

Abstract
Dystonia is a movement disorder characterized by involuntary and repetitive co-contractions of agonist and antagonist muscles. Dystonia 6 (DYT6) is an autosomal dominant dystonia caused by loss-of-function mutations in the zinc finger transcription factor THAP1. We have generated Thap1 knock-out mice with a view to understanding its transcriptional role. While germ-line deletion of Thap1 is embryonic lethal, mice lacking one Thap1 allele—which in principle should recapitulate the haploinsufficiency of the human syndrome—do not show a discernable phenotype. This is because mice show autoregulation of Thap1 mRNA levels with upregulation at the non-affected locus. We then deleted Thap1 in glial and neuronal precursors using a nestin-conditional approach. Although these mice do not exhibit dystonia, they show pronounced locomotor deficits reflecting derangements in the cerebellar and basal ganglia circuitry. These behavioral features are associated with alterations in the expression of genes involved in nervous system development, synaptic transmission, cytoskeleton, gliosis and dopamine signaling that link DYT6 to other primary and secondary dystonic syndromes.
Introduction
Dystonia is characterized by involuntary repetitive co-contraction of agonist and antagonist muscles. It can occur as an isolated (primary) movement disorder or in occurrence with (secondary) other brain pathology (1–3). After essential tremor and Parkinsonism, it is the third most common movement disorder, yet its neurological underpinnings are unknown (2,3). There are, however, clues at the anatomical level. For instance, when dystonia occurs secondary to other central nervous system pathology—as in trauma, demyelination or strokes—the basal ganglia and cerebellum (CB) are typically affected (2,4–6). Involvement of these anatomical regions is also a theme in other neurodegenerative syndromes where dystonia is one among several other clinical features (such as Parkinson’s disease, Huntington’s disease, Corticobasal syndromes and the Spinocerebellar ataxias) (7–10). These regional network observations have found support in functional brain imaging of primary dystonia syndromes—using positron emission tomography and magnetic resonance imaging (11–15).
However, beyond clues to circuit dysfunction, the cellular and pathological basis has yet to be resolved. As with other neurological conditions, the hope is that genetic syndromes with precise molecular defects will provide insights applicable to the full spectrum of sporadic and acquired dystonic disorders. In this context, we have been intrigued by the primary dystonia DYT6 that is caused by a mutation in a C2CH zinc finger transcription factor—THAP1 [thanatos-associated (THAP) domain-containing apoptosis-associated protein 1] (16). Because DYT6 results from a mutation in a transcription factor, a likely proximate mechanism for this dystonia is an alteration in the gene expression profile.
DYT6 is inherited in an autosomal dominant manner with incomplete penetrance (of ~60%) and symptoms first appearing in the cervical and cranial regions during adolescence (17). Approximately 90 mutations have been described throughout the gene, most of which tend to cluster in the DNA-binding and coiled-coil domain of THAP1 (18,19). These mutations include point mutations, truncations and deletions, all of which are expected to interfere with its transcriptional function—and indeed this is what is observed in functional cell-based studies (20–22). There is so far no evidence that any of these mutations cause toxicity by gain of novel or native functions of the protein. Moreover, mutations have also been observed close to the start site, including the initiating methionine, which would suggest that absence of expression from one allele is sufficient to cause DYT6 (23). For this reason, we decided to study the role of Thap1 in mice by utilizing a loss-of-function approach. Implementing a floxed gene-trap strategy, we made constitutive and conditional Thap1 null mice. Focusing on the striatum (STR, which is a component of the basal ganglia circuitry) and CB, we discovered significant changes in gene expression that help explain the complex pathophysiology underlying the disease with ramifications for other genetic dystonic syndromes.
Results
Generation and characterization of Thap1 conditional knockout mice
We generated constitutive Thap1 null mice using gene trap deletion (deleting exons 2 and 3; see Fig. 1A for schema) (24). Heterozygous mating of Thap1 gene trap null mice (henceforth called Thap1+/−) did not produce any viable homozygous null pups (Thap1−/−; Supplementary Material, Fig. S1A). These results are similar to the findings from another strategy for making Thap1 null mice (exon 2 deletion) (25).
Figure 1.

Generation of conditional Thap1 KO mouse using cre-recombinase with the nestin-promoter. (A) Schematic of KO first gene construct (top) and crossed with either a cre-deleter strain to generate the Thap1 null allele (bottom left) or a Flp-deleter strain to generate the Thap1 floxed allele (bottom right). (B) Thap1 expression based on lacZ co-expression in a P50 Thap1+/− mouse brain (scale bar: 1 mm) and panel below of STR and CB (scale bar: 250 μm). (C) Time course of Thap1 based on lacZ co-expression in Thap1+/− mouse brains starting at P0 (scale bar: 1 mm). (D) Quantitative RT-PCR of cerebellar, cortical, striatal and liver (control) tissue for Thap1 mRNA in wild-type and Thap1 cKO mice normalized to Gapdh (n = 4, two-way ANOVA, F(3,20) = 5.059; **P < 0.01).
Mice lacking one copy of Thap1 should in principle recapitulate the haploinsufficiency or loss of function in this autosomal dominant disease resulting from one mutant allele. We therefore performed behavioral phenotyping on Thap1+/− mice. However, we failed to observe not only dystonic posturing but also other behavioral or motor deficits (such as motor incoordination on rotarod testing; Supplementary Material, Fig. S1B and C). In the course of studying these mice, we found that Thap1+/− mice did not show the expected reduction in Thap1 mRNA levels compared to wild-type littermates (Supplementary Material, Fig. S1D; the level of Thap1 protein cannot be measured since there are no reliable Thap1 antibodies) (26). This suggests that there is autoregulation of Thap1 at the level of mRNA. These findings are also consistent with recent studies demonstrating that Thap1 binds its own promoter and represses its expression (27). The lack of reduction in Thap1 mRNA that we observed differs from a previously characterized Thap1 exon 2 deleted mouse (25). These discordant findings could be explained by variation in genetic background (our mice are in a pure C57Bl/6 background, while the exon 2 deleted mice were in a mixed genetic background of C57Bl/6 and SV129) (25). The influence of genetic background on phenotype may also help explain why only a subset of human carriers of the mutation display the disease phenotype, where some patients might suffer from the disease because this autoregulation is lost (mutations in THAP1 are ~60% penetrant) (17).
As of yet, there is no Thap1 antibody suitable for immunohistochemical studies (26). We therefore took advantage of the lacZ cassette that encodes β-galactosidase to perform a detailed analysis of the spatiotemporal expression pattern of Thap1. We observed Thap1 expression throughout the adult brain (P50), including the STR and the CB. β-galactosidase expression is seen as early as P0 with the levels of expression stabilizing at P21, assuming adult levels (Fig. 1B and C). The protein expression is consistent with mRNA expression data (from in situ hybridization deposited at the Allen Brain Atlas) (28). Within the CB the β-galactosidase staining is particularly prominent in Purkinje cells (Fig. 1B).
In the absence of a phenotype displayed by the Thap1+/− mice, we turned to a conditional approach to deplete Thap1 in a tissue-specific manner. Since we were interested in the role of Thap1 in the nervous system, we mated gene-trap mice that have loxP sites to mice expressing cre-recombinase under a nestin promoter (nestin is expressed in all neuronal and glial precursor cells by E11) (29). These nestin conditional knockout mice (cKO) were viable, fertile and display virtually no expression of Thap1 mRNA in the brain. The residual Thap1 mRNA (<5% of wild-type levels in the cortex, CB and STR) presumably is expressed by non-cre expressing cells, such as microglia and endothelial cells. Since nestin does not express in the liver there was no statistically significant reduction in the levels of Thap1 in the liver (negative control; Fig. 1D).
Thap1 cKO mice do not show signs of spontaneous dystonia. This is not entirely surprising since, with a few exceptions, mice engineered to recapitulate human dystonias do not show dystonia (29–34). Nevertheless, we subjected these mice to behavioral tests to elucidate the role of Thap1—an important question in itself. We performed detailed locomotor testing to determine whether Thap1 affects functions sub-served by the CB and basal ganglia: two brain regions implicated in dystonia. In the cerebellar motor learning test—the rotarod test—Thap1 cKO mice display a decreased latency to fall compared to wild-type littermates (mice tested at 3 and 6 months of age; Fig. 2A). On the other hand, in the open-field test—a broad measure to determine any signs of bradykinetic or hyperkinetic behavior that might reflect basal ganglia dysfunction—there were no major ambulatory deficits displayed by Thap1 cKO mice. They did, however, spend a greater percentage of time in the periphery of the chamber compared to wild-type littermates (male WT: 72.9 ± 2.1%, male cKO: 84.9 ± 0.8%, female WT: 72.0 ± 1.9%, female cKO: 83.0 ± 2.7%; P < 0.05); furthermore, Thap1 cKO mice crossed between the inner and outer zones of the chamber fewer times (male WT: 58 ± 4 crossings, male cKO: 31 ± 2 crossings, female WT: 55 ± 2 crossings, female cKO: 43 ± 3 crossings; P < 0.05; Fig. 2B). Increased time spent in the periphery is indicative of anxiety and is consistent with the finding that dystonia patients display an increased propensity to be anxious (30,31). Finally, we also assayed the ability of mice to hold onto a grip test apparatus. We found that Thap1 cKO mice display a small but statistically significant decrease in their ability to grab the apparatus (male WT: 104 ± 5 gf, male cKO: 88 ± 6 gf, female WT: 103 ± 6 gf, female cKO: 86 ± 7gf; P < 0.05; Fig. 2C). Since there were no visible correlates of muscle weakness—such as an altered gait or muscle wasting—it is possible that this inability to grip stems from the aberrant contraction of agonists and antagonists in the forepaws. In addition to these behavioral findings, we also observed a reduction in body weight, size and clasping (described in a recent study using a similar conditional approach; Supplementary Material, Fig. S2).
