

Abstract
It is highly demanded and still a big challenge to develop an effective formulation for immunochemotherapy against advanced tumors. We have previously reported a PEG-NLG-based immunostimulatory nanocarrier (PEG2k-Fmoc-NLG919) for codelivery of an IDO1 inhibitor (NLG919) and a chemotherapeutic agent (paclitaxel, PTX). Although antitumor immune responses were enhanced with PTX-loaded nanocarrier, the accumulation of myeloid-derived suppressor cells (MDSCs) was also significantly increased, which may limit the overall efficacy of therapy. In the present work, we developed an improved dual-functional nanocarrier (PEG5k-Fmoc-NLG2) to co-load PTX and sunitinib (SUN, a multi-target receptor tyrosine kinase inhibitor) for improved cancer immunochemotherapy. We found that the recruited MDSCs negatively impacted the overall antitumor activity of PTX-loaded PEG-NLG nanocarrier. Mechanistic study suggests that this is likely attributed to the PTX-mediated induction of a number of chemokines that are involved in the recruitment of MDSCs. We have further shown that the induction of these chemokines was drastically blocked by SUN. Codelivery of PTX and SUN via PEG5k-Fmoc-NLG9192 nanocarrier led to a further improvement in the therapeutic efficacy with a concomitant reduction in MDSCs.
Keywords: myeloid-derived suppressor cells (MDSCs); immunotherapy; indoleamine 2,3-dioxygenase 1 (IDO1); sunitinib
Graphical Abstract
Introduction
The immune system plays an important role in cancer initiation and progression 1. Various mechanisms can be developed by cancer cells to escape from immune surveillance 2, 3, such as immune checkpoints, which refer to inhibitory receptors expressed on immune effector cells or immune inhibitory ligands on tumor cells 1, 4. Various immune checkpoint molecules have been discovered such as programmed cell death protein 1 (PD-1), cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and others 5–7. Blockade of PD-1 or CTLA-4 using monoclonal antibodies (mAbs) has led to many exciting preclinical and clinical results, as shown by enhanced and sustained endogenous antitumor immune responses, resulting in durable tumor control 8, 9. Despite the successes in some cancer patients, the overall clinical efficacy of checkpoint blockade is still limited due to the fact that other suppressive mechanisms are also involved in immune escape such as increased expression of indoleamine 2,3-dioxygenase (IDO) 10, 11. Another reason is that the efficacy of this treatment is closely associated with pre-existing antitumor immune responses 1, 12. Chemotherapy drugs can trigger immunogenic cell death and initiate antitumor immune responses 13, 14. Checkpoint blockade can then further enhance and sustain such antitumor immunity. Therefore, one ideal strategy will be to induce immunogenic tumor cell death while simultaneously blocking multiple tumor immunosuppressive pathways. Immunochemotherapy that combines an immune-modulating agent with a chemotherapeutic drug represents one such strategy 15–22. However, its success largely depends on an effective strategy to codeliver two different therapeutic agents to the tumors. We have previously developed an immunostimulatory nanomicellar carrier (PEG2k-Fmoc-NLG) assembled from PEG conjugates with the IDO inhibitor NLG919 16. IDO is overexpressed in cancer cells and neighboring immune cells, and facilitates the degradation of tryptophan (Trp) and accumulation of its metabolites, leading to inhibition of the proliferation and function of effector T cells, but increases in the number of regulatory T cells 23, 24. We showed that PEG2k-Fmoc-NLG alone could inhibit the growth of tumor through upregulating T cell immune responses in vivo. Furthermore, PEG2k-Fmoc-NLG could form a micellar nanocarrier that was capable of selective codelivery of paclitaxel (PTX) to tumor tissues. Intravenous administration of PTX formulated in PEG2k-Fmoc-NLG nanocarrier significantly enhanced antitumor response in murine breast cancer and melanoma models 16. However, treatment with PTX/PEG2k-Fmoc-NLG was also associated with untoward effects such as increased infiltration of myeloid-derived suppressor cells (MDSCs) compared to carrier alone (PEG2k-Fmoc-NLG) group.
MDSCs play a key role in maintaining a highly immunosuppressive tumor microenvironment (TEM) 25. There are two major subpopulations: monocytic MDSCs and granulocytic MDSCs26, 27. Despite their similarities in morphology to monocytes and neutrophils, MDSCs behave quite differently and are highly immunosuppressive 28, 29. MDSCs can induce immunosuppression through increased expression of inducible nitric oxide synthase (iNOS) and arginase I (Arg1), leading to inhibition of T-cell function 30, 31. In addition, MDSCs can enhance the proliferation of tumor cell and facilitate tumor metastasis and angiogenesis 32, 33. In addition, they can promote drug resistance34. Increased tumor-infiltrating MDSCs numbers are often related with high tumor burden and metastasis and contribute to poor prognosis 35. Targeting of MDSCs is currently been explored as a new therapeutic approach to improve the responses to various treatments such as chemotherapy and immunotherapy 36, 37.
Sunitinib (SUN) is a small-molecule inhibitor that targets multiple receptor tyrosine kinases (RTK) 38. It is a FDA-approved drug for the treatment of metastatic renal cell carcinoma and gastrointestinal stromal tumors (GIST) 39. SUN is effective in blocking a variety of receptor tyrosine kinases, including Fms-like tyrosine kinase receptor (Flt), platelet-derived growth factor receptor (PDGFR), vascular endothelial growth factor receptor 2 (VEGFR2), and ckit 40, 41. In addition to its direct effects on tumor cells and tumor angiogenesis, SUN has been shown to reduce the level and function of MDSCs and Treg cells both systemically and in the TME 42, 43. SUN is capable of augmenting IFNγ production of tumor-infiltrating T cell and downregulating the expression of CTLA4, PD1 and PD-L1 44, 45. The unique properties of SUN suggest its potential in further improving the therapeutic efficacy of PTX/PEG2k-Fmoc-NLG.
In this work, we first examined whether depletion of MDSCs could further improve the therapeutic effect of PTX/PEG2k-Fmoc-NLG-based combination therapy. The underlying mechanism of recruitment of MDSCs following treatment with PTX/PEG2k-Fmoc-NLG was also investigated. Finally, an improved dual-functional nanocarrier (PEG5k-Fmoc-NLG2) was developed to evaluate the therapeutic potential of codelivery with both PTX and SUN.
Materials and methods
Materials
Paclitaxel (PTX) and Sunitinib (SUN) were bought from TSZ Chem and LC Laboratories, respectively. α-Fmoc-ε-Boc-lysine, α, ɛ-Di-Boc-lysine, triethylamine (TEA), trifluoroacetic acid (TFA) and N, N’-dicyclohexylcarbodiimide (DCC) were purchased from Acros Organic. Monomethoxy PEG5K, Triton X-100 and 4-dimethylaminopyridine (DMAP) were bought from Sigma Aldrich. Cell culture and animal study were similarly operated as previously reported [16].
