AF087999, which has been proposed to regulate mRNAs in trans through Staufen binding, lies within the 3′ UTR of the RBM4 gene. There is little evidence supporting AF087999 as an independent transcript, leaving it unresolved whether it is a standalone lncRNA or a misannotated UTR fragment28.
Amongst the most frequent use of lncRNA annotations is as a reference for quantifying and identifying differentially expressed genes and transcripts in RNA-seq experiments. Quantifier programs, such as RSEM29 or Kallisto30, take annotation files as an input together with mapped RNA-seq reads and attempt to estimate abundances of lncRNA transcripts. This is a challenging problem, particularly for lowly-expressed transcripts31. Inaccuracies or omissions in lncRNA annotations will propagate to transcript abundance estimates. For example, an excessively long 3′ exon annotation will lead to artificially low expression estimates, given that measures such as fragments per kilobase per million mapped (FPKM) are scaled to the annotated length of transcripts32.
Accurate estimates of lncRNA transcription start sites (TSSs) are of particular importance for studies of lncRNA promoters or CRISPR–Cas screens, which depend on targeting Cas9 molecules to gene promoters6,19. Such studies should only examine transcripts with confident 5′ ends, which may be achieved by using independent evidence such as CAGE data to exclude unvalidated TSSs27,33–35.
Biomedical applications for lncRNA annotations are of growing importance. The recent availability of cancer genomes has enabled searches for driver lncRNAs, whose mutations are positively selected for during tumorigenesis36,37. Predictions are critically dependent on lncRNA annotation quality. Similarly, diagnostic screening and genome-wide association studies (GWAS) depend on making accurate inferences of the functional impact of trait-associated mutations38. Such mutations are often assumed to be regulatory when they fall outside exonic regions. Truncated lncRNA annotations could therefore lead to the misinterpretation of mutations that actually fall inside a lncRNA exon and act through the mature lncRNA transcript, for example, by modulating a microRNA response element, as in the case of lnc-LAMC2-1:1 (REF.39). Finally, the identification of lncRNA biomarkers, such as PCA3 for the detection of prostate tumours40, uses RNA-seq quantified against lncRNA annotations. In cases where the analysis output is a diagnosis, annotation quality can thus have a direct impact on patient outcomes.
Additional examples of the diverse uses for lncRNA annotations include evolutionary phylogenies24, analysis of splicing regulation and conservation41, identification of small ORFs (sORFs)42, lncRNA-specific gene properties25 and RNA modifications43. Finally, the success of the nascent field of lncRNA functional domain prediction will depend in large part on the availability of comprehensive and complete lncRNA annotations (BOX 1).
The ecosystem of annotations
Thanks to ongoing efforts over the past two decades (BOX 2), a range of lncRNA annotation resources obtained by different methods are presently available. Contemporary annotation efforts are principally based either on automated transcriptome assemblies from short reads or on manual annotation of existing cDNA and EST libraries (FIG. 2). Recent years have seen considerable efforts in consolidating lncRNA collections, with attention shifting from quantity to quality and a premium placed on 5′ and 3′ completeness. In this section, we review presently available annotations, grouped by method.
Box 2 |. The evolution of lncRNA collections.
The first hint at the volume of long non-coding RNAs (lncRNAs) populating our genome came from genomic microarray technology. Starting in 2002, tiled microarrays with increasing density and genomic span revealed extensive transcription outside of then-known gene loci129. However, the exact sequence and hence protein-coding potential, of those transcripts could not be resolved with this technology. The sequences of these unannotated transcripts were first resolved by massive cDNA sequencing undertaken by the Functional Annotation of the Mammalian genome (FANTOM) consortium130,131. The consortium used a combination of cap analysis of gene expression (CAGE), which can identify transcription start sites (TSSs) by sequencing the 3′ end of cDNAs (that is, the 5′ end of RNAs), and ditag sequencing (also known as paired-end tag sequencing), which is capable of identifying both TSSs and polyadenylation sites. Approximately one-third of cDNAs did not contain identifiable protein-coding sequences; in other words, they were lncRNAs. This data set facilitated the first studies demonstrating purifying evolutionary selection on lncRNAs as a population, implying that at least a subset is functional rather than “transcriptional noise”4.
lncRNA genes were also identified indirectly through their patterns of histone modifications23. Reasoning that lncRNA genes may carry similar combinations of histone 3 lysine 4 trimethylation (H3K4Me3) and histone 3 lysine 36 trimethylation (H3K36Me3) modifications — known markers of active protein-coding genes — researchers identified approximately 1,000 long intergenic non-coding RNAs (lincRNAs) in human and mouse23,132. These lincRNA genes exhibited low steady-state expression levels compared with mRNAs, now known to be a general property of lncRNAs.
Growing volumes of publicly available cDNA sequences opened the way to accurate lncRNA annotations, similar to those for protein-coding genes. The first catalogue of 5,446 human lncRNA loci was generated largely on the basis of cDNAs filtered by an open reading frame (ORF) prediction tool and a pipeline based on the protein basic local alignment search tool (BLASTP)133.
The advent of RNA sequencing (RNA-seq) democratized lncRNA annotation. Using only a sequencer and off-the-shelf computational tools, any laboratory was able to identify thousands of lncRNA loci in their favourite cell type. A central requirement for this approach is transcriptome assembly, whereby computational algorithms are used to reconstruct the underlying transcript structures responsible for observed RNA-seq reads67 (FIG. 2a). Reference-based methods that make use of read-to-genome alignments to infer transcript structures tend to be more accurate than de novo methods68. Foremost amongst reference-based assemblers are Cufflinks67 and, more recently, StringTie69. In the first attempt to apply RNA-seq to lncRNA annotation, Cabili et al.134 assembled RNA sequences from a variety of human tissues to yield a total of 4,662 lncRNA loci. This study discovered another fundamental property of lncRNAs: high tissue-specificity and cell type-specificity.
Fig. 2 |. Annotation strategies for lncRNAs.
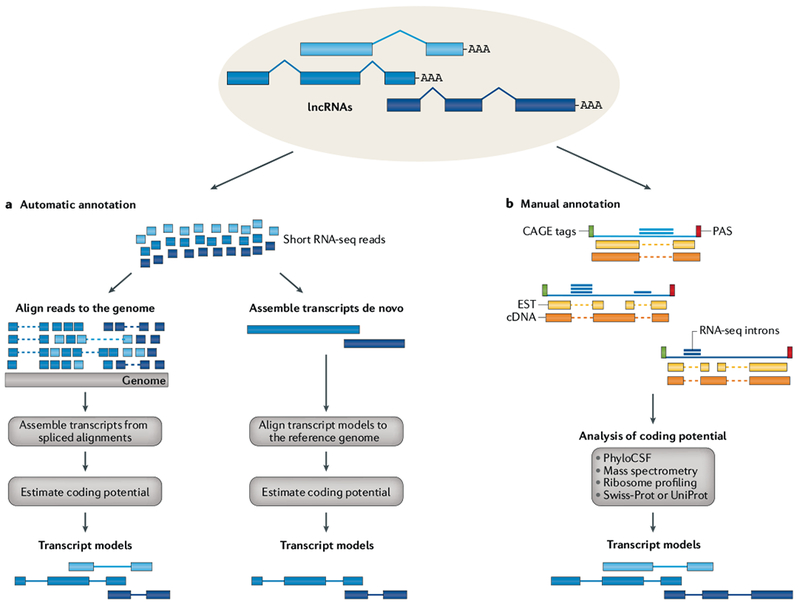
a | Automatic annotation based on RNA sequencing (RNA-seq) may follow two distinct strategies that differ in how the genome reference is used. The align-then-assemble strategy (left) aligns reads to the reference genome to reveal possible splicing events and then assembles reads into transcript models. The assemble-then-align strategy (right) builds transcript models de novo, directly from the RNA-seq reads, and then aligns them to the reference genome to determine their exon–intron structure. De novo transcriptome assembly has more explorative potential than alignment-based assembly but tends to have worse performance68. b | In manual annotation, human annotators employ various sources of data to build transcript models. Expressed sequence tags (ESTs) and cDNA form the primary evidence for transcript models and are often supplemented with RNA-seq reads to validate introns, cap analysis of gene expression (CAGE) clusters to identify 5′ ends45 and poly(A)-position profiling by sequencing (3P-seq) to identify polyadenylation sites (PASs)47. A key step in the annotation process is to assess the protein-coding potential of transcripts, usually on the basis of a combination of methods. lncRNA, long non-coding RNA.
Annotations based on transcriptome assembly using short reads.
Short-read RNA-seq experiments produce hundreds of millions of reads, providing a deep sampling of even large mammalian transcriptomes. These reads can be used to annotate transcripts from known and novel genes, both coding and non-coding. However, the fact that reads are much shorter than typical mRNAs and lncRNAs means that they must be bioinformatically assembled to infer the structure of the underlying transcript (FIG. 2a). Despite drawbacks inherent in this approach (discussed below), RNA-seq has facilitated the creation of large lncRNA catalogues.
