

Summary
Objective:
To characterize the phenotypic spectrum associated with GNAO1 variants and establish genotype‐protein structure‐phenotype relationships.
Methods:
We evaluated the phenotypes of 14 patients with GNAO1 variants, analyzed their variants for potential pathogenicity, and mapped them, along with those in the literature, on a three‐dimensional structural protein model.
Results:
The 14 patients in our cohort, including one sibling pair, had 13 distinct, heterozygous GNAO1 variants classified as pathogenic or likely pathogenic. We attributed the same variant in two siblings to parental mosaicism. Patients initially presented with seizures beginning in the first 3 months of life (8/14), developmental delay (4/14), hypotonia (1/14), or movement disorder (1/14). All patients had hypotonia and developmental delay ranging from mild to severe. Nine had epilepsy, and nine had movement disorders, including dystonia, ataxia, chorea, and dyskinesia. The 13 GNAO1 variants in our patients are predicted to result in amino acid substitutions or deletions in the GNAO1 guanosine triphosphate (GTP)‐binding region, analogous to those in previous publications. Patients with variants affecting amino acids 207‐221 had only movement disorder and hypotonia. Patients with variants affecting the C‐terminal region had the mildest phenotypes.
Keywords: developmental and epileptic encephalopathy, GNAO1, mosaicism, movement disorders
1 |. INTRODUCTION
GNAO1, or guanosine nucleotide‐binding protein G(o) subunit α, is a gene that has been associated with neurodevelopmental disorders, including early onset developmental and epileptic encephalopathy (DEE),1 developmental delay without epilepsy, and a range of movement disorders.2–16 GNAO1 encodes a G‐protein α subunit that, along with dimerized β and γ subunits, forms a heterotrimeric G‐protein complex. G‐protein α subunits contain guanosine triphosphate (GTP)‐binding sites and dissociate from both the G‐protein–coupled receptor and the β‐γ dimer of the complex when activated. Alpha subunits are generally classified as stimulatory, inhibitory, or other (o). GNAO1 encodes an “o” type α subunit; beyond GTP binding, it has a less well‐established role in signaling.10 GNAO1 is highly expressed in the central nervous system and is involved in neuronal excitability and neurotransmission.2,10
Here, we report 14 previously unpublished patients with heterozygous, de novo GNAO1 variants classified as pathogenic or likely pathogenic, including one variant shared by two siblings attributable to parental mosaicism. Two key components of our in silico variant analyses were the comparison of variants in our cohort with those present in the general population and the variant location on the predicted GNAO1 protein structure. Both analyses demonstrate the importance of the GTP‐binding region of the protein. We expand and add evidence to the previously reported phenotypic spectrum of GNAO1 encephalopathy with the addition of our 14 patients. In addition, we identify a region of the GNAO1 protein that correlates with a movement disorder–dominated phenotype without epilepsy.
2 |. MATERIALS AND METHODS
Research was conducted with approval from the institutional review board at Boston Children’s Hospital (BCH). Patient ascertainment included the Epilepsy Genetics Program, Genetics and Genomics Clinic, and Movement Disorders Clinic at BCH, the National Institutes of Health Undiagnosed Diseases Network, and the Australian Epilepsy Research Centre. Written informed consent was obtained from the parents or legal guardians of all patients included.
Deep phenotyping was performed, including review of medical records, electroencephalogram (EEG) reports, magnetic resonance imaging (MRI), and imaging reports when available, by our multidisciplinary team consisting of pediatric neurologists, including those with training in epilepsy and movement disorders, geneticists, a neuroradiologist, and a genetic counselor.
All 14 individuals had GNAO1 variants identified through clinical testing, either by whole exome sequencing (WES; Patients 1‐6, 8, 9, 13, and 14) or by targeted next generation sequencing epilepsy panels (Patients 7 and 10‐12). Parental testing was completed for all but one patient. In the case of two siblings with the same variant, each reported as de novo, we reanalyzed parental WES data for evidence of parental mosaicism using the BCH Connect analysis platform. We conducted a thorough review of the literature (https://www.ncbi.nlm.nih.gov/pubmed/?term=gnao1, accessed October 2017) and ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/?term=GNAO1, accessed March 2018) to identify additional cases for phenotypic comparison, to evaluate whether a given variant was previously reported in the literature or in ClinVar, and to evaluate for potential pathogenicity. Additionally, in silico predictions regarding pathogenicity were assessed using a combination of predictors, including the Alamut software suite (http://www.interactive-biosoftware.com/alamut-visual) and PolyPhen‐2 (http://genetics.bwh.harvard.edu/pph2/). We compared the presence and location of variants in patients, including those in the literature and ClinVar, to those in the population databases ExAC (http://exac.broadinstitute.org/) and gnomAD (http://gnomad.broadinstitute.org/).17 Variant classifications were determined based on the guidelines of the American College of Medical Genetics and Genomics (ACMGG).18
Using the GNAO1 protein sequence (canonical transcript ENST00000262494, CCDS10757, NM_020988.2), we analyzed the position of the 13 variants in this report along with 41 additional variants identified through our literature search (Table S1), totaling 54 disease‐associated variants. For the following analyses, we excluded the intronic variant c.723+1G>A, because it cannot be annotated into the protein sequence.15 To assess variant pathogenicity, we evaluated the missense variant tolerance ratio (MTR) based on regional depletion in the general population.19 The MTR is a statistically normalized measure of how often a coding region “tolerates” missense variation based on the variant burden observed in gnomAD (n = 138 632). We then evaluated the amino acid gene‐family paralog conservation (Parazscore, https://doi.org/10.1101/159780) to assess for enrichment of patient‐related variants compared to gnomAD variants in conserved regions. The Parazscore leverages amino acid conservation across gene‐family members assuming that conserved sites are more likely to be important for protein function and thus more likely to be present in patients than controls. Statistical comparison between the variant counts of patients versus gnomAD was conducted using a two‐sided Fisher’s exact test with nominal two‐sided P values < 0.05 considered significant.
We assessed genotype‐phenotype correlations by comparing the locations of variants and the phenotypic features associated with each variant, with a focus on epilepsy and movement disorders. A model of the GNAO1 protein structure was generated on a template from the Protein Data Bank20 identifier 3C7K,21 using SWISS‐MODEL22 and mapped on complex structures using deconStruct.23 Illustrations were generated using PyMol.24
2.1 |. Data availability
Anonymized data will be made available by request from any qualified investigator.