Synthesis of PEG5K-Fmoc-NLG2 conjugate
Monomethoxy PEG5K (1 equiv.), α-Fmoc-ɛ-Boc-lysine (3 equiv.), DCC (6 equiv.) and DMAP (1 equiv.) were mixed in dichloromethane (DCM) and stirred for 2 days (RT). After filteration, the solution was precipitated in cold ethanol/ether twice to yield PEG5K-Lys(Fmoc)-Boc. The Boc group was removed in the mixture of DCM/TFA (1:1, v/v) for 2 h at RT, and then precipitated in cold ethanol/ether to give PEG5K-Lysine(Fmoc)-NH2. PEG5K-Lys(Fmoc)-Lys(Boc2) was then synthesized by mixing PEG5K-lysine(Fmoc)-NH2 (1 equiv.), α, ɛ-Di-Boc-lysine (5 equiv.), DCC (5 equiv.) and DMAP (1 equiv.) in DCM at RT for 2 days. After deprotection of Boc group in DCM/TFA (1:1, v/v), PEG5K-Lys(Fmoc)-Lys(NH2)2 was obtained and then mixed with NLG919 (5 equiv.), DCC (5 equiv.) and DMAP (1 equiv.) in DCM. After stirring at RT for 2 days, the mixture was precipitated in cold ethanol/ether for three times, yielding the final product PEG5K-Fmoc-NLG2.
Preparation of PTX+SUN/PEG5K-Fmoc-NLG2 micelles
PTX&SUN co-loaded PEG5K-Fmoc-NLG2 micelles were prepared via a film-hydration method46. Briefly, PTX (5 mg/mL in DCM), SUN (2 mg/mL in ethanol), and PEG5K-Fmoc-NLG2 (50 mg/mL in DCM) were mixed at designated ratios in a glass tube. After completely removing the organic solvent, PBS was added and a clear solution of PTX+SUN/PEG5K-Fmoc-NLG2 micelles was obtained. PTX/PEG5K-Fmoc-NLG2, SUN/PEG5K-Fmoc-NLG2, and drug-free PEG5K-Fmoc-NLG2 micelles were similarly prepared. The critical micelle concentration (CMC), particle size and morphologies of these micelles as well as drug loading capacity (DLC) and efficiency (DLE) were measured according to previous publication47.
In vitro IDO inhibition assay
The in vitro effect of PEG5K-Fmoc-NLG2 in inhibiting IDO activity was evaluated by IDO assay. HeLa cells were seeded in a 96-well plate (5 × 103 cells/well). After 24 h, the cells were incubated with recombinant human IFN-γ (50 ng/mL). Then immediately cells were treated with PEG5K-Fmoc-NLG2 or free NLG919 at various concentrations of NLG919 ranging from 50 nM ~ 20 µM. After incubation for 48 h, the supernatants were transferred into new wells and incubated with 30% trichloroacetic acid (75 µL) for 30 min (50 °C). The supernatants were collected, followed by the addition of Ehrlich reagent. After incubation for 10 min at RT, the mixture was measured by a plate reader at 490 nm. For HPLC-MS detection, the plate was first centrifuged at 12,500 rpm and then the supernatants (100 µL) were taken from each well for kynurenine and tryptophan quantification.
T-cell proliferation study
T-cell proliferation mediated by various treatments was evaluated by a co-culture study of lymphocyte and Panc02 cell 48. Splenocyte suspensions were harvested from BALB/c mice and passed through the nylon wool columns. After lysing red blood cells, splenocytes were pre-stained with 5-(and 6)-carboxyfluorescein diacetate (CFSE)). IFN-γ (50 ng/mL) was added into the Panc02 cells to induce IDO expression. After being irradiated at 6000 rad, the IFN-γ stimulated Panc02 cells (1×105 cells/well) were co-cultured with splenocytes (5×105 cells/well) in a 96-well plate. Then the cells were treated with various concentrations of PEG5K-Fmoc-NLG2 and NLG919, respectively, followed by the addition of anti-CD3 (100 ng/mL) and mouse recombinant IL-2 (10 ng/mL). After incubation for 72 h, the number of T cells (CD8+ and CD4+) was determined by FACS analysis.
In vitro MTT assay
4T1.2 murine breast cancer cell lines (1.5 × 103 cells/well) were seeded in 96-well plates and incubated for 24 h. Then cells were treated with PTX, Carrier alone, PTX/PEG5k-Fmoc-NLG2, SUN/PEG5k-Fmoc-NLG2 or PTX+SUN/PEG5k-Fmoc-NLG2 in various concentrations. After treatment for 72 h, the cell viabilities were measured by MTT assay as previously reported [16].
Trp/Kyn ratio in plasma and tumor tissues
The Kyn/Trp ratios in plasma or tumor tissues were determined by HPLC-MS/MS. BALB/c mice bearing 4T1.2 tumors (~50mm3) were i.v. injected with PBS, PEG5K-Fmoc-NLG2, PTX/PEG5K-Fmoc-NLG2, SUN/PEG5K-Fmoc-NLG2, PTX+SUN/PEG5K-Fmoc-NLG2 (PTX dosage: 10 mg/kg; SUN dosage: 10 mg/kg) once every 3 days for 5 times. The plasma and tumor tissues were collected at 24 h after the last treatment. Methanol was added into plasma samples at ratio of plasma/methanol 1/2.5 (v/v) and the mixture was centrifuged at 12,500 rpm for 15 min. The supernatant was analyzed by HPLC-MS for Kyn and Trp quantification. Tumor tissues were homogenized in H2O, and then acetonitrile was added to the homogenates (1:1, v/v). After centrifugation, the supernatants were collected, and proteins in supernatants were further precipitated by adding equal volumes of methanol. After centrifugation, the supernatants were collected for HPLC-MS detection.
Quantification of tumor-infiltrating lymphocytes
Tumor-bearing mice were i.v. administrated with various agents once every 3 days for 5 times. Spleen and tumors were collected at 24 h after the last treatment, and then the single cell suspensions were harvested and stained with various antibodies (CD8, CD4, Granzyme B, IFN-γ, FoxP3, CD11b and Gr-1) for FACS evaluation.
Tissue distribution
Taxol, SUN malate, PTX/PEG5K-Fmoc-NLG2, SUN/PEG5K-Fmoc-NLG2, PTX+SUN/PEG5K-Fmoc-NLG2 micelles (PTX dosage: 10 mg/kg; SUN dosage: 10 mg/kg) were i.v. injected into 4T1.2 tumor-bearing mice. The mice were sacrificed at 24 h, and the samples were similarly extracted from major organs/tissues as described before [16], followed by HPLC analysis of PTX and SUN concentration.
In vivo anti-tumor activity
For combination therapy of PTX loaded micelle and Gr-1 antibody, groups of five female Balb/C mice were administered with Saline, control IgG, PTX-loaded PEG5K-Fmoc-NLG2 micelles, PTX-loaded PEGG5K-Fmoc-NLG2 micelles +control IgG or PTX-loaded PEGG5K-Fmoc-NLG2 micelles+Gr-1 antibody for 5 times (10 mg PTX/kg, 100ug Gr-1 antibody 100µg/mouse i.p.). The tumor volumes and body weights were monitored.
For in vivo antitumor activity of SUN and PTX combination, female BALB/c mice bearing 4T1.2 tumors were treated with saline, PEG5K-Fmoc-NLG2, PTX/PEG5K-Fmoc-NLG2, SUN/PEG5K-Fmoc-NLG2, PTX/SUN/PEG5K-Fmoc-NLG2 (PTX dosage: 10 mg/kg; SUN dosage: 10 mg/kg) through intravenous injection every three days for 5 times. Tumor volumes were followed as described above. At the end of the experiment, tumors were collected for hematoxylin and eosin (H&E) analysis. Additionally, the survival of different mice groups (n = 8) were evaluated in a separate study following previous protocol 49.
The populations of various immune cells in the tumors were determined by FACS after various treatments 50. According to the previous protocol, combinations of antibodies were used for intracellular and extracellular staining, respectively 16.