The MiTranscriptome annotation combines 6,503 data sets, heavily weighted to 27 cancer types, to automatically annotate 58,648 lncRNA genes using a two-stage assembly strategy44. At the time of its creation, 54% of loci were not present in any other available resource.
Several studies are taking steps to improve the completeness of annotations. The Functional Annotation of the Mammalian genome (FANTOM) CAGE-associated transcriptome (CAT) meta-assembly combines both published sources and in-house short-read assemblies45. What sets this collection apart is its use of CAGE tags, which mark transcript TSSs, to identify 5′-complete transcript models. The resulting 27,919 gene loci are more complete at the 5′ end compared with other annotations, as judged by independent evidence, such as histone 3 lysine 4 trimethylation (H3K4me3) and DNase I hypersensitivity sites (DHSs)45. One drawback of CAGE is that, similar to other RNA-dependent methods, its signal scales with expression46; hence, lowly-expressed transcripts are more weakly represented.
The BIGTranscriptome catalogue comprises transcripts that are complete at both the 5′ and 3′ ends47. It employs a new method, CAFE, which is capable of inferring strands of unstranded RNA-seq reads. Consequently, CAFE overcomes strand ambiguity, which particularly affects genic transcript models generated from unstranded data sets, such as those from the Human Body Map (HBM) or the Genotype-Tissue Expression (GTEx) project48. CAGE and poly(A)-position profiling by sequencing (3P-seq) were used to assess 5′-end and 3′-end completeness, respectively45,49. Combining 169 RNA-seq data sets, BIGTranscriptome comprises 1,725 novel full-length lncRNA loci.
Annotations based on manual curation.
Gene annotation remains one of the few high-throughput scientific activities where humans still outperform computers. In manual annotation, a team of human annotators systematically assembles transcriptomic and genomic evidence into gene models according to defined protocols. By inspection of high-quality transcript evidence, principally from ESTs and cDNA databases, annotators can create fairly confident annotations, free from many of the artefacts inherent in automated approaches (FIG. 2b).
The most widely used manual annotation is GENCODE2,50, which stands out thanks to its extensive experimental validation and integration into the Ensembl annotation set2. Whereas the main GENCODE protein-coding gene annotation is created by merging the output from two pipelines, one manual and one automated, the lncRNA annotation is almost entirely manual. Individual transcript models are annotated and grouped together on the basis of genomic overlap of exons and splice sites into gene loci.
Unsurprisingly, manual annotation is much slower than automated approaches. Nevertheless, GENCODE annotations, released at 6-month intervals, have grown rapidly since 2012 (BOX 3; FIG. 3a). Moreover, single-exon models and transcript models supported by transcriptomic data from long-read sequencing are now being introduced (discussed below).
Box 3 |. Using GENCODE lncRNA annotations.
Availability
The GENCODE annotation of long non-coding RNAs (lncRNAs) is released in alignment with versioned Ensembl updates. Mouse releases are prefixed M; the most recent human release is GENCODE v27 and for mouse vM15. Full GENCODE annotations are available in GTF and GFF3 formats from the Ensembl and University of California Santa Cruz genome browsers and from gencodegenes.org. Separate files containing only lncRNA transcripts are also available. The GENCODE site also houses a full archive of previous releases and their statistics.
Biotypes
A full description of all GENCODE lncRNA biotypes is presented in the HAVANA annotation guidelines135. More recently, biotypes relating to other genomic features have been added and are being populated; for example, a ‘bidirectional-promoter lncRNA’ describes a locus where a lncRNA lies on the opposite strand to a protein-coding gene and there is evidence — for example, from cap analysis of gene expression (CAGE) data — that their transcription start sites lie within a window of 200 bp.
Biotype labels should be employed with caution as they tend to exhibit considerable inertia. As they are defined with reference to nearby protein-coding gene structures, any changes in those structures can lead to a change in the biotype. If users require up-to-date biotype information, it is recommended to regenerate them, for example, by using the lncrna.annotator script or the classifier module within FEELnc136.
GENCODE Comprehensive versus GENCODE Basic
GENCODE Comprehensive comprises the entire annotation of transcript models. As lncRNA annotations become increasingly complex, a need arises for a simplified annotation: GENCODE Basic. GENCODE Basic contains at least one transcript for every gene locus, ensuring full gene representation. For protein-coding loci, all coding transcripts with full-length coding DNA sequence (that is, ATG to stop codon) are included in the Basic set. For complex lncRNA loci, the Basic set is generated by including the minimal set of transcripts that capture >80% of the splice sites.
Fig. 3 |. Comparison of leading lncRNA annotations.
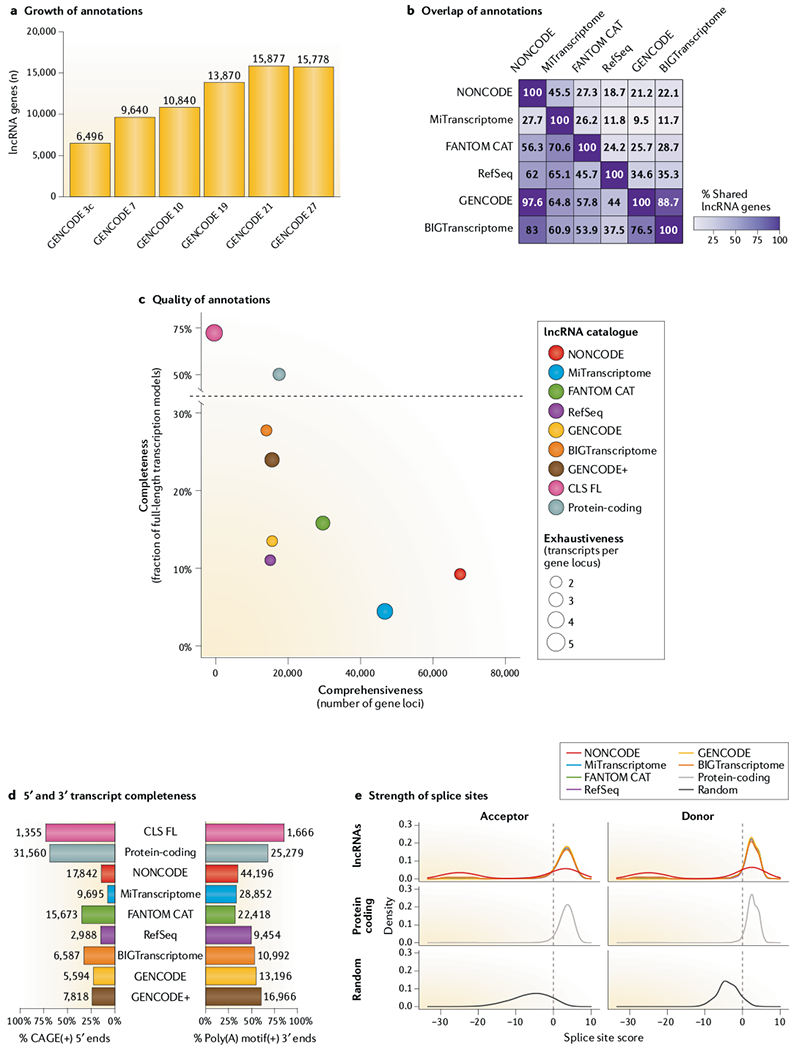
a | Growth of GENCODE long non-coding RNA (lncRNA) collection over time, in terms of gene loci. Only reference releases are included. b | Overlap between annotations at the gene level, based on a medium-stringency definition. Values represent the percentage of gene loci in the annotation of each row that overlap the annotation in each column. Overlap is defined as at least 60% of the span of the shorter gene on the same strand. Only genes with at least one multiexonic transcript were included. See TABLE 1 for details. c | Comparison of quality metrics between annotations. x-axis: comprehensiveness, or the total number of gene loci; y-axis: completeness, or percentage of transcript structures whose start is supported by a robust phase 1/2 Functional Annotation of the Mammalian genome (FANTOM) cap analysis of gene expression (CAGE) cluster (n = 201,802) within ±50 bases and whose end contains a canonical polyadenylation motif154 within a window of 10–50 bp upstream. Circle diameters reflect exhaustiveness, or mean number of transcripts per gene. GENCODE+ is the union of GENCODE version 20 with non-anchor-merged capture long-read sequencing (CLS) transcript models. Protein-coding is a set of confident GENCODE protein-coding transcripts as described in REF.27. d | As for part c, but separately for 5′ and 3′ completeness. e | The distribution of predicted splice junction strength for splice site acceptors and donors in each lncRNA catalogue, as calculated by the GeneID software155. The plots show non-redundant splice sites from lncRNA annotations sets (top), confident GENCODE protein-coding transcripts (middle), and 500,000 randomly selected GC|GT donors + AG acceptors with no evidence of splicing in any of the annotation sets under study (bottom). For each non-canonical splice site not scored by GeneID, a random score between −30 and −20 was assigned.