3 |. RESULTS
3.1 |. Genetic variants in GNAO1
We identified 13 unique heterozygous GNAO1 variants in 14 patients (Table 1, Figure 1). Variants were reported as de novo in 13 patients after parental testing; one did not have parental testing performed. Parental mosaicism was suggested in one family by the same GNAO1 G40E variant present in two siblings, Patients 3 and 4.
TABLE 1.
Pathogenicity predictions of de novo variants in GNAO1
| Gender | Patient 1 Male |
Patient 2 Female |
Patient 3 Male |
Patient 4 Female |
Patient 5 Male |
Patient 6 Female |
Patient 7 Female |
Patient 8 Female |
Patient 9 Male |
Patient 10 Female |
Patient 11 Male |
Patient 12 Female |
Patient 13 Female |
Patient 14 Female |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age, y | 8 | 2 | 20 | 15 | 10 | 2 | 2 | 15 | 4 | 6 | 2 | 5 | 13 | 10 |
| Variant | c.118G>C (p.G40R) | c.118G>T (p.G40W) | c.119G>A (p.G40E) | c.119G>A (p.G40E) | c.620C>A (p.S207Y) | c.625C>T (p.R209C) | c.626G>A (p.R209H) | c.662C>A (p.A221D) | c.692A>G (p.Y231C) | c.818A>T (D273V) | c.836T>A (p.I279N) | c.871T>A (p.Y291N) | c.1030_ 1032delATT (p.I344del) | c.1046_1055del10ins10 (p.R349_G352delinsQGCA) |
| Conservation | High | High | High | High | High | High | High | High | High | High | High | High | N/A | N/A |
| Align‐GVGD | C15 | C15 | C0 | C0 | C65 | C65 | C25 | C65 | C65 | C15 | C0 | C0 | N/A | N/A |
| SIFT | Deleterious | Deleterious | Deleterious | Deleterious | Deleterious | Deleterious | Deleterious | Deleterious | Deleterious | Deleterious | Deleterious | Deleterious | N/A (in‐Frame deletion) | N/A |
| Mutation Taster | Disease causing | Disease causing | Disease causing | Disease causing | Disease causing | Disease causing | Disease causing | Disease causing | Disease causing | Disease causing | Disease causing | Disease causing | N/A | N/A |
| PolyPhen‐2 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 0.989 | 0.999 | 0.993 | 1.0 | 0.997 | N/A | N/A |
| ClinVar (number of cases) | Pathogenic (1); uncertain significance (1) | Pathogenic (1) | Pathogenic (1) | Pathogenic (1) | Pathogenic (1) | Pathogenic/likely pathogenic (3) | Pathogenic (2); uncertain significance (1) | N/A | Likely pathogenic (1) | N/A | Pathogenic (1) | Likely pathogenic (1) | N/A | N/A |
| ExAC AF | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| gnomAD AF | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
GNAO1 variants were annotated based on transcript NM_020988.2.
AF, allele frequency; N/A, not available.
FIGURE 1.
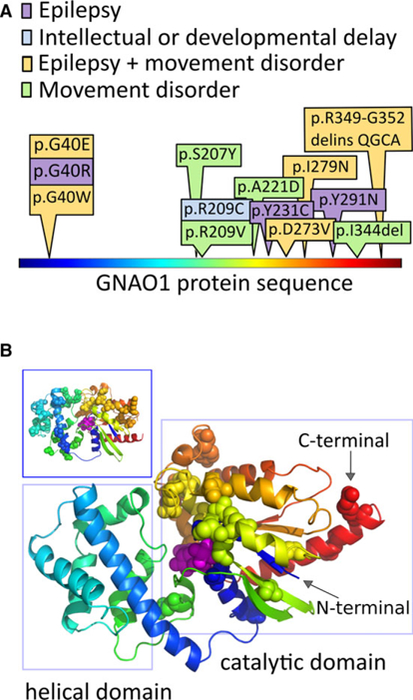
Patient variants in GNAO1 affect the guanosine triphosphate–binding region. Disease‐associated amino acid residues in the GNAO1 protein localize to the catalytic domain. A, The protein is shown in linear representation, colored using a “rainbow” scheme, starting with blue at the N‐terminal, and ending with red at the C‐terminal. The amino acid substitutions altered by the GNAO1 variants in our series are depicted here, with colors depicting the neurological features present in each case. B, The positions of disease‐associated variants, from this series and the literature, are shown as spheres on the model of the GNAO1 structure. The GNAO1 substrate is also shown as spheres, colored magenta. The same coloring scheme is used in A and B. Patient variants cluster in the region of the catalytic domain (right rectangle), which starts at the very N‐terminal aspect of the sequence, weaves into the helical domain, and returns back to complete the catalytic domain structure. The long helix on the N‐terminal is not shown, because its position in the active conformation of Gα is uncertain, and it carries no reported disease‐associated amino acid positions. Inset (upper left-hand box): The distribution of missense variant amino acids in ExAC17 covers both structural domains of the protein and does not overlap with the disease‐associated amino acid changes in our series
Five variants previously reported in the literature, in other patients, are present in Patient 1 (p.G40R),4,15 Patient 6 (p.R209C),12 Patient 7 (p.R209H),6,7,9,11 Patient 9 (p.Y231C),4 and Patient 11 (p.I279N).2 Variants in our cohort affecting the previously implicated GNAO1 G40 amino acid site4,15 are present in Patient 2 (p.G40W) and Patients 3 and 4 (p.G40E). We identified six novel variants localized to previously unreported sites in the protein: c.620C>A (p.S207Y), c.662C>A (p.A221D), c.818A>T (p.D273V), c.871T>A (p.Y291N), c.1030_1032delATT (p.I344del), and c.1046_1055del10ins10 (p.R349_G352delinsQGCA). All GNAO1 variants are annotated based on transcript NM_020988.2.