Results
Combination of PTX/PEG2k-Fmoc-NLG and Gr-1 antibody enhances the overall therapeutic efficacy
Our early study showed that PTX/PEG2k-Fmoc-NLG was effective in inhibiting tumor growth in both breast cancer and melanoma models [16]. However, we also noticed significant increases in MDSCs in the tumors following treatment with PTX/PEG2k-Fmoc-NLG [16]. To examine whether the increased numbers of MDSCs have any impact on the antitumor activity of PTX/PEG2k-Fmoc-NLG, a similar experiment was performed in mice that received anti-Gr-1 antibody or control IgG. In consistent with previous study [16], PTX/PEG2k-Fmoc-NLG demonstrated significant antitumor activity in 4T1.2 tumor model (Fig. 1A). Anti-Gr-1 antibody alone also showed a modest antitumor activity (Fig. 1A). However, combined treatment of PTX/PEG2k-Fmoc-NLG with anti-Gr-1 antibody resulted in the best tumor inhibitory effect among the 6 groups (Fig. 1A). There were no significant changes in body weight for all groups (Fig. 1B).
Fig. 1.
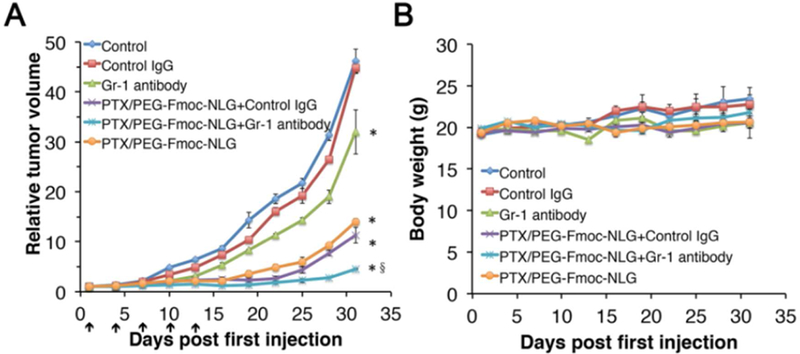
Relative tumor volume (A) and body weights (B) of of PTX/PEG-Fmoc-NLG micelles in combination of Gr-1 antibody in 4T1.2 model. *P<0.05, compared to control, §P<0.01, compared to PTX/PEG-Fmoc-NLG.
Fig. 2A, B showed that PTX/PEG2k-Fmoc-NLG treatment significantly increased the number of GMDSCs but the MMDSCs were not significantly affected, which was consistent with our previous study. With treatment of anti-Gr-1 antibody, the number of GMDSCs was decreased significantly (Fig. 2A, B). Moreover, combined therapy of PTX/PEG2k-Fmoc-NLG and anti-Gr-1 antibody led to significant increases in not only the number of total CD8+ and CD4+ TILs (Tumor-infiltrating lymphocytes), but also the number of IFN-γ+ TILs compared to PTX/PEG-Fmoc-NLG or Gr-1 antibody treatment (Fig. 1C–F). There was also a slight increase in the percentage of granzyme B+ CD8+ T cells in the combination group compared to the group receiving each therapy alone (Fig. 2G).
Fig. 2.

Flow cytometry analysis of various immune cells in tumor tissues after treatments, including Gr-1highCD11b+ granulocytic (G-MDSC) (A) and Gr-1intCD11b+ monocytic (M-MDSC) MDSC subsets (B), tumor infiltrating CD4+ T cells (C), CD8+ T cells(D), IFN+CD4+ T cells (E), IFN-γ+ CD8+ T cells (F) and granzyme B+ CD8+ T cells (G) in 4T1.2 tumor bearing mice. *p<0.05.
Roles of PTX-induced chemokines in the recruitment of MDSCs
The trafficking of different immune cells including MDSCs is governed by the respective chemokines involved. We next examined whether the expression profile of chemokines that are involved in MDSCs recruitment was altered following PTX treatment. As shown in Fig. 3A, treatment of tumor-bearing mice with Taxol, a Cremophor/ethanol formulation of PTX, significantly increased the mRNA expression levels of a number of chemokines examined including CCL2, CXCL1, CXCL2 and CXCL12. To define a potential role of each induced chemokine in the recruitment of MDSCs, we stably knockdowned the expression of CCL2 or CXCL12 in 4T1.2 cells using a shRNA expression plasmid targeting each chemokine. Preliminary data showed that knockdown of CCL2 or CXCL12 alone showed minimal impact on the recruitment of MDSCs following treatment of PTX/PEG2k-Fmoc-NLG (data not shown), suggesting that recruitment of MDSCs may involve the cooperation of multiple chemokines involved.
Fig. 3.

(A) Expression of genes with a role in recruitment of MDSCs in 4T1.2 cells after treatment with PTX. For each gene, expression values are normalized and plotted as log-transformed values (row-scaled and colored on a white-red scale to show the difference between samples). (B–I) Expression of genes with a role in recruitment of MDSCs in 4T1.2 cells (B–E) and tumor tissues (F–I) after treatment with PTX, SUN or combination.
SUN is a tyrosine kinase inhibitor that has been demonstrated to decrease the numbers and function of MDSCs both systemically and in the TME. We hypothesize that SUN may inhibit the recruitment of MDSCs through, at least partially, inhibiting the production of the chemokines involved. To test this hypothesis, we evaluated the effect of SUN on basal and PTX-induced expression of several chemokines in vitro and in vivo. As shown in Fig. 3B–I, SUN showed significant inhibition of both the basal and PTX-induced expression of all of the tested chemokines in vitro and in vivo.
Synthesis and biophysical characterization of PEG5K-Fmoc-NLG2
The above data suggest that incorporation of SUN into PTX/PEG-NLG micelles may represent an effective approach to further improve their therapeutic efficacy through inhibiting the recruitment and function of MDSCs. However, PEG2k-Fmoc-NLG only showed limited effectiveness in co-formulating PTX and SUN in a preliminary study (data not shown). This has led us to the synthesis of PEG5k-Fmoc-NLG2 that is similar to PEG2k-Fmoc-NLG but has an enlarged drug-interactive pocket. The synthesis scheme of PEG5k-Fmoc-NLG2 was presented in Fig. 4A. The chemical structure of PEG5k-Fmoc-NLG2 was confirmed by NMR (Supplementary Fig. 1).
Fig. 4.

(A) Synthesis scheme of PEG5k-Fmoc-NLG2. (B) In vitro IDO inhibition assay of PEG5k-Fmoc-NLG2. HeLa cells were stimulated with 50ng/ml of IFN-γ and treated with free NLG919 or PEG5k-Fmoc-NLG2 conjugate. Kynurenine concentration in supernatants was measured 48h after incubation by Ehrlich’s reagent. Data represent means ± s.e.m. (C-D) PEG5k-Fmoc-NLG2 reversed T cell growth inhibition mediated by IDO-expressing Panc02 cancer cells. Panc02 cells were stimulated with 50ng/ml of IFN-γ, irradiated and co-cultured with mouse splenocytes. IL-2, anti-CD3 were used to stimulate T cell growth. Free NLG919 or PEG-NLG conjugate were added to co-cultured cells and proliferation of (C) CD4+ and (D) CD8+ T cells were examined by flow cytometer.