Newly created transcript models are assessed for protein-coding potential (BOX 4) and whether they are likely to be functional or pseudogenic. Where there is no evidence of coding potential from mass spectrometry data, orthologues or paralogues in reference databases such as UniProt51, structural or functional protein domains identified by Pfam52 or conservation data such as PhyloCSF53, a locus is defined as noncoding. lncRNAs from the literature are assessed with equal stringency. Although much of the annotation of lncRNAs was completed during first-pass manual annotation across the whole human genome, targeted (re)annotation of missing or truncated lncRNAs is now underway.
Box 4 |. Are lncRNAs really non-coding?
The extent to which protein-coding capacity is a qualitative (binary) or quantitative (gradual) property of RNAs has long been debated137. Recently, functional small peptides have been identified in transcripts previously annotated as long non-coding (lncRNAs)42,138. More broadly, ribosome profiling139,140 and bioinformatic141 studies have claimed that a large proportion of annotated lncRNAs encode proteins.
However, these findings are not yet conclusive. Ribosome interaction itself is suggestive, but not direct, evidence of coding potential142,143. For bioinformatic identification, a large fraction of purported, novel coding transcripts are likely to be false positives, arising from inadequate statistical approaches that do not correctly account for technical and biological noise144–146. For example, of the ∼350 best-supported novel open reading frames (ORFs) proposed by Mackowiak et al.141 and manually reviewed by GENCODE, only 35 could be verified (A.F., unpublished observation). Together with the presently low number of cases for which peptides have directly been observed, this observation means that it may be premature to suppose that most lncRNAs are translated into functional peptides.
This is not to say that annotations should not be rigorously screened to flag “transcripts of unknown coding potential” (REF.134). A variety of tools exist to predict protein-coding regions in RNA sequences, which may be classified amongst those using intrinsic sequence properties (for example, Coding-Potential Assessment Tool (CPAT)147), similarity to known proteins (for example, Coding Potential Calculator (CPC)148) and evolutionary signatures of protein evolution (for example, PhyloCSF53). The latter tool is considered to have the greatest sensitivity, particularly for short peptides136,149 but identifies only evolutionarily conserved peptides and is computationally intensive. More direct evidence comes from mass spectrometry, although low sensitivity and the short length of potential peptides complicates their identification94,150–152, and care must be taken to correctly estimate false-positive predictions153.
Most annotation pipelines integrate one or several of these approaches136. For GENCODE, in addition to comparing putative ORFs within lncRNAs to entries in reference protein databases, such as UniProt and Pfam, all lncRNAs are routinely tested using both PhyloCSF and dedicated proteogenomics filtering. Manual re-examination can lead to reclassification of dubious lncRNAs. However, this is fairly infrequent: a stringent proteogenomics workflow to reprocess >52 million spectra revealed more than 1,400 putative novel protein-coding genes, but only 16 were confirmed following detailed reanalysis and just 8 fell in annotated lncRNA loci94.
All new transcripts and genes are assigned stable identifiers on their creation. All updates to annotation are captured in an increment to the version of the gene and transcript identifier (that is, “ENSGXXXXXXX.2”). For example, when extension or trimming of a transcript is undertaken in light of new data or where new data emerges to strongly support changing the biotype of a locus (BOX 3), updates will be made and a version increment applied.
Owing to the quality deriving from its manual annotation, regularly updated versions, long-term support, well-defined and consistent source data, identifier stability and integration into Ensembl50, GENCODE has been adopted by most large-scale genomics projects, including the Encyclopedia of DNA Elements (ENCODE)54 (for which it was originally created), GTEx project48, International Cancer Genome Consortium (ICGC)55, Blueprint56, Epigenome Roadmap57 and FANTOM45. The use of stable Ensembl identifiers simplifies the integration of data across projects and releases. However, the inherent weakness of GENCODE is its relatively small size: 15,778 genes in human (version 27) and 11,975 in mouse (version M15). Of note, the mouse annotation project was commenced later, accounting for the difference in size with human.
Another manual gene annotation resource, Reference Sequence (RefSeq), was created and is maintained by the National Center for Biotechnology Information (NCBI) and covers multiple species, including human58. Consisting of a mixture of manual and automated annotations, RefSeq is created using a variety of evidence, including cDNAs, ESTs and RNA-seq. Entries carry unique and stable identifiers and are associated with metadata summarizing their annotation history. Of relevance in this context are non-coding RNA annotations with accessions ‘NR_’ and ‘XR_’, which refer to manually curated models (NR) and products of an automated pipeline based on Illumina data (XR), respectively. Thus, the RefSeq annotation process is similar to GENCODE, with the exception of usage of RNA-seq. Along with GENCODE, RefSeq is one of the most widely used lncRNA annotations59.
Integrative annotations.
A number of other lncRNA collections are worthy of note. NONCODE has, since 2005, integrated annotations from a mixture of manual literature searches and other annotations3. The latest version, NONCODE (version 5), is to our knowledge the single largest present collection, describing 96,308 lncRNA gene loci in human alone (as of November 2017). It also has data for 15 species other than human and mouse.
RNACentral is a large-scale resource of non-coding RNA sequences, integrating various other databases, which lists 116,292 lncRNA sequences at the time of writing60. It is based on sequences, rather than annotations, making the total number of lncRNA loci unclear.
Finally, LNCipedia and LncRNAWiki stand out in their usefulness for integrating functional data. LNCipedia holds a database of 48,028 carefully filtered lncRNA genes from a range of sources61. Users may access information on peptide mapping, coding potential, RNA folding and microRNA recognition. Similarly, LncRNAWiki holds a variety of useful information, including disease association and putative small peptides, and is an invaluable resource of manually curated functional information for hundreds of lncRNAs62.
How good are lncRNA annotations?
Overlap between annotations.
Annotations tend to have low overlap (FIG. 3b). For example, the two largest annotations, MiTranscriptome and NONCODE, have just 27.7% and 45.5% of genes in common, respectively. Not surprisingly, NONCODE encompasses more than 97% of GENCODE, which it incorporates. What is perhaps unexpected is the poor overlap that is observed between the two manual annotations, GENCODE and RefSeq (34.6% and 44%, respectively). Overall, the low overlap points to much scope for merging of annotations to improve comprehensiveness.
Quality metrics for annotations.
An ideal annotation would be a record of every locus expressed at any point in time from the genome of a given species. An important requirement for future lncRNA mapping projects is the development of standards for assessing quality that go beyond anecdotal examples. For the present discussion, we make the following definitions of annotation quality: (a) comprehensiveness — the fraction of all gene loci that are included; (b) exhaustiveness — the fraction of all transcripts from each locus that are known; (c) completeness — the fraction of transcript models that cover the entire length, from start to end, of the physical RNA molecule. Obviously, comprehensiveness and exhaustiveness are impossible to define, as we do not know the total number of lncRNA genes or transcripts. Nevertheless, we can at least compare proxies for these metrics between annotations (TABLE 1) to get a comparative picture. By contrast, a minimum bound can be placed on completeness, owing to the availability of independent evidence for transcript 5′ and 3′ boundaries.
Based on these three metrics, we have compared the discussed lncRNA annotations (FIG. 3c). Most striking is the general anti-correlation between comprehensiveness and completeness. In other words, there is a trade-off between quality and size: smaller annotations tend to have higher completeness (although this remains low in absolute terms) and vice versa. Amongst the smaller annotations, BIGTranscriptome is the leader in terms of completeness, although with low numbers of annotated transcripts per gene. The two manual annotations, GENCODE and RefSeq, have comparable profiles. For the larger annotations, MiTranscriptome has just 4.4% of complete (full-length) transcript models (FIG. 3c), which is most likely the result of its dependence on transcriptome assembly. NONCODE beats MiTranscriptome in size and completeness but with lower exhaustiveness. FANTOM CAT represents a compromise between completeness and comprehensiveness. Of note, we find substantially lower 5′ completeness than originally reported45, which is due to the use of more stringent CAGE cut-off thresholds: only robust CAGE clusters (FANTOM5 phase 1/2 robust (n = 201,802)) were considered, and FANTOM5 phase 2 unfiltered CAGE clusters (n = 4,218,430) were discarded owing to their seemingly high background rate.
Data are also displayed for protein-coding genes as a reference, with the assumption that their annotation is of the highest quality. The protein-coding gene annotation should be comprehensive, as not many are expected to remain undiscovered63. We also included a recently generated set of full-length lncRNA transcript models produced using capture long-read sequencing (CLS) technology (discussed below)27. These models display high completeness, in part because their 5′ ends were defined using the same CAGE data as used here for evaluation. Incorporation of CLS models into GENCODE resulted in an improved annotation, GENCODE+ (TABLE 1), with dramatically higher completeness. It is noteworthy that GENCODE+ has a slightly reduced gene count as a result of unifying artefactually separate gene models in existing annotations.
One important caveat of this analysis is that CAGE clusters used for 5′-end definition are expression-dependent and only available for a defined set of tissues. This likely accounts, at least in part, for the fact that protein-coding genes have apparent 5′ completeness <100% (FIG. 3d) and will also underestimate completeness of lower expressed lncRNAs. However, it is also possible that some protein-coding gene annotations remain incomplete.
