

Abstract
The 26S proteasome is the major regulated protease in eukaryotes and is responsible for degrading ubiquitinated substrates. It consists of a barrel-shaped 20S core peptidase and one or two 19S regulatory particles, which recognize, unfold, and translocate substrates into the core. The regulatory particle can be further divided into two multi-subunit complexes: the base and the lid. Here we present protocols for expressing the Saccharomyces cerevisiae base and lid recombinantly in Escherichia coli and purifying the assembled subcomplexes using a tandem affinity purification method. The purified complexes can then be reconstituted with 20S core to form fully functional proteasomes. Furthermore, we describe a method for incorporating the unnatural amino acid p-azido-L-phenylalanine into the recombinant complexes at any residue position, allowing for non-disruptive site-specific modifications of these large assemblies. The use of recombinant proteins allows for complete mutational control over the proteasome regulatory particle, enabling detailed studies of the mechanism by which the proteasome processes its substrates. The ability to then specifically modify residues in the regulatory particle opens the door to a wide range of previously impossible biochemical and biophysical studies. The techniques described below for incorporating unnatural amino acids into the proteasomal subcomplexes should be widely transferable to other recombinant proteins, whether individually purified or in larger multi-subunit assemblies.
Keywords: 26S proteasome, Recombinant expression, Macromolecular complex, Tandem affinity purification, Unnatural amino acid incorporation, Click chemistry, p-Azido-L-phenylalanine
1. Introduction
The 26S proteasome is the major protease in eukaryotic cells, where it is not only responsible for protein quality control and homeostasis through the degradation of misfolded, damaged, and obsolete polypeptides but also controls a myriad of vital cellular processes by the specific turnover of regulatory proteins [1–3]. Due to this role as one of the most critical proteome regulators, elucidating the proteasome’s structure, mechanisms of action, and regulation has been of prime interest in the ubiquitin-proteasome field. However, the high complexity of this 2.5 MDa molecular machine with more than 34 different subunits, its compositional heterogeneity, and the critical dependence of eukaryotic cell viability on fully functional proteasomes have previously limited in vitro studies and mutational analyses. To circumvent these limitations of working with endogenous proteasomes, we developed systems for the heterologous expression of two proteasomal subcomplexes, the lid and the base, from Saccharomyces cerevisiae in Escherichia coli and the incorporation of unnatural amino acids to allow specific fluorescence labeling and advanced spectroscopic studies of proteasome function.
At the center of the 26S proteasome is the 20S core peptidase that is capped on one or both ends by the 19S regulatory particle, which recognizes appropriate substrates by their covalently attached ubiquitin modifications, mechanically unfolds them, and translocates the unfolded polypeptides through a narrow axial pore into the 20S core for proteolytic cleavage [4–6]. This regulatory particle can be further subdivided into the base and lid subcomplexes [7, 8], which also represent the intermediates of proteasome assembly in vivo [9] and thus can be used as stable building blocks together with the 20S core peptidase to reconstitute the 26S holoenzyme in vitro. The base contains six distinct AAA+ ATPases, Rpt1-Rpt6, that form a heterohexameric ring and constitute the molecular motor of the proteasome. It also includes three non-ATPases, in yeast called Rpn1, Rpn2, and Rpn13, with Rpn1 and Rpn13 functioning as ubiquitin receptors [10,11]. The assembly of the base proceeds through the initial formation of modules containing pairs of Rpt subunits [9], which then associate to form the ATPase hexamer in the order Rpt1-Rpt2-Rpt6-Rpt3-Rpt4- Rpt5 [12], with Rpn1 bound to Rpt1 and Rpt2 and Rpn2 plus Rpn13 associated with Rpt6 and Rpt3. Correct pairing of Rpt subunits in the three modules as well as their proper assembly into the base subcomplex is controlled by four assembly factors, Rpn14, Hsm3, Nas2, and Nas6 [13–15]. While Nas2 is released during this process [12], Nas6, Rpn14, and Hsm3 remain bound to the C-terminal tails of Rpt subunits until they are displaced by the base docking with the 20S core peptidase [16].