IDO enzyme inhibition effect
The IDO enzyme inhibitory activity of PEG5k-Fmoc-NLG2 was evaluated on Hela cells by examining the changes in the concentrations of Trp and Kyn in cell culture medium. HeLa cells were stimulated with IFN-γ to induce IDO expression, and then treated with various concentrations of carrier or free NLG919. The amounts of Trp and Kyn in culture medium were examined by a colorimetric assay. Fig. 4B shows that free NLG919 could effectively inhibit the IDO enzyme activity as shown by a decreased conversion of Trp/Kyn. The free drug had an EC50 of 0.3 µM while the PEG5k-Fmoc-NLG2 could also maintain the IDO inhibition activity with an EC50 of 1.4 µM.
T cell proliferation-enhancing effect
As we showed before, T cell proliferation was significantly inhibited when lymphocytes were co-cultured with IDO-expressing tumor cells. We then examined whether PEG5k-Fmoc-NLG2 could enhance T-cell proliferation through inhibiting the IDO activity in tumor cells. As shown in Fig. 4C–D, co-culture of IDO+ panc02 cells with splenocytes isolated from BALB/c mice significantly inhibited T-cell proliferation. When the co-culture was treated with NLG919 or PEG5k-Fmoc-NLG2, the T cell proliferation could be significantly restored. PEG5k-Fmoc-NLG2 was slightly less effective compared to free NLG919.
Preparation and characterizations of drug-loaded PEG5k-Fmoc-NLG2 micelles
PEG5k-Fmoc-NLG2 micelles co-loaded with PTX and SUN were prepared via a film hydration method. Drug-free micelles and micelles loaded with PTX or SUN alone were similarly prepared as controls. Through HPLC test, the drug loading content for PTX and SUN was 5.6 % and 8.0%, respectively. The drug loading efficiency was 98.3% and 96.4% for PTX and SUN, respectively. The sizes and morphologies of blank and drug-formulated micelles were examined by DLS (dynamic light scattering) and TEM (transmission electron microscopy), respectively (Fig. 5A–D). The DLS measurements showed that the mean diameter of blank micelles was 122 nm and the average particle sizes of PTX/PEG5k-Fmoc-NLG2 and PTX+SUN/PEG5k-Fmoc-NLG2 were 125 nm and 126 nm, respectively. TEM showed that both blank and drug-loaded micelles exhibited a spherical morphology. The sizes of the particles on TEM were approximately 40 nm, which were smaller than those obtained from the DLS measurements. Fig. 5E shows that PEG5k-Fmoc-NLG2 micelles have a low critical micelle concentration (CMC) of 0.161µM.
Fig. 5.

Size distribution of blank PEG5k-Fmoc-NLG2 micelles (A) and PTX+SUN loaded micelles (B) were examined by a zeta sizer. TEM morphologies of blank PEG5k-Fmoc-NLG2 micelles (C) and PTX+SUN loaded micelles (D). Measurement of CMC of PEG5k-Fmoc-NLG2 micelles (E).
Tab. 1 compares the biophysical properties of PEG5k-Fmoc-NLG2 and PEG2k-Fmoc-NLG micelles loaded with PTX or co-loaded with PTX and SUN. All drug-loaded micelles were prepared at a carrier/drug molar ratio of 2.5/1. It is apparent that micelles loaded with PTX alone were more stable than the micelles co-loaded with PTX and SUN. It is also clear that PEG5k-Fmoc-NLG2-based micellar system is more stable than PEG2k-Fmoc-NLG system in either formulating one single drug (PTX) or co-formulating two drugs (PTX and SUN).
Table 1.
Characterizations and comparison of two PEG-NLG nanocarriers
| Micelles | CMS (µM) | EC50 (µM) | Size (nm) | Stability (RT) |
|---|---|---|---|---|
| PEG2K-Fmoc-NLG | 0.737 | 3.4 | 93.9±0.6 | -- |
| PEG2K-Fmoc-NLG:PTX2.5:1 | -- | -- | 93.9±0.3 | 60h |
| PEG2K-Fmoc-NLG:(PTX+Sun)2.5:1 | -- | -- | 105±0.5 | 48h |
| PEG2K-Fmoc-NLG2 | 0.161 | 1.4 | 122±0.5 | -- |
| PEG2K-Fmoc-NLG2:PTX2.5:1 | -- | -- | 125±0.3 | 1week |
| PEG2K-Fmoc-NLG2:(PTX+Sun)2.5:1 | -- | -- | 126±0.6 | 96h |
Note: Stability means that during the follow-up period, there were no precipitation or size changes at RT.
Release kinetics of PTX & SUN from PEG5k-Fmoc-NLG2 micelles
The release kinetics of PTX and SUN from PTX+SUN/PEG5k-Fmoc-NLG2 in PBS buffer (pH 7.4) was studied through a dialysis method and the results were shown in Supplementary Fig. 2A&B. Taxol, SUN malate, and PEG5k-Fmoc-NLG2 micelles loaded with PTX or Sun alone were used as controls. As shown in Supplementary Fig. 2A&B, Taxol or SUN showed a rapid release with more than 70% and 60% of drug released within 4 h, respectively. However, either PTX or SUN that was formulated in the micelles exhibited a sustained release profile. PTX release from PTX+SUN/PEG5k-Fmoc-NLG2 micelles was similar to that from PTX/PEG5k-Fmoc-NLG2, while SUN release from PTX+SUN/PEG5k-Fmoc-NLG2 was slightly slower compared to SUN/PEG5k-Fmoc-NLG2. Overall, PTX and SUN showed comparable release kinetics from co-formulated micelles.
In vitro cytotoxicity
The cytotoxicity of blank and drug-loaded PEG5k-Fmoc-NLG2 micelles against 4T1.2 tumor cells was shown in Supplementary Fig. 2C. The PEG5k-Fmoc-NLG2 carrier alone exhibited negligible cytotoxicity at various polymer concentrations ranged from 0.07 to 17.5 µg/mL. The IC50s of SUN/ PEG5k-Fmoc-NLG2 and SUN malate were 5.14 and 7.78 µM, respectively. PTX/PEG5k-Fmoc-NLG2 (IC50=103 ng/mL) showed comparable cytotoxicity compared to free PTX (IC50=112 ng/ml). The slightly increased cytotoxicity of SUN-loaded micelles might be due to the enhanced delivery of SUN to 4T1.2 cells by micelle delivery system. PTX+SUN/PEG5k-Fmoc-NLG2 showed the best in vitro cytotoxicity, which could be due to the combined tumor cell killing effect of PTX and SUN.
In vivo biodistribution of PTX+SUN formulated micelles
The distribution, accumulation and elimination of administered drugs in main organs and tumors could directly affect their therapeutic efficacy as well as the toxicity. To test whether PEG5k-Fmoc-NLG2 could specifically deliver SUN and PTX to tumors, in vivo biodistribution studies were performed in 4T1.2 tumor-bearing mice. Groups of 3 mice were intravenously injected with SUN+PTX/PEG5k-Fmoc-NLG2, PTX/PEG5k-Fmoc-NLG2, SUN/PEG5k-Fmoc-NLG2, Taxol or SUN malate at the same dosage of SUN and PTX (10mg/kg). Major organs and tumor tissues were collected after 24h and PTX or SUN concentrations were measured by HPLC. As shown in Fig. 6A&B, more PTX and SUN were found in tumors for SUN+PTX/PEG5k-Fmoc-NLG2 compared to Taxol or SUN malate. Meanwhile there was reduced accumulation of PTX or SUN in normal tissues such as heart, liver and kidney following i.v. administration of SUN+PTX/PEG5k-Fmoc-NLG2. There were no significant differences between single drug formulation and the co-loaded micelles.
