

Abstract
Cardiovascular toxicities associated with immune checkpoint inhibitors (ICIs) have been reported in case series but have been underappreciated due to their recent emergence, difficulties in diagnosis and non-specific clinical manifestations. ICIs are antibodies that block negative regulators of the T cell immune response, including cytotoxic T lymphocyte-associated protein-4 (CTLA-4), programmed cell death protein-1 (PD-1), and PD-1 ligand (PD-L1). While ICIs have introduced a significant mortality benefit in several cancer types, the augmented immune response has led to a range of immune-related toxicities, including cardiovascular toxicity. ICI-associated myocarditis often presents with arrhythmias, may co-exist with myositis and myasthenia gravis, can be severe, and portends a poor prognosis. In addition, pericardial disease, vasculitis, including temporal arteritis, and non-inflammatory heart failure, have been recently described as immune-related toxicities from ICI. This narrative review describes the epidemiology, diagnosis, pathophysiology, and treatment of cardiovascular toxicities of ICI therapy, highlighting recent developments in the field in the past year.
Keywords: Cardio-oncology, Immune checkpoint inhibitors, Myocarditis, Vasculitis, Pericarditis, Cardiovascular toxicity
This article is part of the Spotlight Issue on Cardio-oncology.
1. Introduction
Over the past decade, cancer treatment has been revolutionized by the development of immunotherapy, a diverse set of strategies that treat cancer by generating or augmenting an immune response against cancer. Several branches of immunotherapy have emerged: immune-cell-targeted monoclonal antibody therapy, adoptive cellular therapy, non-specific cytokines, cancer vaccines, and immune checkpoint inhibitors (ICIs).1 ICIs have changed the treatment landscape for patients with a variety of cancer types by achieving unprecedented rates of durable anti-tumour response. Two prominent scientists who made seminal discoveries in this area were recently awarded the 2018 Nobel Prize in Physiology or Medicine.
When inhibitory receptors expressed on T lymphocytes, such as cytotoxic T lymphocyte-associated protein-4 (CTLA-4) and programmed cell death protein-1 (PD-1), bind to their corresponding ligands on tumour cells, such as programmed cell death 1 ligand-1 (PD-L1), the cellular immune response is ‘turned off’.2 Exploiting this process, tumour cells up-regulate expression of PD-L1 to escape recognition and evade destruction by the immune system.3 ICIs block these immune checkpoints, ‘turning back on’ the cellular immune response against tumour cells. ICIs have shown remarkable results in treating advanced metastatic cancers including melanoma, non-small cell lung cancer (NSCLC), renal cell carcinoma, head and neck squamous cell carcinoma, urothelial cancer, refractory Hodgkin’s lymphoma, and malignancies with microsatellite instability.2 Increasingly, ICI are being used in combination in order to increase anti-tumour activity.
The systemic augmentation of immune responses by ICIs, especially when used in combination, leads to a range of immune-related toxicities including colitis, hepatitis, pneumonitis, thyroiditis, myositis, hypophysitis, and dermatitis.4 These immune-mediated toxicities are largely reversible and can typically be controlled with administration of glucocorticoid therapy.5 However, emerging case reports have raised awareness of cardiovascular complications from ICI therapy.6–9 The aim of this review is to describe cardiovascular toxicities of ICI therapy, particularly myocarditis, that clinicians may encounter. Secondly, this review aims to discuss less-recognized forms of ICI-related cardiovascular toxicities including pericardial disease and vasculitis.
2. Physiology
2.1 T cell activation
In T cell-mediated immunity, neoantigens released by dead cancer cells are captured by antigen presenting cells (APCs) such as dendritic cells and presented on major histocompatibility complex (MHC) molecules (Figure 1).10 CD8+ T cells and CD4+ T cells recognize the neoantigen-MHC I and -MHC II complexes, respectively, become activated and migrate to the tumour bed, where they recognize cancer cells via the interaction between the neoantigen-MHC complex and T cell receptors (TCR) on the T cell. Activation of a T cell requires two signals: (1) First, the TCR recognizes a specific peptide presented by MHC on an APC.10 This binding initiates an intracellular signalling cascade in the T cell that is modulated by (2) a molecule on the surface of the T cell that can be co-stimulatory or co-inhibitory. CD28 is a co-stimulatory molecule, while PD-1 and CTLA-4 are co-inhibitory molecules. Cytokines and other molecules refine the function of T cells (particularly CD4+ T cells) by promoting their differentiation into T helper 1 (TH1), T helper 2 (TH2), or regulatory T (Treg) cells.
Figure 1.

Cell surface receptors and ligands at immune checkpoints. Neoantigens from cancer cells are captured by APCs such as dendritic cells and presented on MHC molecules. CD8+ T cells and CD4+ T cells recognize the neoantigen-MHC I and -MHC II complexes, respectively, in the lymph node (left half of figure), become activated and migrate to the tumour bed (right half of figure). Top left: The process of T cell activation can be modulated by co-stimulatory signals from binding of B7 with CD28. Alternatively, it can be modulated by co-inhibitory signals from the binding between cytotoxic T lymphocyte-associated protein-4 (CTLA-4) and programmed cell death protein-1 (PD-1) with their respective ligands, B7 and PD-1 ligand 1 (PD-L1). Binding of CTLA-4 with B7 inhibits RACα serine/threonine-protein kinase (AKT) via activation of type II serine threonine phosphatase 2A (PP2A). Binding of PD-1 with PD-L1 or PD-L2 leads to AKT inhibition via SRC homology 2 domain-containing tyrosine phosphatase 1 and 2 (SHP1 and SHP2) inhibition of phosphoinositide 3-kinase (PI3K). Inhibition of AKT results in a range of downstream effects including reduced effector T cell proliferation, T cell metabolism, cell survival factors and increased regulatory T cell proliferation. Top right: Tumour cells can detect high levels of interferon-γ (IFN-γ) in the tumour environment via IFN-γ receptors (IFN-γR). IFN-γR signals through Janus kinase (JAK) 1 and 2, which phosphorylate and thereby activate signal transducers and activators of transcription (STAT), which dimerize and lead to promotion of transcription of PD-L1. Increased cell surface expression of PD-L1 on tumour cells enacts a ‘brake’ on the ability of T cells to mount an attack on the tumour cells. This ‘brake’ can be released by a class of anti-tumour drugs known as ICIs. Bottom: Immune checkpoints can be blocked by monoclonal antibodies against CTLA-4 (ipilimumab), PD-1 (nivolumab, pembrolizumab, and cemiplimab) and PD-L1 (atezolizumab, avelumab, and durvalumab). Blocking of these immune checkpoints restores the T cell immune response against tumour cells. Tumour necrosis factor-α, granzyme B, and interferon-γ are released, resulting in tumour cell death.
2.2 CTLA-4 and PD-1
PD-1 is a co-inhibitory molecule expressed on T cells, activated natural killer cells, B cells, monocytes, and immature Langerhans’ cells. PD-1 can bind to the ligands PD-L1 and PD-L2. While PD-L2 expression is restricted to APCs such as macrophages and dendritic cells, PD-L1 is expressed by both lymphoid and non-lymphoid cells, including cardiac and endothelial cells.11 Both PD-L1 and PD-L2 are up-regulated by cytokines including interferons, tumour necrosis factor-alpha, and vascular endothelial growth factor (VEGF). When PD-1 binds to PD-L1 or PD-L2, the cytoplasmic domain of PD-1 is phosphorylated, recruiting SHP1 and SHP2 (Src homology 2 domain-containing protein tyrosine phosphatases) to dephosphorylate and inactivate ZAP70 (ζ-chain associated protein 70) and PI3K (phosphoinositide 3-kinase), leading to dampening of T cell migration and proliferation.
CTLA-4 is a co-inhibitory molecule that is expressed on conventional and regulatory T cells. CTLA-4 outcompetes CD28 (a co-stimulatory receptor) in binding to CD80 (also known as B7.1) or CD86 (also known as B7.2), with a 10-fold higher affinity than CD28 does. Like PD-1, CTLA-4 also inhibits the AKT pathway, but does so via type II serine threonine phosphatase 2 A (PP2A) rather than SHP1 and SHP2.
PD-1, PD-L1, and CTLA-4 are known as immune checkpoints as they are negative regulators of immune activation. Their binding is required for T cells to remain self-tolerant and for modulating the duration and degree of immune responses in peripheral tissues to attenuate peripheral tissue damage. This is underscored by genetic knockout models of PD-1 or CTLA-4 which manifest as systemic autoimmunity.12,13
2.3 Immune checkpoints in cancer
Cancer cells can disarm the T cell response by up-regulating PD-L1 expression. Sensing the presence of tumour-infiltrating T cells via an increase in interferon gamma (IFN-γ) concentrations in the tumour micro-environment, cancer cells can reactively up-regulate PD-L1 expression.3 This up-regulation occurs as binding of IFN-γ to IFN-γ receptors causes JAK1/2 (Janus kinase)14 to phosphorylate and activate STAT (signal transducers and activators of transcription) proteins to turn on interferon regulatory factor 1 (IRF-1), which binds to the promoter of PD-L1, leading to increased surface expression of PD-L1 on cancer cells.15,16 Binding of the up-regulated PD-L1 to PD-1 on CD8+ cells diminishes the T cell-mediated immune response. This allows cancer cells to evade attack from the immune system.