Figure 2.
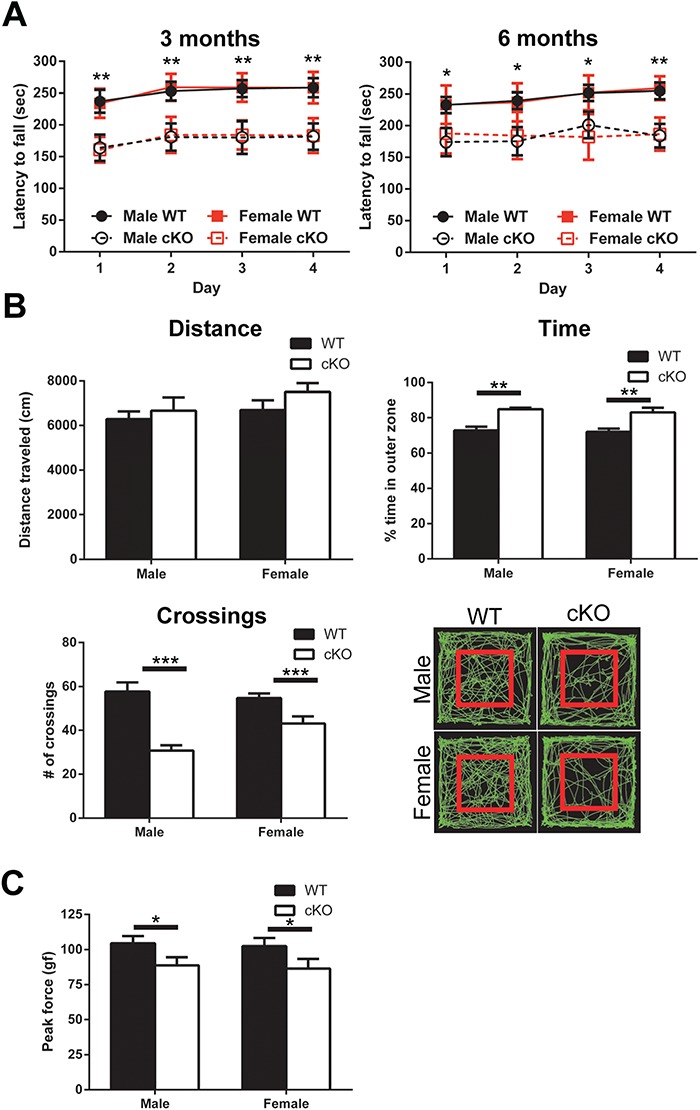
Motor behavior phenotyping of Thap1 cKO mice. (A) Accelerating rotarod at 3 and 6 months of age on 4 consecutive days (n = 12–14, repeated measures two-way ANOVA, 3 months: F(3,192) = 71.66 and 6 months: F(3,192) = 34.65; *P < 0.05 and **P < 0.01). (B) Metrics of open field assay on 4-month-old mice (n = 10, two-way ANOVA, total distance traveled: F(1,34) = 0.2418; time in outer zone: F(1,34) = 29.01; **P < 0.01; number of crossings between zones: F(1,34) = 34.39; ***P < 0.001); and representative traces, where red square designates boundary between inner and outer zones. (C) Forelimb grip strength of 4-month-old wild-type and Thap1 cKO mice (n = 10, two-way ANOVA, F(1,34) = 6.83; *P < 0.05).
Differential gene expression in Thap1 cKO
Since Thap1 is a transcription factor, we next turned to elucidating the transcriptional targets of Thap1 in the cKO mice. We performed RNA-Seq—the most rigorous method to interrogate gene expression. We focused on the STR and CB of 6-month-old mice, comparing cKO to wild-type littermates. We found 230 genes differentially expressed in the STR (185 downregulated and 45 upregulated) and 1189 genes to be differentially expressed in the CB (793 downregulated and 396 upregulated; Fig. 3A–C). Of these, 101 genes were differentially expressed in both CB and STR (Fig. 3C).
Figure 3.
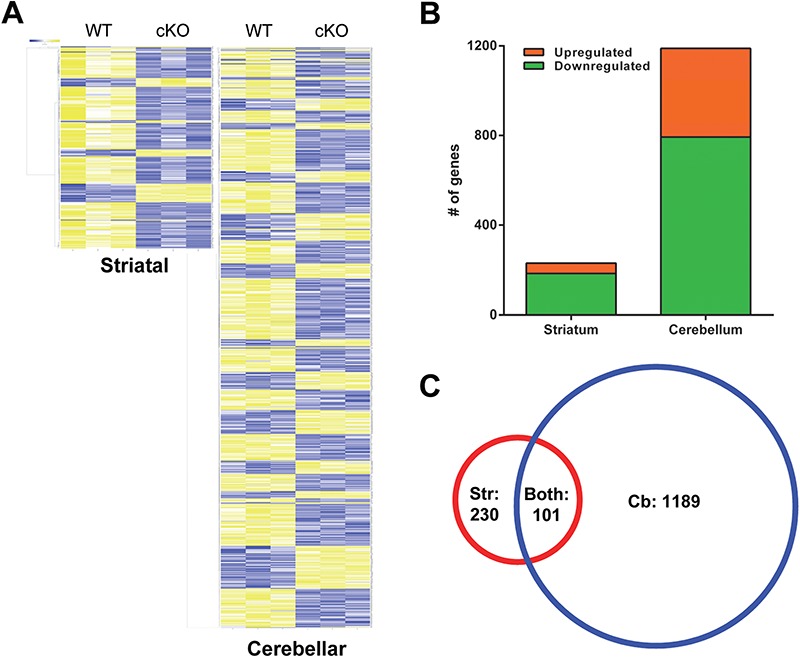
Differential gene expression in striatal and cerebellar tissue of 6-month-old Thap1 cKO mice. (A) Heatmaps of differentially expressed genes by RNA-Seq in striatal and cerebellar tissues of Thap1 cKO and wild-type mice with yellow indicating increased expression and blue decreased. (B) Breakdown of upregulated and downregulated genes with 230 for striatal and 1189 total genes in cerebellar tissues. (C) Venn diagrams demonstrating overlap of differentially expressed genes in both tissues, 101 genes.
To understand the biological functions of the differentially expressed genes, we performed gene ontology (GO) analyses. The top enriched biological processes in the STR revolved around the regulation of the cytoskeleton, nervous system development, and gliogenesis (Fig. 4A; for the full list of terms, see Supplementary Material, Table S2; for the list of cytoskeletal genes, see Supplementary Material, Table S3; for the list of genes involved in nervous system development, see Supplementary Material, Table S4; and for the list of genes involved in gliogenesis, see Supplementary Material, Table S5). We confirmed genes involved in oligodendroglial function identified in a recent microarray study performed on Thap1 cKO mice (this study was limited to approximately 19 000 genes on the array) (32). The overlap in the two gene sets is shown in Supplementary Material, Table S6.
Figure 4.
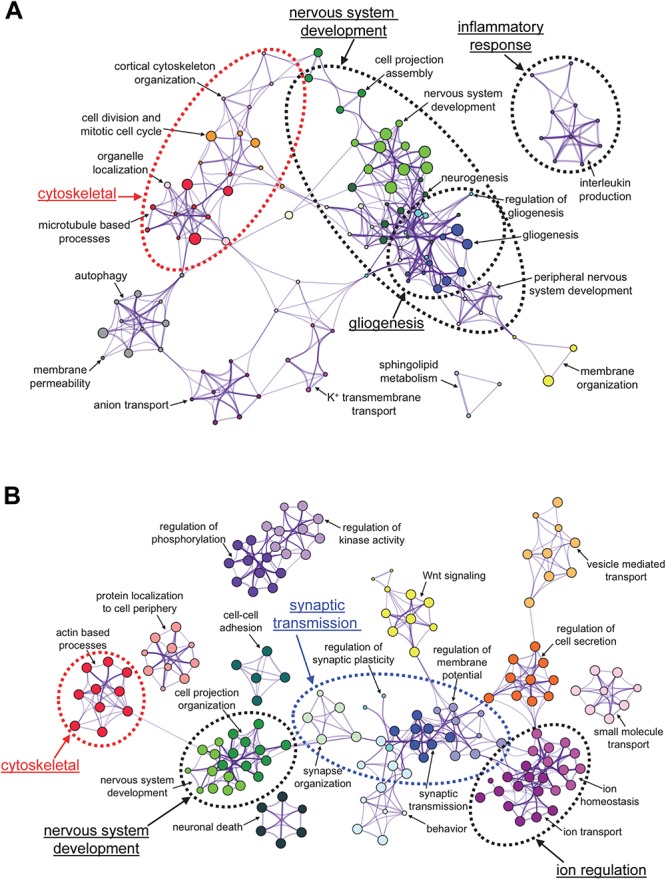
GO networks for genes that show altered levels in the STR (A) and CB (B) in the Thap1 cKO mice.