Fig. 6.
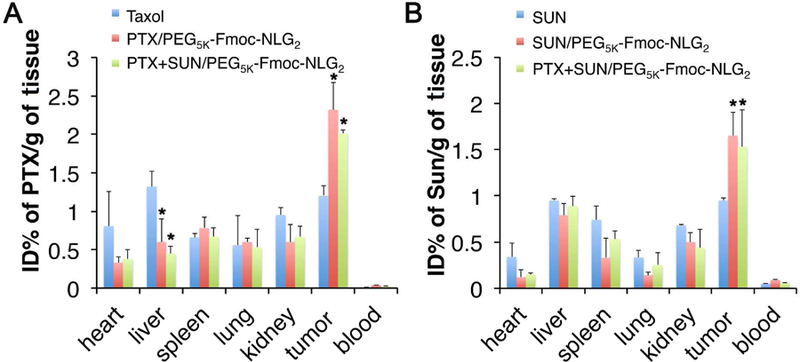
Biodistribution of different PTX and Sun formulations in 4T1.2 tumor-bearing mice. Tissue and organs were harvested 24 h after i.v. administration of (A) Taxol, PTX/PEG5K-Fmoc-NLG2 mixed micelles or PTX+Sun/PEG5K-Fmoc-NLG2 mixed micelles at a PTX dose of 10 mg kg−1 or (B) Sun, Sun/PEG5K-Fmoc-NLG2 mixed micelles or PTX+Sun/PEG5K-Fmoc-NLG2 at a Sun dose of 10 mg kg−1. PTX and Sun concentrations were measured by HPLC-MS. *P<0.05 (vs control, N=3).
In vivo antitumor efficacy
The antitumor activity of PTX+SUN/PEG5k-Fmoc-NLG2 was determined in a murine breast cancer model (4T1.2). Various formulations were given i.v. and tumor growth was followed once every three days (Fig. 7). As shown in Fig. 7A, all treatments including carrier alone exhibited significant antitumor effect compared to control group (saline treatment). PTX+SUN/PEG5k-Fmoc-NLG2 exhibited the best antitumor activity, much more effective than PTX/PEG5k-Fmoc-NLG2 or Sun/PEG5k-Fmoc-NLG2. The tumor inhibition indexes of carrier alone, PTX/PEG5k-Fmoc-NLG2, SUN/PEG5k-Fmoc-NLG2 and PTX+SUN/PEG5k-Fmoc-NLG2 group were 53.4%, 67.4%, 43.8% and 80.9%, respectively. No significant changes in body weights were observed among all groups, suggesting that all of the micellar formulations were safe at the dose tested.
Fig. 7.

(A) Enhanced in vivo antitumor activity of PTX+Sun/PEG5k-Fmoc-NLG2 mixed micelles compared to Sun formulated PEG5k-Fmoc-NLG2 (#P < 0.05, N = 5), PTX formulated in PEG5k-Fmoc-NLG2 micelles (&P < 0.05, N = 5) or PEG5k-Fmoc-NLG2 blank micelles. *P < 0.05 (vs control, N = 5). Data represent means ± s.e.m. (B) Histological analyses (H&E) of lung and tumor tissues collected at day 19 in the in vivo therapeutic study. (D-E) Kynurenine concentrations in (C) blood and (D) tumor following various treatments. *P < 0.05 (vs control). (E) Survival rate of various treatment groups. **P < 0.01 (vs control), &&P < 0.01 (vs PTX+Sun/PEG5k-Fmoc-NLG2).
Tumors were harvested at the end of therapeutic study and tumor tissue slices were stained with hematoxylin and eosin (H&E) (Fig. 7B). Because of high proliferation rate, most of the tumor cells in control group had large nuclei. Compared to control group, tumor slices in other treatment groups showed shrunk nuclei and cell necrosis, particularly the one treated with PTX+SUN/PEG5k-Fmoc-NLG2.
Tumor tissues and blood were also examined for Kyn (nM) concentrations as an indicator of IDO inhibition effect. As shown in Fig. 7 C&D, treatment with either PEG5k-Fmoc-NLG2 alone or the drug-loaded micelles significantly reduced the Kyn concentrations in both tumor tissues and blood.
The survival curves of the mice treated with different formulations were shown in Fig. 7E. Control groups showed a relative short survival time of around 22 days. Compared to control group, the mice treated with carrier alone, PTX loaded micelles and SUN loaded micelles showed longer survival times, with a median survival time of 30, 46, and 40 days, respectively. The mice treated with PTX+SUN co-formulated micelles showed the longest survival time (56 days).
Changes of immune cell populations in the TME
Tumor tissues from mice receiving different treatments were harvested and analyzed for the tumor infiltrating immune cell populations using flow cytometry. Fig. 8A & S3A show that higher numbers of CD4+ and CD8+ TIL were present in the tumors treated with PEG5k-Fmoc-NLG2 alone or the drug-loaded micelles. Strikingly, treatment with PTX+SUN/PEG5k-Fmoc-NLG2 resulted in the highest percentages of CD4+ and CD8+ T cells in the TME among all of the groups (p<0.001 vs. PBS; p<0.05 vs. Taxol and PTX/PEG5k-Fmoc-NLG2). It was also noted that treatment with PTX+SUN/PEG5k-Fmoc-NLG2 led to a dramatic increase in the percentage of IFN-γ+CD8+ T cells but not IFN-γ+CD4+ T compared to carrier alone, Taxol or PTX/PEG5k-Fmoc-NLG2 (Fig. 8B,C& S3B,C). In addition, the percentage of GZMB+CD8+ TIL was significantly increased in the PTX+SUN/PEG5k-Fmoc-NLG2 group compared to Taxol or PTX/PEG5k-Fmoc-NLG2 (Fig. 8D&S3D). Consistent with our previous studies, treatment with Taxol or PTX/PEG5k-Fmoc-NLG2 led to an increase in GMDSCs in the tumors (Fig. 8E & S3E). Incorporation of SUN into PTX/PEG5k-Fmoc-NLG2 resulted in significant decreases in both GMDSCs and MMDSCs. The numbers of GMDSCs and MMDSCs in tumors treated with SUN+PTX/PEG5k-Fmoc-NLG2 were similar to those treated with SUN monotherapy.
Fig. 8.

The percentages of immune cells in tumor tissues were analyzed by flow cytometry. 4T1.2 tumor bearing mice were received various treatments of Taxol, PEG5K-Fmoc-NLG2, PTX/PEG5K-Fmoc-NLG2 mixed micelles, Sun/PEG5K-Fmoc-NLG2 mixed micelles or PTX+Sun/PEG5K-Fmoc-NLG2 mixed micelles at a PTX dosage of 10 mg/kg and SUN dosage of 10 mg/kg for 3 times at every 3 days and tumoral T-cell infiltration, including (A) CD4+ and CD8+, (B) IFN-γ+CD4+ T cells, (C) IFN-γ+CD8+ T cells and (D) granzyme B-positive CD8+ T cells, were measured by flow cytometry. (E) Gr-1highCD11b+ granulocytic (G-MDSC) and Gr-1intCD11b+ monocytic (M-MDSC) were analyzed by flow cytometry. *P < 0.05.
Discussion
We have recently developed a combination therapy that is based on targeted delivery of a chemotherapeutic agent (e.g., PTX) via a PEG-NLG919-based immunostimulatory nanocarrier. In this work, we showed the therapeutic efficacy of this approach could be further improved via incorporation of another agent that is targeted at MDSCs.