The use of proxies for comprehensiveness (numbers of loci) and exhaustiveness (transcripts per gene) makes the key assumption that no false-positive annotations exist. This assumption is probably incorrect and will affect some annotations more than others. In particular, assembly-based collections may hold substantial numbers of false-positive transcripts. Inspection of splice junctions supports the idea that certain annotations, particularly NONCODE, suffer from high rates of false-positive structures (FIG. 3e).
Overall, this analysis illustrates the strengths and weaknesses of contemporary annotations. It highlights the great scope for improving lncRNA annotations, first, by increasing their completeness to levels observed for protein-coding genes and, second, by improving their comprehensiveness by merging diverse available resources.
Sources of incompleteness.
The lack of completeness in existing lncRNA annotations may be traced to several historical and technical factors. cDNA molecules often tend to be 5′ truncated, owing to a combination of RNA degradation and the tendency of reverse transcriptase molecules to disengage before reaching the 5′ end of the template RNA, often as a result of RNA secondary structures64. In short-read RNA-seq, a range of processes create non-uniformity in read coverage, particularly at the 5′ and 3′ ends65. Together, these factors introduce a tendency for short-read assemblies and cDNA libraries, upon which most annotations are based, to be 5′ and 3′ incomplete34,35,45,66.
More generally, the assembly of transcriptomes from short reads is inherently challenging. Assembly programs, including the widely used Cufflinks67, have high error rates. Whereas exons are identified with reasonable sensitivity, their assembly into correct transcripts is particularly difficult68. Simulations across a range of assembly programs demonstrate a mean sensitivity of only 41% in assembling expressed genes, dropping to 21% at the transcript level68. The majority of such transcript models lacks one or more exons. Assemblies are sensitive to gene expression levels and coverage uniformity68, which has a particular impact on lowly-expressed lncRNAs. However, even when controlling for expression, transcriptome assemblies are less sensitive for lncRNAs compared with mRNAs for unknown reasons68. More recent assemblers such as StringTie and Scallop run far faster than Cufflinks and have demonstrably better sensitivity and specificity, but resultant assemblies remain far from ideal69,70. In a study using StringTie to assemble synthetic spliced RNAs, it was found that for the correct assembly of >50% of its nucleotides, a transcript must be expressed at a level equivalent to 23 FPKMs — far in excess of the average lncRNA or even mRNA66. These issues will result in low comprehensiveness, exhaustiveness and completeness of annotations based on transcriptome assemblies.
Another issue that probably has an impact on comprehensiveness is of historical nature: the material used for the generation of cDNA libraries has been biased towards adult tissues, tumour samples and cell lines2,44. Thus, modern annotations may omit much of the wealth of lncRNAs likely to be expressed during embryogenesis, development and childhood48. Similarly, certain lncRNAs may only be expressed in rare subpopulations of cells within a tissue or even cell culture71 and thus are likely to be missed owing to the low apparent expression in bulk cell samples.
In summary, present annotations are likely to fall short in all the quality metrics described, leaving thousands of gene loci, transcripts and exonic nucleotides unmapped.
Emerging technologies in lncRNA annotation
Advances in key technologies for targeting and sequencing lncRNA transcripts promise to directly address the two principal challenges facing lncRNA annotations of low target abundance and incomplete transcript models.
Long-read sequencing technologies.
Pacific Biosciences’ (PacBio) technology employs zero-mode waveguides to sequence single circularized DNA or cDNA molecules. Around 40,000 reads are produced by each lane, with an average ∼1.5 kb length27,72. This length is several-fold longer than the average exon, meaning that the exon connectivity of complete or almost complete transcript structures can be resolved. A recent study in human showed that, for transcripts up to ∼1.5–2.0 kb, the majority of reads yields full-length transcript structures, falling short on average 47 nt and 6 nt from the annotated 5′ and 3′ sites, respectively72.
Raw PacBio sequencing reads tend to have relatively high error rates72. To mitigate this issue, consensus reads of insert (ROIs) are assembled from multiple passes of the same circular template molecule. Resulting per-base sequencing errors are moderate, approximately twofold greater than for Illumina and with a tendency for nucleotide deletions72. At this rate, the majority of reads can be mapped with high confidence73. Despite its advantages, widespread adoption of PacBio is hindered by its cost and low throughput. Given the low representation of lncRNAs within the cellular transcriptome, pure PacBio sequencing would be an inefficient method to map lncRNA loci72. Perhaps its greatest drawback is its sequencing preference for short templates in a mixture. This limitation creates the need to size-select cDNA libraries, introducing a length-dependent bias in the sequenced transcripts27,72.
Nanopore-based technologies read single molecules in real time74; nucleic acid molecules are translocated at a controlled rate through a membrane-bound protein nanopore. Changes to electric currents through nanopores are used to infer the identity of each nucleotide. This technology has reached the mainstream market with Oxford Nanopore’s MinION technology74, which is capable of returning ∼5 million reads per flow cell, at a cost of ∼€500.
Nanopore has a range of advantages over other sequencing approaches. By dispensing with the amplification or enzymatic modification of target molecules, important sources of bias are avoided. cDNA molecules can be directly sequenced with minimal preparation75, and it may even be feasible to identify chemically modified bases76. A recent report describes direct sequencing of RNA (as opposed to cDNA) from a variety of samples, with read lengths of up to 7.5 kb and sequencing accuracy of 80%77. Reads are free from biases regarding template length or GC content, which affect other technologies78. Most importantly in the present context, nanopore sequencing yields reads of lengths that are virtually unlimited and that far exceed known lncRNAs and mRNAs78. These beneficial properties of throughput, read length and cost make nanopore technology highly appealing in the context of gene annotation.
RNA capture sequencing.
lncRNAs tend to be expressed approximately one order of magnitude lower than mRNA and represent about 1–2% of polyA+ RNA in a cell27,72,79,80. This creates a considerable hurdle for annotation, because lncRNA molecules are simply less likely to be sampled at a given depth of sequencing. One solution is to boost their apparent concentration in a cDNA library using oligonucleotide capture in a technique known as RNA CaptureSeq81,82. Custom libraries of tiled complementary oligonucleotide probes are used to enrich a population of desired targets in solution. This approach boosts the representation of lncRNA sequences to >25%, improving sequencing coverage by tens of fold compared with a conventional, uncaptured sample81. To date, this approach has been used successfully in human83 and mouse84 tissues.
RNA CaptureSeq demonstrates powerfully increased sensitivity compared with conventional, unbiased sequencing methods, typically discovering novel transcripts and gene loci expressed at far less than one copy per cell83. Often, adjacent and erroneously separate annotations are merged, or annotated loci are extended with new exonic sequence84. However, previous studies have largely relied on short-read Illumina sequencing coupled to Cufflinks transcriptome assembly81,83,84. Consequently, resulting annotations suffer from the same uncertainties and weaknesses as discussed above and have low 5′ and 3′ coverage66,68.
The dependence of RNA CaptureSeq on short reads has recently been overcome by coupling it to PacBio technology in a method termed CLS27,85. By using long reads, CLS avoids the issues associated with short reads and transcript assembly, enabling the identification of full-length transcript models. By integrating CAGE data and fragments of poly(A) tails contained in PacBio reads, CLS can assess the completeness of transcript models at 5′ and 3′ ends, respectively (FIG. 4). The use of template-switching reverse transcriptase technology to generate almost full-length cDNAs can boost 5′ completeness further64. Short reads sequenced from the same samples can be used to assess the accuracy of splice site predictions86. As such, CLS marries the enhanced sequencing coverage provided by capture to transcript model confidence afforded by long reads. In the first report of this method, 2 million reads each in human and mouse across a panel of tissues and cells yielded novel full-length transcript models from 947 previously annotated human lncRNA loci27. Although the annotation complexity of the probed regions was approximately doubled, there was no sign of saturation of splice junctions, indicating that much more sequencing depth will be required to establish definitive gene structures in detected loci. A similar conclusion was reached in a study using essentially the same strategy to survey the transcriptional landscape of chromosome 21 in human testis85.
Fig. 4 |. Integrating capture and long-read sequencing with annotation pipelines.