We also performed a KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway analysis that expands upon the GO analysis and allows for a more comprehensive overview of dysregulated pathways by including disease terms and druggable pathways (33). The top KEGG pathway is dopaminergic synaptic signaling; other terms include amphetamine addiction, cocaine addiction and cholinergic signaling (Supplementary Material, Table S7). The list of genes involved in dopamine (DA) signaling is further expanded in Supplementary Material, Table S8. This includes Drd2, which is also the gene mutated in another dystonic syndrome DYT11b (34,35), encodes one of two DA receptors expressed by striatal medium spiny neurons (MSNs) and plays a critical role in the indirect pathway of the basal ganglia.
In the CB, the top GO pathways were similar to the STR with a few additional terms, including `synaptic transmission’ and `ion homeostasis’ (Fig. 4B; for enriched terms, see Supplementary Material, Table S9; for the list of synaptic genes, see Supplementary Material, Table S10; for the list of genes involved in ion homeostasis, see Supplementary Material, Table S11). Intriguingly, the KEGG pathway analysis of this list identified several pathways involved in neurodegenerative disorders (Supplementary Material, Table S12).
The alterations in gene expression could result from alterations in bonafide Thap1 targets; alternatively, some of the observed gene expression changes could be secondary or downstream events. In order to address this issue, we compared our gene lists to previously performed Thap1 ChIP-Seq experiments on human K562 cells and mouse ES cells (GEO: GSM803408 and GSE86911) (36). We identified 77 genes in the STR and 284 genes in the CB that are bound by Thap1 and are likely to be direct targets of Thap1 (Supplementary Material, Fig. S3A; for the full list, see Supplementary Material, Table S13); of these, 62 genes are shared. We performed GO pathway analysis on this subset of genes (Supplementary Material, Fig. S3B; for the full list of terms, see Supplementary Material, Table S14). Intriguingly, the terms relating to the cytoskeleton are enriched in this shared target list. We also confirmed 20 of these genes by quantitative reverse transcriptase polymerase chain reaction (RT-PCR; 10 upregulated and 10 downregulated at the earlier time point of 3 months, which corresponds to our initial behavioral analysis; Supplementary Material, Fig. S4A and B).
Transcriptomic dysregulation of disease genes
There are 23 hereditary dystonic (DYT) syndromes with known gene mutations [for a recent review, see (37)]. We asked whether there is an overlap between our RNA-Seq results and the expression of genes mutated in these other dystonic syndromes. We found 13 DYT genes dysregulated in the STR and 12 in the CB in our list, which we confirmed by quantitative RT-PCR (Table 1). Intriguingly, we also found alterations in three genes in spontaneous mouse models of dystonia (Table 2). These dystonia genes—both human and mouse—align with the enriched GO terms (cytoskeleton, synaptic transmission and DA signaling; Fig. 4). Our results point to convergence at a molecular level of dystonic syndromes in mice and humans.
Table 1.
Quantitative RT-qPCR of dystonia genes in the STR and CB of Thap1 cKO mice
| Gene (dystonia) | Protein (function) | STR | P-value | CB | P-value |
|---|---|---|---|---|---|
| Tor1a (DYT1) | TorsinA (protein interactions in nuclear envelope and endoplasmic reticulum) | 99.8 ± 3.1% | ns | 94.6 ± 3.8% | ns |
| Hpca (DYT2) | Hippocalcin (neuron specific calcium binding protein) | 96.2 ± 2.0% | ns | 91.4 ± 3.4% | ns |
| Taf1 (DYT3) | TATA-box binding protein associated factor 1 (transcription factor) | 92.6 ± 3.3% | ns | 91.4 ± 4.7% | ns |
| Tubb4a # (DYT4)* | Tubulin beta 4A (nervous system enriched microtubule subunit) | 70.7 ± 2.0% | 0.0002 | 75.2 ± 1.9% | <0.0001 |
| Gch1 ∧ (DYT5a) | GTP cyclohydrolase 1 (biopterin biosynthetic enzyme) | 73.2 ± 3.1% | 0.0002 | 121.9 ± 7.9% | ns |
| Th ∧ (DYT5b) | Th(catecholamine synthesis) | 130.8 ± 5.6% | 0.0003 | 85.1 ± 2.3% | 0.0005 |
| Spr ∧ (DYT5c) | Sepiapterin reductase (biopterin biosynthetic enzyme) | 94.1 ± 4.2% | ns | 116.5 ± 7.5% | ns |
| Thap1 (DYT6)* | THAP domain containing protein 1 (zinc finger transcription factor) | 2.42 ± 0.19% | <0.0001 | 3.14 ± 0.15% | <0.0001 |
| Pnkd/Mr1 (DYT8)* | Myofibrillogenesis regulator 1 (synaptic protein?) | 83.9 ± 3.8% | 0.0037 | 88.8 ± 5.8% | ns |
| Slc2a1 & (DYT9/18)* | GLUT1 (glucose transporter) | 79.6 ± 2.1% | <0.0001 | 90.5 ± 3.7% | ns |
| Prrt2 & (DYT10)* | Proline-rich transmembrane protein 2 (SNARE complex interacting protein) | 123.2 ± 2.6% | 0.0002 | 141.3 ± 9.1% | 0.0205 |
| Sgce # (DYT11a) | Epsilon sarcoglycan (dystrophin-glycoprotein complex component, linking ECM and cytoskeleton) | 78.5 ± 3.4% | 0.0017 | 98.3 ± 6.3% | ns |
| Drd2 ∧ (DYT11b)* | Dopamine receptor 2 (dopamine neurotransmitter receptor) | 77.5 ± 1.8% | <0.0001 | 79.9 ± 2.5% | <0.0001 |
| Reln # (DYT11c)* | Reelin (extracellular matrix protein) | 78.5 ± 7.0% | 0.0319 | 66.3 ± 5.2% | 0.0087 |
| Atp1a3 & (DYT12) | Na+/K+ ATPase alpha 3 (ion transporter) | 77.4 ± 4.0% | 0.0034 | 122.3 ± 10.5% | ns |
| Prkra (DYT16) | Protein kinase interferon inducible dsRNA dependent activator (stress response kinase) | 103.7 ± 6.2% | ns | 124.8 ± 6.5% | 0.0185 |
| Ciz1 (DYT23a) | CDKN1A-interacting zinc finger protein 1 (zinc finger DAN binding protein) | 86.7 ± 5.5% | ns | 132.1 ± 7.1% | 0.0264 |
| Cacna1b & (DYT23b)* | Cav2.2 (N-type voltage gated calcium channel) | 92.1 ± 13.6% | ns | 64.3 ± 3.9% | 0.0011 |
| Ano3 & (DYT24)* | Anoctamin 3 (calcium activated chloride channel?) | 77.1 ± 4.8% | 0.0199 | 118.9 ± 6.7% | 0.0367 |
| Gnal ∧ (DYT25)* | Guanine-nucleotide binding protein alpha (stimulatory G protein) | 73.7 ± 5.2% | 0.049 | 91.9 ± 6.1% | ns |
| Kctd17 # (DYT26) | Potassium channel tetramerization domain containing protein 17 (substrate adaptor) | 142.1 ± 14.7% | 0.029 | 118.8 ± 6.2% | 0.441 |
| Col6a3 # (DYT27) | Collagen type VI alpha 3 (ECM collagen subunit) | 100.6 ± 13.0% | ns | 71.2 ± 7.0% | 0.0195 |
| Kmt2b (DYT28) | Lysine-specific methyltransferase 2B (histone methyltransferase) | 89.7 ± 10.6% | ns | 74.6 ± 7.1% | 0.0290 |
Cytoskeletal and extracellular matrix genes are indicated by #, dopamine signaling genes are indicated by ∧ and synaptic genes are indicated by &; genes with * indicates also differentially expressed in RNA-Seq. Statistical significance determined using unpaired Student’s t-test with values representing the average percent change normalized to Gapdh ± SEM, n = 8.
Table 2.
Quantitative RT-PCR of mouse dystonia genes
| Gene (mouse) | Protein (function) | STR | P-value | CB | P-value |
|---|---|---|---|---|---|
| Itpr1 (Itpr1 18p del) | Inositol 1,4,5-triphosphate receptor 1 (intracellular calcium channel) | 86.8 ± 4.3% | ns | 132.6 ± 8.6% | 0.0074 |
| Dst # (dystonia musculorum) | Dystonin (cytoskeletal linker protein) | 87.2 ± 3.6% | ns | 72.8 ± 4.5% | 0.0161 |
| Cacna1a & (tottering) | Cav2.1 (L-type voltage gated calcium channel) | 77.5 ± 4.1% | 0.0069 | 68.1 ± 3.0% | 0.0012 |
Genes denoted with # are cytoskeletal/ECM and & are synaptic genes. Statistical significance determined using unpaired Student’s t-test with values representing the average percent change normalized to Gapdh ± SEM, n = 8.