MDSCs have been implicated in tumor progression and drug responses 51. Targeting the pre-existing MDSCs has demonstrated potential as a new strategy to improve the outcome of various therapies including immunochemotherapy 52, 53. In this study, we have shown that the recruitment of MDSCs can be further induced following treatment with PTX/PEG2k-Fmoc-NLG. The effect on MDSC recruitment is likely attributed to PTX as PEG2k-Fmoc-NLG alone exhibited a negative effect on tumor-infiltrating MDSCs. The fact that PTX/PEG2k-Fmoc-NLG caused more MDSCs recruitment than Taxol is likely attributed to more effective delivery of PTX into tumors via PEG2k-Fmoc-NLG. A negative role of MDSCs in the overall antitumor activity of PTX/PEG2k-Fmoc-NLG was clearly established by the data that elimination of MDSCs function led to a significant improvement in the therapeutic efficacy (Fig. 1A).
The underlying mechanism for the PTX-induced recruitment of MDSCs is not completely understood; however, our preliminary study showed that PTX causes increased expression of a number of chemokines that have been shown to play a role in the recruitment of MDSCs 54, 55 (Fig. 3A). Stable knockdown of CCL2 or CXCL12 alone via shRNA expression plasmid showed minimal impact on MDSCs recruitment following treatment with PTX/PEG2k-Fmoc-NLG (data not shown), suggesting that more than one chemokine is involved in the recruitment of MDSCs, at least in our experimental setting.
SUN is a FDA-approved multi-targeted receptor RTK inhibitor for the treatment of various types of cancers 56. In addition to its direct effects on tumor cells and tumor angiogenesis, SUN can decrease the numbers and function of MDSCs both systemically and in the TME 42. Recent studies have revealed several possible mechanisms for SUN regarding their effects on the functions and recruitment of MDSCs 43. It has been demonstrated that SUN could inhibit several RTK expressed on tumor cells and MDSCs, such as VEGFR, Flt3 and c-kit 57. The blockade of these receptors could inhibit MDSCs function and reverse the immune tolerance status in several tumor models 51, 58. SUN might work through inhibition of VEGF signaling, which has been reported to enhance the expansion and accumulation of MDSCs 41, 57. Furthermore, through inhibition of Flt3 and other myelopoietic factors, SUN can effectively inhibit MDSCs expansion 59, 60. Another possible mechanism is that SUN decreases the accumulation of MDSCs via inhibiting STAT3 and c-Kit or directly inducing the apoptosis of MDSCs 34. It is possible that other mechanisms may also be implicated in the inhibition of MDSCs by SUN 61. Data from this work showed that SUN could inhibit both basal and PTX-induced chemokines from tumor cells both in vitro and in vivo, suggesting that SUN could also inhibit MDSCs accumulation through blocking the production of related chemokines (Fig. 3). More studies are needed to better appreciate the underlying mechanisms in the future.
PEG5k-Fmoc-NLG2 represents an improved carrier for co-delivery of SUN and PTX. It has a lower CMC value and forms more stable mixed micelles with PTX and SUN compared to PEG2K-Fmoc-NLG (Tab. 1). The might be due to the expanded drug-interactive pocket, which contributes to the loading of both PTX and SUN through hydrophobic interaction and π−π stacking. In addition, a relatively larger PEG in our improved system (5k vs. 2k) may provide better steric hindrance to prevent nonspecific interactions with serum proteins in the blood circulation.
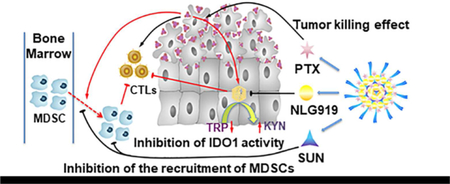
Fig. 9 shows our proposed mechanism of action for our combination therapy. Systemic administration of our drugs-loaded nanoparticles shall lead to simultaneous delivery of PTX, SUN, and PEG-modified NLG919 prodrug to tumor tissues. The rapid release of the physically loaded PTX and SUN will lead to synergistic killing of tumor cells as well as the induction of antitumor immune response through the facilitated release and the subsequent presentation of released tumor antigens. The released SUN can also inhibit the recruitment of MDSCs through inhibition of related chemokines as well as other mechanisms. At the same time, active NLG919 will be released slowly from the prodrug-based carrier during a prolonged period of time, helping to sustain an immune-active tumor microenvironment through inhibiting the activity of upregulated IDO 1. As a result, our approach has led to significantly improved antitumor activity as shown in Fig. 7A, B. Our data also demonstrated that the SUN+PTX/PEG-Fmoc-NLG significantly increases tumoral infiltrating CD8+ T cells while reducing intra-tumor MDSCs and Tregs, creating a more immunogenic microenvironment for antitumor therapy (Fig. 8).
Fig. 9.

Schematic illustration of PTX+SUN co-loaded micelles.
Conclusions
We have shown that targeted delivery of PTX via a PEG-NLG-based immunostimulatory nanocarrier induces recruitment of MDSCs, which represents an important feedback mechanism that limits the effectiveness of the immunochemotherapy. Codelivery of SUN and PTX via an improved nanocarrier, PEG5k-Fmoc-NLG2, effectively decreases the infiltrating MDSCs, leading to more active tumor immune microenvironment and further improved antitumor activity.
Supplementary Material
Acknowledgements
This work was supported by NIH grants R01CA174305, R01CA219399, and RO1CA223788, and a grant from Shear Family Foundation.
Footnotes
Supporting Information
NMR spectra of PEG5K-Fmoc-NLG2, In vitro release of PTX and SUN from the micelles, Cytotoxicity of PEG5k-Fmoc-NLG2 based various formulations, and representative flow cytometry gatings of immune cell subsets in tumor tissues after various treatments.