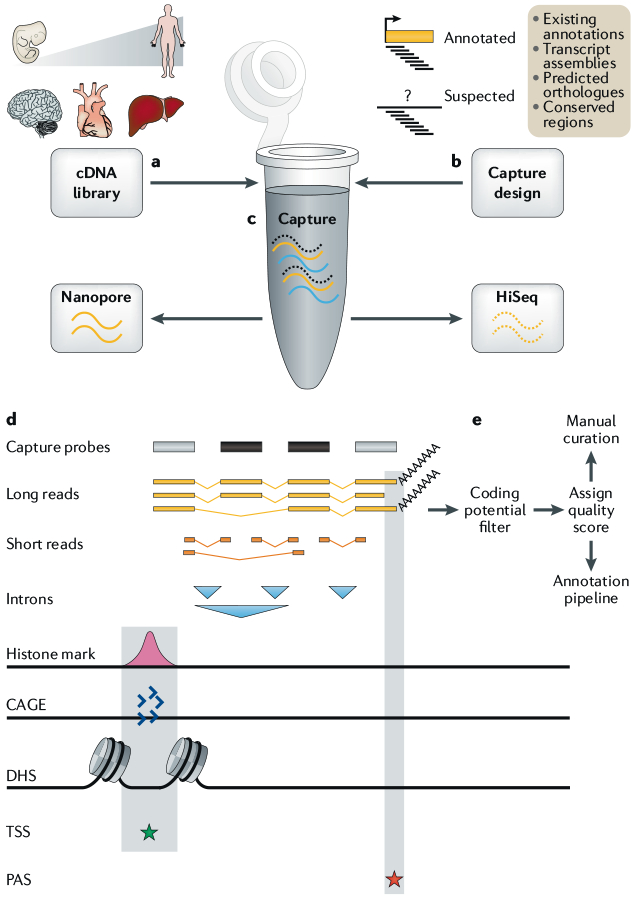
a | Full-length cDNA libraries are prepared from a variety of tissues across the human lifespan. b | Target annotations are prepared from a variety of known and suspected long non-coding RNA (lncRNA) loci and used to design capture probes (black bars). c | Solution-phase oligonucleotide capture is performed, and enriched cDNA libraries are sequenced by long-read nanopore and short-read Illumina technologies. d | The resulting long reads are collapsed to produce non-redundant transcript models. The completeness and accuracy of these models are assessed using various evidence: introns (blue triangles) by short reads; transcription start site (TSS; green star) by promoter histone modifications, cap analysis of gene expression (CAGE) clusters and DNase I hypersensitivity sites (DHSs); and polyadenylation site (PAS; red star) by long-read-encoded poly(A) tails. e | With this information, transcript models are graded for completeness, checked for protein-coding potential and passed to annotators for either direct incorporation into annotation pipelines (for complete models) or further manual curation (incomplete models).
Although the speed and cost of the CLS approach make it a substantial step towards more comprehensive lncRNA annotations, it suffers from some weaknesses that must first be resolved. The lengths of full-length transcript models are limited by PacBio reads, leaving many targeted transcripts incomplete. Also, sequencing depths are insufficient to saturate targeted loci. The incorporation of nanopore sequencing technology in the CLS workflow should help to overcome these barriers.
Towards complete lncRNA annotations
With the tools of long-read sequencing and RNA capture in hand, we may now envisage an eventual complete lncRNA annotation: maps of the entire universe of lncRNAs expressed throughout the lifetime of an organism, beginning with Homo sapiens.
A roadmap.
The most obvious route to complete annotation lies in the systematic application of CLS coupled to nanopore sequencing (FIG. 4). Capture library designs would have two main components. First, in order to complete existing annotations, the entire catalogue of known lncRNAs would be targeted27. Second, in order to map unknown lncRNAs, suspected loci lying outside of annotated exons would be probed. These would come from two main sources: first, loci with a high confidence for lncRNA production, such as physical evidence from RNA-seq-derived assemblies and introns44,87, and second, regions with more speculative evidence, such as predicted lncRNA orthologues from other species24, bioinformatic predictions88, GWAS regions89 or small RNAs with presumed long precursors90.
Capture libraries would be used to probe diverse organ and tissue panels across the human lifespan from embryos to aged adults, thus going beyond the adult organ panels and tumours that tend to dominate present data sets91,92. Given that organs are complex mixtures of common and rare cell types, it will also be beneficial to probe purified cell populations93. Technology permitting, this may eventually be extended to sampling single-cell transcriptomes of rare types that would be missed in bulk preparations71. Finally, the majority of transcriptome studies to date have been performed on individuals of European ancestry, making future sampling across different human populations a priority.
Such an ambitious project would entail considerable logistical and economic challenges. As recognized by ENCODE54, a practical first step would be to focus on complete collections of lncRNA in defined cell types or organs. These might entail complex organs or cell lines of particular scientific or biomedical relevance, such as ENCODE cell lines80.
Captured cDNA libraries would be sequenced using nanopore technology, up to a rationally chosen depth, defined below. The accuracy of the 5′ end, the 3′ end and splice junctions would be validated using independent data sets, in addition to bioinformatic and experimental screening for protein-coding capacity53,94. With this level of quality, resulting transcript models can be added to existing annotations with low levels of scrutiny by human annotators, minimizing the delay between sequencing and public availability.
How do we know when to stop?.
A number of considerations must guide decisions regarding resource allocation in annotation projects. First, we must take care to focus efforts on collecting lncRNAs of biological relevance. Unfortunately, we remain far from having reliable methods for distinguishing functional lncRNAs from transcriptional noise. Although imposing a minimum expression threshold is an obvious path, the discovery of apparently functional lncRNAs with expression of <<1 copy per cell20 would argue against imposing a hard expression cut-off. Nevertheless, to maximize usefulness in downstream applications such as RNA-seq, it is sometimes helpful to eliminate unnecessary complexity arising from growing numbers of transcript isoforms. This has prompted the creation of simplified annotations such as GENCODE’s Basic annotation (BOX 3).
A question of singular importance to the design of annotation projects is: is the lncRNA population finite, and if so, how many transcripts and loci does it comprise? Or conversely, is an effort at complete annotation doomed by the fact that the transcriptome is infinite, owing to pervasive transcription or unlimited combinatorial splicing85? Certainly, after a decade of research, we are little closer to assigning an upper bound to the first question. Recent CLS studies finished sequencing before saturating even already known lncRNA loci27, while a recent study claims that lncRNA genes explore astronomical numbers of available splicing combinations85. Furthermore, present upper estimates of lncRNA numbers are biased towards adult cell types, raising the possibility of existence of untold numbers of developmentally regulated lncRNAs.
A further source of complexity could be ‘personal’ transcriptomes — lncRNAs that are unique to individuals or populations95,96. Such transcripts might arise from individual-specific genomic regions that are not represented in the reference or else shared genomic regions that are active in certain individuals thanks to processes such as transposon insertion97,98 or trans-acting factors99. Even if the size of every individual personal transcriptome is small, summed across the entire population it could be enormous. Efforts to map personal genomes and transcriptomes are underway with the ENCODE Tissue Expression (EnTEx) project amongst others100. Personal lncRNAs, if they exist, may explain individual-specific phenotypes and features and could be of crucial importance to personalized medicine.
However, there is evidence supporting the finiteness of the lncRNA transcriptome. Simulations performed on relatively shallow CLS sequences from an admittedly limited range of tissues exhibited a decreasing rate of discovery with depth27, indicating that lncRNA transcript complexity tends towards an asymptote. Deveson et al. seem to have exhaustively mapped all exons on chromosome 21 in testis85. Similarly, in analyses of nearly the entire volume of public RNA-seq data, the number of splice sites almost reached a plateau87. Finally, a more focused study in B cells also found evidence for an upper threshold in lncRNA isoform diversity101. Therefore, although lncRNA transcripts are highly complex and challenging to exhaustively map, a full map of at least their exons and splice sites is tractable.
Nevertheless, in any large-scale annotation project involving third-generation sequencing at depth, it will be imperative to periodically monitor the rate of novel transcript discovery in each tissue sample as a function of sequencing depth. This will indicate when transcriptome complexity has been saturated and hence when sequencing resources should be reallocated to other samples.
Conclusions and perspective
lncRNA annotations are a fundamental resource for basic research and also have growing importance for practical applications such as personalized medicine102. Although it has been argued, quite reasonably, that many lncRNAs may represent non-functional noise, the growing number of clearly documented counter-examples suggests that at least a substantial fraction of transcripts is functional in the strictest sense of enhancing organismal fitness.
The rapidly growing volume of the annotated lncRNA transcriptome will bring benefits but also new challenges, particularly in making this information available in a way that maximizes usefulness without sacrificing genuine biological complexity.
At present, lncRNA annotations lag far behind those for protein-coding genes, to an extent not often appreciated by individual researchers. However, there is now an opportunity to create complete annotations, at least in certain well-defined cell types. This will not only open new vistas into the molecular biology of the cell, disease mechanisms and diagnostics, but also enable us to answer fundamental questions about the functionality of lncRNAs.
Acknowledgements
R.J. acknowledges the support of the Swiss National Science Foundation through the National Centres for Competence in Research (NCCR) ‘RNA & Disease’ and the Medical Faculty of the University Hospital and University of Bern. The authors thank J. Carlevaro-Fita (University of Bern) for help with data analysis and J. Harrow (Illumina), J. Mudge (European Bioinformatics Institute), P. Flicek (European Bioinformatics Institute) and I. Jungreis (Massachusetts Institute of Technology) for fruitful discussions and feedback. A.F. is supported by the Wellcome Trust (WT098051 and WT108749/Z/15/Z), the National Human Genome Research Institute (NHGRI) (U41HG007234, 2U41HG007234) and the European Molecular Biology Laboratory. Work described in this publication was supported by the National Human Genome Research Institute of the US National Institutes of Health (grants U41HG007234, U41HG007000 and U54HG007004) and the Wellcome Trust (grant WT098051 to R.G.). Work in the laboratory of R.G. was supported by the National Human Genome Research Institute (awards U54HG007000, R01MH101814 and U41HG007234), the Spanish Ministry of Economy and Competitiveness, ‘Centro de Excelencia Severo Ochoa 2013-2017’ and CERCA Programme/Generalitat de Catalunya. The authors thank the following individuals for administrative support: R. Garrido (Centre for Genomic Regulation) and S. Roesselet and D. Re (both at the University of Bern).