The lid subcomplex includes six PCI (proteasome-CSN-initiation factor 3) domain-containing subunits, Rpn3, Rpn5, Rpn6, Rpn7, Rpn9, and Rpn12, the small peptide Sem1, and two MPN (Mpr1-Pad1 N-terminal) domain-containing subunits, Rpn8 and Rpn11, with the latter representing the essential Zn2+-dependent deubiquitinase of the proteasome [17–19]. A helical bundle formed by the C-terminal helices of the eight globular lid subunits functions as a hub to control the ordered lid assembly [20], which is independent of any additional factors [9]. Another ubiquitin receptor subunit, Rpn10, is not stably associated with the isolated lid or base but interacts with both subcomplexes in the assembled regulatory particle.
Given this knowledge about the in vivo formation of lid and base, we created recombinant expression systems in E. coli with three compatible plasmids coding for either the nine lid subunits or the nine subunits of the base plus the four essential assembly factors, as described below (Subheadings 3.1 and 3.2). While the lid subunits associate without additional chaperones, the base subunits assemble efficiently only in the presence of the assembly factors. Subcomplexes are isolated by tandem affinity purification using tags on two subunits of the lid and the base followed by size-exclusion chromatography. The use of affinity tags on two separate subunits ensures that only fully assembled complexes are purified and thus corrects for differences in the expression levels or stability of individual subunits.
One of the advantages of these heterologous expression systems is that they can be easily combined with techniques for the incorporation of unnatural amino acids (UAAs) [21]. By co-expressing a synthetase/tRNA pair that has been evolved to recognize one of the amber/ochre/opal codons, an UAA can be placed anywhere within the expressed proteins. These UAAs allow for the incorporation of a variety of useful chemical properties into proteins, including reactive groups for cross-linking [22], photocaged amino acids such as lysine [23], and handles for orthogonal labeling [21]. The protocol below (Subheading 3.3) describes how to incorporate the UAA p-azido-L-phenylalanine (AzF) at a UAG codon that has been introduced at a specific position in the base or lid subcomplex. The azide group of the AzF can then be conjugated to an alkyne through a cycloaddition reaction, often referred to as “click” chemistry, allowing for the site-specific modification of the complex. Incorporation of AzF into the base will be used as an example below, though the protocol can be easily adapted to the lid or any other desired recombinant protein.
The vector used for UAA incorporation, pAM87, was constructed based on a synthetase variant (pAzFRS.2.t1) evolved by the Isaacs lab and a vector (pUltra) designed by the Schultz lab that also encodes the UAG-recognizing tRNA [24, 25]. Proteins with the UAG codon inserted in the desired position are expressed in E. coli strains carrying pAM87 with AzF added to the growth media. Once the protein is purified, the azide group of the incorporated AzF can be modified with an alkyne-containing molecule using Cu-free click chemistry [26]. In the protocol below, the base is modified with a fluorophore linked to dibenzocyclooctyne (DBCO), a strained cyclooctyne that allows for mild, copper-free conjugation [27]. Though designed to be bio-orthogonal, it is known that DBCO reacts with exposed cysteines [28]. To suppress this reaction, free thiols are protected with Ellman’s reagent before the labeling. Once the labeling reaction is complete, the cysteines are restored to their native state through reduction with dithiothreitol (DTT).
2. Materials
2.1. Reagents and Equipment for Base and Lid Expression and Purification
Electrocompetent E. coli BL21 Star (DE3) cells [29].
Electroporation apparatus.
Expression plasmids (see Table 1).
1000 × ampicillin: 300 mg/mL ampicillin sodium in 50% EtOH.
1000 × kanamycin: 50 mg/mL kanamycin monosulfate in water.
1000 × chloramphenicol: 25 mg/mL chloramphenicol in 100% EtOH.
LB-antibiotic plates: 1% (w/v) tryptone, 0.5% (w/v) yeast extract, 0.5% (w/v) NaCl, 1.5% (w/v) agar. Autoclave, cool to 60 °C, or lower in a water bath before adding appropriate antibiotics to 1× from 1000 × stocks.
dYT liquid media: 1.6% (w/v) tryptone, 1% (w/v) yeast extract, 0.5% (w/v) NaCl. Autoclave.
Terrific broth (TB): 47 g of Novagen terrific broth (Millipore) in 1 L of water +4 mL of glycerol. Autoclave.