References
- 1.Pardoll DM The blockade of immune checkpoints in cancer immunotherapy. Nature Reviews Cancer 2012, 12, (4), 252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim R; Emi M; Tanabe K Cancer immunoediting from immune surveillance to immune escape. Immunology 2007, 121, (1), 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yamaguchi T; Sakaguchi S In Regulatory T cells in immune surveillance and treatment of cancer, Seminars in cancer biology, 2006; Elsevier: pp 115–123. [DOI] [PubMed] [Google Scholar]
- 4.Topalian SL; Drake CG; Pardoll DM Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer cell 2015, 27, (4), 450–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leach DR; Krummel MF; Allison JP Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, (5256), 1734–1736. [DOI] [PubMed] [Google Scholar]
- 6.Freeman GJ; Long AJ; Iwai Y; Bourque K; Chernova T; Nishimura H; Fitz LJ; Malenkovich N; Okazaki T; Byrne MC Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. Journal of Experimental Medicine 2000, 192, (7), 1027–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sharma P; Allison JP Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell 2015, 161, (2), 205–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Topalian SL; Hodi FS; Brahmer JR; Gettinger SN; Smith DC; McDermott DF; Powderly JD; Carvajal RD; Sosman JA; Atkins MB Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. N Engl j Med 2012, 2012, (366), 2443–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hirano F; Kaneko K; Tamura H; Dong H; Wang S; Ichikawa M; Rietz C; Flies DB; Lau JS; Zhu G Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer research 2005, 65, (3), 1089–1096. [PubMed] [Google Scholar]
- 10.Munn DH Blocking IDO activity to enhance anti-tumor immunity. Frontiers in bioscience (Elite edition) 2012, 4, 734–745. [DOI] [PubMed] [Google Scholar]
- 11.Yu J; Du W; Yan F; Wang Y; Li H; Cao S; Yu W; Shen C; Liu J; Ren X Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. The Journal of Immunology 2013, 190, (7), 3783–3797. [DOI] [PubMed] [Google Scholar]
- 12.Rosenberg SA; Yang JC; Restifo NP Cancer immunotherapy: moving beyond current vaccines. Nature medicine 2004, 10, (9), 909–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Michaud M; Martins I; Sukkurwala AQ; Adjemian S; Ma Y; Pellegatti P; Shen S; Kepp O; Scoazec M; Mignot G Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 2011, 334, (6062), 1573–1577. [DOI] [PubMed] [Google Scholar]
- 14.Machiels J-PH; Reilly RT; Emens LA; Ercolini AM; Lei RY; Weintraub D; Okoye FI; Jaffee EM Cyclophosphamide, doxorubicin, and paclitaxel enhance the antitumor immune response of granulocyte/macrophage-colony stimulating factor-secreting whole-cell vaccines in HER-2/neu tolerized mice. Cancer research 2001, 61, (9), 3689–3697. [PubMed] [Google Scholar]
- 15.Weber JS; D’Angelo SP; Minor D; Hodi FS; Gutzmer R; Neyns B; Hoeller C; Khushalani NI; Miller WH; Lao CD Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. The Lancet Oncology 2015, 16, (4), 375–384. [DOI] [PubMed] [Google Scholar]
- 16.Chen Y; Xia R; Huang Y; Zhao W; Li J; Zhang X; Wang P; Venkataramanan R; Fan J; Xie W An immunostimulatory dual-functional nanocarrier that improves cancer immunochemotherapy. Nature communications 2016, 7, 13443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fritsch K; Kasenda B; Schorb E; Hau P; Bloehdorn J; Möhle R; Löw S; Binder M; Atta J; Keller U High-dose methotrexate-based immuno-chemotherapy for elderly primary CNS lymphoma patients (PRIMAIN study). Leukemia 2017, 31, (4), 846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yin Y; Hu Q; Xu C; Qiao Q; Qin X; Song Q; Peng Y; Zhao Y; Zhang Z Co-delivery of Doxorubicin and Interferon-gamma by Thermosensitive Nanoparticles for Cancer Immunochemotherapy. Mol. Pharm 2018. [DOI] [PubMed]
- 19.Kumar A; Sirohi VK; Anum F; Singh PK; Gupta K; Gupta D; Saraf SA; Dwivedi A; Chourasia MK Enhanced apoptosis, survivin down-regulation and assisted immunochemotherapy by curcumin loaded amphiphilic mixed micelles for subjugating endometrial cancer. Nanomedicine 2017, 13, (6), 1953–1963. [DOI] [PubMed] [Google Scholar]
- 20.Jiang L; Ding Y; Xue X; Zhou S; Li C; Zhang X; Jiang X Entrapping multifunctional dendritic nanoparticles into a hydrogel for local therapeutic delivery and synergetic immunochemotherapy. Nano Research, 1–12.
- 21.Yu S; Zhang D; He C; Sun W; Cao R; Cui S; Deng M; Gu Z; Chen X Injectable thermosensitive polypeptide-based CDDP-complexed hydrogel for improving localized antitumor efficacy. Biomacromolecules 2017, 18, (12), 4341–4348. [DOI] [PubMed] [Google Scholar]
- 22.Sun J; Chen Y; Huang Y; Zhao W; Liu Y; Venkataramanan R; Lu B; Li S Programmable co-delivery of the immune checkpoint inhibitor NLG919 and chemotherapeutic doxorubicin via a redox-responsive immunostimulatory polymeric prodrug carrier. Acta Pharmacol. Sin 2017, 38, (6), 823–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holmgaard RB; Zamarin D; Li Y; Gasmi B; Munn DH; Allison JP; Merghoub T; Wolchok JD Tumor-expressed IDO recruits and activates MDSCs in a Treg-dependent manner. Cell reports 2015, 13, (2), 412–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Munn DH; Mellor AL Indoleamine 2, 3-dioxygenase and tumor-induced tolerance. Journal of Clinical Investigation 2007, 117, (5), 1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Highfill SL; Cui Y; Giles AJ; Smith JP; Zhang H; Morse E; Kaplan RN; Mackall CL Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Science translational medicine 2014, 6, (237), 237ra67–237ra67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Giallongo C; Tibullo D; Parrinello NL; La Cava P; Di Rosa M; Bramanti V; Di Raimondo C; Conticello C; Chiarenza A; Palumbo GA Granulocyte-like myeloid derived suppressor cells (G-MDSC) are increased in multiple myeloma and are driven by dysfunctional mesenchymal stem cells (MSC). Oncotarget 2016, 7, (52), 85764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nagaraj S; Gabrilovich DI Tumor escape mechanism governed by myeloid-derived suppressor cells. Cancer research 2008, 68, (8), 2561–2563. [DOI] [PubMed] [Google Scholar]
- 28.Umemura N; Saio M; Suwa T; Kitoh Y; Bai J; Nonaka K; Ouyang G-F; Okada M; Balazs M; Adany R Tumor-infiltrating myeloid-derived suppressor cells are pleiotropic-inflamed monocytes/macrophages that bear M1-and M2-type characteristics. Journal of leukocyte biology 2008, 83, (5), 1136–1144. [DOI] [PubMed] [Google Scholar]
- 29.Peranzoni E; Zilio S; Marigo I; Dolcetti L; Zanovello P; Mandruzzato S; Bronte V Myeloid-derived suppressor cell heterogeneity and subset definition. Current opinion in immunology 2010, 22, (2), 238–244. [DOI] [PubMed] [Google Scholar]
- 30.Greten TF; Manns MP; Korangy F Myeloid derived suppressor cells in human diseases. International immunopharmacology 2011, 11, (7), 802–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu C-Y; Wang Y-M; Wang C-L; Feng P-H; Ko H-W; Liu Y-H; Wu Y-C; Chu Y; Chung F-T; Kuo C-H Population alterations of L-arginase-and inducible nitric oxide synthase-expressed CD11b+/CD14−/CD15+/CD33+ myeloid-derived suppressor cells and CD8+ T lymphocytes in patients with advanced-stage non-small cell lung cancer. Journal of cancer research and clinical oncology 2010, 136, (1), 35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murdoch C; Muthana M; Coffelt SB; Lewis CE The role of myeloid cells in the promotion of tumour angiogenesis. Nature Reviews Cancer 2008, 8, (8), 618–631. [DOI] [PubMed] [Google Scholar]