Surprisingly, Tor1a (DYT1) was not one of the differentially expressed genes even though Tor1a has been shown to be repressed by Thap1 in cell-based assays (36,38). The absence of differences in Tor1a in our cKO mice could suggest potential redundancy in the regulation of Tor1a transcription at the organismal level (alternatively, some of the previous studies should still be treated with caution). However, we observe overlap of stress response, nervous system development and cell morphogenesis between transcriptomic studies of DYT1 (HEK cells overexpressing mutant Tor1a), DYT6 (haploinsufficient Thap1 neonates and ES cells) and DYT23 (Ciz1 KO) models, suggesting there are convergent mechanisms at play (Supplementary Material, Table S15) (36,39–41).
Pathological characterization of Thap1 cKO
There was no obvious neurodegeneration in Thap1 cKO mice as determined by immunostaining with the neuronal marker, NeuN, and Nissl staining (Fig. 5A and B). However, given our RNA-Seq results, we performed a detailed histochemical analysis focusing on both neuronal and glial pathology that might stem from alterations in genes involved in the cytoskeleton, synapse formation, DA signaling and gliogenesis.
Figure 5.
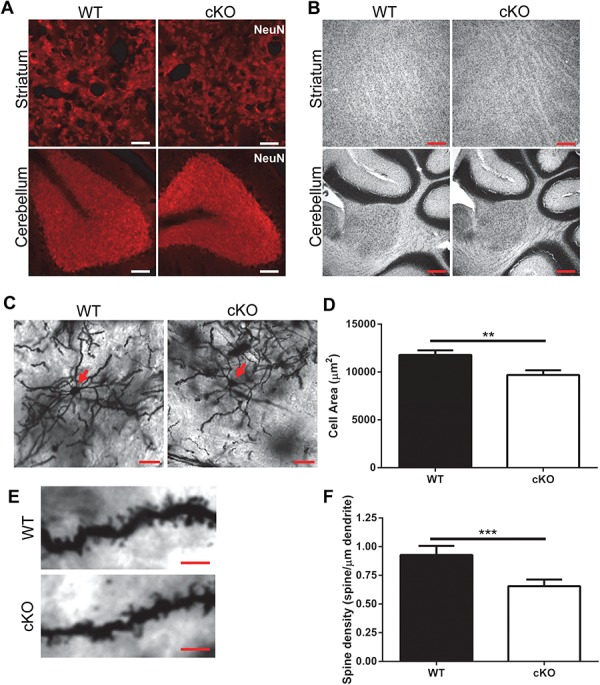
Decreased dendritic arborization and spine density in striatal MSNs in Thap1 cKO mice. (A) NeuN immunostaining in STR and CB of wild-type and Thap1 cKO mice (scale bar: 50 μm). (B) Nissl staining of STR and CB of wild-type and Thap1 cKO mice (scale bar: 200 μm). (C) Representative images of Golgi-Cox stain of striatal MSNs, red arrow pointing to soma (scale bar: 20 μm). (D) Quantification of cell size. Cells were sampled from 10 independent sections per mouse (n = 3 pairs of mice, error bars SEM, unpaired t-test; **P < 0.001). (E) Representative images of spine morphology (scale bar 5: μm) and (F) quantification of spine density, average with error bars representing SEM, 10 cells per section with 10 sections per mouse were quantified from 3 mice of each genotype (n = 3, unpaired t-test; ***P < 0.0001).
As might be expected from cytoskeletal and synaptic dysfunction, we found a decrease in cell size, dendritic arborization, and spine density of neurons as determined by the Golgi-Cox staining of striatal MSNs shown in Figure 5C–F. Consistent with our RNA-Seq results, we found a decrease in the expression of the DA receptor 2 (D2R) by quantitative RT-PCR and western blot (Table 1, Fig. 6A). There was no change in the protein level of DA type 1 receptor (D1R; Fig. 6A). Since D2R activity is responsible for inhibiting basal ganglia output through the indirect pathway, the specific reduction in the D2R would be expected to cause a surplus of movement and make mice prone to dystonia (42).
Figure 6.
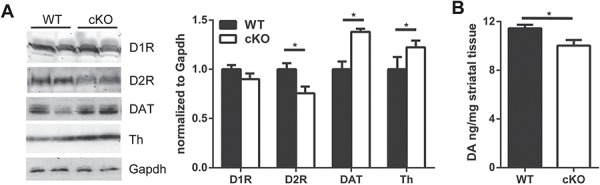
Changes in dopaminergic proteins in striatal tissue of Thap1 cKO mice. (A) Western blot analysis of striatal tissue lysates from 4-month-old Thap1 cKO and wild-type mice with Gapdh loading control. Densitometric quantification normalized to Gapdh levels (n = 7, two-way ANOVA, F(3,48) = 27.40 with Sidak’s multiple comparisons test; *P < 0.05). (B) DA levels in striatal tissue measured by HPLC in 4-month-old Thap1 cKO and wild-type mice (n = 6, unpaired t-test; *P < 0.05).
We also found a decrease in DA levels using high-performance liquid chromatography (HPLC; WT: 11.4 ± 0.3 ng/mg, cKO: 10.0 ± 0.4 ng/mg; P < 0.05; Fig. 6B). This reduction appears to result from an increase in the activity of synaptic DA transporter, DAT, which is responsible for the removal of DA from the synaptic cleft (Fig. 6A). In fact, there is an upregulation of tyrosine hydroxylase (Th) levels, the rate-limiting enzyme controlling its synthesis (Fig. 6A). This suggests that the decrease in DA levels is caused by increased removal rather than a lack of synthesis.
With regard to glial pathology, we observed a reduction in oligodendrocyte markers by western blot with myelin basic protein (MBP) and myelin oligodendrocyte glycoprotein and immunohistochemistry on tissue with MBP (Supplementary Material, Fig. S5). These results confirm recent work implicating Thap1 in oligodendrocyte maturation (32). We also observed increased astrocytic gliosis as evidenced by the upregulation of astrocytic glial fibrillary acidic protein (Gfap) staining by immunohistochemistry and confirmed by western blot (Fig. 7A–D).
Figure 7.
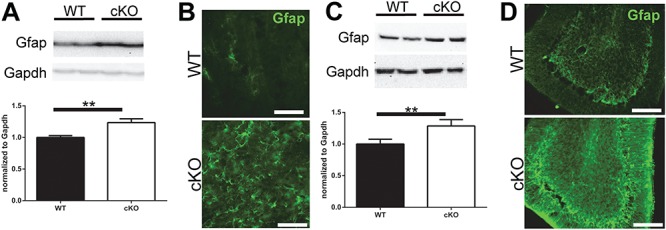
Reactive gliosis in Thap1 cKO mouse striatal and cerebellar tissue. Western blot protein expression and quantification of Gfap in (A) striatal and (C) cerebellar tissue (n = 7, paired t-test; P < 0.01). Representative images of Gfap immunostaining in STR (B) and CB (C) of Thap1 cKO and wild-type mice at 4 months (scale bar: 50 μm).
Discussion
DYT6 is caused by a loss of function of THAP1, a protein involved in transcriptional regulation, and its haploinsufficiency in humans leads to dystonia. We generated mice lacking Thap1 with the aim of understanding the gene networks Thap1 controls and to further elucidate the role of Thap1 in the nervous system. Thap1 haploinsufficient mice cannot be studied as a model of the human disease because of autoregulation and a lack of reduction of Thap1 mRNA levels. We therefore generated mice that lack Thap1 in the nervous system.
While Thap1 conditional null mice do not display spontaneous dystonia, they demonstrate robust behavioral and pathological phenotypes. These alterations are accompanied by gene expression changes that revolve around broad classes of genes in neurons and glia. In neurons, the genes misregulated encode proteins that regulate neuronal growth and synaptic transmission, particularly implicating the dopaminergic network. In glia, the genes encode proteins involved in regulating gliosis, cytokine signaling and myelination. These gene expression changes are associated with important pathological changes: a reduction in dendritic arborization, gliosis and a decrease in myelination. Overall, our results demonstrate that Thap1 is an important transcription factor in the nervous system.
Intriguingly, many of the genes that are mutated in other dystonic syndromes are also misregulated in Thap1 conditional null mice. There are also similarities in pathology between our mouse model and other models of human DYT syndromes. Both of these findings suggest points of convergence of pathological pathways in dystonia. For instance, decreased spine density is seen in mouse models of DYT1 (heterozygous mutant Tor1a knock-in), DYT11 (Reln haploinsufficiency) and DYT25 (Gnal haploinsufficiency) (43–47); defects in neuronal morphology are seen in mouse models of DYT1 (heterozygous mutant Tor1a knock-in), DYT4 (Tubb4a mutant), DYT11 (Reln haploinsufficiency) and DYT27 (Col6a3 deletion) (43–45,48–50); defects in myelination are seen in mouse models of DYT4 (Tubb4a mutant) and DYT6 (Thap1 KO) (32,51); and gliosis is seen in mouse models of DYT1 (conditional Tor1a KO, inducible Tor1a KO), DYT9/18 (Slc2a1 haploinsufficiency) and DYT23 (Ciz1 KO) (52–55). In addition to these changes, there are derangements in DA signaling, for which there is considerable evidence of involvement in other dopaminergic syndromes. For instance, we see a reduction in DA levels that is most severe in mouse models of the DA responsive dystonias DYT5a/b (Gch1 deficient and Th mutant mice) but is also observed in mouse models of DYT1 (mutant transgenic and heterozygous mutant Tor1a knock-in), DYT8 (mutant Pnkd), DYT23 (Cacna1b KO) and DYT25 (Gnal haploinsufficiency) (47,56–61). Lastly, we also see a reduction in the level of D2R, a key receptor in the indirect pathway, which is also decreased in other dystonia mouse models (DYT1, 8, 11a) and mutated in DYT11b (34,60,62,63).
