

Abstract
A new study identifies loss-of-function mutations in HAVCR2, which encodes TIM-3, in patients with a rare cutaneous T cell lymphoma associated with aberrant immunological activation. These mutations lead to loss of the TIM-3 immunological checkpoint, thus promoting inflammation and malignancy.
Subcutaneous panniculitis-like T cell lymphoma (SPTCL) is a rare cutaneous T cell lymphoma that is often associated with aberrant immunological activation referred to as hemophagocytic lymphohistiocytosis (HLH). Affected individuals typically exhibit primarily subcutaneous adipose tissue infiltrates of pleomorphic α/β CD8+ T cells and myeloid cells, thus leading to rimming of adipocytes. Approximately 20% of patients develop autoimmune disorders. There is no standardized therapy, and despite SPTCL being a lymphoma, immunosuppressive regimes including cyclosporin A appear to be more effective treatments than chemotherapy or radiotherapy. In this issue, Gayden et al.1 report the discovery of germline mutations in HAVCR2 in the majority of patients with SPTCL from two different ancestries; these mutations result in loss-of-function missense variants in the cell-surface molecule T cell–immunoglobulin mucin-3 (TIM-3).
This work is the first description of a genetic disease with loss-of-function mutations in HAVCR2, which encodes TIM-3. Interestingly, these mutations result in T cell malignancy and autoimmunity. TIM-3 was initially described as a negative regulator of type 1 immunity2. Studies with TIM-3-deficient mice have shown that TIM-3 signaling is required for the induction of antigen-specific immunological tolerance and that TIM-3 blockade enhances the development of autoimmunity3. In addition to its expression on TH1 and TC1 cells, TIM-3 is highly expressed on myeloid cells4, thus implicating its role in regulating both innate and adaptive immunity. The idea that TIM-3 may have an important regulatory role in multiple cell types of the immune system is strengthened by the high expression of TIM-3 on ‘exhausted’ T cells in cancers5 and chronic viral infections6; its coexpression with other checkpoint molecules; and its expression on CD4+Foxp3+ regulatory T (Treg) cells in both humans and mice7,8 and on myeloid-derived suppressor cells9. However, TIM-3 does not possess either an ITIM or an ITSM motif, signature domains of classical inhibitory molecules, thus leading some to argue that it may behave as an activating molecule in certain conditions10.
TIM-3 biology
TIM-3 binds multiple ligands, including phosphatidylserine11, expressed on the surfaces of apoptotic cells; CEACAM-1, expressed by activated T cells12; and Galectin-9 (Gal-9), a lectin that binds N-linked sugars in the immunoglobulin variable (IgV) domain of TIM-3 through its two carbohydrate-recognition domains13. Interestingly, the authors found that 60% of patients with SPTCL have one of two mutations in HAVCR2, and both these mutations affect the part of the IgV domain of TIM-3 that is critical for Gal-9 binding. Staining patterns in patient biopsies and other biochemical experiments carried out with mutant constructs show that loss of the Gal-9-binding residues in TIM-3 leads to limited plasma membrane expression and intracellular aggregation, thereby resulting in cytoplasmic accumulation of TIM-3 and a lack of expression on immune cells in TIM-3-mutant individuals (Fig. 1). A debate still exists on whether Gal-9 is a bona fide ligand for TIM-3 (ref. 14), and the exact mechanism by which Gal-9 regulates tolerance induction by TIM-3 remains unclear. Gal-9 lacks a signal peptide required for transport through the endoplasmic reticulum–Golgi15 and consequently requires trafficking machinery for its secretion and biological activity. On the basis of the Gal-9 structure and the observations made in the present study, the association of Gal-9 with TIM-3 might be speculated to support trafficking of the complex to the cell surface. This trafficking may further facilitate clustering and orientation of TIM-3 in a manner that promotes binding to other ligands, such as phosphatidylserine and CEACAM-1, on the opposite face of the IgV domain. The latter interaction in turn facilitates TIM-3 signaling through phosphorylation of key tyrosine residues in its tail, thereby triggering the immunoregulatory function of TIM-3. It will be important to generate tools to decipher the exact mechanism by which Gal-9 and CEACAM-1 mediate TIM-3 activation. Further studies must be pursued with TIM-3-mutant mice representing the mutations found in patients with SPTCL, as well as with constitutively active TIM-3-mutant mice bearing a phosphomimetic alteration in the TIM-3 tail, to investigate exactly how TIM-3 mediates immunomodulatory responses.