Glossary
- Long non-coding RNAs (lncRNAs)
RNA transcripts ≥200 nucleotides long that do not encode any identifiable peptide product.
- Annotation
Catalogue of gene loci comprising detailed and hierarchical information on their genomic coordinates and that of their constituent transcript isoforms and exons, all of which are assigned unique and stable identifiers.
- Transcriptome assembly
The use of bioinformatic algorithms to reconstruct gene and transcript models based on short sequence reads.
- Manual annotation
The creation of gene and transcript models by human annotators based on RNA and protein evidence and according to defined protocols.
- Biotype
An annotation label referring to the genomic classification, processing or other characteristics of a locus or transcript intended to provide insights into biological function.
- Expressed sequence tags (ESTs)
An early transcriptomic method in which short fragments of transcribed regions, often from 5′ or 3′ ends, are identified through sequencing of cDNA.
- Cap analysis of gene expression (CAGE)
A cap-trapping and sequencing method that is considered a gold standard for mapping RNA 5′ ends.
- Transcript models
Abstract descriptions of a transcription event, defining the genomic location of the start point, the end point and splice junctions.
- Fragments per kilobase per million mapped (FPKM)
One of the principal units of RNA abundance in the context of RNA sequencing experiments, defined as the number of sequenced fragments per kilobase of annotation per million mapped fragments.
- Oligonucleotide capture
A method for enriching cDNA libraries with sequences of interest using solution-phase hybridization to tiled, labelled oligonucleotide probes.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Liu G, Mattick J & Taft RJ A meta-analysis of the genomic and transcriptomic composition of complex life. Cell Cycle 12, 2061–2072 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Derrien T et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 22, 1775–1789 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fang S et al. NONCODEV5: a comprehensive annotation database for long non-coding RNAs. Nucleic Acids Res. 46, D308–D314 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study presents the latest instalment of the long-running NONCODE annotation, which was amongst the first ncRNA annotations and currently represents the most extensive collection.
- 4.Ponjavic J, Ponting CP & Lunter G Functionality or transcriptional noise? Evidence selection within long noncoding RNAs. Genome Res. 17, 556–565 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study initially demonstrated that lncRNA exons and promoters are under purifying evolutionary selection and hence provided strong evidence that, as a gene class, they are functional.
- 5.Pegueroles C & Gabaldón T Secondary structure impacts patterns of selection in human lncRNAs. BMC Biol. 14, 60 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhu S et al. Genome-scale deletion screening of human long non-coding RNAs using a paired-guide RNA CRISPR–Cas9 library. Nat. Biotechnol. 34, 1279–1286 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wen K et al. Critical roles of long noncoding RNAs in Drosophila spermatogenesis. Genome Res. 26, 1233–1244 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li L & Chang HY Physiological roles of long noncoding RNAs: insight from knockout mice. Trends Cell Biol. 24, 594–602 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sauvageau M et al. Multiple knockout mouse models reveal lincRNAs are required for life and brain development. eLife 2, e01749 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ip JY et al. Gomafu lncRNA knockout mice exhibit mild hyperactivity with enhanced responsiveness to the psychostimulant methamphetamine. Sci. Rep. 6, 27204 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen G et al. LncRNADisease: a database for long-non-coding RNA-associated diseases. Nucleic Acids Res. 41, D983–D986 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amândio AR, Necsulea A, Joye E, Mascrez B & Duboule D Hotair is dispensible for mouse development. PLoS Genet. 12, e1006232 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Quek XC et al. lncRNAdb v2.0: expanding the reference database for functional long noncoding RNAs. Nucleic Acids Res. 43, D168–D173 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; For many years, this publication was the reference resource for manually curated, experimentally validated functional lncRNAs.
- 14.Sheik Mohamed J, Gaughwin PM, Lim B, Robson P & Lipovich L Conserved long noncoding RNAs transcriptionally regulated by Oct4 and Nanog modulate pluripotency in mouse embryonic stem cells. RNA 16, 324–337 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Loewer S et al. Large intergenic non-coding RNA-RoR modulates reprogramming of human induced pluripotent stem cells. Nat. Genet. 42, 1113–1117 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huarte M et al. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell 142, 409–419 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ng S-Y, Johnson R & Stanton LW Human long non-coding RNAs promote pluripotency and neuronal differentiation by association with chromatin modifiers and transcription factors. EMBO J. 31, 522–533 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ounzain S et al. CARMEN, a human super enhancer-associated long noncoding RNA controlling cardiac specification, differentiation and homeostasis. J. Mol. Cell. Cardiol. 89, 98–112 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Liu SJ et al. CRISPRi-based genome-scale identification of functional long noncoding RNA loci in human cells. Science 355, aah7111 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper provides a map of hundreds of proliferation-altering lncRNAs across seven human cell lines, representing an invaluable resource of functional genes.
- 20.Seiler J et al. The lncRNA VELUCT strongly regulates viability of lung cancer cells despite its extremely low abundance. Nucleic Acids Res. 45, 5458–5469 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study presents an intriguing example of an extremely lowly expressed lncRNA that yields a reproducible cellular phenotype after knockdown, thereby challenging the notion that expression cut-off thresholds can be used to discriminate functional lncRNAs.
- 21.Yang L, Duff MO, Graveley BR, Carmichael GG & Chen L-L Genomewide characterization of non-polyadenylated RNAs. Genome Biol. 12, R16 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carrieri C et al. Long non-coding antisense RNA controls Uchl1 translation through an embedded SINEB2 repeat. Nature 491, 454–457 (2012). [DOI] [PubMed] [Google Scholar]
- 23.Guttman M et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 458, 223–227 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hezroni H et al. Principles of long noncoding RNA evolution derived from direct comparison of transcriptomes in 17 species. Cell Rep. 11, 1110–1122 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haerty W & Ponting CP Unexpected selection to retain high GC content and splicing enhancers within exons of multiexonic lncRNA loci. RNA 21, 320–332 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mason MK et al. Retinoic acid-independent expression of Meis2 during autopod patterning in the developing bat and mouse limb. Evodevo 6, 6 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lagarde J et al. High-throughput annotation of full-length long noncoding RNAs with capture long-read sequencing. Nat. Genet. 49, 1731–1740 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study describes the method of CLS for mapping full-length transcript models in human and mouse samples.
- 28.Gong C & Maquat LE lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3′ UTRs via Alu elements. Nature 470, 284–288 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li B & Dewey CN RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bray NL, Pimentel H, Melsted P & Pachter L Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Kanitz A et al. Comparative assessment of methods for the computational inference of transcript isoform abundance from RNA-seq data. Genome Biol. 16, 150 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Conesa A et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 17, 13 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marques AC et al. Chromatin signatures at transcriptional start sites separate two equally populated yet distinct classes of intergenic long noncoding RNAs. Genome Biol. 14, R131 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alam T et al. Promoter analysis reveals globally differential regulation of human long non-coding RNA and protein-coding genes. PLoS ONE 9, e109443 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Melé M et al. Chromatin environment, transcriptional regulation, and splicing distinguish lincRNAs and mRNAs. Genome Res. 27, 27–37 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lanzós A et al. Discovery of cancer driver long noncoding RNAs across 1112 tumour genomes: new candidates and distinguishing features. Sci. Rep. 7, 41544 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Juul M et al. Non-coding cancer driver candidates identified with a sample-and position-specific model of the somatic mutation rate. eLife 6, e21778 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tan JY et al. cis -acting complex-trait-associated lincRNA expression correlates with modulation of chromosomal architecture. Cell Rep. 18, 2280–2288 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Gong J et al. A functional polymorphism in lnc-LAMC2–1:1 confers risk of colorectal cancer by affecting miRNA binding. Carcinogenesis 37, 443–451 (2016). [DOI] [PubMed] [Google Scholar]
- 40.de Kok JB et al. DD3(PCA3), a very sensitive and specific marker to detect prostate tumors. Cancer Res. 62, 2695–2698 (2002). [PubMed] [Google Scholar]
- 41.Tilgner H et al. Deep sequencing of subcellular RNA fractions shows splicing to be predominantly co-transcriptional in the human genome but inefficient for lncRNAs. Genome Res. 22, 1616–1625 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Anderson DM et al. A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell 160, 595–606 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou KI et al. N6-methyladenosine modification in a long noncoding RNA hairpin predisposes its conformation to protein binding. J. Mol. Biol. 428, 822–833 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Iyer MK et al. The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet. 47, 199–208 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This publication describes MiTranscriptome, the largest annotation to date based on transcriptome assembly using thousands of tumour RNA-seq samples.