3.1.1 |. Assessment of pathogenicity of GNAO1 variants
Ten of the 13 unique variants have been reported in ClinVar, adding support for their pathogenicity (Table 1); five were classified as pathogenic, one with conflicting classification as pathogenic versus likely pathogenic, two as likely pathogenic, and two as pathogenic versus uncertain significance (Table 1). The two variants with conflicting classifications as pathogenic or uncertain significance (Patient 1 with p.G40R4,15 and Patient 7 with p.R209H6,7,9,11) were de novo variants resulting in different amino acid substitutions at residues previously reported as pathogenic in multiple patients; thus, we classified them as pathogenic in accordance with ACMGG variant classification criteria.18 All variants were additionally evaluated for pathogenicity using Alamut. The three variants not listed in ClinVar were classified as pathogenic based on in silico predictions and in accordance with ACMGG criteria (Table 1).18
3.1.2 |. Siblings with apparently de novo GNAO1 variants due to parental mosaicism
Patients 3 and 4 were a 20‐year‐old brother and a 15‐year‐old sister. Within 2 hours of life, Patient 3 presented with episodes of apnea and lip smacking. Subsequently, at 2 months, he developed infantile spasms and hypsarrhythmia as well as focal motor seizures with left eye twitching, left head turning, and left arm extension. His sister also presented with seizures, initially described as bilateral tonic–clonic, on her first day of life. She eventually developed multiple seizure types including focal motor seizures, described as left eye twitching, tonic seizures, and infantile spasms. Additional phenotypic features are summarized in Table 2. Family history was notable for febrile seizures in the father, which we consider likely to be unrelated to the siblings’ presentation given their GNAO1 findings. Extensive genetic and metabolic testing for both siblings was initially nondiagnostic. Initially, a mitochondrial disorder was suspected based on muscle biopsy in Patient 4 suggesting a defect in complex IV of the respiratory chain that was later considered to be a secondary finding. Genetic evaluation included a normal karyotype, chromosomal microarray, and infantile epilepsy gene panel in 2015. WES performed for both siblings and their parents showed a pathogenic variant in GNAO1 at c.119G>A (p.G40E) that appeared to be de novo in each sibling, leading to the presumption of either germline or gonadal parental mosaicism. Reevaluation of parental WES data revealed the variant allele (c.119G>A) in one parent in three of 150 reads (2%) consistent with germline mosaicism. This level of mosaicism is, however, below the current reporting threshold of Clinical Laboratory Improvement Amendments–certified laboratories.
TABLE 2.
Genotype and phenotype of 14 patients with disease‐associated variants in GNAO1
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | Patient 6 | Patient 7 | Patient 8 | Patient 9 | Patient 10 | Patient 11 | Patient 12 | Patient 13 | Patient 14 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age, y/sex | 8/M | 2/F | 20/M | 15/F | 10/M | 2/F | 2/F | 15/F | 4/M | 6/F | 2/M | 5/F | 13/F | 10/F |
| Variant | c.118G>C (p.G40R) | c.118G>T (p.G40W) | c.119G>A (p.G40E) | c.119G>A (p.G40E) | c.620C>A (p.S207Y) | c.625C>T (p.R209C) | c.626 G>A (p.R209H) | c.662C>A (p.A221D) | c.692A>G (p.Y231C) | c.818A>T (D273V) | c.836T>A (p.I279N) | c.871T>A (p.Y291N) | c.1030_1032delATT (p.I344del) | c.1046_1055del10ins10 (p.R349_G352delins QGCA) |
| Novel/reported | Reported4,15 | Novel | Novel | Novel | Novel | Reported12 | Reported6,7,9,11 | Novel | Reported5 | Novel | Reported2 | Novel | Novel | Novel |
| Initial symptom (age) | Seizures (2.5 mo) | Seizures (5 wk) | Seizures (15 h) | Seizures (2 h) | Developmental delay (infancy) | Hypotonia (6 mo) | Developmental delay (6 mo) | Developmental delay (9 mo) | Seizure (5 d) | Seizures (2 d) | Seizures (1 h) | Seizures (2 mo) | Movement disorder (12 mo) | Developmental delay (6 mo) |
| Epilepsy type | DEE → LGS | DEE | DEE | DEE | N/A | N/A | N/A | N/A | DEE | DEE | DEE | DEE | N/A | Focal seizures |
| Onset | Spasms | Focal seizures | Spasms | GTC | N/A | N/A | N/A | N/A | Myoclonic seizures | GTC | Spasms | Focal seizures | N/A | Focal seizures |
| Other seizure types | Tonic, atonic, myoclonic seizures, GTC, spasms | N/A | Myoclonic, tonic seizures, GTC, spasms | Spasms | N/A | N/A | N/A | N/A | Focal, myoclonic seizures | N/A | Focal seizures | N/A | N/A | |
| Movement disorder | Ataxia | N/A | N/A | Left arm dystonia | Dystonia | N/A | Dystonia, ataxia | Dystonia | N/A | Dyskinesia | Chorea, akathisia | N/A | Dystonia, chorea | Facial dyskinesia, tremor |
| Cognition | ID | ID | ID | ID | Normal | ID | MID | MID | ID | ID | ID | ID | MID | ID |
| Motor | Nonambulatory | Delay | Nonambulatory | Nonambulatory | Delay | Delay | Delay | Delay | Delay | Nonambulatory | Delay | Nonambulatory | Delay | Oromotor apraxia |
| Speech | Nonverbal | Nonverbal | Nonverbal | Nonverbal | Delay | Delay | Nonverbal | Dysarthria | Delay | Nonverbal | Delay | Nonverbal | Delay | Delay, dysarthria |
| Tone | Hypotonia | Hypotonia | Hypotonia | Hypotonia | Hypotonia | Hypotonia | Hypotonia | Hypotonia | Hypotonia | Hypotonia | Hypotonia | Hypotonia | Hypotonia | Hypotonia |
| EEG | 3 y: slow spike and wave, multifocal spikes | 9 mo: right temporal seizures, focal spikes, focal slowing | 2 mo: hypsarrhythmia; 14 y: generalized onset of tonic seizures and epileptic spasms, generalized slowing | 9 d: multifocal spikes; 14 y: focal spikes and waves, absence of normal awake and sleep features | N/A | 15 mo: slow posterior dominant rhythm | 13 mo: normal | 11 y: normal; 15 y: abnormal during sleep, frequent sharp waves | Neonatal: multifocal epileptiform sharp waves; 20 mo: frequent bioccipital spikes | Abnormal epileptiform activity | Neonatal: multifocal sharp waves with high‐ amplitude bursts (not burst suppression); 8 mo: modified hypsarrhythmia | 1 y: multifocal spikes, focal seizures; 3 y: intermittent posterior slowing | 9 y: normal | 7 y: sleep‐ activated posterior temporal and occipital spikes |
| MRI | 4 mo: mildly prominent bifrontal subarachnoid spaces | 8 mo: bilateral mesial temporal sclerosis, diffuse parenchymal atrophy, delayed myelination | 2 y: status posttemporal lobectomy, left cerebral atrophy | 15 mo: nonspecific signal increase in globi pallidi, normal myelination | 9 y: normal | 1 y: generalized thinning of corpus callosum, relative paucity of deep white matter | 13 mo: frontal lobe volume loss | 15 y: normal | 2 y: prominent subarachnoid spaces | Normal | 2.5 y: moderate to progressive atrophy with delayed myelination | 2.5 y: normal | 8 y: normal | 9 y: normal |
| Other features | Swallowing dysfunction, chronic respiratory dysfunction | Gastroesophageal reflux | Swallowing dysfunction, cortical visual impairment, scoliosis | Astigmatism, myopia, alternating exotropia, scoliosis | N/A | N/A | N/A | Neonatal feeding difficulties | Spastic quadriplegia | Swallowing dysfunction | Intermittent exotropia | Hyperopia, esotropia | Single seizure at age 4 y | Swallowing dysfunction |
All variants are confirmed de novo except Patient 10 (D273V), for whom parental testing has not been possible.