2.5 L Ultra Yield baffled shaking flask (Thomson).
0.5 M IPTG: 0.5 M isopropyl-beta-D-thiogalactoside in water.
100× PMSF: 0.2613 g phenylmethylsulfonyl fluoride in 15 mL ethanol (safety note: protease inhibitors are hazardous and should only be handled after taking appropriate safety precautions).
1000× aprotinin: 1 mg/mL aprotinin in 25% methanol/75% H2O (safety note: protease inhibitors are hazardous and should only be handled after taking appropriate safety precautions).
1000 × pepstatin = 1 mg/mL pepstatin A in 90% methanol/ 10% acetic acid (safety note: protease inhibitors are hazardous and should only be handled after taking appropriate safety precautions).
1000 × leupeptin = 1 mg/mL leupeptin hemisulfate in H2O (safety note: protease inhibitors are hazardous and should only be handled after taking appropriate safety precautions).
Benzonase nuclease.
NiA buffer: 60 mM HEPES, pH 7.6,100 mM NaCl, 100 mM KCl, 10 mM MgCl2, 20 mM imidazole, 5% glycerol. Cool to 4 °C, adjust the pH, and then filter using a 0.22 μm filter.
NiB buffer: NiA buffer + 250 mM imidazole. Add dry imidazole to NiA buffer, and then readjust pH to 7.6.
Ultrasonic homogenizer with horn attachment.
125 mL metal beaker (Polar Ware).
5 mL HisTrap FF crude (GE).
5 M NaOH.
0.5 M ATP: Dissolve 9.08 g ATP disodium trihydrate in 15 mL of water. Adjust pH to 7.0 with 5 M NaOH, and then add water to a final volume of 30 mL. Aliquot and store at –20 °C.
Bradford reagent.
2× SDS loading buffer: 100 mM Tris-HCL, pH 6.8,4% (w/v) sodium dodecyl sulfate (SDS), 0.2% (w/v) bromophenol blue, 20% (w/v) glycerol. Add 10% (v/v) dithiothreitol (DTT) just before using.
Anti-FLAG M2 agarose (Sigma).
Gravity drip column.
TBS: 50 mM Tris–HCl, pH 7.6, 150 mM NaCl.
3× FLAG peptide: Dissolve peptide (MDYKDHDGDYKDH-DIDYKDDDDK) in TBS. Adjust pH to 7.6 using 5 M NaOH. Then adjust final concentration of FLAG peptide to 20 mg/ mL using TBS.
100 kDa cutoff Ultra-15 and Ultra-0.5 centrifugal filter concentrator (Amicon).
Spin-X 0.22 0.5 mL cellulose acetate centrifuge filter (Corning).
Superose 6 Increase 10/300 GL size-exclusion column (GE).
GF Buffer: 30 mM HEPES, pH 7.6, 50 mM NaCl, 50 mM KCl, 10 mM MgCl2, 5% glycerol. Cool to 4 °C, adjust the pH, and then filter with a 0.22 μm filter.
0.5 M TCEP: Add 5.73 g tris(2-carboxyethyl)phosphine-HCl to 30 mL of water. Adjust pH to 7.0 with 5 M NaOH, and then adjust final volume to 40 mL with water. Aliquot and store at –20 °C.
1 mg/mL BSA: Add 10 mg of bovine serum albumin (fatty acid free) to 10 mL of GF buffer. Adjust the concentration to 15 μM by measuring the A280 and using an extinction coefficient of43,824 M–1 cm–1.
Amylose resin (NEB).
0.5 M maltose: Add 18 g maltose monohydrate to water, adjust to 100 mL final volume, and then filter sterilize with a 0.22 μm filter into a sterile bottle.
Human rhinovirus 3C protease (Thermo Fisher).
8% NuPAGE SDS-PAGE gels (Thermo Fisher).
10% NuPAGE SDS-PAGE gels (Thermo Fisher).
20 × NuPAGE MOPS running buffer (Thermo Fisher).
Coomassie blue stain: 0.25% Coomassie blue R-250, 45% methanol, 45% water, 10% glacial acetic acid. Filter before using.
Coomassie destain: 20% methanol, 10% glacial acetic acid.
UV spectrophotometer.