It is still unclear which of these pathological consequences of Thap1 loss in mice are most relevant to the human syndrome. Clearly, one impediment to addressing this issue is that Thap1 conditional null mice, while displaying a behavioral phenotype, do not display dystonia per se. It might be possible to more closely mirror the human disease by stressing these mice with pharmacological agents, particularly with DA-receptor blocking agents or cholinergic agonists, as has been done with other dystonia mouse models of DYT1, 5, 8, 12 and 25 (47,57,59,60,64,65). Another method to improve our model might be to deplete Thap1 acutely in adult mice using a temporally inducible cre-recombinase approach. This approach is proving useful in mouse models of other dystonias (DYT1 and 12), where compensatory developmental pathways are thought to prevent the later expression of dystonic posturing (53,66). Regardless, it is intriguing that our pathological findings—the reduction in D2R receptor levels (by raclopride binding), gliosis and the oligodendrocytic hypomyelination—are all observed in autopsy studies from human patients (67–70).
In sum, our comprehensive and unbiased transcriptomic studies have led us to discover that Thap1 plays a critical role in the regulation of gene expression in the nervous system. The cellular pathways for the cytoskeleton, nervous system development, synaptic transmission and gliogenesis are dysregulated by loss of Thap1 and many of these pathways are also found to be affected in other dystonia models. Our results suggest that Thap1 is acting as a unifying factor amongst the various dystonias, providing greater insight into the shared pathogenesis. Continued investigation of the cellular pathways and neuronal circuits involved in DYT6 is pivotal for the systemic understanding of dystonia pathogenesis and the development of therapeutic platforms for these ineffectively treated disorders.
Materials and Methods
Mice
Mice were housed in a specific pathogen-free facility, and all experiments were performed in compliance with the National Institutes of Health’s Guide for the Care and Use of Laboratory Animals, and Northwestern University’s Institutional Animal Care and Use Committee. Mice were euthanized by isoflurane inhalation unless otherwise noted.
Thap1 null mice were generated using an insertional mutagenesis gene-trap approach [Knock-Out Mouse Project; ES clone #126393; C57Bl/6 background] (24). The gene-trap cassette was inserted after exon 1 of Thap1 [the gene-trap cassette contains a splice acceptor site with the enzymatic reporter β-galactosidase (En2SA-IRES-lacZ), a neomycin cassette for antibiotic selectivity driven by the human beta actin promoter (hβactP-neo-pA) and loxP and FRT sites for DNA recombination (Fig. 1A)]. Mice generated from these ES cells (called the KO first mice) were mated to the Ella-cre deleter mice (The Jackson Laboratory, 003724; C57Bl/6 background) (71), causing a germline deletion of the DNA cassette between the loxP sites, and subsequently resulting in mice referred to as Thap1 null mice. Thap1 null mice generate an mRNA in which exon 1 (that only encodes the first 24 amino acids of Thap1) is spliced to the engrailed 2 splice acceptor site (En2SA) upstream of an internal ribosomal entry site and lacZ sequence (encoding for the enzymatic reporter β-galactosidase) with a polyadenylated 3′ tail (pA).
KO first mice were also mated with CAG-FLPe Deleter mice (RIKEN, RBRC01834, C57Bl/6 background) (72)) permitting a reversion to a functional wild-type allele while maintaining loxP sites flanking exons 2 and 3 for later conditional deletion (Thap1 floxed mice) through the removal of the DNA cassette between the FRT sites. Conditional Thap1 KO mice (Thap1 cKO) were generated by mating Thap1 floxed mice with a nestin-cre expressing mouse line (Jackson Laboratory, 003771, C57Bl/6 background) (29).
Mice were genotyped by PCR using tail-extracted DNA (taken at P10-P14). PCR sequence for genotyping were as follows:
Thap1 WT-F: 5′-TAAAGATAAGCCCTAACACGAAGGTTGGCTAG-3′;
Thap1 WT-R: 5′-AGATTAGTTCTGGTTACCAATCTCCCTCAGG-3′;
FRT/loxP-F: 5′-CTCTAGAAAGTATAGGAACTTCGTCGAGATAACTTCG-3′;
LacZ-F: 5′-GTCGCTACCATTACCAGTTGGTCTGGTGTC-3′;
Thap1 3′ UTR-R: 5’-CCAGGTCCCAACTTTTCTTTCTTTAGAAGGTTG-3′;
Cre-F: 5′-ACACCAAAATTTGCCTGCATTACCGGTCG-3′;
Cre-R: 5′-CTAGAGCCTGTTTTGCACGTTCACCG-3′.
Behavioral assays
Behavioral and pathological assays were performed by researchers blinded to the experimental genotype.
Rotarod testing
Accelerating Rota-Rod (Ugo Basile) was performed at 3 and 6 months of age. Mice were placed on the rotarod accelerating from a speed of 4–40 rpm over a 5 min period, and the latency to fall was recorded (73,74). Mice were tested four times per day over four consecutive days. The average performances for each day were plotted.
Open field testing
The 4-month-old mice were placed in the center of a 56 × 56 cm arena in a noise reduced, dark chamber for 10 min. The location of the mouse in the arena and distance traveled were analyzed (using LimeLight Software; Actimetrics). Statistical significance was determined using two-way ANOVA to assess the impact of genotype on total distance traveled, percentage of time in the outer zone and number of crossings between inner and outer zones.
Forelimb grip strength
The 4-month-old mice were trained to grab a horizontal waffle grid attached to a recording device (Grip-Strength Meter; Ugo Basile); they were then slowly pulled by the tail until they released their grip. The average grip strength over three trials was plotted.
Transcriptomic assays
RNA isolation
Striatal and cerebellar brain regions were isolated using microdissection. RNA was isolated from tissues using the RNeasy Plus Universal Mini Kit (cat# 73404, Qiagen). The tissues were homogenized in QIAzol Lysis Reagent and then treated with gDNA Eliminator Solution and chloroform for subsequent phase separation. After centrifugation, the aqueous phase was removed and loaded onto a spin column for washing and elution of RNA.
Quantitative RT-PCR
Single-strand cDNA was reverse transcribed from the purified mRNA using SuperScript III First Strand Synthesis System (cat# 18080–051, Invitrogen). The quantitative PCR (qPCR) was carried out using iTaq Universal SYBR Green Supermix (cat#172–5122, Bio-Rad). The qPCR cycling parameters for amplification and quantification were 95°C for 5 s then 60°C for 30s repeated 40 times with a melt curve protocol (65–95°C with a heating rate of 0.5°C per second). qPCR (normalized to Gapdh) levels was carried out in a CFX Connect Real-Time PCR Detection System (Bio-Rad) and analyzed using CFX Manager Software (Bio-Rad). The primers used for qPCR are listed in Supplementary Material, Table S1.
RNA-Seq
mRNA-Seq was conducted in the Northwestern University NUSeq Core Facility. The quality of the RNA was checked using Agilent Bioanalyzer 2100 (all samples had RNA integrity numbers of <9.5). The Illumina TruSeq Stranded mRNA Library Preparation Kit was used to prepare sequencing libraries from 800 ng of total RNA (the RNA quantity was determined with Qubit fluorometer). This procedure includes mRNA purification and fragmentation, cDNA synthesis, 3′ end adenylation, Illumina adapter ligation, library PCR amplification and validation. Illumina NextSeq 500 Sequencer was used to sequence the libraries with the production of single-end, 75 bp reads. The quality of DNA reads was evaluated using FastQC and reads of poor quality or those aligning to rRNA sequences were filtered. The cleaned reads were aligned to the Mus musculus genome (mm10; obtained from UCSC (University of California Santa Cruz; http://genome.ucsc.edu)) using STAR (Spliced Transcripts Alignment to a Reference) (75). Read counts for each gene were calculated using HTSeq-count (76). Normalization and differential expression were determined using DESeq2 (77). The statistical cutoff for determining differentially expressed genes was an false discovery rate-adjusted P-value less than 0.05. GO pathway analysis was performed on both gene lists using MetaScape (http://metascape.org) (78) to identify pathways that are enriched with genes that are upregulated and downregulated. Enriched GO pathways were visualized using Cytoscape (79). KEGG pathway analyses were performed using DAVID (Database for Annotation, Visualization and Integrated Discovery, National Institute of Allergy and Infectious Disease, NIH) (80,81).
High-performance liquid chromatography
Mice were euthanized by CO2 inhalation and striatal tissue was obtained by microdissection. The tissue was placed on dry ice and processed for HPLC as previously described (82). Briefly, dissected brain regions were placed in a buffer containing 0.2 m perchloric acid, 0.05% Na2EDTA, 0.1% Na2S2O5 and isoproterenol (internal standard) to extract monoamine neurotransmitters. Monoamine lysates were placed in a refrigerated automatic sampler (model WPS-3000TSL) until being separated by a reversed-phase C18 column with a flow rate of 0.6 ml/min using a Dionex Ultimate 3000 HPLC system (pump ISO-3100SD, Thermo Scientific, Bannockburn, IL). Electrochemical detection was achieved using a CoulArray model 5600A coupled with an analytical cell (microdialysis cell 5014B) and a guard cell (model 5020). Data acquisition and analysis were performed using Chromeleon 7 and ESA CoulArray 3.10 HPLC Software; all results were normalized to the wet-weight of the analyzed tissue.