2.4 Immune checkpoint inhibitors
Because immune checkpoint molecules exert their effect via ligand-receptor interactions, they can be readily blocked by specific monoclonal antibodies called ICIs, thus permitting the T cell-mediated immune response to proceed against cancer cells.2 A total of seven ICIs have been approved for use in patients with various cancers including melanoma, NSCLC, and renal cell carcinoma (Table 1). These checkpoint inhibitors include inhibitors of CTLA-4, including ipilimumab; inhibitors of PD-1, including nivolumab, pembrolizumab, and cemiplimab; and inhibitors of PD-L1, including atezolizumab, avelumab, and durvalumab (Figure 2).17 Combination therapies have also been approved for use, including ipilimumab and nivolumab.18 Other immune checkpoints currently being studied for therapeutic potential are T cell immunoglobulin and mucin-containing protein 3 (TIM-3),19 lymphocyte-activated gene-3 (LAG-3),20 T cell immunoreceptor with Ig and ITIM domains (TIGIT),21 B and T lymphocyte attenuator (BTLA),22 V-domain Ig suppressor of T cell activation (VISTA, also known as PD-1 homologue, or PD-1H),23 and others.
Table 1.
ICIs and their Federal Drug Administration (FDA)-approved indications as of December 2018
| Checkpoint | Checkpoint inhibitor | Approved uses |
|---|---|---|
| CTLA-4 | Ipilimumab (Yervoy) |
|
| PD-1 | Nivolumab (Opdivo) |
|
| Pembrolizumab (Keytruda) |
|
|
| Cemiplimab (Libtayo) | Cutaneous squamous cell carcinoma (metastatic) | |
| PD-L1 | Atezolizumab (Tecentriq) |
|
| Avelumab (Bavencio) |
|
|
| Durvalumab (Imfinzi) |
|
|
| Checkpoints under investigation and that have not undergone FDA approval | ||
| TIM-3, LAG-3, TIGIT, BTLA, VISTA (PD-1H) | ||
CTLA-4, cytotoxic T lymphocyte-associated protein-4; PD-1, programmed cell death protein-1; PD-L1, programmed cell death 1 ligand-1; TIM-3, T cell immunoglobulin and mucin-containing protein 3; LAG-3, lymphocyte-activated gene-3; TIGIT, T cell immunoreceptor with Ig and ITIM domains; BTLA, B and T lymphocyte attenuator; VISTA, also known as PD-1 homologue, or PD-1H, V-domain Ig suppressor of T cell activation. Approved indications are up to date as of December 2018.
Figure 2.

Approved ICIs and indications. Monoclonal antibodies against CTLA-4 (ipilimumab), PD-1 (nivolumab, pembrolizumab, and cemiplimab) and PD-L1 (atezolizumab, avelumab, and durvalumab) have been approved by the FDA to treat patients with a variety of advanced and metastatic cancers. The period of clinical development is illustrated from the date of the first patient dosed (green dot), and until FDA approval for specific indications (brown dot). Each month of the year is represented by a vertical line (yellow). This figure extends earlier work by Ribas and Wolchok2 with indications approved by the FDA in 2018. RCC, renal cell carcinoma; MSI-H, high microsatellite instability; MMR-D, mismatch repair deficient; NSCLC, non-small cell lung cancer; CHL, classical Hodgkin’s lymphoma; HNC, head and neck cancer; CRC, colorectal cancer; HCC, hepatocellular carcinoma; GEJ, gastroesophageal junction; PMBCL, primary mediastinal large B cell lymphoma; SqCC, squamous cell carcinoma.
While blocking immune checkpoints can achieve tremendous tumour regression in some patients, the systemic activation of autoreactive T cells can damage off-target host tissues, causing a range of toxicities such as colitis, hepatitis, pneumonitis, thyroiditis, myositis, hypophysitis, and dermatitis.5 Conspicuously, cardiovascular toxicity has been underestimated and omitted from some major reviews of adverse effects of ICIs until recently.
3. Overview of immune-related adverse events from ICIs
In the largest study of immune-related cardiovascular adverse events to date, using VigiBase, the World Health Organization’s (WHO) global database of individual drug case safety reports, patients who received ICI had a reporting odds of myocarditis that was 11 times that of patients who did not receive ICI.24 Most remarkably, ICI treatment was also associated with other inflammatory cardiovascular adverse effects that have previously been underappreciated, particularly pericardial diseases and vasculitis (Table 2). Within vasculitis, temporal arteritis was highly over-represented. Note that in this pharmacovigilance study, the number of temporal arteritis reports among all ICI monotherapy drug case safety reports (the numerator) was compared with the number of temporal arteritis reports among all-class drug case safety reports (the denominator).
Table 2.
Reports of cardiovascular adverse events from ICIs in a pharmacovigilance study
| Reported drug-related adverse effect | Among immunotherapy reports (IMU; n: 31, 321) |
Among FULL database reports (n: 12, 455, 401) | PD1 vs. CTLA4 ROR and 95% CI | COMB vs. MONO ROR and 95% CI | IMU vs. FULL database ROR and 95% CI | ||
|---|---|---|---|---|---|---|---|
| MONO (n: 28, 909) |
COMB (n: 2, 412) | ||||||
| MONO-PD1 (n: 20, 643) | MONO-CTLA4 (n: 8, 266) | ||||||
| Myocarditis | 84 (0.41%) | 6 (0.07%) | 32 (1.3%) | 4, 454 (0.04%) | 5.62 [2.46-12.88] | 4.31 [2.86-6.38] | 11.21 [9.36-13.43] |
| Pericardial diseases | 74 (0.36%) | 13 (0.16%) | 8 (0.33%) | 10, 009 (0.08%) | 2.28 [1.27-4.12] | 1.1 [0.53-2.24] | 3.8 [3.08-4.62] |
| Vasculitis | 56 (0.27%) | 18 (0.22%) | 8 (0.33%) | 20, 987 (0.2%) | 1.25 [0.73-2.12] | 1.3 [0.62-2.67] | 1.56 [1.25-1.94] |
| Reports within vasculitis | |||||||
| Temporal arteritis | 7 (0.03%) | 10 (0.12%) | 1 (0.04%) | 568 (<0.01%) | 0.28 [0.11-0.74] | 0.71 [0.07-3.94] | 12.99 [8.12-20.77] |
| Polymyalgia rheumatica | 14 (0.07%) | 1 (0.01%) | 1 (0.04%) | 1254 (0.01%) | 5.61 [0.74-42.66] | 0.8 [0.08-4.62] | 5.13 [3.13-8.40] |
A recent study of 12 455 401 individual drug case safety reports (ICSRs) in the World Health Organization’s (WHO) VigiBase pharmacovigilance database found higher reporting of cardiovascular adverse events in users of ICIs compared with the full database. This table displays the reporting odds ratios (ROR) and 95% confidence intervals (CI), comparing selected cardiovascular adverse events in overall immunotherapy (IMU) vs. full database (FULL); combined immunotherapy (COMB) vs. mono-immunotherapy (MONO); mono-immunotherapy with anti-PD-1/PD-L1 (PD1) vs. mono-immunotherapy with anti-CTLA-4 (CTLA4) from VigiBase (time period: January 2008 to January 2018). Significant over-reporting is in marked in bold font, after Bonferroni adjustment for multiple tests within immunotherapy subgroups (p≤0.05/10 tests), which is P≤0.005). First reports of ICSRs associated with ICI started in 2008. Note that the database is a database of reports of adverse events from any system from any drug, not limited to ICIs. As this is a drug case safety report database, the database is not reflective of the general population. Modified with permission (Data from Salem, 2018).
Overall immunotherapy (IMU): Any individual case safety report related to nivolumab, pembrolizumab, atezolizumab, avelumab, durvalumab, ipilimumab or tremelimumab.
Mono-immunotherapy (MONO): Any individual case safety report related to any of the following seven drugs only when used alone (monotherapy): • Anti-PD-1/PD-L1 monotherapy: any of nivolumab, pembrolizumab, atezolizumab, avelumab, durvalumab used alone (MONO-PD1) • Anti-CTLA-4 monotherapy: ipilimumab or tremelimumab alone (MONO-CTLA4) • Combination immunotherapy (COMB): Any individual case safety report related to at least one drug from anti-PD-1/PD-L1 inhibitors combined to an anti-CTLA-4. ICSRs, individual case safety reports.
Non-inflammatory cardiovascular toxicities have been reported in individual cases. These include Takotsubo-like syndrome with both apical6,25,26 and basal variants27; asymptomatic non-inflammatory left-ventricular dysfunction28; myocardial infarction29; and coronary vasospasm.30 Arrhythmias have also emerged as a sign of cardiotoxicity in patients receiving ICI.24 However, arrhythmias are common in the cancer population and were shown to co-occur with other immune-related adverse events, suggesting that they may not necessarily be a direct effect of ICI itself.24 This is particularly true with acute thyrotoxicosis secondary to ICI-mediated thyroiditis and atrial fibrillation. Case reports of ICI-associated third-degree atrioventricular block and conduction disease were often assessed to be secondary to myocarditis involving the conduction system.7,31,32
Given the prevalence and clinical importance of these events, this review will focus on the triad of myocarditis, pericardial disease, and vasculitis as the predominant cardiovascular toxicities of ICI (Figure 3). Notably, cases of ICI-associated myocarditis, pericarditis, and vasculitis rarely overlap.24
Figure 3.