DEE, developmental and epileptic encephalopathy; EEG, electroencephalogram; F, female; GTC, generalized tonic–clonic seizures; ID, intellectual disability; LGS, Lennox–Gastaut syndrome; M, male; MID, mild intellectual disability; MRI, magnetic resonance imaging; N/A, not available.
3.2 |. Analysis of GNAO1 variants in patients compared to controls
Based on the linear amino acid sequence of GNAO1 (canonical transcript ENST00000262494, CCDS10757, NM_020988.2), we compared the position of the exonic variants in 54 patients with the variants identified in the gnomAD reference database.17 Collectively, our 14 patients plus 40 from the literature comprise 30 unique variants affecting 23 different amino acid positions (Figure 2), whereas gnomAD variants affect 95 different amino acids. To further assess variant pathogenicity, we implemented MTR scores and Parazscores for variant interpretation. As expected, both MTR scores (P = 4.407e‐04) and Parazscores (P = 1.756e‐5) from patient variants were significantly lower than those observed in gnomAD controls, indicating population and evolutionary constraint of patient variants, respectively (Figure 2). Furthermore, within the amino acid boundaries of the GTP‐binding domain, we observed high intolerance for missense mutations (ie, lower MTR scores) and strong conservation across family members (ie, greater Parazscores). Taken together, these predictions strongly support the pathogenicity of GNAO1 variants in our cohort, and for patient variants found within the GTP‐binding domain.
FIGURE 2.
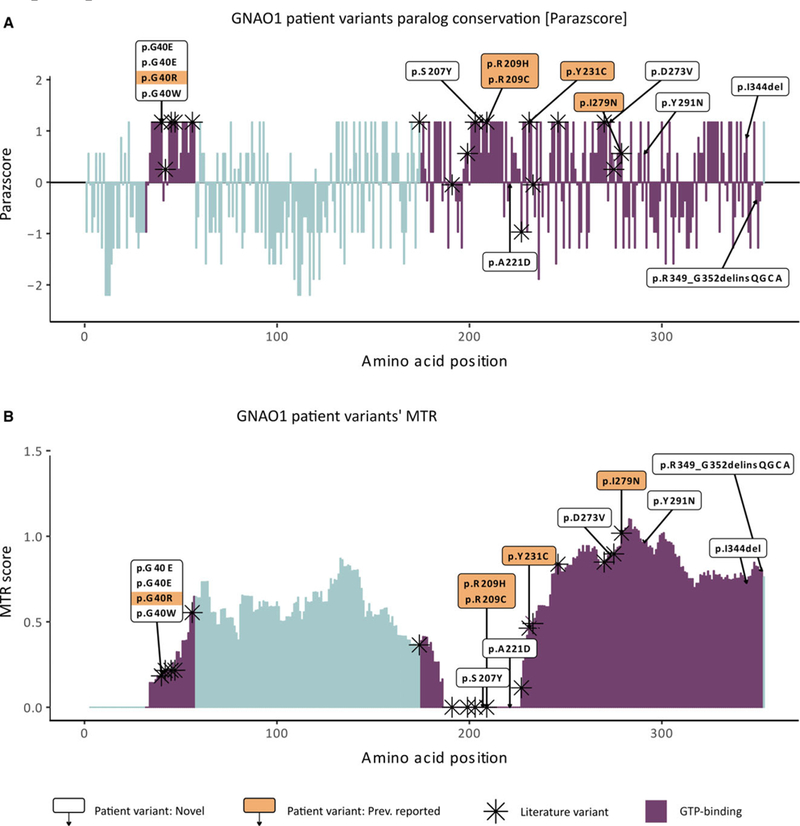
GNAO1 patient variant evolutionary conservation and population constrained assessment. A, GNAO1 patient variants’ paralog conservation score (Parazscore) across the linear protein sequence. Comparing the amino acid sequence of the GNAO1 protein to that of paralogous proteins in its gene family, the gene family‐wise paralog conservation is shown for each amino acid of GNAO1 protein sequence. Parazscore values range from negative values, representing less conservation at a given amino acid position, to positive values, representing high conservation, with the highest value indicating that identical amino acids are present in all related proteins. B, GNAO1 patient variants’ missense tolerance ratio (MTR).19 The score visualizes the tolerance to missense across the GNAO1 protein sequence based on depletion of variants in population controls from the gnomAD database (ExAC v2).17 MTR values range from 0 (extremely intolerant to missense variant amino acids) to 1.5 (for positions tolerant to missense variant amino acids). For both graphs, patient variant‐related amino acid substitutions not previously reported in the literature (white boxes, n = 9) and patient variant‐related amino acid substitutions previously reported in the literature (shaded boxes, n = 5) are labeled alongside amino acids altered by GNAO1 variants in the literature (asterisks, n = 40). Altogether, 23 amino acid positions are affected. Amino acids belonging to the guanosine triphosphate (GTP)‐binding domain are marked in blue and represent the vast majority of amino acids affected in patients. Intronic variant c.723+1G>A was not included.15 All patient variants fall within the boundaries of the GTP‐binding domain (blue bars)
3.3 |. Phenotypes associated with GNAO1 variants
Our 14 patients ranged in age from 2 to 20 years; five were male, and nine were female (Table 2). Nine of 14 (64%) had epilepsy, nine of 14 (64%) had movement abnormalities, and five of 14 (36%) had both epilepsy and movement abnormalities. A diagnosis of epilepsy was reported in nine patients. Eight patients had early onset DEE, with seizure onset at <3 months, epileptiform activity on EEG, and developmental stagnation or regression (Patients 1‐4 and 9‐12). The EEGs of all 9 patients with epilepsy showed epileptiform activity, including hypsarrhythmia in two 2 patients (Table 2). Patient 13 had a single afebrile seizure at 4 years of age and a normal EEG. Of the four patients without a history of epilepsy or seizure, three had an EEG; one was normal, one showed background slowing, and one was initially normal but later showed frequent focal spikes during sleep (Table 2).