Quartz cuvette, 20 μL sample volume (Hellma).
Table 1.
Expression plasmids used in this protocol
| Plasmid # | Description | Vector backbone |
Antibiotic marker |
|---|---|---|---|
| pAM81 [32] | Rpnl, Rpn2, Rpn13 | pETDuet | Ampicillin |
| pAM82 [32] | FLAG-Rpt1, Rpt2, 6xHis-Rpt3, Rpt5, Rpt6, Rpt4 | pCOLADuet Kanamycin | |
| pAM83 [32] | RIL rare tRNAs, Nas6, Nas2, Hsm3, Rpn14 | pACYCDuet | Chloramphenicol |
| pAM85 (modified from [33]) | Rpn9, Rpnll, Rpn8, MBP-HRV-Rpn6, Rpn5 | pETDuet | Ampicillin |
| pAM86 (modified from[33]) | 6xHis-HRV-Rpn12, Rpn7, Rpn3 | pCOLADuet Kanamycin | |
| pAM80 [33] | Seml, Hsp90 | pACYCDuet | Chloramphenicol |
| pAM87 (constructed from [24, 25]) | pAzFRS.2.tl, UAG tRNA | pCDF | Spectinomycin |
| pAM82 (modified from [32]) | pAM82 with the UAG codon inserted at the desired location | pCOLADuet | Kanamycin |
2.2. Reagents for Expression and Purification of Multi-subunit Complexes Incorporating p-Azido-L-Phenylalanine
1000× spectinomycin: 100 mg/mL spectinomycin dihydrochloride pentahydrate in water.
Unnatural amino acid incorporation plasmid (see Table 1).
10× phosphate buffer: 0.17 M KH2PO4, 0.72 M K2HPO4. Autoclave.
UAA media: Add 24 g yeast extract, 20 g of tryptone, and 10 mL of glycerol to 880 mL of water. Autoclave. Prepare 1 L of UAA media by adding 100 mL of 10 × phosphate buffer to 900 mL of media.
p-Azido-L-phenylalanine (Amatek Chemical).
5 mM DTNB: Add 5 mg of 5,5′-dithiobis-(2-nitrobenzoic acid) to 5 mL of GF buffer. Make fresh.
1 M DTT: Add 1.54 g dithiothreitol to 10 mL of water. Aliquot and store at –20 °C.
40 mM fluorophore-DBCO: Dissolve fluorophore conjugated to dibenzocyclooctyne (DBCO) in DMSO to 40 mM (Click Chemistry Tools).
3. Methods
3.1. Expression and Purification of the Base
3.1.1. E. coli Transformation and Expression of the Base
Prepare electrocompetent E. coli BL21 Star (DE3) cells, and then transform them with 50 ng each of pAM81, pAM82, and pAM83 (see Fig. 1 and Table 1 for more information on the plasmids). Plate on LB + ampicillin/kanamycin/chloramphenicol. Grow at 37 °C overnight (see Note 1).
Select a single colony from the plate to inoculate 50 mL of dYT medium containing ampicillin (300 μg/mL), chloramphenicol (25 μg/mL), and kanamycin (50 μg/mL). Grow overnight at 30 °C, shaking at 180 rpm.
Pellet the overnight culture in a conical tube by centrifuging at 3000 rcf for 4 min, pour offthe supernatant, and resuspend the pellet of cells in 50 mL of fresh dYT. Repeat this wash step once. Resuspend the pellet in 25 mL of dYT and divide evenly between three 2.5 L baffled flasks, each containing 1 L of TB + ampicillin (300 μg/mL), chloramphenicol (25 μg/ mL), and kanamycin (50 μg/mL) (see Notes 2 and 3).
Grow cells at 37 °C in a shaker at 180 rpm for 3–5 h, until OD600 reaches 0.7. Turn down the temperature to 30 °C, wait 30 min, and then induce protein expression by adding 1 mL of 0.5 M IPTG (0.5 mM final concentration) to each flask.
Incubate cells for 5 h at 30 °C, shaking at 180 rpm, and then change temperature to 16 ° C and let induce overnight.