Cardiovascular toxicity from ICIs. Cardiovascular toxicity from ICIs can come in the form of myocarditis, pericardial disease, or vasculitis. These presentations are almost never overlapping. Less commonly, it can also come in the form of Takotsubo-like syndrome and other non-inflammatory left-ventricular dysfunction. Angiography of Takotsubo-like syndrome reproduced with permission from Elsevier from Anderson and Brooks.25
4. ICI-associated myocarditis
4.1 Epidemiology
4.1.1 Incidence and timeline
While myocarditis was not prospectively assessed in randomized controlled trials of ICIs, existing retrospectively assessed literature estimates the incidence of ICI-associated myocarditis to be between 0.27%7 and 1.14%.33 Most cases occur early, with a, median time to onset of toxicity of 30 days (inter-quartile range [IQR]: 18–60) for myocarditis, which is approximately after the first or second ICI infusion.24
4.1.2 Combination immunotherapy as the predominant risk factor
Receipt of combination ICI therapy (e.g. a CTLA-4 inhibitor combined with a PD-1 inhibitor) is the most well-established risk factor for ICI-associated myocarditis.7 In a retrospective database analysis, the combination of nivolumab and ipilimumab conferred a 4.74-fold risk of developing myocarditis compared with treatment with nivolumab alone.7 Myocarditis resulting from combination ICI therapy is also more likely to be severe (grade 3 or 4) compared with myocarditis arising from ICI monotherapy.7 Myocarditis from combination ICI therapy is also associated with increased rates of co-occurring myasthenia gravis and myositis, and has a higher fatality rate compared with myocarditis from ICI monotherapy.34 It remains unclear whether the incidence of myocarditis is higher when ICIs are combined with other non-ICI cancer therapies. In a trial of 55 patients receiving avelumab and axitinib (a VEGF inhibitor), 1 patient (2%) developed fatal myocarditis.35 A pre-clinical study in mice showed that PD-1 blockade in combination with cardiac irradiation increases the risk of myocarditis and mortality beyond the risk from cardiac irradiation alone.36 Although the specific mechanism for toxicity from combination immunotherapy remains to be elucidated, it is possible that non-immune myocyte injury may lead to exposure of cardiac antigens and subsequent T cell responses which is unmasked upon ICI administration. This hypothesis remains to be tested.
4.1.3 Other risk factors
Apart from combination ICI therapy, few other risk factors for ICI-associated myocarditis have been established. There appears to be no age predisposition, with cases occurring in patients across a wide age range (20–90 years).24,26,33,37 Interestingly, toxicity reports indicate a potential male predominance of this phenomenon (66.7%),24 but this does not necessarily represent a predisposition of males compared with females, as males were over-represented at baseline in both ICI use and in clinical trial enrolment.38 In the future, a critical issue will be to identify the patients at greatest risk of myocarditis. Theoretical risk factors include other treatments (e.g. use of other cardiotoxic anti-neoplastic agents such as anthracyclines), underlying cardiovascular disease with or without previous myocardial injury (e.g. prior myocardial infarction, heart failure, myocarditis, or previous cancer therapy-induced left-ventricular dysfunction), underlying autoimmune disease (e.g. lupus, rheumatoid arthritis, sarcoidosis, Dressler’s syndrome), tumour-related factors (cardiac antigens expressed in tumour, activation of T cell clones directed at [self] cardiac antigens), concurrent immune-related toxic effects (ICI-related skeletal myositis), and genetic factors.39
4.1.4 Prognosis
Current data suggest that the prognosis of ICI-associated myocarditis is poor. In the aforementioned retrospective pharmacovigilance study, the incidence of fatality in ICI-associated myocarditis was 50%.24 Among myocarditis cases, fatality was more frequent with ICI combination therapy (65.6%) than ICI monotherapy (44.4%).24 This unfavourable prognosis may partly be due to reporting bias since early cases of ICI-associated myocarditis cases have been fulminant. This pattern of reporting bias may also extend to other cardiovascular toxicities.
4.2 Presentation
4.2.1 Overview of manifestations
Clinical presentation of myocarditis includes signs of acute heart failure (although the degree of systolic dysfunction can vary), manifesting clinically as chest pain, shortness of breath, pulmonary oedema, and even cardiogenic shock.40 It can also present with arrhythmias which can lead to syncope and sudden death.40 ICI-associated myocarditis can present similarly.7 However, early data suggest a higher risk of arrhythmias, including heart block (Figure 4) and both atrial and ventricular arrhythmias.33 In addition, about half of the patients have no evidence of reduced ejection fraction.33 In ICI-associated myocarditis, serum cardiac biomarkers such as cardiac troponin and creatine kinase-muscle/brain (CK-MB) are almost always elevated, although the positive predictive value of each varies, as detailed below.33 Evidence of myocardial inflammation can often be seen on cardiac magnetic resonance imaging (cMRI), cardiac PET/CT, or most specifically on endomyocardial biopsy. Early histopathological examination revealed an infiltrate of abundant CD4+ and CD8+ T cell and CD68+ macrophages in the myocardium, the cardiac conduction system, and skeletal muscle (Figure 5).7
Figure 4.

Electrocardiogram findings from ICI-associated myocarditis. In one patient, ECG initially showed (A) sinus bradycardia with first degree atrioventricular block, leading to (B) Sinus bradycardia with complete atrioventricular block and left bundle branch block, leading to (C) ventricular tachycardia, resulting in death. The patient was a 65-year-old woman who received one dose of nivolumab combination therapy with ipilimumab.
Figure 5.

Histologic findings from ICI-associated myocarditis. (A–C) Photomicrographs of the myocardium, specifically, the interventricular septum. (A) Dense mononuclear cell infiltrate with extensive myocyte damage and necrosis, consistent with lymphocytic myocarditis, H&E stained section. (B) Abundance of CD3-positive T cells, CD3 immunohistochemical stain. (C) Prominent CD68-positive macrophage infiltrate, CD68 immunohistochemical stain. (D–E) are photomicrographs of skeletal muscle. (D) Dense mononuclear cell infiltrate and evidence of myocyte necrosis and damage with nuclear internalization, demonstrating myositis, H&E stained section. (E) Abundance of CD-3 positive T cells, CD3 immunohistochemical stain. (F) Photomicrograph of metastatic focus of melanoma with a lymphocytic infiltrate, H&E stained section. All histology displayed at 40× original magnification. All scale bars in this figure denote 50 µm. The patient was a 65-year-old woman who received one dose of nivolumab combination therapy with ipilimumab.
4.2.2 Subtleties of ICI-associated myocarditis manifestations
Some patterns have emerged from the two largest cohorts of ICI-associated myocarditis assembled to date.33,37
Notably, a normal ejection fraction does not rule out ICI myocarditis. Echocardiography may show diffuse left-ventricular systolic function, changes in cardiac chambers, geometry or regional wall motion abnormalities (WMAs). Preservation of normal cardiac dimensions may be suggestive of an acute process, whereas remodelling and dilatation suggest a chronic process. In a case control study, although left-ventricular systolic dysfunction was common, severe systolic dysfunction (left-ventricular ejection fraction <35%) was present in less than half of cases. Therefore, ICI-associated cardiotoxicity should still be kept in mind in patients with mild to normal left-ventricular systolic dysfunction (35–55%).33 In a case series, rescue and recovery of left-ventricular systolic dysfunction was shown to be possible, with complete recovery of left-ventricular function observed in half of the surviving patients in one cohort.37
cMRI is superior to echocardiography as it provides better tissue characterization both with and without gadolinium contrast. Features of myocarditis on cMRI include oedema, necrosis, and scar formation, detailed previously as the Lake Louise Criteria.41 In the case control study, cMRI was able to detect myocardial oedema in one-third of cases, suggesting that cMRI might lack the sensitivity to detect myocarditis, although this may also reflect cases of ICI-mediated non-inflammatory LV dysfunction mislabeled as myocarditis.33 In patients where MRI is contraindicated or not available, a cardiac FDG-PET-CT is an alternative imaging modality that can be used to assess for inflammation.
Myocarditis is often accompanied by elevations in serum biomarkers of myocardial injury and stress. Biomarkers are not highly specific for myocarditis, but are useful as one element of a comprehensive set of tests for ICI-associated myocarditis. In the case control study, troponin was elevated in 94% of ICI-associated myocarditis cases, and NT-proBNP was elevated in 66% of cases.33 In the case series, troponin was elevated in 46% of cases, and brain natriuretic peptide (BNP) or N-terminal proBNP levels were increased in 100% of cases.37 However, biomarker data were only available for 14 out of 30 patients in this latter study. At present, troponin is considered more specific for myocarditis whereas BNP is a marker of LV strain which can be elevated in non-inflammatory LV dysfunction and other causes of acute cardiac stress. BNP also may chronically elevated in many cancer patients due to cancer-related inflammation and would be poorly specific.42 Of note, cardiac troponin T and creatine phosphokinase may also be elevated from myositis; for this reason troponin I is preferred for establishing cardiac injury.43 Elevated serum biomarkers may also have prognostic implications. In the case control study, higher levels of serum cardiac troponin T were associated with a greater risk of major cardiovascular events.33
Myocarditis may be associated with additional toxicities. If the inflammatory infiltrates encroach upon the conduction system, electrocardiogram (ECG) may show intraventricular conduction delay, PR interval prolongation, and eventually complete heart block.7 Early data suggest that ICI-associated myocarditis can present with various forms of arrhythmia including atrial fibrillation, ventricular arrhythmias and conduction disease.37 Among patients with ICI myocarditis, 25% had concomitant myositis and 10–11% had concomitant myasthenia gravis.24,34 Patients receiving ICI who present with myositis or myasthenia gravis should be assessed for ICI-associated myocarditis.