Western blots
Striatal and cerebellar brain regions were isolated using microdissection and then the tissues were homogenized using a dounce homogenizer in RIPA lysis buffer (150 mm NaCl, 0.1% SDS, 50 mm Tris pH 7.6, 1 mm EDTA, 0.5% sodium deoxycholate, 1% NP-40) with protease inhibitors (Sigma). Samples were sonicated for 10 s on ice and centrifuged at 12,000g for 15 min at 4°C, heated at 95°C for 10 min with Laemmli’s Sample Buffer (Bio-Rad). A total of 20 μg of lysate was loaded per well in a 10% SDS-PAGE gel. The blots were probed with primary antibodies: 1:2000 anti-mouse Gapdh (#14C10, Cell Signaling); 1:1000 anti-rabbit Th (#AB152, Millipore); 1:500 anti-rabbit D2R (#AB5084P, Millipore); 1:1000 anti-rabbit D1R (#ab20066, Abcam); 1:1000 anti-rabbit DAT (#AB1591P, Millipore); 1:1000 anti-rabbit Gfap (#ab7260, Abcam); 1:1000 anti-mouse Mbp (#ab62631, Abcam); 1:1000 anti-rabbit Mog (#ab32760, Abcam) in 5% milk in TBST (0.1% Triton X-100). Secondary antibodies conjugated to horse radish peroxidase (goat anti-rabbit #1706515, goat anti-mouse #1706516; Bio-Rad) were applied at a concentration of 1:5000–1:10 000 in 5% milk in TBST. Blots were imaged using SuperSignal West Pic Chemiluminescence Detection kit (ThermoScientific) and signals were detected using a ChemiDoc XRS System equipped with Quantity One software (Bio-Rad). To visualize individual lanes, we performed staining with Ponceau S (Sigma-Aldrich). This allowed us to attribute each chemiluminescent image to its specific lane. Average intensity was determined by normalizing to Gapdh levels (quantified with ImageJ-based software; National Institutes of Health).
X-gal staining for β-galactosidase activity
X-gal staining was performed as described (74). Briefly, brains were isolated then fixed 4°C overnight with 0.2% paraformaldehyde (PFA) in PIPES buffer (0.1 M PIPES, pH 6.9, 2 mm MgCl2 and 5 mm EGTA). The following day, the brains were equilibrated in 30% sucrose in phosphate buffered saline (PBS) supplemented with 2 mm MgCl2 and embedded in optimal cutting temperature (OCT) medium (Fisher). Para-sagittal sections of 60 μm were cut using a cryostat (Microm M505, ThermoScientific) and post-fixed with 2% paraformaldehyde in PIPES buffer on ice for 10 min. The sections were then incubated with rinse buffer (100 mm sodium phosphate, pH 7.4, 2 mm MgCl2, 0.1% sodium deoxycholate and 0.2% NP-40) on ice for 10 min and stained with staining solution (rinse buffer containing 5 mm potassium ferricyanide, 5 mm potassium ferrocyanide, 1 mg/mL X-gal) at 37°C for 48 h. The stained slices were then rinsed in PBS supplemented with 2 mm MgCl2 and mounted onto glass slides using Vectashield (Vector Laboratories). Sections were imaged on TissueGnostics microscope and imaging software.
Immunostaining
Mice were anesthetized using isoflurane and perfused with 4% PFA in PBS, after which the brains were removed and fixed overnight in 4% PFA. The brains were then placed in 30% sucrose in PBS prior to being embedded in OCT. Sagittal sections of 40 μm were cut using a cryostat (Microm M505, ThermoScientific) and immunostained as described (73,74). Briefly, brain sections were washed in PBS, permeabilized with PBS plus 0.5% Triton X-100 (PBST) and blocked with 5% normal goat serum in PBST. Sections were incubated with primary antibodies overnight at 4°C in blocking solution, 1:500 anti-rabbit NeuN (#ab177487, Abcam), 1:1000 anti-rabbit Gfap (#Z0334, Dako) and 1:500 anti-mouse Mbp (#ab62631, Abcam). They were washed in PBST and incubated with fluorophore-conjugated secondary antibodies, 1:1000 Alexa488 goat anti-rabbit, Alexa488 goat anti-mouse and Alexa594 goat anti-rabbit (Invitrogen), in blocking solution for 1 h at room temperature. Sections were washed and mounted on slides using Prolong Diamond Antifade Mountant with DAPI (Invitrogen). Images were captured using a CTR6500 confocal microscope (Leica) equipped with Leica LAS AF software.
Golgi-Cox staining
Golgi-Cox staining was performed using the FD Rapid GolgiStain Kit (Cat# PK401, FD NeuroTechnologies). The brains were removed, rinsed in double-distilled water and placed in equal-part solutions A and B. The solution was removed and replaced after 24 h, after which the brains were incubated in the staining solution (for 2 weeks at room temperature in the dark). The brains were then transferred to solution C for 3 days and flash frozen in isopentane. They were sectioned into 200 μm para-sagittal sections using a cryostat (Microm M505, ThermoScientific) and mounted on gelatin coated slides. The sections were then processed following the manufacturer’s protocol. Sections were imaged on a Nikon Ti2 Widefield microscope and image analysis was done using FIJI (National Institutes of Health).
Availability of data and materials
All materials will be made available upon request, and RNA sequencing data have been deposited in GEO under accession GSE123880.
Supplementary Material
Acknowledgements
We thank members of the Opal laboratory for their intellectual input. We also thank the Northwestern University Behavioral Phenotyping Core for helping us with the behavioral assays; NUSeq Core Facility that is supported by the Northwestern University Center for Genetic Medicine, Feinberg School of Medicine; and Shared and Core Facilities of the University’s Office for Research for helping us with the transcriptomic studies; and Northwestern University Nikon Imaging Center for the microscopy help.
Conflict of Interest statement. None declared.
Funding
U.S. National Institutes of Health NINDS (National Institute of Neurological Disorders and Stroke; F31 NS095461, R01 NS082351, R01 NS062051) and Northwestern University Dr. John N. Nicolson Fellowship.
References
- 1. Fahn S. (1988) Concept and classification of dystonia. Adv Neurol, 50,–8. [PubMed] [Google Scholar]
- 2. Breakefield X.O., Blood A.J., Li Y., Hallett M., Hanson P.I. and Standaert D.G. (2008) The pathophysiological basis of dystonias. Nat. Rev. Neurosci., 9, 222–234. [DOI] [PubMed] [Google Scholar]
- 3. Petrucci S. and Valente E.M. (2013) Genetic issues in the diagnosis of dystonias. Front Neurol., 4,–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Trost M., Carbon M., Edwards C., Ma Y., Raymond D., Mentis M.J., Moeller J.R., Bressman S.B. and Eidelberg D. (2002) Primary dystonia: is abnormal functional brain architecture linked to genotype? Ann. Neurol., 52, 853–856. [DOI] [PubMed] [Google Scholar]
- 5. Neychev V.K., Fan X., Mitev V.I., Hess E.J. and Jinnah H.A. (2008) The basal ganglia and cerebellum interact in the expression of dystonic movement. Brain, 131, 2499–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Phukan J., Albanese A., Gasser T. and Warner T. (2011) Primary dystonia and dystonia-plus syndromes: clinical characteristics, diagnosis, and pathogenesis. Lancet Neurol., 10, 1074–1085. [DOI] [PubMed] [Google Scholar]
- 7. Moghimi N., Jabbari B. and Szekely A.M. (2014) Primary dystonias and genetic disorders with dystonia as clinical feature of the disease. Eur. J. Paediatr. Neurol., 18, 79–105. [DOI] [PubMed] [Google Scholar]
- 8. Louis E.D., Anderson K.E., Moskowitz C., Thorne D.Z. and Marder K. (2000) Dystonia-predominant adult-onset Huntington disease: association between motor phenotype and age of onset in adults. Arch. Neurol., 57, 1326–1330. [DOI] [PubMed] [Google Scholar]
- 9. Gaalen J., Giunti P. and Warrenburg B.P. (2011) Movement disorders in spinocerebellar ataxias. Mov. Disord., 26, 792–800. [DOI] [PubMed] [Google Scholar]
- 10. Wickremaratchi M.M., Knipe M.D.W., Sastry B.S.D., Morgan E., Jones A., Salmon R., Weiser R., Moran M., Davies D., Ebenezer L. et al. (2011) The motor phenotype of Parkinson's disease in relation to age at onset. Mov. Disord., 26, 457–463. [DOI] [PubMed] [Google Scholar]
- 11. Carbon M., Su S., Dhawan V., Raymond D., Bressman S. and Eidelberg D. (2004) Regional metabolism in primary torsion dystonia: effects of penetrance and genotype. Neurology, 62, 1384–1390. [DOI] [PubMed] [Google Scholar]
- 12. Carbon M. and Eidelberg D. (2009) Abnormal structure-function relationships in hereditary dystonia. Neuroscience, 164, 220–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Carbon M., Argyelan M. and Eidelberg D. (2010) Functional imaging in hereditary dystonia. Eur. J. Neurol., 17, 58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Neychev V.K., Gross R.E., Lehéricy S., Hess E.J. and Jinnah H.A. (2011) The functional neuroanatomy of dystonia. Neurobiol. Dis., 42, 185–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Niethammer M., Carbon M., Argyelan M. and Eidelberg D. (2011) Hereditary dystonia as a neurodevelopmental circuit disorder: Evidence from neuroimaging. Neurobiol. Dis., 42, 202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fuchs T., Gavarini S., Saunders-Pullman R., Raymond D., Ehrlich M.E., Bressman S.B. and Ozelius L.J. (2009) Mutations in the THAP1 gene are responsible for DYT6 primary torsion dystonia. Nat. Genet., 41, 286–288. [DOI] [PubMed] [Google Scholar]