Spin down the culture in three 1 L centrifuge bottles for 15 min at 3500 rcf, pour off the supernatant, and resuspend the cell pellets in cold NiA buffer + protease inhibitors and Benzonase (1 μg/mL aprotinin, 1 μg/mL pepstatin, 1 μg/mL leupeptin, 174 μg/mL PMSF, 50 units of Benzonase). The final volume of the combined, resuspended pellets should be 75 mL. The pellets can be transferred to 50 mL conical tubes and stored at –80 °C or carried on directly to the purification.
Fig. 1.
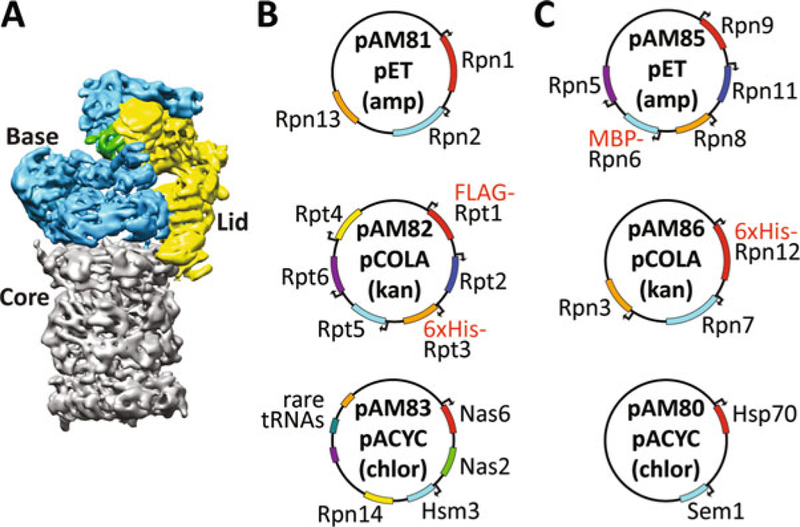
(a) The 26S proteasome consists of three subcomplexes, the 20S core (gray), the base (blue, containing the AAA+ motor), and the lid (yellow, containing the deubiquitinating enzyme Rpn11 shown in green). (b, c) Maps of the plasmids used for expression of the base (b) and lid (c). Each plasmid was constructed by insertion of the respective proteasome-subunit genes from Saccharomyces cerevisiae S288C into a duet vector (pETDuet, pCOLADuet, pACYCDuet). The majority of genes are expressed with their own T7 promoter (indicated by an arrow at the beginning of the ORF), but there is only one T7 terminator per plasmid
3.1.2. Purification of the Base
If using frozen pellets, thaw them in a water bath. All remaining purification steps should be performed at 4 °C. Combine the lysate into one 125 mL metal beaker, and use an ultrasonic homogenizer to sonicate the lysate on ice for 3 min at 75% amplitude, using a cycle of 15 s on and 60 s off (see Note 4).
Centrifuge the lysate for 30 min at 26,000 rcf in a pre-chilled rotor at 4 °C.
Prepare a 5 mL HisTrap charged with Ni2+ by washing the resin with 15 mL of NiA buffer (see Note 5).
Flow the lysate supernatant at 1 mL/min over the HisTrap. Take 1 μL of lysate supernatant, and then take another 1 μL of HisTrap flow-through for gel samples.
Wash the HisTrap with 50 mL of NiA + 1 mM ATP. All following buffers should contain 1 mM ATP to ensure the stability of the complex (see Note 6).
Elute the protein from the HisTrap with 20 mL of NiB buffer +1 mM ATP. Take 2 μL of the eluate for a gel sample.
Prepare 5 mL of anti-FLAG M2 agarose in a gravity drip column by washing with NiA + 1 mM ATP (see Notes 7 and 8).
Flow the nickel eluate over the anti-FLAG resin 4–5 times, exposing the protein to the resin for at least 30 min. Take 2 μL of the flow-through for a gel sample to ensure good depletion of FLAG-Rpt1.
Wash the anti-FLAG column 2× with 25 mL of NiA + 1 mM ATP.
Elute with 12 mL of NiA + 1 mM ATP + 0.15 mg/mL 3× FLAG peptide. Take 5 μL of FLAG eluate for a gel sample (see Note 9).
Concentrate FLAG elution to 400 μL in a 15 mL 100 kDa cutoff spin concentrator by spinning at 2000 rcf at 4 °C (see Note 10).