4.2.3 Diagnostic categories
ICI-associated myocarditis can be difficult to diagnose given the multitude of tests needed to exclude alternative diagnoses. In a proposed consensus statement (Bonaca et al., unpublished results), ICI-associated myocarditis is defined as definite myocarditis, probable myocarditis, or possible myocarditis based on evidence from different diagnostic modalities (Table 3). The gold standard for diagnosis of general myocarditis is histopathological evidence on endomyocardial biopsy or autopsy,44 although false negatives may occur from sampling error.45 If biopsy cannot be obtained or is inconclusive, diagnosis may be made by a combination of cardiac biomarkers, cardiac imaging, and symptoms, as detailed in the table. Isolated troponinemia, in the absence of symptoms, is no longer sufficient for diagnosing myocarditis, according to the fifth edition of the Common Terminology Criteria for Adverse Events.46
Table 3.
Different modalities for diagnosing ICI-associated myocarditis, as proposed by Bonaca et al. (unpublished results)
| Modality | Tissue pathology on biopsy or autopsy (gold standard) | cMRI | New WMA on echocardiogram | New elevated biomarker beyond baseline |
|---|---|---|---|---|
| Definite myocarditis is any of: | Pathology sufficient | cMRI plus:
|
WMA plus:
|
|
| Probable myocarditis is any of: |
|
WMA plus:
|
||
| Possible myocarditis is any of: | Non-diagnostic cMRI without syndrome, biomarker, or ECG | WMA plus:
|
Biomarker plus:
|
Myocarditis can be diagnosed by one of several modalities, in decreasing order of superiority: tissue pathology on biopsy or autopsy; cMRI; echocardiogram showing new WMA; or elevated biomarkers. In each of these modalities, positive findings must be supported with a combination of objective laboratory findings, physical exam, and pertinent history. Tissue pathology diagnostic of myocarditis is the gold standard and, by itself, establishes a diagnosis of definite myocarditis.
cMRI positive for myocarditis is considered definite myocarditis if accompanied by biomarker elevations and positive ECG findings. If positive cMRI is accompanied by neither physical exam and history findings, biomarker elevations, nor ECG findings, the diagnosis is probable myocarditis. If cMRI is suggestive of myocarditis but non-diagnostic, the diagnosis can still be probable myocarditis if there are physical exam and history findings, elevated biomarkers, or ECG findings. On the other hand, if the non-diagnostic suggestive cMRI is accompanied by none of these, the diagnosis would be limited to possible myocarditis.
Using echocardiography, new WMA not explained by another diagnosis is considered definite myocarditis if it is accompanied by physical exam and history findings, elevated biomarkers, ECG findings, and negative angiography or other testing to exclude coronary artery disease. New WMA with physical exam and history findings and either elevated biomarkers or ECG findings are consistent with probable myocarditis. New WMA with either physical exam and history findings or ECG findings is consistent with possible myocarditis. If biomarkers are the lone studies available and positive, the diagnosis of possible myocarditis can still be made if there are physical exam and history findings and ECG findings.
cMRI, cardiac magnetic resonance imaging; WMA, wall motion abnormality.
ICI-associated myocarditis can additionally be clinically categorized as either fulminant, clinically significant, or subclinical. Fulminant myocarditis refers to myocarditis with concomitant haemodynamic and/or electrical instability; subclinical myocarditis refers to myocarditis that was not recognized or treated, with no evidence of clinical consequence.
Evidence of a causal relationship may be established using the nine Bradford Hill criteria.47 However, many of these criteria, such as removal and re-challenge of the agent, are often not feasible in patients. The determination of whether myocarditis is related to ICI therapy should be made by an assessment of temporality and consideration of alternative exposures and explanations for acute cardiac dysfunction.
4.2.4 Mechanisms of toxicity
At present, there are several proposed mechanisms by which end-organs experience immune-related toxicity from ICIs. ICIs may cause end-organ damage via direct ICI binding to CTLA4 expressed on these tissues; by permitting the T cell response, which may inadvertently recognize antigens in off-target tissues with high homology to tumour antigens; by increasing levels of circulating cytokines in off-target tissues; or by promoting the formation of autoantibodies against off-target tissues.48 Although not demonstrated in the cardiovascular system yet, susceptibility to checkpoint blockade toxicity may be modulated by the composition of microbiota.49 Data from animal models and human studies provide insights into underlying mechanisms for ICI-related cardiovascular toxicity.
Pre-clinical models using transgenic mice suggest a critical role for immune checkpoints, including CTLA-4 and PD-1/PD-L1 signalling, in the myocardium. Inflammation is especially deleterious in this context, due to the myocardium’s lack of redundancy and inability to regenerate.50 The integrity of PD-1, PD-L1, and CTLA-4 signalling is critical to downregulating excessive immune responses in the myocardium. Notably, the specific presentation of myocarditis in mice depends on the genetic background of the mice. PD-1 deficient mice (in BALB/c genetic background) developed dilated cardiomyopathy and premature mortality51 as a result of high-titre IgG autoantibodies to cardiac troponin I, augmenting the voltage-dependent L-type calcium current of normal cardiomyocytes.52 Mice (in C3H/He background) given PD-1 or PD-L1 inhibitors experienced greater myocardial injury from coxsackievirus B3, whereas mice given PD-1 or PD-L1 inductors experienced reduced myocardial injury from coxsackievirus B3.53 PD-1 deficient mice (in MRL background) developed high-titre autoantibodies to cardiac myosin and died from lymphocytic myocarditis characterized by myocardial infiltration of CD4+ and CD8+ T cells and myeloid cells.54 CTLA-4-deficient mice (in BALB/c and C57BL/6 backgrounds) developed lymphoproliferative myocarditis in the context of systemic inflammation and fatal multi-organ failure.55 However to date autoantibodies have not been detected in patients with ICI-mediated myocarditis, and there is no evidence at the present time that it is a B-cell-dependent inflammatory pathophysiology. These animal studies are further detailed by Grabie and colleagues56 in this Spotlight Issue.
Furthermore, PD-L1 was found to be expressed on the cell surface of injured cardiomyocytes, but not on skeletal muscle cells or tumour cells, in a case report of two patients with fatal fulminant ICI-associated myocarditis.7 PD-L1 is up-regulated in the myocardium as a cytokine-induced protective mechanism to curtail to T cell-mediated inflammation in states of cardiac stress and disease.57 During time of cardiac injury, cardiomyocyte death may expose cardiac antigens to T lymphocytes, and up-regulation of immune checkpoints (such as PD-1/PD-L1) may serve as an inhibitory signal for T cell invasion. ICIs abrogate this protective measure of the heart. Recent investigations have revealed the unexpected role of PD-1/PD-L1, CTLA-4, and other immune checkpoints in the pathogenesis of atherosclerosis.58 Investigations in the coming years will provide deeper insight into the mechanism of immune checkpoint cardiovascular toxicity.
4.3 Treatment
4.3.1 Treatment principles
The treatment strategies for ICI-related cardiovascular complications are tripartite: holding ICI to prevent further toxicity, immunosuppression to alleviate inflammatory changes, and supportive therapy to address cardiac complications. Because of the complexities involved and the limited data available, management of ICI cardiovascular toxicity should be carried out via a discussion between the primary treatment team, oncologist, and cardiologist or cardio-oncologist, if available. It is also imperative to establish the diagnosis of myocarditis as definitively as possible. These patients are best served in a coronary care unit or level 2 high-dependency unit for continuous ECG and haemodynamic monitoring.39,59 Of note, current treatment strategies have been extrapolated from standard-of-care treatment paradigms for patients with these conditions in non-ICI settings. Clinicians should bear in mind that these treatment regimens are empiric and have not yet been put to test in any prospective cohort or randomized controlled trial.
Immunosuppressive treatment for ICI-associated myocarditis requires high-dose corticosteroids, for which the specific regimen varies by practice. Current American Society for Clinical Oncology (ASCO)/NCCN (National Cancer Control Network) guidelines advise 1–2 mg/kg of prednisone intravenously or orally, with consideration in refractory cases for intravenous methylprednisolone at 500–1000 mg.59 A recent review suggests that administration of intravenous methylprednisolone at 500–1000 mg daily until the patient is clinically stable, followed by oral prednisolone at 1 mg/kg daily tapered over 4–6 weeks.39 For cases that are unresponsive to corticosteroids, both recommendations suggest consideration of mycophenolate mofetil or infliximab.39,59 It should be noted that infliximab may be potentially linked to worsening heart failure and is contraindicated in patients with moderate-to-severe heart failure.60 Given the histological similarities between ICI-associated myocarditis and cardiac transplantation rejection, anti-transplant rejection medications [such as anti-thymocyte globulin (ATG)] have been used in one case.61 Rituximab, a CTLA-4 analogue, is an experimental treatment that may be considered in select cases in which the toxicity is related to receipt of a CTLA-4 inhibitor.