- 45.Hon C-C et al. An atlas of human long non-coding RNAs with accurate 5′ ends. Nature 543, 199–204 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carninci P et al. High-efficiency full-length cDNA cloning by biotinylated CAP trapper. Genomics 37, 327–336 (1996). [DOI] [PubMed] [Google Scholar]
- 47.You B-H, Yoon S-H & Nam J-W High-confidence coding and noncoding transcriptome maps. Genome Res. 27, 1050–1062 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study first attempted the automated annotation of full-length transcripts using CAGE and 3 P-seq data.
- 48.Mele M et al. The human transcriptome across tissues and individuals. Science 348, 660–665 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jan CH, Friedman RC, Ruby JG & Bartel DP Formation, regulation and evolution of Caenorhabditis elegans 3′UTRs. Nature 469, 97–101 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harrow J et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 22, 1760–1774 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; This report represents the reference publication for the GENCODE annotation of protein-coding and non-coding genes.
- 51.Apweiler R et al. UniProt: the Universal Protein knowledgebase. Nucleic Acids Res. 32, 115D–119 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sonnhammer E, Eddy SR, Birney E, Bateman A & Durbin R Pfam: multiple sequence alignments and HMM-profiles of protein domains. Nucleic Acids Res. 26, 320–322 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lin MF, Jungreis I & Kellis M PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics 27, i275–i282 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dunham I et al. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hudson TJ et al. (Chairperson) International network of cancer genome projects. Nature 464, 993–998 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Adams D et al. BLUEPRINT to decode the epigenetic signature written in blood. Nat. Biotechnol. 30, 224–226 (2012). [DOI] [PubMed] [Google Scholar]
- 57.Kundaje A et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pruitt KD et al. RefSeq: an update on mammalian reference sequences. Nucleic Acids Res. 42, D756–D763 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.O’Leary NA et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–D745 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.The RNAcentral Consortium. RNAcentral: a comprehensive database of non-coding RNA sequences. Nucleic Acids Res. 45, D128–D134 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Volders P-J et al. An update on LNCipedia: a database for annotated human lncRNA sequences. Nucleic Acids Res. 43, D174–D180 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ma L et al. LncRNAWiki: harnessing community knowledge in collaborative curation of human long non-coding RNAs. Nucleic Acids Res. 43, D187–D192 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ezkurdia I et al. Multiple evidence strands suggest that there may be as few as 19,000 human protein-coding genes. Hum. Mol. Genet. 23, 5866–5878 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhu YY, Machleder EM, Chenchik A, Li R & Siebert PD Reverse transcriptase template switching: a SMART approach for full-length cDNA library construction. Biotechniques 30, 892–897 (2001). [DOI] [PubMed] [Google Scholar]
- 65.Hansen KD, Brenner SE & Dudoit S Biases in Illumina transcriptome sequencing caused by random hexamer priming. Nucleic Acids Res. 38, e131–e131 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hardwick SA et al. Spliced synthetic genes as internal controls in RNA sequencing experiments. Nat. Methods 13, 792–798 (2016). [DOI] [PubMed] [Google Scholar]; A groundbreaking study using artificial spliced RNAs from a simulated genome as a gold standard by which to evaluate the sensitivity and specificity of transcriptome assembly methods.
- 67.Trapnell C et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Steijger T et al. Assessment of transcript reconstruction methods for RNA-seq. Nat. Methods 10, 1177–1184 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; A key resource benchmarking the ability of a range of transcriptome assembly tools to recall annotated exons and transcripts, highlighting their overall poor performance.
- 69.Pertea M et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shao M & Kingsford C Scallop enables accurate assembly of transcripts through phasing-preserving graph decomposition. Preprint at bioRxiv, 123612 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liu SJ et al. Single-cell analysis of long non-coding RNAs in the developing human neocortex. Genome Biol. 17, 67 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sharon D, Tilgner H, Grubert F & Snyder M A single-molecule long-read survey of the human transcriptome. Nat. Biotechnol. 31, 1009–1014 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; An early detailed view of human transcriptome sequencing using PacBio long-read technology, which established benchmarks for error rates, read lengths and sensitivity in detecting known and novel transcripts.
- 73.Weirather JL et al. Comprehensive comparison of Pacific Biosciences and Oxford Nanopore Technologies and their applications to transcriptome analysis. F1000Research 6, 100 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jain M, Olsen HE, Paten B & Akeson M The Oxford Nanopore MinION: delivery of nanopore sequencing to the genomics community. Genome Biol. 17, 239 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Byrne A et al. Nanopore long-read RNAseq reveals widespread transcriptional variation among the surface receptors of individual B cells. Nat. Commun. 8, 16027 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Smith AM, Jain M, Mulroney L, Garalde DR & Akeson M Reading canonical and modified nucleotides in 16S ribosomal RNA using nanopore direct RNA sequencing. Preprint at bioRxiv, 132274 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Garalde DR et al. Highly parallel direct RNA sequencing on an array of nanopores. Nat. Methods 15, 201–206 (2018). [DOI] [PubMed] [Google Scholar]; An early glimpse of unlimited-length direct RNA-seq using nanopore technology.
- 78.Oikonomopoulos S, Wang YC, Djambazian H, Badescu D & Ragoussis J Benchmarking of the Oxford Nanopore MinION sequencing for quantitative and qualitative assessment of cDNA populations. Sci. Rep. 6, 31602 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Housman G & Ulitsky I Methods for distinguishing between protein-coding and long noncoding RNAs and the elusive biological purpose of translation of long noncoding RNAs. Biochim. Biophys. Acta 1859, 31–40 (2016). [DOI] [PubMed] [Google Scholar]
- 80.Djebali S et al. Landscape of transcription in human cells. Nature 489, 101–108 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mercer TR et al. Targeted sequencing for gene discovery and quantification using RNA CaptureSeq. Nat. Protoc. 9, 989–1009 (2014). [DOI] [PubMed] [Google Scholar]
- 82.Mercer TR et al. Targeted RNA sequencing reveals the deep complexity of the human transcriptome. Nat. Biotechnol. 30, 99–104 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; Description of the RNA CaptureSeq method, identifying novel isoforms of deeply-studied protein-coding and lncRNA genes.
- 83.Clark MB et al. Quantitative gene profiling of long noncoding RNAs with targeted RNA sequencing. Nat. Methods 12, 339–342 (2015). [DOI] [PubMed] [Google Scholar]
- 84.Bussotti G et al. Improved definition of the mouse transcriptome via targeted RNA sequencing. Genome Res. 26, 705–716 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Deveson IW et al. Universal alternative splicing of noncoding exons. Cell Syst. 6, 245–255. e5 (2018). [DOI] [PubMed] [Google Scholar]
- 86.Tilgner H, Grubert F, Sharon D & Snyder MP Defining a personal, allele-specific, and single-molecule long-read transcriptome. Proc. Natl Acad. Sci. USA 111, 9869–9874 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nellore A et al. Human splicing diversity and the extent of unannotated splice junctions across human RNA-seq samples on the Sequence Read Archive. Genome Biol. 17, 266 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; Describes intropolis, a large-scale data set of splice junctions from essentially all short-read RNA-seq experiments to date, which suggests that the number of splice junctions in the human genome can be exhaustively mapped.
- 88.Seemann SE et al. The identification and functional annotation of RNA structures conserved in vertebrates. Genome Res. 27, 1371–1383 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; A rigorous data set of evolutionarily conserved structures in lncRNA exons, sure to be of value in future efforts to map their functional elements.
- 89.Bartonicek N et al. Intergenic disease-associated regions are abundant in novel transcripts. Genome Biol. 18, 241 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Saini HK, Griffiths-Jones S & Enright AJ Genomic analysis of human microRNA transcripts. Proc. Natl Acad. Sci. USA 104, 17719–17724 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jaffe AE et al. Developmental regulation of human cortex transcription and its clinical relevance at single base resolution. Nat. Neurosci. 18, 154–161 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gerrard DT et al. An integrative transcriptomic atlas of organogenesis in human embryos. eLife 5, e15657 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ahn RS et al. Transcriptional landscape of epithelial and immune cell populations revealed through FACS-seq of healthy human skin. Sci. Rep. 7, 1343 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wright JC et al. Improving GENCODE reference gene annotation using a high-stringency proteogenomics workflow. Nat. Commun. 7, 11778 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; A description of how large-scale peptidomic data sets can be used at controlled false-discovery rates to identify misidentified protein-coding transcripts amongst lncRNA annotations.