Hypotonia was reported in all patients. Movement disorders were present in nine patients; five had severe dystonia (Patients 4, 5, 7, 8 and 13), two had ataxia (Patients 1 and 7), two had dyskinesia (Patients 10 and 14), two had chorea (Patients 11 and 13), one had akathisia (Patient 11), and one had tremors (Patient 14). Patient 14 had intermittent facial dyskinesia, swallowing dysfunction, and dysarthria, which has been reported in four patients with a severe phenotype.
Occipitofrontal circumference (OFC) data were available for 13 of 14 patients. One patient had microcephaly, Patient 3 with OFC 41 cm at 10 months (2.8‰). Brain magnetic resonance (MR) images were reviewed for 10 patients; MRI reports only were available for the remaining patients. The MR images were either normal or had nonspecific abnormalities, including diffuse atrophy (six patients) and abnormal myelination (two patients). One MR image showed globus pallidus/dentate signal increase, possibly related to vigabatrin use, and one MR image showed signs of mesial temporal sclerosis (Table 2).
All patients had developmental delay and showed a broad spectrum of severity. Five patients showed profound impairment and were nonverbal and nonambulatory. Patients 5, 13, and 14 had the mildest developmental abnormalities. Patient 5 presented with delayed speech and motor development but was reported to have met motor and language milestones and to have normal cognition at 10 years. Patient 13 presented with language delay, speaking single words at 2.5 years and full sentences at 4 years, and mild intellectual disability. Patient 14 presented with oromotor apraxia and moderate intellectual disability. At 11 years, she could recognize colors, some words and numbers, and understand early math concepts but is making very slow academic progress.
We compared the phenotypes of our 14 patients with those of 41 individuals with disease‐associated GNAO1 variants in the literature (Table S1).2–16,25 Striking phenotypic patterns emerged, despite incomplete phenotype information in some patients.
Epilepsy and movement disorders are common phenotypic features. Forty‐two of 55 individuals (76%) had movement disorders, 35 of 55 (64%) had epilepsy, and 23 of 55 (42%) had both. The range of movement disorders includes chorea, dystonia, dyskinesia, stereotypies, and ataxia. The 35 individuals with epilepsy most often had DEE (24/35, 69%), including infantile spasms with hypsarrhythmia; seven individuals had focal seizures only, two had generalized seizures only, one had both generalized and focal seizures, one had infantile spasms only, and three reports did not contain enough information as to the type of epilepsy to allow further classification.
All patients had developmental delay in our cohort and the cases in the literature, with a wide range of severity, including our patients with mild delay. Hypotonia was commonly reported (76%), and microcephaly was reported in 20% of those cases for which data were available. MRI was reported to be normal in 20 of 55 individuals; 16 showed atrophy (both generalized and focal), 12 had corpus callosum abnormalities (most often a thin corpus callosum), seven showed delayed myelination, three had mild hypoplasia of the caudate nuclei, two had bilateral globus pallidus abnormalities (hypointense signal in a case with p.E246K6 and hyperintense signal in Patient 4 with p.G40E), and one had a left frontal astrocytoma thought to be unrelated to the GNAO1 variant.15
3.4 |. Relationship of phenotype with amino acid position and protein domain
Given the high sequence similarity of GNAO1 to crystallized forms of Gα proteins, its structure is predicted to consist of two domains: one helical and one catalytic with a GTPase‐binding pocket. Mapping the disease‐associated variants reported to date onto this model structure demonstrates that disease‐associated variants are located preferentially in the catalytic domain. This is even more intriguing when taking into account that the two globular domains of GNAO1 are formed by nonconsecutive regions of the protein sequence (Figure 1). In contrast, variants present in the general, unaffected population can be found in both the helical and catalytic domains (Figure 1B, inset).
Evaluating the disease‐associated variants on the predicted protein structure, the variants associated with movement disorders exclusively are found outside the catalytic pocket (Figure S2), between AA207 and AA221. Reviewing our patients and those in the literature, most patients with variants affecting AA207‐AA246 present with movement disorder but not with epilepsy, with the exception of two patients with GNAO1 p.Y231C (Patient 9, this report) and p.A227V.12 Conversely, any variant in or near the catalytic pocket resulted in epilepsy, either alone or in association with movement disorders.
The most commonly affected amino acids R209 and G40 are both located in the catalytic domain. Examining the phenotypes associated with variants at R209 (p.R209C12 in Patient 6 and p.R209H6,7,9,11 in Patient 7), only three of 10 individuals (30%) were reported to have seizures, one generalized tonic–clonic and two focal. By comparison, variants at G40 produce a far more severe phenotype (p.G40R4,15 in Patient 1, p.G40W in Patient 2, and p.G40E in Patients 3 and 4). The G40 site has been implicated in seven patients with epilepsy, including six with DEE. Furthermore, three of these seven individuals are reported to have both a movement disorder and epilepsy classified as DEE.4,15,25 Six of these seven also had developmental delay and/or intellectual disability, hypotonia, and abnormal findings on MRI.
4 |. DISCUSSION
GNAO1 encephalopathy has a broad phenotypic spectrum, most commonly presenting with seizures and less frequently with movement disorders. Developmental delay and hypotonia occur in nearly all cases, although they led less frequently to medical attention. The severe phenotype, occurring in 44% of cases, comprises an early onset profound DEE, with seizure onset soon after birth in 24 of 55 cases.2–5,8,10,12,14–16,26
MRI studies have not shown major structural abnormalities in patients with GNAO1 variants. Progressive diffuse atrophy and delayed myelination are noted in some cases with thinning of the corpus callosum.2,5,6,8,10,12,14–16 For patients with GNAO1-related movement disorders, treatments such as deep brain stimulation have been undertaken9,13,15 and require neuroimaging. Therefore, longitudinal assessment of the MRI features of patients with movement disorders will likely be available in the future.