Given the prevalence of conduction disease in ICI-associated myocarditis, patients should be monitored closely and percutaneous pacers may be placed at the patient’s bedside in anticipation of a bradyarrhythmia, perhaps caused by infiltration of the conduction system by T cells. Clinicians should have a low threshold for introducing pacing since atrioventricular block is common in the setting of myocarditis.
The safety of ICI re-challenge following ICI-related cardiovascular toxicity is unknown. The threshold for ICI re-challenge should be much higher after ICI-associated myocarditis compared with after ICI-associated pericardial disease or vasculitis, given the high fatality rate of ICI-associated myocarditis.
4.3.2 Surveillance and stepwise management
Surveillance for ICI-related cardiovascular toxicity is important given its severe and life-threatening nature, but there are currently no prospective trial data on which to base a formal surveillance algorithm. Instead, we shall detail the protocol used at Vanderbilt (Figure 6). Prior to initiating ICI therapy, we obtain a cardiac history and assess cardiovascular risk factors in all patients. In patients at increased risk of myocarditis, such as those receiving combination immunotherapy, we perform screening with cardiac troponin levels and an ECG prior to ICI initiation. After ICI initiation, we perform surveillance with troponin levels on a weekly basis for 6 weeks. since fulminant myocarditis occurs a median of 30 days after initiation of ICI.24 Patients with troponinemia prior to ICI initiation are assumed to have non-ICI-related injury. Patients with troponinemia after ICI initiation are worked up as follows.
Figure 6.

Management of ICI-associated myocarditis at Vanderbilt University Medical Center. Depicted is the screening and surveillance algorithm used at Vanderbilt for patients with increased risk of developing ICI-associated myocarditis, namely, patients on combination immunotherapy. ATG, anti-thymocyte globulin; CCU, coronary care unit; CHF, congestive heart failure; CK, creatine kinase; CK-MB, creatine kinase-muscle/brain; cMRI, cardiac magnetic resonance imaging; EKG, electrocardiogram; ICI, immune checkpoint inhibitor; IV, intravenous; MI, myocardial infarction. Icons from Servier Medical Art and the Noun Project are shared under a Creative Commons Attribution 3.0 Unported License.
In patients with elevated troponin on surveillance, we check a 12-lead ECG and other biomarkers such as CK-MB and serum CK for evidence of concomitant myositis. Patients are referred to a cardiologist or cardio-oncologist, who may obtain an echocardiogram to assess for WMAs or cMRI to further evaluate for structural cardiac abnormalities. Some providers obtain an echocardiogram prior to ICI initiation in high-risk patients in order to document baseline cardiac function. If work-up shows no evidence of myocarditis or cardiac dysfunction, then a serum troponin is rechecked. If troponinemia is isolated and asymptomatic, we consider resuming ICI therapy if the troponin returns to normal within 2 weeks.
If the patient complains of symptoms (such as chest pain, dyspnoea, palpitations, presyncope, or syncope), or if the above studies are abnormal, we hold the ICI and admit the patient to the hospital floor with cardiac monitoring and a cardiology consultation. Myocardial infarction and other aetiologies should be ruled out. We will consider endomyocardial biopsy to confirm findings on echocardiogram or cMRI. We will start intravenous methylprednisolone 2 mg/kg/day. If there is no improvement in 24 h, or the patient is unstable (hypotension, arrhythmia, or sudden decrease in ejection fraction), we will transfer the patient to the cardiovascular intensive care unit and begin intravenous methylprednisolone at 1 g daily. If the patient shows signs of improvement in 24 h, we will taper the methylprednisone over a minimum of 4 weeks. If the patient continues to be unstable, secondary agents are considered, including ATG, tacrolimus, infliximab, mycophenolate mofetil, and rituximab.
5. ICI-associated pericardial disease
ICI-associated pericardial disease can present as pericarditis, 9,62 pericardial effusion,9,63,64 or cardiac tamponade.62,63,65 Perhaps due to under-recognition as an adverse effect of ICI, pericardial disease has been limited to case reports and has not been subject to a systematic review. Pericarditis can present as chest pain relieved by forward position on exam, new pericardial effusion on echocardiogram, new PR depression and diffuse saddle-shaped ST elevation on ECG, or active pericardial inflammation on cMRI (Figure 7) or cardiac PET/CT.66 In non-ICI settings, pericarditis is diagnosed by the presence of at least two of the following criteria: sharp pleuritic chest pain relieved by sitting up and leaning forward; presence of a pericardial friction rub on auscultation; widespread ST segment elevation on ECG; and new or worsening pericardial effusion on echocardiogram.66 It remains to be investigated whether the prevalence of these manifestations differs between general pericarditis and ICI-associated pericarditis. Pericarditis and pericardial effusion can lead to cardiac tamponade, which is a life-threatening complication.
Figure 7.
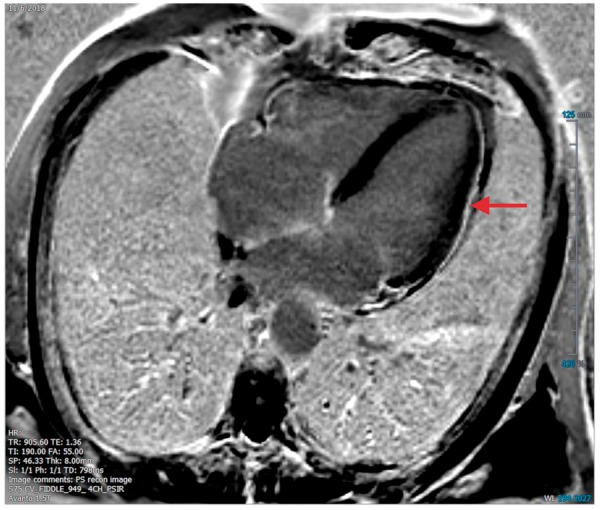
Imaging findings from ICI-associated pericardial disease. cMRI of the heart demonstrating focal myocardial delayed gadolinium enhancement in the mid-lateral wall and mild late enhancement of the pericardium along the lateral wall suggestive of myopericarditis. The patient is a 70-year-old male who had received six doses enoblituzumab (an IgG antibody against B7-H3).
There are no available estimates of the incidence of ICI-associated pericardial disease. A systemic evaluation of 95 cases of ICI-associated myocarditis presented to the WHO database of presumed ICI-associated pericardial disease suggests a median time to onset of toxicity of 30 days (IQR: 8.5–90), after a median of two ICI administrations.24 As in myocarditis, toxicity reports have been more common in males than females for pericardial disorders (60.0% male), 24 but as with myocarditis, this may simply reflect the demographics of ICI use and clinical trial enrolment.38 There is no age predisposition.24 The incidence of fatality was 21.1% in ICI-associated pericardial disease,24 although it remains difficult to assess the direct contribution of pericardial disease to this fatality rate.
To date, there have been no specific studies investigating the mechanism of pericardial disease from ICIs. This is further limited by the lack of available mouse models for pericardial disease.
In addition to interrupting ICI administration, current practice is to initiate immunosuppression with prednisone 1 mg/kg daily (or equivalent) with subsequent taper,39 although no specific tapering protocol has been established. For cardiac supportive therapy, colchicine and NSAIDs should be administered based on guidelines in non–ICI-associated pericarditis.66 Emergency pericardiocentesis and haemodynamic support should be considered if cardiac tamponade is also present.
6. ICI-associated vasculitis
ICI-associated vasculitis can affect vessels of any size,67–73 but has most commonly been reported in large vessels such as temporal (giant cell) arteritis and aortitis, according to a systematic review of case reports.74 The over-representation of temporal arteritis reports among ICI users was also recently identified in a pharmacovigilance study.24 Temporal arteritis is an autoimmune and autoinflammatory disease of the aorta and its branches. It presents with headache, jaw claudication, transient monocular visual loss (amaurosis fugax) or diplopia, and systemic symptoms such as fatigue, fever, and weight loss. Patients have elevated inflammatory markers such as erythrocyte sedimentation rate and C-reactive protein.75 The gold standard for diagnosing temporal arteritis is temporal artery biopsy, which would demonstrate CD4+ T cells and macrophages organized in granulomas (Figure 8). Colour Doppler ultrasonography of the temporal artery is a non-invasive alternative, but is not definitive. It remains to be investigated whether the predictive values of manifestations differ between general vasculitis and ICI-associated vasculitis.
Figure 8.

Histologic findings from ICI-associated vasculitis. (A) Photomicrograph of myocardium with lymphocytic myocarditis and vasculitis, with obliteration of the lumen of this small artery. H&E stained section, 200× original magnification, with scale bar denoting 50 µm. (B) Photomicrograph of small artery with fibrinoid necrosis and perivascular chronic inflammation. H&E stained section, 400× original magnification, with scale bar denoting 20 µm.