- 95.Gonzalez-Porta M, Calvo M, Sammeth M & Guigo R Estimation of alternative splicing variability in human populations. Genome Res. 22, 528–538 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kornienko AE et al. Long non-coding RNAs display higher natural expression variation than protein-coding genes in healthy humans. Genome Biol. 17, 14 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kelley D & Rinn J Transposable elements reveal a stem cell-specific class of long noncoding RNAs. Genome Biol. 13, R107 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kapusta A et al. Transposable elements are major contributors to the origin, diversification, and regulation of vertebrate long noncoding RNAs. PLoS Genet. 9, e1003470 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kasowski M et al. Variation in transcription factor binding among humans. Science 328, 232–235 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Consortium GTEx. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 45, 580–585 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sen R, Doose G & Stadler P Rare splice variants in long non-coding RNAs. Non-Coding RNA 3, 23 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nguyen Q & Carninci P Expression specificity of disease-associated lncRNAs: toward personalized medicine. Curr. Top. Microbiol. Immunol. 394, 237–258 (2016). [DOI] [PubMed] [Google Scholar]
- 103.Kanehisa M et al. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 42, D199–D205 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ashburner M et al. Gene Ontology: tool for the unification of biology. Nat. Genet. 25, 25–29 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kibbe WA et al. Disease Ontology 2015 update: an expanded and updated database of human diseases for linking biomedical knowledge through disease data. Nucleic Acids Res. 43, D1071–D1078 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yu G et al. BRWLDA: bi-random walks for predicting lncRNA-disease associations. Oncotarget 8, 60429–60446 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhang J, Zhang Z, Wang Z, Liu Y & Deng L Ontological function annotation of long non-coding RNAs through hierarchical multi-label classification. Bioinformatics 10.1093/bioinformatics/btx833 (2017). [DOI] [PubMed] [Google Scholar]
- 108.Guo X et al. Long non-coding RNAs function annotation: a global prediction method based on bi-colored networks. Nucleic Acids Res. 41, e35 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ning S et al. Lnc2Cancer: a manually curated database of experimentally supported lncRNAs associated with various human cancers. Nucleic Acids Res. 44, D980–D985 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Carlevaro-Fita J et al. Unique genomic features and deeply-conserved functions of long non-coding RNAs in the Cancer LncRNA Census (CLC). Preprint at bioRxiv, 152769 (2017). [Google Scholar]
- 111.Kaewsapsak P, Shechner DM, Mallard W, Rinn JL & Ting AY Live-cell mapping of organelle-associated RNAs via proximity biotinylation combined with protein-RNA crosslinking. eLife 6, e29224 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mas-Ponte D et al. LncATLAS database for subcellular localisation of long noncoding RNAs. RNA 23, 1080–1087 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Benoit Bouvrette LP et al. CeFra-seq reveals broad asymmetric mRNA and noncoding RNA distribution profiles in Drosophila and human cells. RNA 24, 98–113 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cabili MN et al. Localization and abundance analysis of human lncRNAs at single-cell and single-molecule resolution. Genome Biol. 16, 20 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lubelsky Y & Ulitsky I Sequences enriched in Alu repeats drive nuclear localization of long RNAs in human cells. Preprint at bioRxiv, 189746 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Carlevaro-Fita J, Das M, Polidori T, Navarro C & Johnson R Ancient exapted transposable elements promote nuclear enrichment of long noncoding RNAs. Preprint at bioRxiv, 189753 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zhang B et al. A novel RNA motif mediates the strict nuclear localization of a long noncoding RNA. Mol. Cell. Biol. 34, 2318–2329 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Marín-Béjar O et al. The human lncRNA LINC-PINT inhibits tumor cell invasion through a highly conserved sequence element. Genome Biol. 18, 202 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Guttman M & Rinn JL Modular regulatory principles of large non-coding RNAs. Nature 482, 339–346 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Smola MJ et al. SHAPE reveals transcript-wide interactions, complex structural domains, and protein interactions across theXistlncRNA in living cells. Proc. Natl Acad. Sci. USA 113, 10322–10327 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Fang R, Moss WN, Rutenberg-Schoenberg M & Simon MD Probing Xist RNA structure in cells using Targeted Structure-Seq. PLoS Genet. 11, e1005668 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hawkes EJ et al. COOLAIR antisense RNAs form evolutionarily conserved elaborate secondary structures. Cell Rep. 16, 3087–3096 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Xue Z et al. A G-rich motif in the lncRNA Braveheart interacts with a zinc-finger transcription factor to specify the cardiovascular lineage. Mol. Cell 64, 37–50 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lee S et al. Noncoding RNA NORAD regulates genomic stability by sequestering PUMILIO proteins. Cell 164, 69–80 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Li J-H, Liu S, Zhou H, Qu L-H & Yang J-H starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein–RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 42, D92–D97 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Paraskevopoulou MD et al. DIANA-LncBasev2: indexing microRNA targets on non-coding transcripts. Nucleic Acids Res. 44, D231–D238 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Buske FA, Bauer DC, Mattick JS & Bailey TL Triplex-Inspector: an analysis tool for triplex-mediated targeting of genomic loci. Bioinformatics 29, 1895–1897 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kelley DR, Hendrickson DG, Tenen D & Rinn JL Transposable elements modulate human RNA abundance and splicing via specific RNA-protein interactions. Genome Biol. 15, 537 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kapranov P et al. Large-scale transcriptional activity in chromosomes 21 and 22. Science 296, 916–919 (2002). [DOI] [PubMed] [Google Scholar]
- 130.Carninci P et al. The transcriptional landscape of the mammalian genome. Science 309, 1559–1563 (2005). [DOI] [PubMed] [Google Scholar]
- 131.Carninci P et al. Targeting a complex transcriptome: the construction of the mouse full-length cDNA encyclopedia. Genome Res. 13, 1273–1289 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Khalil AM et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl Acad. Sci. USA 106, 11667–11672 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Jia H et al. Genome-wide computational identification and manual annotation of human long noncoding. RNA genes. RNA 16, 1478–1487 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Cabili M et al. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 25, 1915–1927 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.HAVANA Annotation Guidelines, Version24. Wellcome Sanger Institute ftp://ftp.sanger.ac.uk/pub/project/havana/Guidelines/Guidelines_March_2016.pdf (2016).
- 136.Wucher V et al. FEELnc: a tool for long non-coding RNA annotation and its application to the dog transcriptome. Nucleic Acids Res. 45, gkw1306 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Dinger ME, Pang KC, Mercer TR & Mattick JS Differentiating protein-coding and noncoding RNA: challenges and ambiguities. PLoS Comput. Biol. 4, e1000176 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Huang J-Z et al. A peptide encoded by a putative lncRNA HOXB-AS3 suppresses colon cancer growth. Mol. Cell 68, 171–184. e6 (2017). [DOI] [PubMed] [Google Scholar]
- 139.Ingolia NT, Lareau LF & Weissman JS Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 147, 789–802 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ruiz-Orera J, Messeguer X, Subirana JA & Alba MM Long non-coding RNAs as a source of new peptides. eLife 3, e03523 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Mackowiak SD et al. Extensive identification and analysis of conserved small ORFs in animals. Genome Biol. 16, 179 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Guttman M, Russell P, Ingolia NT, Weissman JS & Lander ES Ribosome profiling provides evidence that large noncoding RNAs do not encode proteins. Cell 154, 240–251 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Carlevaro-Fita J, Rahim A, Guigó R, Vardy LA & Johnson R Cytoplasmic long noncoding RNAs are frequently bound to and degraded at ribosomes in human cells. RNA 22, 867–882 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Banfai B et al. Long noncoding RNAs are rarely translated in two human cell lines. Genome Res. 22, 1646–1657 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Verheggen K et al. Noncoding after all: biases in proteomics data do not explain observed absence of lncRNA translation products. J. Proteome Res. 16, 2508–2515 (2017). [DOI] [PubMed] [Google Scholar]; One of several studies that carefully examines proteomic evidence for productive translation of lncRNAs.
- 146.Bruford EA, Lane L & Harrow J Devising a consensus framework for validation of novel human coding loci. J. Proteome Res. 14, 4945–4948 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wang L et al. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 41, e74 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kong L et al. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 35, W345–W349 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]; A pioneering bioinformatic tool for the discrimination of protein-coding and non-coding transcripts, in this case using an alignment-free sequence-feature and homology strategy.
- 149.Nelson BR et al. A peptide encoded by a transcript annotated as long noncoding RNA enhances SERCA activity in muscle. Science 351, 271–275 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ma J et al. Discovery of human sORF-encoded polypeptides (SEPs) in cell lines and tissue. J. Proteome Res. 13, 1757–1765 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Gibb EA et al. Activation of an endogenous retrovirus-associated long non-coding RNA in human adenocarcinoma. Genome Med. 7, 22 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Gascoigne DK et al. Pinstripe: a suite of programs for integrating transcriptomic and proteomic datasets identifies novel proteins and improves differentiation of protein-coding and non-coding genes. Bioinformatics 28, 3042–3050 (2012). [DOI] [PubMed] [Google Scholar]
- 153.Ezkurdia I et al. The potential clinical impact of the release of two drafts of the human proteome. Expert Rev. Proteom. 12, 579–593 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Lopez F, Granjeaud S, Ara T, Ghattas B & Gautheret D The disparate nature of “intergenic” polyadenylation sites. RNA 12, 1794–1801 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Blanco E, Parra G & Guigó R Using geneid to identify genes. Curr. Protoc. Bioinformatics 10.1002/0471250953.bi0403s18 (2007). [DOI] [PubMed] [Google Scholar]