- 17. Paudel R., Hardy J., Revesz T., Holton J.L. and Houlden H. (2012) Review: genetics and neuropathology of primary pure dystonia. Neuropathol. Appl. Neurobiol., 38, 520–534. [DOI] [PubMed] [Google Scholar]
- 18. Blanchard A., Ea V., Roubertie A., Martin M., Coquart C., Claustres M., Béroud C. and Collod-Béroud G. (2011) DYT6 dystonia: review of the literature and creation of the UMD Locus-Specific Database (LSDB) for mutations in the THAP1 gene. Hum. Mutat., 32, 1213–1224. [DOI] [PubMed] [Google Scholar]
- 19. Charlesworth G., Bhatia K.P. and Wood N.W. (2013) The genetics of dystonia: new twists in an old tale. Brain, 136, 2017–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Clouaire T., Roussigne M., Ecochard V., Mathe C., Amalric F. and Girard J.-P. (2005) The THAP domain of THAP1 is a large C2CH module with zinc-dependent sequence-specific DNA-binding activity. Proc. Natl. Acad. Sci. U. S. A., 102, 6907–6912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bessière D., Lacroix C., Campagne S., Ecochard V., Guillet V., Mourey L., Lopez F., Czaplicki J., Demange P., Milon A. et al. (2008) Structure-function analysis of the THAP zinc finger of THAP1, a large C2CH DNA-binding module linked to Rb/E2F pathways. J. Biol. Chem., 283, 4352–4363. [DOI] [PubMed] [Google Scholar]
- 22. LeDoux M.S., Xiao J., Rudzińska M., Bastian R.W., Wszolek Z.K., Van Gerpen J.A., Puschmann A., Momčilović D., Vemula S.R. and Zhao Y. (2012) Genotype-phenotype correlations in THAP1 dystonia: molecular foundations and description of new cases. Parkinsonism Relat. Disord., 18, 414–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bressman S.B., Raymond D., Fuchs T., Heiman G.A., Ozelius L.J. and Saunders-Pullman R. (2009) Mutations in THAP1 (DYT6) in early-onset dystonia: a genetic screening study. Lancet Neurol., 8, 441–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Skarnes W.C., Rosen B., West A.P., Koutsourakis M., Bushell W., Iyer V., Mujica A.O., Thomas M., Harrow J., Cox T. et al. (2011) A conditional knockout resource for the genome-wide study of mouse gene function. Nature, 474, 337–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ruiz M., Perez-Garcia G., Ortiz-Virumbrales M., Méneret A., Morant A., Kottwitz J., Fuchs T., Bonet J., Gonzalez-Alegre P., Hof P.R. et al. (2015) Abnormalities of motor function, transcription and cerebellar structure in mouse models of THAP1 dystonia. Hum. Mol. Gen., 24, 7159–7170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ortiz-Virumbrales M., Ruiz M., Hone E., Dolios G., Wang R., Morant A., Kottwitz J., Ozelius L.J., Gandy S. and Ehrlich M.E. (2014) Dystonia type 6 gene product Thap1: identification of a 50 kDa DNA-binding species in neuronal nuclear fractions. Acta Neuropathol. Commun., 2, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Erogullari A., Hollstein R., Seibler P., Braunholz D., Koschmidder E., Depping R., Eckhold J., Lohnau T., Gillessen-Kaesbach G., Grünewald A. et al. (2014) THAP1, the gene mutated in DYT6 dystonia, autoregulates its own expression. Biochim. Biophys. Acta, 1839, 1196–1204. [DOI] [PubMed] [Google Scholar]
- 28. Lein E.S., Hawrylycz M.J., Ao N., Ayres M., Bensinger A., Bernard A., Boe A.F., Boguski M.S., Brockway K.S., Byrnes E.J. et al. (2007) Genome-wide atlas of gene expression in the adult mouse brain. Nature, 445, 168–176. [DOI] [PubMed] [Google Scholar]
- 29. Tronche F., Kellendonk C., Kretz O., Gass P., Anlag K., Orban P.C., Bock R., Klein R. and Schütz G. (1999) Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat. Genet., 23, 99–103. [DOI] [PubMed] [Google Scholar]
- 30. Kuyper D.J., Parra V., Aerts S., Okun M.S. and Kluger B.M. (2011) Nonmotor manifestations of dystonia: a systematic review. Mov. Disord., 26, 1206–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Torres J.A.K.L. and Rosales R.L. (2017) Nonmotor symptoms in dystonia. Int. Rev. Neurobiol., 134, 1335–1371. [DOI] [PubMed] [Google Scholar]
- 32. Yellajoshyula D., Liang C.-C., Pappas S.S., Penati S., Yang A., Mecano R., Kumaran R., Jou S., Cookson M.R. and Dauer W.T. (2017) The DYT6 dystonia protein THAP1 regulates myelination within the oligodendrocyte lineage. Dev. Cell, 42, 52–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Du J., Yuan Z., Ma Z., Song J., Xie X. and Chen Y. (2014) KEGG-PATH: Kyoto encyclopedia of genes and genomes-based pathway analysis using a path analysis model. Mol. Biosyst., 10, 2441–2447. [DOI] [PubMed] [Google Scholar]
- 34. Klein C., Brin M.F., Kramer P., Sena-Esteves M., Leon D., Doheny D., Bressman S., Fahn S., Breakefield X.O. and Ozelius L.J. (1999) Association of a missense change in the D2 dopamine receptor with myoclonus dystonia. Proc. Natl. Acad. Sci. U. S. A., 96, 5173–5176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Beukers R.J., Booij J., Weisscher N., Zijlstra F., Amelsvoort T.A.M.J. and Tijssen M.A.J. (2009) Reduced striatal D2 receptor binding in myoclonus-dystonia. Eur. J. Nucl. Med. Mol. Imaging, 36, 269–274. [DOI] [PubMed] [Google Scholar]
- 36. Aguilo F., Zakirova Z., Nolan K., Wagner R., Sharma R., Hogan M., Wei C., Sun Y., Walsh M.J., Kelley K. et al. (2017) THAP1: role in mouse embryonic stem cell survival and differentiation. Stem Cell Rep., 9, 92–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lohmann K. and Klein C. (2017) Update on the genetics of dystonia. Curr. Neurol. Neurosci. Rep., 17, 26. [DOI] [PubMed] [Google Scholar]
- 38. Kaiser F.J., Osmanoric A., Rakovic A., Erogullari A., Uflacker N., Braunholz D., Lohnau T., Orolicki S., Albrecht M., Gillessen-Kaesbach G. et al. (2010) The dystonia gene DYT1 is repressed by the transcription factor THAP1 (DYT6). Ann. Neurol., 68, 554–559. [DOI] [PubMed] [Google Scholar]
- 39. Baptista M.J. (2003) Microarray analysis reveals induction of heat shock proteins mRNAs by the torsion dystonia protein, TorsinA. Neurosci. Lett., 343, 5–8. [DOI] [PubMed] [Google Scholar]
- 40. Zakirova Z., Fanutza T., Bonet J., Readhead B., Zhang W., Yi Z., Beauvais G., Zwaka T.P., Ozelius L.J., Blitzer R.D. et al. (2018) Mutations in THAP1/DYT6 reveal that diverse dystonia genes disrupt similar neuronal pathways and functions. PLoS Genet., 14, e1007169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Xiao J., Vemula S.R., Xue Y., Khan M.M., Kuruvilla K.P., Marquez-Lona E.M., Cobb M.R. and LeDoux M.S. (2016) Motor phenotypes and molecular networks associated with germline deficiency of Ciz1. Exp. Neurol., 283, 110–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kravitz A.V., Freeze B.S., Parker P.R.L., Kay K., Thwin M.T., Deisseroth K. and Kreitzer A.C. (2010) Regulation of parkinsonian motor behaviours by optogenetic control of basal ganglia circuitry. Nature, 466, 622–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Song C.-H., Bernhard D., Bolarinwa C., Hess E.J., Smith Y. and Jinnah H.A. (2013) Subtle microstructural changes of the striatum in a DYT1 knock-in mouse model of dystonia. Neurobiol. Dis., 54, 362–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Song C.-H., Bernhard D., Hess E.J. and Jinnah H.A. (2014) Subtle microstructural changes of the cerebellum in a knock-in mouse model of DYT1 dystonia. Neurobiol. Dis., 62, 372–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang L., Yokoi F., Jin Y.-H., DeAndrade M.P., Hashimoto K., Standaert D.G. and Li Y. (2011) Altered dendritic morphology of Purkinje cells in Dyt1 ΔGAG knock-in and purkinje cell-specific Dyt1 conditional knockout mice. PLoS One, 6, e18357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Iafrati J., Orejarena M.J., Lassalle O., Bouamrane L. and Chavis P. (2014) Reelin, an extracellular matrix protein linked to early onset psychiatric diseases, drives postnatal development of the prefrontal cortex via GluN2B-NMDARs and the mTOR pathway. Mol. Psychiatry, 19, 417–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pelosi A., Menardy F., Popa D., Girault J.-A. and Hervé D. (2017) Heterozygous gnal mice are a novel animal model with which to study dystonia pathophysiology. J. Neurosci., 37, 6253–6267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Curiel J., Rodríguez Bey G., Takanohashi A., Bugiani M., Fu X., Wolf N.I., Nmezi B., Schiffmann R., Bugaighis M., Pierson T. et al. (2017) TUBB4A mutations result in specific neuronal and oligodendrocytic defects that closely match clinically distinct phenotypes. Hum. Mol. Gen., 26, 4506–4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Won S.J., Kim S.H., Xie L., Wang Y., Mao X.O., Jin K. and Greenberg D.A. (2006) Reelin-deficient mice show impaired neurogenesis and increased stroke size. Exp. Neurol., 198, 250–259. [DOI] [PubMed] [Google Scholar]