There are no available estimates of the incidence of ICI-associated vasculitis. Median time to onset of toxicity is 55 days (IQR: 21–98), after a median of three ICI administrations.24 Like in myocarditis, toxicity reports have been more common in males than females for vasculitis (58.4% male),24 and there is no age predisposition.24,38 The incidence of fatality is 6.1% in ICI-associated vasculitis.24
The mechanism by which this phenomenon occurs may involve checkpoint pathway deficiency in temporal arteries predisposing the walls of these arteries to autoimmune attack.76 In temporal arteritis, affected temporal arteries have deficient expression of PD-1 and PD-L1.77 PD-1 and PD-L1 deficiency allows aggregation of T cells and production of cytokines leading to arterial wall inflammation and pathogenic remodelling.76 It is likely that the use of ICIs might indeed reproduce a similar immune environment, predisposing to temporal arteritis that can even lead to blindness.24,78
In addition to interrupting ICI administration, immunosuppressive treatment should be initiated without delay for temporal artery biopsy scheduling, due to the risk of visual loss. Current practice is to initiate immunosuppression according to existing consensus statements for temporal arteritis treatment. According to existing consensus statements, patients with threatened or established visual loss should initiate immunosuppression with intravenous methylprednisolone at 500–1000 mg daily for 3 days before switching to oral corticosteroids.79 Patients without visual loss should initiate immunosuppression with oral prednisone at 40–60 mg, given as a single daily dose.79 Steroid tapering can be done at 2-week intervals in decrements in accordance to existing consensus statements for temporal arteritis treatment.79
7. Conclusion
In summary, ICIs are associated with cardiovascular toxicities such as myocarditis, pericardial disease, and vasculitis such as temporal arteritis. These immune-related adverse effects require prompt immunosuppressive treatment and ICI discontinuation, particularly considering high fatality rates.80 Given that there are almost 600 000 patients in the USA alone eligible for ICI therapy, and considering that the use of ICI is projected to rise in the coming years,81 it is crucial that deeper collaborations be forged among cardiology, oncology, and immunology in clinical practice and basic science82 in order to better recognize and understand the cardiovascular toxicities of ICI therapies.
Conflict of interest: E.J.L. is a consultant for Bristol-Myers Squibb (BMS), Novartis, EMD Serono, Array BioPharma, Macrogenics, Merck, and Millennium, and receives research Funding from BMS, Merck, and Sysmex. E.J.L. has a patent application pending, entitled ‘Method of preventing organ transplant rejections using agonists to the PD-1 checkpoint pathway’. J.N. receives research funding from Merck, AstraZeneca, and Medimmune; consulting fees from AstraZeneca, Medimmune, BMS, Genentech, and Roche; and honoraria from AstraZeneca, Medimmune, and BMS. C.G.T. has grants from Federico II University-Ricerca d'Ateneo and personal fees from speaker honoraria from Alere, unrelated to this paper. C.G.T. also holds Canadian patent no. 2,613,477: ‘Thiol Sensitive Positive Inotropes’, issued on 3 December, 2013, with royalties paid. A.R.L. has received speaker, advisory board or consultancy fees and/or research grants from Pfizer, Novartis, Servier, Amgen, Clinigen Group, Takeda, Roche, Eli Lily and Boehringer Ingelheim. D.B.J. sits on advisory boards for Bristol-Myers-Squibb, Array Biopharma, Incyte, Merck, and Genoptix, and receives research funding from BMS and Incyte. J.M. receives consulting fees from Pfizer, Takeda/Millennium, Ariad, Bristol-Myers Squibb, Acceleron, Vertex, Incyte, and Verastem.
Funding
This work was supported by NIH R56HL141466 (to J.M.).
References
- 1. Farkona S, Diamandis EP, Blasutig IM.. Cancer immunotherapy: the beginning of the end of cancer? BMC Med 2016;14:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ribas A, Wolchok JD.. Cancer immunotherapy using checkpoint blockade. Science 2018;359:1350–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, Chen S, Klein AP, Pardoll DM, Topalian SL, Chen L.. Colocalization of inflammatory response with B7-H1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med 2012;4: 127ra37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Johnson DB, Chandra S, Sosman JA.. Immune checkpoint inhibitor toxicity in 2018. JAMA 2018;320:1702–1703. [DOI] [PubMed] [Google Scholar]
- 5. Friedman CF, Proverbs-Singh TA, Postow MA.. Treatment of the immune-related adverse effects of immune checkpoint inhibitors: a review. JAMA Oncol 2016;2:1346–1353. [DOI] [PubMed] [Google Scholar]
- 6. Geisler BP, Raad RA, Esaian D, Sharon E, Schwartz DR.. Apical ballooning and cardiomyopathy in a melanoma patient treated with ipilimumab: a case of takotsubo-like syndrome. J ImmunoTher Cancer 2015;3:4.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Johnson DB, Balko JM, Compton ML, Chalkias S, Gorham J, Xu Y, Hicks M, Puzanov I, Alexander MR, Bloomer TL, Becker JR, Slosky DA, Phillips EJ, Pilkinton MA, Craig-Owens L, Kola N, Plautz G, Reshef DS, Deutsch JS, Deering RP, Olenchock BA, Lichtman AH, Roden DM, Seidman CE, Koralnik IJ, Seidman JG, Hoffman RD, Taube JM, Diaz LA, Anders RA, Sosman JA, Moslehi JJ.. Fulminant myocarditis with combination immune checkpoint blockade. N Engl J Med 2016;375:1749–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Läubli H, Balmelli C, Bossard M, Pfister O, Glatz K, Zippelius A.. Acute heart failure due to autoimmune myocarditis under pembrolizumab treatment for metastatic melanoma. J ImmunoTher Cancer 2015;3:11.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yun S, Vincelette ND, Mansour I, Hariri D, Motamed S, Late onset ipilimumab-induced pericarditis and pericardial effusion: a rare but life threatening complication. Case Rep Oncol Med2015;2015:794842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Murphy KM, Weaver C, Janeway’s Immunobiology. 9th ed New York, NY: W. W. Norton & Company; 2016. [Google Scholar]
- 11. Eppihimer MJ, Gunn J, Freeman GJ, Greenfield EA, Chernova T, Erickson J, Leonard JP.. Expression and regulation of the PD-L1 immunoinhibitory molecule on microvascular endothelial cells. UMIC 2002;9:133–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Klocke K, Sakaguchi S, Holmdahl R, Wing K.. Induction of autoimmune disease by deletion of CTLA-4 in mice in adulthood. Proc Natl Acad Sci USA 2016;113:E2383–E2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nishimura H, Nose M, Hiai H, Minato N, Honjo T.. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 1999;11:141–151. [DOI] [PubMed] [Google Scholar]
- 14. Mehta NN. Potential cardiovascular implications of Janus kinase inhibitors in immune mediated diseases. Cardiovasc Res 2018;114:e81–e83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, Grasso CS, Hugo W, Sandoval S, Torrejon DY, Palaskas N, Rodriguez GA, Parisi G, Azhdam A, Chmielowski B, Cherry G, Seja E, Berent-Maoz B, Shintaku IP, Le DT, Pardoll DM, Diaz LA, Tumeh PC, Graeber TG, Lo RS, Comin-Anduix B, Ribas A.. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov 2017;7:188–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sucker A, Zhao F, Pieper N, Heeke C, Maltaner R, Stadtler N, Real B, Bielefeld N, Howe S, Weide B, Gutzmer R, Utikal J, Loquai C, Gogas H, Klein-Hitpass L, Zeschnigk M, Westendorf AM, Trilling M, Horn S, Schilling B, Schadendorf D, Griewank KG, Paschen A.. Acquired IFNγ resistance impairs anti-tumor immunity and gives rise to T-cell-resistant melanoma lesions. Nat Commun 2017;8:15440.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.FDA. Hematology/Oncology (Cancer) Approvals & Safety Notifications. U.S. Food and Drug Administration, Center for Drug Evaluation and Research; 2018. [Google Scholar]
- 18. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, Ferrucci PF, Hill A, Wagstaff J, Carlino MS, Haanen JB, Maio M, Marquez-Rodas I, McArthur GA, Ascierto PA, Long GV, Callahan MK, Postow MA, Grossmann K, Sznol M, Dreno B, Bastholt L, Yang A, Rollin LM, Horak C, Hodi FS, Wolchok JD.. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 2015;373:23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yan J, Zhang Y, Zhang J-P, Liang J, Li L, Zheng L.. Tim-3 expression defines regulatory T cells in human tumors. PLoS One 2013;8:e58006.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Huang C-T, Workman CJ, Flies D, Pan X, Marson AL, Zhou G, Hipkiss EL, Ravi S, Kowalski J, Levitsky HI, Powell JD, Pardoll DM, Drake CG, Vignali DAA.. Role of LAG-3 in regulatory T cells. Immunity 2004;21:503–513. [DOI] [PubMed] [Google Scholar]