Fig. 1 |. TIM-3 deficiency leads to uncontrolled immunological activation resulting in T cell malignancy and autoinflammation.
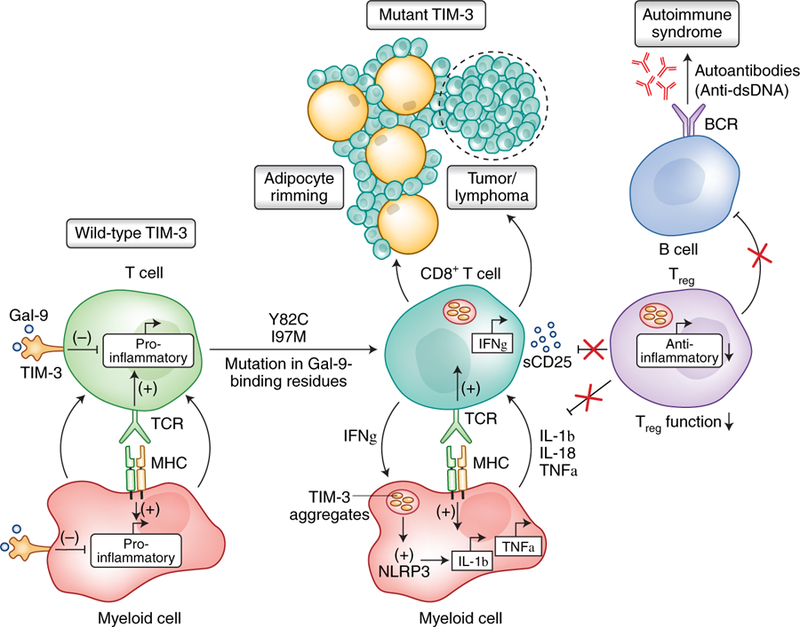
In unaffected individuals, Gal-9-driven TIM-3 signaling in immune cells tunes and shapes activating signals, thereby limiting autoimmunity and maintaining immunological homeostasis. Two mutant variants in the galectin-binding residues of TIM-3 (Y82C and I97M) observed in patients with SPTCL induce TIM-3 misfolding, thereby preventing its plasma membrane expression. This lack of expression leads to persistent immune activation, owing to a lack of intrinsic TIM-3 signaling in CD8 T cells, and impaired Treg function failing to regulate these overactive T cells. In myeloid cells, the misfolded TIM-3 molecules induce activation of the NLRP3 inflammasome, thus resulting in release of proinflammatory cytokines (IL-1β and IL-18). As in T cells, the lack of intrinsic TIM-3 signaling in myeloid cells results in hyperactive myeloid cells with enhanced production of TNFα. Together, these effects propagate a positive feed-forward mechanism that further amplifies inflammatory responses and results in the production of autoantibodies from B cells and induction of uncontrolled proliferation of effector T cells, thus culminating in adipose tissue infiltration of lymphocytes and T cell lymphoma. TCR, T cell receptor; BCR, B cell receptor; MHC, major histocompatibility complex; sCD25, soluble IL-2 receptor; dsDNA, double-stranded DNA.