- 50. Zech M., Lam D.D., Francescatto L., Schormair B., Salminen A.V., Jochim A., Wieland T., Lichtner P., Peters A., Gieger C. et al. (2015) Recessive mutations in the α3 (VI) collagen gene COL6A3 cause early-onset isolated dystonia. Am. J. Hum. Genet., 96, 883–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Duncan I.D., Bugiani M., Radcliff A.B., Moran J.J., Anido C.L., Duong P., August B.K., Wolf N.I., Knaap M.S. and Svaren J. (2017) A mutation in the Tubb4a gene leads to microtubule accumulation with hypomyelination and demyelination. Ann. Neurol., 81, 690–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Liang C.-C., Tanabe L.M., Jou S., Chi F. and Dauer W.T. (2014) TorsinA hypofunction causes abnormal twisting movements and sensorimotor circuit neurodegeneration. J. Clin. Invest., 124, 3080–3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Weisheit C.E. and Dauer W.T. (2015) A novel conditional knock-in approach defines molecular and circuit effects of the DYT1 dystonia mutation. Hum. Mol. Gen., 24, 6459–6472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ullner P.M., Di Nardo A., Goldman J.E., Schobel S., Yang H., Engelstad K., Wang D., Sahin M. and De Vivo D.C. (2009) Murine Glut-1 transporter haploinsufficiency: postnatal deceleration of brain weight and reactive astrocytosis. Neurobiol. Dis., 36, 60–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Khan M.M., Xiao J., Patel D. and Ledoux M.S. (2017) DNA damage and neurodegenerative phenotypes in aged Ciz1 null mice. Neurobiol. Aging, 62, 180–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zeng B.-Y., Heales S.J.R., Canevari L., Rose S. and Jenner P. (2004) Alterations in expression of dopamine receptors and neuropeptides in the striatum of GTP cyclohydrolase-deficient mice. Exp. Neurol., 190, 515–524. [DOI] [PubMed] [Google Scholar]
- 57. Rose S.J., Yu X.Y., Heinzer A.K., Harrast P., Fan X., Raike R.S., Thompson V.B., Pare J.-F., Weinshenker D., Smith Y. et al. (2015) A new knock-in mouse model of l-DOPA-responsive dystonia. Brain, 138, 2987–3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Shashidharan P., Sandu D., Potla U., Armata I.A., Walker R.H., McNaught K.S., Weisz D., Sreenath T., Brin M.F. and Olanow C.W. (2005) Transgenic mouse model of early-onset DYT1 dystonia. Hum. Mol. Gen., 14, 125–133. [DOI] [PubMed] [Google Scholar]
- 59. Song C.-H., Fan X., Exeter C.J., Hess E.J. and Jinnah H.A. (2012) Functional analysis of dopaminergic systems in a DYT1 knock-in mouse model of dystonia. Neurobiol. Dis., 48, 66–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lee H.-Y., Nakayama J., Xu Y., Fan X., Karouani M., Shen Y., Pothos E.N., Hess E.J., Fu Y.-H., Edwards R.H. et al. (2012) Dopamine dysregulation in a mouse model of paroxysmal nonkinesigenic dyskinesia. J. Clin. Invest., 122, 507–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Nakagawasai O., Onogi H., Mitazaki S., Sato A., Watanabe K., Saito H., Murai S., Nakaya K., Murakami M., Takahashi E. et al. (2010) Behavioral and neurochemical characterization of mice deficient in the N-type Ca2+ channel alpha1B subunit. Behav. Brain Res., 208, 224–230. [DOI] [PubMed] [Google Scholar]
- 62. Dang M.T., Yokoi F., Cheetham C.C., Lu J., Vo V., Lovinger D.M. and Li Y. (2012) An anticholinergic reverses motor control and corticostriatal LTD deficits in Dyt1 ΔGAG knock-in mice. Behav. Brain Res., 226, 465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zhang L., Yokoi F., Parsons D.S., Standaert D.G. and Li Y. (2012) Alteration of striatal dopaminergic neurotransmission in a mouse model of DYT11 myoclonus-dystonia. PLoS One, 7, e33669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Deandrade M.P., Yokoi F., Groen T., Lingrel J.B. and Li Y. (2011) Characterization of Atp1a3 mutant mice as a model of rapid-onset dystonia with parkinsonism. Behav. Brain Res., 216, 659–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Calderon D.P., Fremont R., Kraenzlin F. and Khodakhah K. (2011) The neural substrates of rapid-onset Dystonia-Parkinsonism. Nat. Neurosci., 14, 357–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Fremont R., Tewari A. and Khodakhah K. (2015) Aberrant Purkinje cell activity is the cause of dystonia in a shRNA-based mouse model of rapid onset dystonia-parkinsonism. Neurobiol. Dis., 82, 200–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Carbon M., Kingsley P.B., Tang C., Bressman S. and Eidelberg D. (2008) Microstructural white matter changes in primary torsion dystonia. Mov. Disord., 23, 234–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Carbon M., Niethammer M., Peng S., Raymond D., Dhawan V., Chaly T., Ma Y., Bressman S. and Eidelberg D. (2009) Abnormal striatal and thalamic dopamine neurotransmission: Genotype-related features of dystonia. Neurology, 72, 2097–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Vemula S.R., Xiao J., Zhao Y., Bastian R.W., Perlmutter J.S., Racette B.A., Paniello R.C., Wszolek Z.K., Uitti R.J., Van Gerpen J.A. et al. (2014) A rare sequence variant in intron 1 of THAP1 is associated with primary dystonia. Mol. Genet. Genomic Med., 2, 261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Paudel R., Li A., Hardy J., Bhatia K.P., Houlden H. and Holton J. (2016) DYT6 dystonia: a neuropathological study. Neurodegener. Dis., 16, 273–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lakso M., Pichel J.G., Gorman J.R., Sauer B., Okamoto Y., Lee E., Alt F.W. and Westphal H. (1996) Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc. Natl. Acad. Sci. U. S. A., 93, 5860–5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kanki H., Suzuki H. and Itohara S. (2006) High-efficiency CAG-FLPe deleter mice in C57BL/6J background. Exp. Anim., 55, 137–141. [DOI] [PubMed] [Google Scholar]
- 73. Cvetanovic M., Kular R.K. and Opal P. (2012) LANP mediates neuritic pathology in Spinocerebellar ataxia type 1. Neurobiol. Dis., 48, 526–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Venkatraman A., Hu Y.-S., Didonna A., Cvetanovic M., Krbanjevic A., Bilesimo P. and Opal P. (2014) The histone deacetylase HDAC3 is essential for Purkinje cell function, potentially complicating the use of HDAC inhibitors in SCA1. Hum. Mol. Gen., 23, 3733–3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M. and Gingeras T.R. (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics, 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Anders S., Pyl P.T. and Huber W. (2015) HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics, 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Love M.I., Huber W. and Anders S. (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol, 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Tripathi S., Pohl M.O., Zhou Y., Rodriguez-Frandsen A., Wang G., Stein D.A., Moulton H.M., DeJesus P., Che J., Mulder L.C.F. et al. (2015) Meta- and orthogonal integration of influenza ‘OMICs’ data defines a role for UBR4 in virus budding. Cell Host Microbe, 18, 723–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Merico D., Isserlin R., Stueker O., Emili A. and Bader G.D. (2010) Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS One, 5, e13984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Huang D.W., Sherman B.T. and Lempicki R.A. (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res., 37,–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Huang D.W., Sherman B.T. and Lempicki R.A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc., 4, 44–57. [DOI] [PubMed] [Google Scholar]
- 82. Langley M.R., Ghaisas S., Ay M., Luo J., Palanisamy B.N., Jin H., Anantharam V., Kanthasamy A. and Kanthasamy A.G. (2018) Manganese exposure exacerbates progressive motor deficits and neurodegeneration in the MitoPark mouse model of Parkinson’s disease: relevance to gene and environment interactions in metal neurotoxicity. Neurotoxicology, 64, 240–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All materials will be made available upon request, and RNA sequencing data have been deposited in GEO under accession GSE123880.