- 21. Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, Tom I, Ivelja S, Refino CJ, Clark H, Eaton D, Grogan JL.. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol 2009;10:48–57. [DOI] [PubMed] [Google Scholar]
- 22. Watanabe N, Gavrieli M, Sedy JR, Yang J, Fallarino F, Loftin SK, Hurchla MA, Zimmerman N, Sim J, Zang X, Murphy TL, Russell JH, Allison JP, Murphy KM.. BTLA is a lymphocyte inhibitory receptor with similarities to CTLA-4 and PD-1. Nat Immunol 2003;4:670–679. [DOI] [PubMed] [Google Scholar]
- 23. Lines JL, Pantazi E, Mak J, Sempere LF, Wang L, O'Connell S, Ceeraz S, Suriawinata AA, Yan S, Ernstoff MS, Noelle R.. VISTA is an immune checkpoint molecule for human T cells. Cancer Res 2014;74:1924–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Salem J-E, Manouchehri A, Moey M, Lebrun-Vignes B, Bastarache L, Pariente A, Gobert A, Spano J-P, Balko JM, Bonaca MP, Roden DM, Johnson DB, Moslehi JJ.. Cardiovascular toxicities associated with immune checkpoint inhibitors: an observational, retrospective, pharmacovigilance study. Lancet Oncol 2018;19:1579–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Anderson RD, Brooks M.. Apical takotsubo syndrome in a patient with metastatic breast carcinoma on novel immunotherapy. Int J Cardiol 2016;222:760–761. [DOI] [PubMed] [Google Scholar]
- 26. Heinzerling L, Ott PA, Hodi FS, Husain AN, Tajmir-Riahi A, Tawbi H, Pauschinger M, Gajewski TF, Lipson EJ, Luke JJ.. Cardiotoxicity associated with CTLA4 and PD1 blocking immunotherapy. J ImmunoTher Cancer 2016;4:50.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ederhy S, Cautela J, Ancedy Y, Escudier M, Thuny F, Cohen A.. Takotsubo-Like syndrome in cancer patients treated with immune checkpoint inhibitors. JACC Cardiovasc Imaging 2018;11:1187–1190. [DOI] [PubMed] [Google Scholar]
- 28. Roth ME, Muluneh B, Jensen BC, Madamanchi C, Lee CB.. Left ventricular dysfunction after treatment with ipilimumab for metastatic melanoma. Am J Ther 2016;23:e1925–e1928. [DOI] [PubMed] [Google Scholar]
- 29. Weinstock C, Khozin S, Suzman D, Zhang L, Tang S, Wahby S, Goldberg KB, Kim G, Pazdur R.. U.S. Food and Drug Administration approval summary: atezolizumab for metastatic non–small cell lung cancer. Clin Cancer Res 2017;23:4534–4539. [DOI] [PubMed] [Google Scholar]
- 30. Nykl R, Fischer O, Vykoupil K, Taborsky M.. A unique reason for coronary spasm causing temporary ST elevation myocardial infarction (inferior STEMI) – systemic inflammatory response syndrome after use of pembrolizumab. Arch Med Sci Atheroscler Dis 2017;2:100–e102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Behling J, Kaes J, Münzel T, Grabbe S, Loquai C.. New-onset third-degree atrioventricular block because of autoimmune-induced myositis under treatment with anti-programmed cell death-1 (nivolumab) for metastatic melanoma. Melanoma Res 2017;27:155.. [DOI] [PubMed] [Google Scholar]
- 32. Gibson R, Delaune J, Szady A, Markham M.. Suspected autoimmune myocarditis and cardiac conduction abnormalities with nivolumab therapy for non-small cell lung cancer. BMJ Case Rep 2016; 2016. pii: bcr2016216228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mahmood SS, Fradley MG, Cohen JV, Nohria A, Reynolds KL, Heinzerling LM, Sullivan RJ, Damrongwatanasuk R, Chen CL, Gupta D, Kirchberger MC, Awadalla M, Hassan MZO, Moslehi JJ, Shah SP, Ganatra S, Thavendiranathan P, Lawrence DP, Groarke JD, Neilan TG.. Myocarditis in patients treated with immune checkpoint inhibitors. J Am Coll Cardiol 2018;71:1755–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Moslehi JJ, Salem J-E, Sosman JA, Lebrun-Vignes B, Johnson DB.. Increased reporting of fatal immune checkpoint inhibitor-associated myocarditis. Lancet 2018;391:933.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Choueiri TK, Larkin J, Oya M, Thistlethwaite F, Martignoni M, Nathan P, Powles T, McDermott D, Robbins PB, Chism DD, Cho D, Atkins MB, Gordon MS, Gupta S, Uemura H, Tomita Y, Compagnoni A, Fowst C, Pietro A, di Rini BI.. Preliminary results for avelumab plus axitinib as first-line therapy in patients with advanced clear-cell renal-cell carcinoma (JAVELIN Renal 100): an open-label, dose-finding and dose-expansion, phase 1b trial. Lancet Oncol 2018;19:451–460. [DOI] [PubMed] [Google Scholar]
- 36. Du S, Zhou L, Alexander GS, Park K, Yang L, Wang N, Zaorsky NG, Ma X, Wang Y, Dicker AP, Lu B.. PD-1 modulates radiation-induced cardiac toxicity through cytotoxic T lymphocytes. J Thorac Oncol 2018;13:510–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Escudier M, Cautela J, Malissen N, Ancedy Y, Orabona M, Pinto J, Monestier S, Grob J-J, Scemama U, Jacquier A, Lalevee N, Barraud J, Peyrol M, Laine M, Bonello L, Paganelli F, Cohen A, Barlesi F, Ederhy S, Thuny F.. Clinical features, management, and outcomes of immune checkpoint inhibitor-related cardiotoxicity. Circulation 2017;136:2085–2087. [DOI] [PubMed] [Google Scholar]
- 38. Conforti F, Pala L, Bagnardi V, Pas TD, Martinetti M, Viale G, Gelber RD, Goldhirsch A.. Cancer immunotherapy efficacy and patients’ sex: a systematic review and meta-analysis. Lancet Oncol 2018;19:737–746. [DOI] [PubMed] [Google Scholar]
- 39. Lyon AR, Yousaf N, Battisti NML, Moslehi J, Larkin J.. Immune checkpoint inhibitors and cardiovascular toxicity. Lancet Oncol 2018;19:e447–e458. [DOI] [PubMed] [Google Scholar]
- 40. Caforio ALP, Pankuweit S, Arbustini E, Basso C, Gimeno-Blanes J, Felix SB, Fu M, Helio T, Heymans S, Jahns R, Klingel K, Linhart A, Maisch B, McKenna W, Mogensen J, Pinto YM, Ristic A, Schultheiss H-P, Seggewiss H, Tavazzi L, Thiene G, Yilmaz A, Charron P, Elliott PM.. Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2013;34:2636–2648. [DOI] [PubMed] [Google Scholar]
- 41. Friedrich MG, Sechtem U, Schulz-Menger J, Holmvang G, Alakija P, Cooper LT, White JA, Abdel-Aty H, Gutberlet M, Prasad S, Aletras A, Laissy J-P, Paterson I, Filipchuk NG, Kumar A, Pauschinger M, Liu P.. Cardiovascular magnetic resonance in myocarditis: a JACC white paper. J Am Coll Cardiol 2009;53:1475–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bando S, Soeki T, Matsuura T, Tobiume T, Ise T, Kusunose K, Yamaguchi K, Yagi S, Fukuda D, Iwase T, Yamada H, Wakatsuki T, Shimabukuro M, Muguruma N, Takayama T, Kishimoto I, Kangawa K, Sata M.. Plasma brain natriuretic peptide levels are elevated in patients with cancer. PLoS One 2017;12:e0178607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hughes M, Lilleker JB, Herrick AL, Chinoy H.. Cardiac troponin testing in idiopathic inflammatory myopathies and systemic sclerosis-spectrum disorders: biomarkers to distinguish between primary cardiac involvement and low-grade skeletal muscle disease activity. Ann Rheum Dis 2015;74:795–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kindermann I, Barth C, Mahfoud F, Ukena C, Lenski M, Yilmaz A, Klingel K, Kandolf R, Sechtem U, Cooper LT, Böhm M.. Update on myocarditis. J Am Coll Cardiol 2012;59:779–792. [DOI] [PubMed] [Google Scholar]
- 45. Hauck AJ, Kearney DL, Edwards WD.. Evaluation of postmortem endomyocardial biopsy specimens from 38 patients with lymphocytic myocarditis: implications for role of sampling error. Mayo Clin Proc 1989;64:1235–1245. [DOI] [PubMed] [Google Scholar]
- 46.National Cancer Institute. Common Terminology Criteria for Adverse Events (CTCAE), Version 5.0 Bethesda: National Cancer Institute; 2017. [Google Scholar]
- 47. Hill AB. The environment and disease: association or causation? Proc R Soc Med 1965;58:295–300. [PMC free article] [PubMed] [Google Scholar]
- 48. Sury K, Perazella MA, Shirali AC.. Cardiorenal complications of immune checkpoint inhibitors. Nat Rev Nephrol 2018;14:571–588. [DOI] [PubMed] [Google Scholar]
- 49. Dubin K, Callahan MK, Ren B, Khanin R, Viale A, Ling L, No D, Gobourne A, Littmann E, Huttenhower C, Pamer EG, Wolchok JD.. Intestinal microbiome analyses identify melanoma patients at risk for checkpoint-blockade-induced colitis. Nat Commun 2016;7:10391.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ertl G, Frantz S.. Healing after myocardial infarction. Cardiovasc Res 2005;66:22–32. [DOI] [PubMed] [Google Scholar]