Spin filter the concentrated protein using a 0.22 μm filter (see Note 11).
Load and run the protein on a pre-equilibrated Superose 6 Increase 10/300 GL size-exclusion column using GF buffer + 0.5 mM TCEP and 0.5 mM ATP (see Fig. 2 for an example size-exclusion trace and a typical purification gel).
Collect the peak indicated in Fig. 2, typically from 12 mL to 14 mL when run at a flow rate of 0.4 mL/min at 4 °C. Concentrate the protein by successive spins in a 0.5 mL 100 kDa cutoff spin concentrator to a final volume of 100 μL. Take 1 μL of concentrated protein for a gel sample.
As the presence of ATP masks the A280 signal for the protein, the Bradford assay is used to measure concentration of the complex with bovine serum albumin (BSA) as a standard. Typical concentrations are 5–15 μM base (see Note 12).
Mix gel samples with 2× SDS loading buffer, and confirm the purity of the final complex by SDS-PAGE.
Aliquot the protein into 8 μL aliquots, flash freeze in liquid nitrogen, and store at –80 °C.
Fig. 2.
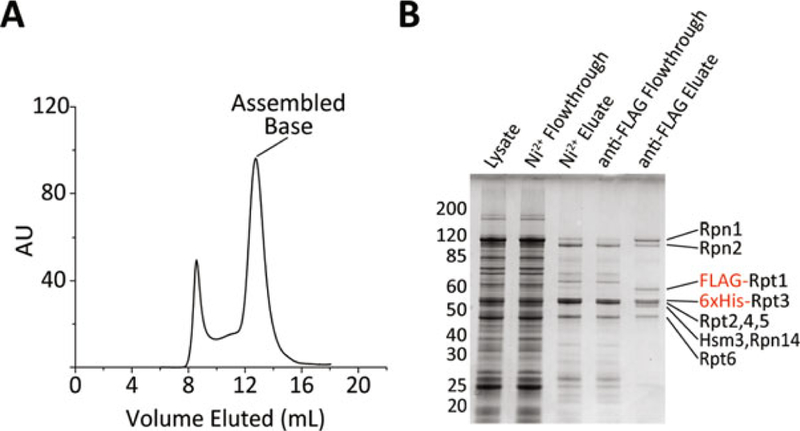
(a) The trace from a typical size-exclusion run of purified base is shown. The peak between the void and the assembled base contains misassembled base and on a gel looks similar to properly assembled base. (b) Gel samples from a typical base purification run on a 10% NuPAGE gel in MOPS buffer and stained using Coomassie blue
3.2. Expression and Purification of the Lid
3.2.1. E. coli Transformation and Expression of the Lid
Prepare electrocompetent E. coli BL21 Star (DE3) cells, and then transform with 50 ng of pAM85, pAM86, and pAM80 (see Fig. 1 and Table 1 for more information on the plasmids). Plate on LB + ampicillin/kanamycin/chloramphenicol. Grow at 37 °C overnight (see Note 1).
Select a single colony from the plate to inoculate 50 mL of dYT medium containing ampicillin (300 μg/mL), chloramphenicol (25 μg/mL), and kanamycin (50 μg/mL). Grow overnight at 30 °C, shaking at 180 rpm.
Pellet the 50 mL of culture in a conical tube by centrifuging at 3000 rcf for 4 min, pour off the supernatant, and resuspend the pellet of cells in 50 mL of fresh dYT. Repeat this wash step once. Resuspend the pellet in 25 mL of dYT, and divide evenly between three 2.5 L baffled flasks, each containing 1 L of TB + ampicillin (300 μg/mL), chloramphenicol (25 μg/ mL), and kanamycin (50 μg/mL) (see Notes 2 and 3).
Grow cells at 37 °C in a shaker at 180 rpm for 3–5 h, until OD600 reaches 0.7. Turn down the temperature to 18 °C, wait 30 min, and then induce protein expression by adding 1 mL of 0.5 M IPTG (0.5 mM final) to each flask. Let the cells induce overnight.