Previous reports mention a slight female predominance in patients with GNAO1‐related neurodevelopmental disorders10 and an apparent excess of sibling pairs identified with apparently de novo GNAO1 pathogenic variants.9,11 In our cohort, nine of 14 (64%) patients are female, and of the 40 cases in the literature identified as male or female, 24 (60%) were female; as more cases continue to accrue, the sex ratio can be more accurately determined. Together with our series, there are now three affected sibling pairs as well as an individual whose brother had previously passed away with the same phenotype.9,11 These variants appeared to occur de novo based on “negative” parental testing. However, their recurrence in siblings leads to the hypothesis of parental mosaicism, either in the germline with low allele frequency undetectable by standard clinical sequencing or restricted to the gametes. In our case, reanalysis of the clinical sequencing data identified parental mosaicism in 2% (3/150) WES reads. The phenomenon of parental mosaicism has been observed in association with other disease‐associated genes, including those associated with neurodevelopmental disorders.27 At present, we do not have evidence to conclude that GNAO1 is more likely to be associated with parental mosaicism than other genes.28 However, the possibility of parental mosaicism needs to be taken into account when providing genetic counseling to families that have one child with a de novo variant in GNAO1. A subsequent affected child must be considered, and if further children are planned, testing with high depth coverage in the parents offers an opportunity to assess this risk.
GNAO1 encodes a G‐protein α subunit highly expressed in the brain. Heterozygous de novo variants are thought to cause a gain of function, as demonstrated in the GNAO1+/G184S mutant mouse model.29 However, recent in vitro studies suggest that the functional consequences of GNAO1 variants depend on their location within the gene.30 The G‐protein α subunit consists of two sections of a P‐loop structure containing a GTP‐binding domain. The amino acids between these domains create a helical insertion domain that isolates and stabilizes the guanine nucleotide upon GTP binding and must be displaced for GTP/guanosine diphosphate dissociation to occur. It is in this critical region that the majority of patient variants are located, both in our patients and in the literature.2
We observed interesting correlations between phenotype and genotype across all reported cases according to amino acid position and protein structure. Not surprisingly, the picture was dominated by pathogenic or likely pathogenic variants affecting the GTP‐binding regions of the protein in all but one of the published variants, with the only variant identified in the helical insertion domain located at AA174,2 a site adjacent to the start of the GTP‐binding protein domain. The localization of patient variants in the GTP‐binding domain is consistent with our current understanding of the Gα function; both the catalytic pocket and interactions with its protein partners are affected through this domain (Figure S1). It is possible that the role of the helical domain is simply to function as the “lid” in the active conformation of the protein, requiring a conserved amino acid sequence. A dysfunctional GNAO1 catalytic pocket may lead to extended GTP hydrolysis time and consequentially the failure of dissociation between Gα and the Gβ‐γ dimer. This in turn, through the failure of ion channel regulation exerted by the Gβ‐γ dimer, may lead to neuronal hyperexcitability and an epilepsy phenotype; this hypothesis will require further functional testing, as pioneered by Feng et al.30
Many of the shared features identified in the current GNAO1 patient population are not specific to any single variant or amino acid position. Epilepsy was seen across variants affecting amino acids throughout the protein, and different types of epilepsy seem to be equally distributed across the gene. Movement disorders were associated with amino acid positions across the gene as well, particularly associated with the AA207‐246 region, which may act as a “hotspot” for movement disorder phenotypes typically, although not always, without epilepsy. In addition, it is worth noting that patients with variants affecting GNAO1 AA40 had particularly severe phenotypic presentations including all features.
As the case number for GNAO1‐associated neurodevelopmental disorders grows, expansion of the clinical phenotypic spectrum continues to emerge. When GNAO1 variants are identified in individuals, it is most often by WES rather than by targeted testing. This likely reflects the lag time between gene discovery and inclusion of a given new gene on targeted multigene panels as well as a lag in clinician awareness for the gene. Although the number of individuals undergoing WES is increasing, we would support clinical testing laboratories adding GNAO1 to their panels for early onset epilepsy, DEE, and movement disorders. Earlier genetic identification of a variant associated with DEE might change management, in that epilepsies arising from variants in signal transduction genes are generally not treated by focal brain resection (as had been pursued in one of our patients). Similarly, recognition of a GNAO1 variant in a patient with a mixed movement disorder should prompt evaluation for therapy with deep brain stimulation, as it has been shown to be effective in a number of cases.9,13 Patients with GNAO1 variants have often been mistakenly diagnosed with secondary movement disorders, which derive much lower benefit from deep brain stimulation,31 in contrast to a GNAO1‐related disorder.
5 |. CONCLUSIONS
The phenotypic spectrum of GNAO1‐related neurodevelopmental disease includes epilepsy and a range of movement disorders, often with epilepsy as the presenting feature. A range of movement disorders are seen in the majority of patients, even if not as the presenting symptoms. Hypotonia and developmental delay were present in all patients in our series with a wide range of severity.
All patients had variants in the GTP‐binding region of GNAO1, highlighting the importance of this region for normal neurodevelopment. We identified a small cluster of variants apparently associated only with movement disorders but not epilepsy. The presence of two siblings with the same pathogenic variant, with parents who initially appear to lack that variant, highlights the importance of considering parental mosaicism when counseling families.
In addition to expanding and refining the phenotypic spectrum of GNAO1‐related neurodevelopmental disease in a cohort rigorously analyzed for variant pathogenicity and phenotypic features, we highlight more generally the importance of considering protein structure‐phenotype correlations, which may help determine disease prognosis in children with early onset neurodevelopmental diagnoses. Our findings highlight the importance of pursuing a genetic etiology in patients with a wide range of presenting symptoms, including patients who were previously diagnosed with acquired etiologies for their multisymptom neurodevelopmental conditions. Particularly when the course of treatment may be altered by the presence of a pathogenic variant in GNAO1—currently deep brain stimulation for movement disorders, but perhaps one day a more targeted treatment addressing the epilepsy and other features of the disorder—early genetic diagnosis should be pursued.
Epilepsia is a member of COPE (Committee on Publication Ethics), and we adhere to its principles (http://publicationethics.org/).
Supplementary Material
Significance:
GNAO1 encephalopathy most frequently presents with seizures beginning in the first 3 months of life. Concurrent movement disorders are also a prominent feature in the spectrum of GNAO1 encephalopathy. All variants affected the GTP‐binding domain of GNAO1, highlighting the importance of this region for G‐protein signaling and neurodevelopment.