- 51. Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, Sasayama S, Mizoguchi A, Hiai H, Minato N, Honjo T.. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science 2001;291:319–322. [DOI] [PubMed] [Google Scholar]
- 52. Okazaki T, Tanaka Y, Nishio R, Mitsuiye T, Mizoguchi A, Wang J, Ishida M, Hiai H, Matsumori A, Minato N, Honjo T.. Autoantibodies against cardiac troponin I are responsible for dilated cardiomyopathy in PD-1-deficient mice. Nat Med 2003;9:1477–1483. [DOI] [PubMed] [Google Scholar]
- 53. Seko Y, Yagita H, Okumura K, Azuma M, Nagai R.. Roles of programmed death-1 (PD-1)/PD-1 ligands pathway in the development of murine acute myocarditis caused by coxsackievirus B3. Cardiovasc Res 2007;75:158–167. [DOI] [PubMed] [Google Scholar]
- 54. Wang J, Okazaki I, Yoshida T, Chikuma S, Kato Y, Nakaki F, Hiai H, Honjo T, Okazaki T.. PD-1 deficiency results in the development of fatal myocarditis in MRL mice. Int Immunol 2010;22:443–452. [DOI] [PubMed] [Google Scholar]
- 55. Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH.. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 1995;3:541–547. [DOI] [PubMed] [Google Scholar]
- 56. Grabie N, Lichtman AH, Padera R.. T cell checkpoint regulators in the heart. Cardiovasc Res 2019;115:869–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Baban B, Liu JY, Qin X, Weintraub NL, Mozaffari MS.. Upregulation of programmed death-1 and its ligand in cardiac injury models: interaction with GADD153. PLoS One 2015;10:e0124059.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kusters PJH, Lutgens E, Seijkens TTP.. Exploring immune checkpoints as potential therapeutic targets in atherosclerosis. Cardiovasc Res 2018;114:368–377. [DOI] [PubMed] [Google Scholar]
- 59. Brahmer JR, Lacchetti C, Schneider BJ, Atkins MB, Brassil KJ, Caterino JM, Chau I, Ernstoff MS, Gardner JM, Ginex P, Hallmeyer S, Holter Chakrabarty J, Leighl NB, Mammen JS, McDermott DF, Naing A, Nastoupil LJ, Phillips T, Porter LD, Puzanov I, Reichner CA, Santomasso BD, Seigel C, Spira A, Suarez-Almazor ME, Wang Y, Weber JS, Wolchok JD, Thompson JA.. Management of immune-related adverse events in patients treated with immune checkpoint inhibitor therapy: American Society of Clinical Oncology Clinical Practice Guideline. J Clin Oncol 2018;36:1714–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kwon HJ, Cot TR, Cuffe MS, Kramer JM, Braun MM.. Case reports of heart failure after therapy with a tumor necrosis factor antagonist. Ann Intern Med 2003;138:807.. [DOI] [PubMed] [Google Scholar]
- 61. Tay RY, Blackley E, McLean C, Moore M, Bergin P, Gill S, Haydon A.. Successful use of equine anti-thymocyte globulin (ATGAM) for fulminant myocarditis secondary to nivolumab therapy. Br J Cancer 2017;117:921–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Almeida DVP, de Gomes JR, Haddad FJ, Buzaid AC.. Immune-mediated pericarditis with pericardial tamponade during nivolumab therapy. J Immunother 2018;41;329–331. [DOI] [PubMed] [Google Scholar]
- 63. Nesfeder J, Elsensohn AN, Thind M, Lennon J, Domsky S.. Pericardial effusion with tamponade physiology induced by nivolumab. Int J Cardiol 2016;222:613–614. [DOI] [PubMed] [Google Scholar]
- 64. Shaheen S, Mirshahidi H, Nagaraj G, Hsueh C-T.. Conservative management of nivolumab-induced pericardial effusion: a case report and review of literature. Exp Hematol Oncol 2018;7:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kushnir I, Wolf I.. Nivolumab-induced pericardial tamponade: a case report and discussion. Cardiology 2017;136:49–51. [DOI] [PubMed] [Google Scholar]
- 66. Adler Y, Charron P, Imazio M, Badano L, Barón-Esquivias G, Bogaert J, Brucato A, Gueret P, Klingel K, Lionis C, Maisch B, Mayosi B, Pavie A, Ristić AD, Sabaté Tenas M, Seferovic P, Swedberg K, Tomkowski W, Achenbach S, Agewall S, Al-Attar N, Angel Ferrer J, Arad M, Asteggiano R, Bueno H, Caforio ALP, Carerj S, Ceconi C, Evangelista A, Flachskampf F.. 2015 ESC Guidelines for the diagnosis and management of pericardial diseases: The Task Force for the Diagnosis and Management of Pericardial Diseases of the European Society of Cardiology (ESC)Endorsed by: the European Association for Cardio-Thoracic Surgery (EACTS). Eur Heart J 2015;36:2921–2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Goldstein BL, Gedmintas L, Todd DJ.. Drug-associated polymyalgia rheumatica/giant cell arteritis occurring in two patients after treatment with ipilimumab, an antagonist of CTLA-4. Arthritis Rheumatol 2014;66:768–769. [DOI] [PubMed] [Google Scholar]
- 68. Manusow JS, Khoja L, Pesin N, Joshua AM, Mandelcorn ED.. Retinal vasculitis and ocular vitreous metastasis following complete response to PD-1 inhibition in a patient with metastatic cutaneous melanoma. J ImmunoTher Cancer 2014;2:41.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Padda A, Schiopu E, Sovich J, Ma V, Alva A, Fecher L.. Ipilimumab induced digital vasculitis. J ImmunoTher Cancer 2018;6:12.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Burel SL, Champiat S, Mateus C, Marabelle A, Michot J-M, Robert C, Belkhir R, Soria J-C, Laghouati S, Voisin A-L, Fain O, Mékinian A, Coutte L, Szwebel T-A, Dunogeant L, Lioger B, Luxembourger C, Mariette X, Lambotte O.. Prevalence of immune-related systemic adverse events in patients treated with anti-programmed cell death 1/anti-programmed cell death-ligand 1 agents: a single-centre pharmacovigilance database analysis. Eur J Cancer 2017;82:34–44. [DOI] [PubMed] [Google Scholar]
- 71. Roy AK, Tathireddy HR, Roy M.. Aftermath of induced inflammation: acute periaortitis due to nivolumab therapy. BMJ Case Rep 2017;2017. pii: bcr-2017-221852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Arellano K, Mosley JC, Moore DC.. Case report of ipilimumab-induced diffuse, nonnecrotizing granulomatous lymphadenitis and granulomatous vasculitis. J Pharm Pract 2018;31:227–229. [DOI] [PubMed] [Google Scholar]
- 73. Brom RRH, van den Abdulahad WH, Rutgers A, Kroesen B-J, Roozendaal C, Groot DJA, de Schröder CP, Hospers GAP, Brouwer E.. Rapid granulomatosis with polyangiitis induced by immune checkpoint inhibition. Rheumatology (Oxford) 2016;55:1143–1145. [DOI] [PubMed] [Google Scholar]
- 74. Daxini A, Cronin K, Sreih AG.. Vasculitis associated with immune checkpoint inhibitors—a systematic review. Clin Rheumatol 2018;37:2579–2584. [DOI] [PubMed] [Google Scholar]
- 75. Buttgereit F, Dejaco C, Matteson EL, Dasgupta B.. Polymyalgia rheumatica and giant cell arteritis: a systematic review. JAMA 2016;315:2442–2458. [DOI] [PubMed] [Google Scholar]
- 76. Watanabe R, Zhang H, Berry G, Goronzy JJ, Weyand CM.. Immune checkpoint dysfunction in large and medium vessel vasculitis. Am J Physiol Heart Circ Physiol 2017;312:H1052–H1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Zhang H, Watanabe R, Berry GJ, Vaglio A, Liao YJ, Warrington KJ, Goronzy JJ, Weyand CM.. Immunoinhibitory checkpoint deficiency in medium and large vessel vasculitis. Proc Natl Acad Sci USA 2017;114:E970–E979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Varricchi G, Galdiero MR, Mercurio V, Bonaduce D, Marone G, Tocchetti CG.. Pharmacovigilating cardiotoxicity of immune checkpoint inhibitors. Lancet Oncol 2018;19:1545–1546. [DOI] [PubMed] [Google Scholar]
- 79. Dasgupta B, Borg FA, Hassan N, Alexander L, Barraclough K, Bourke B, Fulcher J, Hollywood J, Hutchings A, James P, Kyle V, Nott J, Power M, Samanta A.. BSR and BHPR guidelines for the management of giant cell arteritis. Rheumatology (Oxford) 2010;49:1594–1597. [DOI] [PubMed] [Google Scholar]
- 80. Wang DY, Salem J-E, Cohen JV, Chandra S, Menzer C, Ye F, Zhao S, Das S, Beckermann KE, Ha L, Rathmell WK, Ancell KK, Balko JM, Bowman C, Davis EJ, Chism DD, Horn L, Long GV, Carlino MS, Lebrun-Vignes B, Eroglu Z, Hassel JC, Menzies AM, Sosman JA, Sullivan RJ, Moslehi JJ, Johnson DB.. Fatal toxic effects associated with immune checkpoint inhibitors: a systematic review and meta-analysis. JAMA Oncol 2018;4:1721–1728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Webster RM. The immune checkpoint inhibitors: where are we now? Nat Rev Drug Discov 2014;13:883–884. [DOI] [PubMed] [Google Scholar]
- 82. Tocchetti CG, Galdiero MR, Varricchi G.. Cardiac toxicity in patients treated with immune checkpoint inhibitors. J Am Coll Cardiol 2018;71:1765–1767. [DOI] [PubMed] [Google Scholar]