Spin down the culture in three 1 L centrifuge bottles for 15 min at 3500 rcf, pour off the supernatant, and resuspend the pellets in cold NiA buffer + protease inhibitors and Benzonase (1 μg/mL aprotinin, 1 μg/mL pepstatin, 1 μg/mL leupeptin, 174 μg/mL PMSF, 50 units of Benzonase). The final volume of the combined, resuspended pellets should be 75 mL. The pellets can be transferred to 50 mL conical tubes and stored at –80 °C or carried on directly to the purification.
3.2.2. Purification of the Lid
If using frozen pellets, thaw them in a water bath. All remaining purification steps should be performed at 4 °C. Combine the lysate into one 125 mL metal beaker, and use an ultrasonic homogenizer to sonicate the lysate on ice for 3 min at 75% amplitude, using a cycle of 15 s on and 60 s off (see Note 4).
Centrifuge the lysate for 30 min at 26,000 rcf in a pre-chilled rotor at 4 °C.
Prepare a 5 mL HisTrap charged with Ni2+ by washing the resin with 15 mL of NiA buffer (see Note 5).
Flow the lysate supernatant at 1 mL/min over the HisTrap. Take 1 μL of lysate supernatant, and then take another 1 μL of HisTrap flow-through for gel samples.
Wash the HisTrap with 100 mL of NiA.
Elute the protein from the HisTrap with 20 mL of NiB buffer. Take 2 μL of the eluate for a gel sample.
Prepare 7.5 mL of amylose resin in a gravity drip column by washing with 30 mL of NiA.
Flow the nickel eluate over the amylose resin 4–5 times, exposing the protein to the resin for at least 30 min. Take 2 μL of the flow-through for a gel sample to ensure good depletion of MBP-Rpn6.
Wash the amylose column twice with 25 mL of NiA.
Elute with 15 mL of NiA + 10 mM maltose. Take 5 μL of amylose eluate for a gel sample.
To cleave off the MBP fusion from Rpn6 and the 6xHis tag from Rpn12, add 20 μL of 2 units/μL human rhinovirus 3C protease to the eluate. Incubate overnight at 4 °C or for 2–3 h at room temperature. After cleavage, take a 5 μL sample. Confirm complete cleavage through SDS-PAGE analysis.
Concentrate protein to 400 μLin a 15 mL 100 kDa cutoff spin concentrator by spinning at 2000 rcf (see Note 10).
Spin filter the concentrated protein using a 0.22 μm filter.
Load and run the protein on a pre-equilibrated Superose 6 Increase 10/300 GL size-exclusion column using GF buffer + 0.5 mM TCEP (see Fig. 3 for an example of size-exclusion trace and a typical purification gel).
Collect the peak indicated in Fig. 3, typically from 13.5 mL to 15 mL when run at 0.4 mL/min at 4 °C. Concentrate the protein by successive spins in a 0.5 mL 100 kDa cutoff spin concentrator to a final volume of 100–200 μL. Take 1 μL of concentrated protein for a gel sample.
Quantify the concentration by measuring the A280. The extinction coefficient calculated from the sequence is 349,000 M–1 cm–1, and the expected concentration is 10–50 μM (see Note 13).
Mix gel samples with 2× SDS loading buffer, and confirm the purity of the final complex by SDS-PAGE.
Aliquot the protein into 8 μL aliquots, flash freeze in liquid nitrogen, and store at –80 °C.
Fig. 3.