Key Points.
Pathogenic variants in GNAO1 result in early onset epilepsy and/or movement disorders, with seizures as the most common presenting feature
Pathogenic variants in GNAO1 lead to alterations in the GTP-binding region of the GNAO1 protein
Genotype-phenotype correlations have emerged, with one region of the protein associated with a movement disorder and hypotonia phenotype without seizures
Parental mosaicism can lead to siblings carrying the identical GNAO1 variant that appeared to be de novo
ACKNOWLEDGMENTS
We thank the patients and their families for participating in this study.
Funding information
A.R. was supported by a Fellowship of the Belgian American Educational Foundation and by a Fulbright Program grant sponsored by the Bureau of Educational and Cultural Affairs of the United States Department of State and administered by the Institute of International Education. A.P. was supported by the Boston Children’s Hospital Translational Research Program. The Undiagnosed Disease Network was supported by the National Institutes of Health (NIH) Common Fund, through the Office of Strategic Coordination/Office of the NIH Director under award numbers U01HG007690 and U01HG007942. I.E.S. was supported by an Australian National Health and Medical Research Council Program Grant and Practitioner Fellowship.
APPENDIX
UNDIAGNOSED DISEASES NETWORK MEMBERS INCLUDE THE FOLLOWING INDIVIDUALS
David R. Adams, Aaron Aday, Mercedes E. Alejandro, Patrick Allard, Euan A. Ashley, Mahshid S. Azamian, Carlos A. Bacino, Eva Baker, Ashok Balasubramanyam, Hayk Barseghyan, Gabriel F. Batzli, Alan H. Beggs, Babak Behnam, Hugo J. Bellen, Jonathan A. Bernstein, Anna Bican, David P. Bick, Camille L. Birch, Devon Bonner, Braden E. Boone, Bret L. Bostwick, Lauren C. Briere, Elly Brokamp, Donna M. Brown, Matthew Brush, Elizabeth A. Burke, Lindsay C. Burrage, Manish J. Butte, Shan Chen, Gary D. Clark, Terra R. Coakley, Joy D. Cogan, Heather A. Colley, Cynthia M. Cooper, Heidi Cope, William J. Craigen, Precilla D’Souza, Mariska Davids, Jean M. Davidson, Jyoti G. Dayal, Esteban C. Dell’Angelica, Shweta U. Dhar, Katrina M. Dipple, Laurel A. Donnell‐Fink, Naghmeh Dorrani, Daniel C. Dorset, Emilie D. Douine, David D. Draper, Annika M. Dries, David J. Eckstein, Lisa T. Emrick, Christine M. Eng, Gregory M. Enns, Ascia Eskin, Cecilia Esteves, Tyra Estwick, Laura Fairbrother, Liliana Fernandez, Carlos Ferreira, Elizabeth L. Fieg, Paul G. Fisher, Brent L. Fogel, Noah D. Friedman, William A. Gahl, Emily Glanton, Rena A. Godfrey, Alica M. Goldman, David B. Goldstein, Sarah E. Gould, Jean‐Philippe F. Gourdine, Catherine A. Groden, Andrea L. Gropman, Melissa Haendel, Rizwan Hamid, Neil A. Hanchard, Francis High, Ingrid A. Holm, Jason Hom, Ellen M. Howerton, Yong Huang, Fariha Jamal, Yong‐hui Jiang, Jean M. Johnston, Angela L. Jones, Lefkothea Karaviti, David M. Koeller, Isaac S. Kohane, Jennefer N. Kohler, Susan Korrick, Mary Koziura, Donna M. Krasnewich, Joel B. Krier, Jennifer E. Kyle, Seema R. Lalani, C. Christopher Lau, Jozef Lazar, Kimberly LeBlanc, Brendan H. Lee, Hane Lee, Shawn E. Levy, Richard A. Lewis, Sharyn A. Lincoln, Sandra K. Loo, Joseph Loscalzo, Richard L. Maas, Ellen F. Macnamara, Calum A. MacRae, Valerie V. Maduro, Marta M. Majcherska, May Christine V. Malicdan, Laura A. Mamounas, Teri A. Manolio, Thomas C. Markello, Ronit Marom, Martin G. Martin, Julian A. Martínez‐Agosto, Shruti Marwaha, Thomas May, Allyn McConkie‐Rosell, Colleen E. McCormack, Alexa T. McCray, Jason D. Merker, Thomas O. Metz, Matthew Might, Paolo M. Moretti, Marie Morimoto, John J. Mulvihill, David R. Murdock, Jennifer L. Murphy, Donna M. Muzny, Michele E. Nehrebecky, Stan F. Nelson, J. Scott Newberry, John H. Newman, Sarah K. Nicholas, Donna Novacic, Jordan S. Orange, James P. Orengo, J. Carl Pallais, Christina G. S. Palmer, Jeanette C. Papp, Neil H. Parker, Loren D. M. Pena, John A. Phillips III, Jennifer E. Posey, John H. Postlethwait, Lorraine Potocki, Barbara N. Pusey, Chloe M. Reuter, Lynette Rives, Amy K. Robertson, Lance H. Rodan, Jill A. Rosenfeld, Jacinda B. Sampson, Susan L. Samson, Kelly Schoch, Daryl A. Scott, Lisa Shakachite, Prashant Sharma, Vandana Shashi, Rebecca Signer, Edwin K. Silverman, Janet S. Sinsheimer, Kevin S. Smith, Rebecca C. Spillmann, Joan M. Stoler, Nicholas Stong, Jennifer A. Sullivan, David A. Sweetser, Queenie K.‐G. Tan, Cynthia J. Tifft, Camilo Toro, Alyssa A. Tran, Tiina K. Urv, Eric Vilain, Tiphanie P. Vogel, Daryl M. Waggott, Colleen E. Wahl, Melissa Walker, Nicole M. Walley, Chris A. Walsh, Jijun Wan, Michael F. Wangler, Patricia A. Ward, Katrina M. Waters, Bobbie‐Jo M. Webb‐Robertson, Monte Westerfield, Matthew T. Wheeler, Anastasia L. Wise, Lynne A. Wolfe, Elizabeth A. Worthey, Shinya Yamamoto, Yaping Yang, Amanda J. Yoon, Guoyun Yu, Diane B. Zastrow, Chunli Zhao, Allison Zheng.