- 33.Joyce JA; Pollard JW Microenvironmental regulation of metastasis. Nature Reviews Cancer 2009, 9, (4), 239–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Finke J; Ko J; Rini B; Rayman P; Ireland J; Cohen P MDSC as a mechanism of tumor escape from sunitinib mediated anti-angiogenic therapy. International immunopharmacology 2011, 11, (7), 856–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Diaz-Montero CM; Salem ML; Nishimura MI; Garrett-Mayer E; Cole DJ; Montero AJ Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin–cyclophosphamide chemotherapy. Cancer immunology, immunotherapy 2009, 58, (1), 49–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ostrand-Rosenberg S; Sinha P Myeloid-derived suppressor cells: linking inflammation and cancer. The Journal of Immunology 2009, 182, (8), 4499–4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Le HK; Graham L; Cha E; Morales JK; Manjili MH; Bear HD Gemcitabine directly inhibits myeloid derived suppressor cells in BALB/c mice bearing 4T1 mammary carcinoma and augments expansion of T cells from tumor-bearing mice. International immunopharmacology 2009, 9, (7), 900–909. [DOI] [PubMed] [Google Scholar]
- 38.Hojjat-Farsangi M Small-molecule inhibitors of the receptor tyrosine kinases: promising tools for targeted cancer therapies. International journal of molecular sciences 2014, 15, (8), 13768–13801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goodman VL; Rock EP; Dagher R; Ramchandani RP; Abraham S; Gobburu JVS; Booth BP; Verbois SL; Morse DE; Liang CY Approval summary: sunitinib for the treatment of imatinib refractory or intolerant gastrointestinal stromal tumors and advanced renal cell carcinoma. Clinical Cancer Research 2007, 13, (5), 1367–1373. [DOI] [PubMed] [Google Scholar]
- 40.Burstein HJ; Elias AD; Rugo HS; Cobleigh MA; Wolff AC; Eisenberg PD; Lehman M; Adams BJ; Bello CL; DePrimo SE Phase II study of sunitinib malate, an oral multitargeted tyrosine kinase inhibitor, in patients with metastatic breast cancer previously treated with an anthracycline and a taxane. Journal of Clinical Oncology 2008, 26, (11), 1810–1816. [DOI] [PubMed] [Google Scholar]
- 41.Roskoski R Sunitinib: a VEGF and PDGF receptor protein kinase and angiogenesis inhibitor. Biochemical and biophysical research communications 2007, 356, (2), 323–328. [DOI] [PubMed] [Google Scholar]
- 42.Ko JS; Zea AH; Rini BI; Ireland JL; Elson P; Cohen P; Golshayan A; Rayman PA; Wood L; Garcia J Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clinical cancer research 2009, 15, (6), 2148–2157. [DOI] [PubMed] [Google Scholar]
- 43.Ozao-Choy J; Ma G; Kao J; Wang GX; Meseck M; Sung M; Schwartz M; Divino CM; Pan P-Y; Chen S-H The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer research 2009, 69, (6), 2514–2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nishioka C; Ikezoe T; Yang J; Yokoyama A Sunitinib, an orally available receptor tyrosine kinase inhibitor, induces monocytic differentiation of acute myelogenous leukemia cells that is enhanced by 1, 25-dihydroxyvitamin D3. Leukemia 2009, 23, (11), 2171–2173. [DOI] [PubMed] [Google Scholar]
- 45.McDermott DF; Atkins MB PD‐1 as a potential target in cancer therapy. Cancer medicine 2013, 2, (5), 662–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen Y; Zhang X; Lu J; Huang Y; Li J; Li S Targeted delivery of curcumin to tumors via PEG-derivatized FTS-based micellar system. The AAPS journal 2014, 16, (3), 600–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sun J; Chen Y; Li K; Huang Y; Fu X; Zhang X; Zhao W; Wei Y; Xu L; Zhang P; Venkataramanan R; Li S A prodrug micellar carrier assembled from polymers with pendant farnesyl thiosalicylic acid moieties for improved delivery of paclitaxel. Acta Biomater 2016, 43, 282–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hou D-Y; Muller AJ; Sharma MD; DuHadaway J; Banerjee T; Johnson M; Mellor AL; Prendergast GC; Munn DH Inhibition of indoleamine 2, 3-dioxygenase in dendritic cells by stereoisomers of 1-methyl-tryptophan correlates with antitumor responses. Cancer Res 2007, 67, (2), 792–801. [DOI] [PubMed] [Google Scholar]
- 49.Sun J; Liu Y; Chen Y; Zhao W; Zhai Q; Rathod S; Huang Y; Tang S; Kwon YT; Fernandez C Doxorubicin delivered by a redox-responsive dasatinib-containing polymeric prodrug carrier for combination therapy. J. Control. Release 2017, 258, 43–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Broz ML; Binnewies M; Boldajipour B; Nelson AE; Pollack JL; Erle DJ; Barczak A; Rosenblum MD; Daud A; Barber DL Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell 2014, 26, (5), 638–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gabrilovich DI; Nagaraj S Myeloid-derived suppressor cells as regulators of the immune system. Nature reviews immunology 2009, 9, (3), 162–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Roth F; Adriana C; Vella JL; Zoso A; Inverardi L; Serafini P Aptamer-mediated blockade of IL4Rα triggers apoptosis of MDSCs and limits tumor progression. Cancer research 2012, 72, (6), 1373–1383. [DOI] [PubMed] [Google Scholar]
- 53.Djeu J; Wei S Chemoimmunomodulation of MDSCs as a novel strategy for cancer therapy. Oncoimmunology 2012, 1, (1), 121–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Umansky V; Blattner C; Gebhardt C; Utikal J The role of myeloid-derived suppressor cells (MDSC) in cancer progression. Vaccines 2016, 4, (4), 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sawanobori Y; Ueha S; Kurachi M; Shimaoka T; Talmadge JE; Abe J; Shono Y; Kitabatake M; Kakimi K; Mukaida N Chemokine-mediated rapid turnover of myeloid-derived suppressor cells in tumor-bearing mice. Blood 2008, 111, (12), 5457–5466. [DOI] [PubMed] [Google Scholar]
- 56.Patyna S; Laird AD; Mendel DB; O’Farrell A-M; Liang C; Guan H; Vojkovsky T; Vasile S; Wang X; Chen J SU14813: a novel multiple receptor tyrosine kinase inhibitor with potent antiangiogenic and antitumor activity. Molecular cancer therapeutics 2006, 5, (7), 1774–1782. [DOI] [PubMed] [Google Scholar]
- 57.DePrimo SE; Bello CL; Smeraglia J; Baum CM; Spinella D; Rini BI; Michaelson MD; Motzer RJ Circulating protein biomarkers of pharmacodynamic activity of sunitinib in patients with metastatic renal cell carcinoma: modulation of VEGF and VEGF-related proteins. Journal of translational medicine 2007, 5, (1), 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kao J; Ko EC; Eisenstein S; Sikora AG; Fu S; Chen S.-h. Targeting immune suppressing myeloid-derived suppressor cells in oncology. Critical reviews in oncology/hematology 2011, 77, (1), 12–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chao Q; Sprankle KG; Grotzfeld RM; Lai AG; Carter TA; Velasco AM; Gunawardane RN; Cramer MD; Gardner MF; James J Identification of N-(5-tert-butyl-isoxazol-3-yl)-N′-{4-[7-(2-morpholin-4-yl-ethoxy) imidazo [2, 1-b][1, 3] benzothiazol-2-yl] phenyl} urea dihydrochloride (AC220), a uniquely potent, selective, and efficacious FMS-like tyrosine kinase-3 (FLT3) inhibitor. Journal of medicinal chemistry 2009, 52, (23), 7808–7816. [DOI] [PubMed] [Google Scholar]
- 60.Small D FLT3 mutations: biology and treatment. ASH Education Program Book 2006, 2006, (1), 178–184. [DOI] [PubMed] [Google Scholar]
- 61.Wongkajornsilp A; Wamanuttajinda V; Kasetsinsombat K; Duangsa-ard S; Sa-ngiamsuntorn K; Hongeng S; Maneechotesuwan K Sunitinib indirectly enhanced anti-tumor cytotoxicity of cytokine-induced killer cells and CD3+ CD56+ subset through the co-culturing dendritic cells. PloS one 2013, 8, (11), e78980. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.