There is no standardized therapy for SPTCL. However, the authors suggest that TIM-3-mutant SPTCL should be considered an inflammatory condition, on the basis of high levels of inflammatory cytokines (IL-1 and TNF-α) in the sera of the patients with hyperactive T cell responses. They therefore suggest targeting of IL-1 and IFN-γ pathways as a possible therapeutic approach for these patients. In fact, treatment of one patient with IL-1-receptor antagonist resulted in the resolution of clinical symptoms and sustained overall improvement, although a more extensive clinical trial would need to be conducted to develop a standardized anti-inflammatory treatment regimen for these patients.
Therapeutic implications
High TIM-3 expression correlates with the inhibition of effector T cell responses and T cell dysfunction associated with viral infections and cancer5,6. Furthermore, the expression of TIM-3 on tumor-associated myeloid cells has been shown to limit the therapeutic efficacy of DNA vaccination by diminishing the immunogenicity of nucleic acids released from dying tumor cells4. A hallmark of SPTCL is the infiltration of subcutaneous adipose tissue by T lymphocytes, which form a rim around the individual adipocytes. The abundant expression of TIM-3 on adipose tissue Treg cells may partly explain the increased inflammation around adipose tissue in these patients.
In vivo blockade of TIM-3 in preclinical models has demonstrated enhancement of antitumor immunity and decreased tumor burden, thus leading to its investigation as a potential therapeutic for human cancers. Ongoing clinical trials are using antibodies to TIM-3 in combination with blockade of other checkpoint molecules to obtain objective antitumor responses in patients with multiple cancers. These anti-TIM-3 candidates should be assessed for whether they interfere with Gal-9 binding. Furthermore, cancer patients receiving anti-TIM-3 treatment should be carefully monitored for potential autoimmune and autoinflammatory side effects, including signs of HLH, SPTCL and related syndromes, as have been observed in patients with HAVCR2 mutations. A deeper understanding of how loss of TIM-3 expression in patients with HLH/ SPTCL drives autoinflammatory disease together with T cell–derived tumors will be an important area for future studies aimed at understanding the mechanism through which TIM-3 mediates its inhibitory effects.
Footnotes
Competing interests
V.K.K. has patents dealing with intellectual property that have been licensed to Novartis Pharmaceuticals by the Brigham and Women’s Hospital.
Contributor Information
Karen O. Dixon, Ann Romney Center for Neurologic Diseases and Evergrande Center for Immunologic Diseases, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, USA.
Madhumita Das, Ann Romney Center for Neurologic Diseases and Evergrande Center for Immunologic Diseases, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, USA..
Vijay K. Kuchroo, Ann Romney Center for Neurologic Diseases and Evergrande Center for Immunologic Diseases, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, USA..
References
- 1.Gayden T et al. Nat. Genet 10.1038/s41588-018-0251-4 (2018). [DOI] [Google Scholar]
- 2.Monney L et al. Nature 415, 536–541 (2002). [DOI] [PubMed] [Google Scholar]
- 3.Sabatos CA et al. Nat. Immunol 4, 1102–1110 (2003). [DOI] [PubMed] [Google Scholar]
- 4.Chiba S et al. Nat. Immunol 13, 832–842 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fourcade J et al. J. Exp. Med 207, 2175–2186 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jin HT et al. Proc. Natl. Acad. Sci. USA 107, 14733–14738 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yan J et al. PLoS One 8, e58006 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sakuishi K et al. OncoImmunology 2, e23849 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dardalhon V et al. J. Immunol 185, 1383–1392 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Avery L, Filderman J, Szymczak-Workman AL & Kane LP Proc. Natl. Acad. Sci. USA 115, 2455–2460 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nakayama M et al. Blood 113, 3821–3830 (2009). [DOI] [PubMed] [Google Scholar]
- 12.Huang YH et al. Nature 536, 359 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu C et al. Nat. Immunol 6, 1245–1252 (2005). [DOI] [PubMed] [Google Scholar]
- 14.Leitner J et al. PLoS Pathog 9, e1003253 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hughes RC Biochim. Biophys. Acta 1473, 172–185 (1999). [DOI] [PubMed] [Google Scholar]