Footnotes
DISCLOSURE
The authors have no conflicts of interest to report. We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of the article.
REFERENCES
- 1.Scheffer IE, Berkovic S, Capovilla G, et al. ILAE classification of the epilepsies: position paper of the ILAE commission for classification and terminology. Epilepsia 2017;58:512–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nakamura K, Kodera H, Akita T, et al. De novo mutations in GNAO1, encoding a Gαo subunit of heterotrimeric G proteins, cause epileptic encephalopathy. Am J Hum Genet 2013;93:496–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.EuroEPINOMICS-RES Consortium, Epilepsy Phenome/Genome Project, Epi4K Consortium. De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. Am J Hum Genet 2014;95:360–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Law CY, Chang ST, Cho SY, et al. Clinical whole‐exome equencing reveals a novel missense pathogenic variant of GNAO1 in a patient with infantile‐onset epilepsy. Clin Chim Acta 2015;451:292–6. [DOI] [PubMed] [Google Scholar]
- 5.Talvik I, Moller RS, Vaher M, et al. Clinical phenotype of de novo GNAO1 mutation: case report and review of literature. Child Neurol Open 2015;2:2329048X15583717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ananth AL, Robichaux-Viehoever A, Kim YM, et al. Clinical course of six children with GNAO1 mutations causing a severe and distinctive movement disorder. Pediatr Neurol 2016;59:81–4. [DOI] [PubMed] [Google Scholar]
- 7.Dhamija R, Mink JW, Shah BB, Goodkin HP. GNAO1‐associated movement disorder. Mov Disord Clin Pract 2016;3: 615–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gawlinski P, Posmyk R, Gambin T, et al. PEHO syndrome may represent phenotypic expansion at the severe end of the early‐onset encephalopathies. Pediatr Neurol 2016;60:83–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kulkarni N, Tang S, Bhardwaj R, Bernes S, Grebe TA. Progressive movement disorder in brothers carrying a GNAO1 mutation responsive to deep brain stimulation. J Child Neurol 2016;31:211–4. [DOI] [PubMed] [Google Scholar]
- 10.Marce-Grau A, Dalton J, Lopez-Pison J, et al. GNAO1 encephalopathy: further delineation of a severe neurodevelopmental syndrome affecting females. Orphanet J Rare Dis 2016;11:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Menke LA, Engelen M, Alders M, Odekerken VJ, Baas F, Cobben JM. Recurrent GNAO1 mutations associated with developmental delay and a movement disorder. J Child Neurol 2016;31:1598–601. [DOI] [PubMed] [Google Scholar]
- 12.Saitsu H, Fukai R, Ben-Zeev B, et al. Phenotypic spectrum of GNAO1 variants: epileptic encephalopathy to involuntary movements with severe developmental delay. Eur J Hum Genet 2016;24:129–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yilmaz S, Turhan T, Ceylaner S, Gokben S, Tekgul H, Serdaroglu G. Excellent response to deep brain stimulation in a young girl with GNAO1‐related progressive choreoathetosis. Childs Nerv Syst 2016;32:1567–8. [DOI] [PubMed] [Google Scholar]
- 14.Arya R, Spaeth C, Gilbert DL, Leach JL, Holland KD. GNAO1‐associated epileptic encephalopathy and movement disorders: c.607G>A variant represents a probable mutation hotspot with a distinct phenotype. Epileptic Disord 2017;19:67–75. [DOI] [PubMed] [Google Scholar]
- 15.Danti FR, Galosi S, Romani M, et al. GNAO1 encephalopathy: broadening the phenotype and evaluating treatment and outcome. Neurol Genet 2017;3:e143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schorling DC, Dietel T, Evers C, et al. Expanding phenotype of de novo mutations in GNAO1: four new cases and review of literature. Neuropediatrics 2017;48:371–7. [DOI] [PubMed] [Google Scholar]
- 17.Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein‐coding genetic variation in 60,706 humans. Nature 2016;536:285–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Traynelis J, Silk M, Wang Q, et al. Optimizing genomic medicine in epilepsy through a gene‐customized approach to missense variant interpretation. Genome Res 2017;27:1715–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berman HM, Westbrook J, Feng Z, et al. The protein data bank. Nucleic Acids Res 2000;28:235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Slep KC, Kercher MA, Wieland T, Chen CK, Simon MI, Sigler PB. Molecular architecture of Galphao and the structural basis for RGS16‐mediated deactivation. Proc Natl Acad Sci U S A 2008;105:6243–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Biasini M, Bienert S, Waterhouse A, et al. SWISS‐MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res 2014;42:W252–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang ZH, Lee HK, Mihalek I. Reduced representation of protein structure: implications on efficiency and scope of detection of structural similarity. BMC Bioinformatics 2010;11:155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alexander N, Woetzel N, Meiler J. bcl::Cluster: a method for clustering biological molecules coupled with visualization in the Pymol Molecular Graphics System. IEEE Int Conf Comput Adv Bio Med Sci 2011;2011:13–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bruun TUJ, DesRoches CL, Wilson D, et al. Prospective cohort study for identification of underlying genetic causes in neonatal encephalopathy using whole‐exome sequencing. Genet Med 2018;20(5):486–94. [DOI] [PubMed] [Google Scholar]
- 26.Pearson TS, Helbig I. Epileptic encephalopathy, movement disorder, and the yin and yang of GNAO1 function. Neurology 2017;89:754–5. [DOI] [PubMed] [Google Scholar]
- 27.Myers CT, Hollingsworth G, Muir AM, et al. Parental mosaicism in “de novo” epileptic encephalopathies. N Engl J Med 2018;378:1646–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen D-H, Méneret A, Friedman JR, et al. ADCY5‐related dyskinesia: broader spectrum and genotype‐phenotype correlations. Neurology 2015;85:2026–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kehrl JM, Sahaya K, Dalton HM, et al. Gain‐of‐function mutation in GNAO1: a murine model of epileptiform encephalopathy (EIEE17)? Mamm Genome 2014;25:202–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feng H, Sjogren B, Karaj B, Shaw V, Gezer A, Neubig RR. Movement disorder in GNAO1 encephalopathy associated with gain‐of‐function mutations. Neurology 2017;89:762–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koy A, Timmermann L. Deep brain stimulation in cerebral palsy: challenges and opportunities. Eur J Paediatr Neurol 2017;21:118–21. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Anonymized data will be made available by request from any qualified investigator.