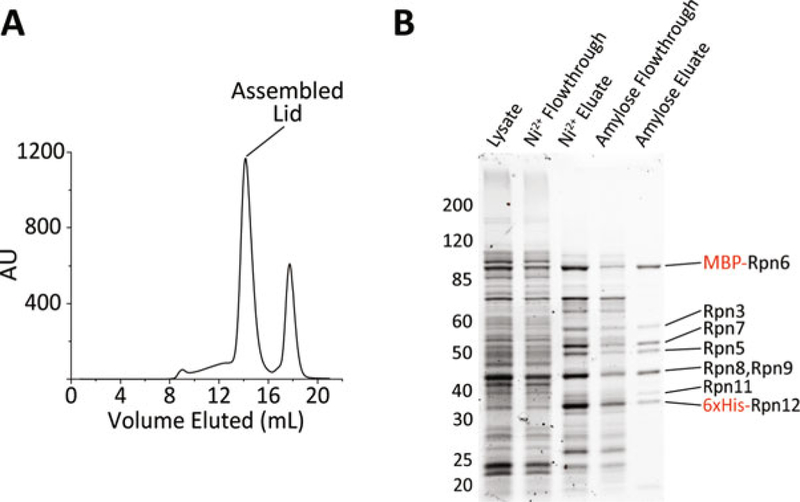
(a) The trace from a typical size-exclusion run of purified lid is shown. The peak after the assembled lid contains mostly cleaved MBP. (b) Gel samples from a typical lid purification run on a 10% NuPAGE gel in MOPS buffer and stained using Coomassie blue
3.3. Expression and Purification of Multi-subunit Complexes Incorporating Unnatural Amino Acids
This protocol will be presented as an abbreviated version of the base protocol presented above, focusing on the unnatural amino acid specific instructions. For more detail on the procedures, see protocol Subheading 3.1. The modifications should be transferable to the purification of other complexes.
3.3.1. E. coli Transformation and Protein Expression
Prepare electrocompetent E. coli BL21 Star (DE3) cells carrying pAM87, which contains the synthetase and tRNA for incorporation of AzF at the UAG amber codon. pAM87 confers resistance to spectinomycin. Please see Note 1 for a protocol.
Use standard molecular cloning techniques to insert the UAG amber codon at the desired location in the base subcomplex (see Notes 14–16).
Transform pAM81, pAM82*, and pAM83 into the pAM87- containing electrocompetent E. coli.
Inoculate 2 × 50 mL cultures of dYT + ampicillin (300 μg/ mL), chloramphenicol (25 μg/mL), kanamycin (50 μg/mL), and spectinomycin (100 μg/mL); grow at 37 °C overnight (see Note 17).
Wash the overnight cultures with fresh dYT twice, resuspend in 25 mL of dYT, and then divide the cells evenly between 6 L of dYT + 0.5× antibiotic (ampicillin (150 ^g/mL), chloramphenicol (12.5 ^g/mL), kanamycin (12.5 μg/mL), and spectinomycin (50 μg/mL)), in 2.5 L baffled flasks (see Notes 2 and 3).
Grow the cultures at 37 °C until OD600 0.7 is reached. Spin down the cultures in clean 1 L centrifuge bottles for 15 min at 3500 rcf at room temperature.
Gently (without vortexing) resuspend the cultures in 1 L total of UAA media, leading to a sixfold concentration of cells. Divide into two 2.5 L baffled flasks to maximize aeration (see Note 18).
Add 0.24 g of AzF to each 500 mL culture (2 mM final), and then shake for 30 min at 30 °C at 180 rpm (see Note 19).
Add 1 mL of 0.5 M IPTG (1 mM final) to each flask to induce expression of both the proteins of interest and the synthetase. Let induce at 30 °C for 5 h and then 16 °C overnight.
Spin down the culture in two 1 L centrifuge bottles for 15 min at 3500 rcf at 4 °C, pour off the supernatant, and resuspend the pellets in cold NiA buffer + protease inhibitors and Benzonase (1 μg/mL aprotinin, 1 μg/mL pepstatin, 1 μg/mL leupeptin, 174 μg/mL PMSF, 50 units of Benzonase). The final volume of the combined, resuspended pellets should be 75 mL. The pellets can be transferred to 50 mL conical tubes and stored at –80 °C or carried on directly to the purification.
3.3.2. Purification of the Base
Follow steps 1–11 of Subheading 3.1.2. In brief, the protein should be purified using Ni2+ affinity and anti-FLAG affinity columns and then concentrated to 400 μL (see Note 20).
To protect surface-exposed cysteines, add DTNB to 150 μM final concentration from a freshly made 5 mM stock, incubate at 22 °C for 10 min (solution will turn yellow), and then cool the protein back down to 4 °C (see Note 21).
Add the DBCO-fluorophore to a final concentration of 300 μM and incubate at 4 °C overnight (see Note 22).
Quench the reaction with 1 mM AzF from a 100 mM stock, then add DTT to a final concentration of 5 mM from a 1 M stock to de-protect the cysteines and resolve any inter-protein disulfide bonds.
Follow steps 12–16 of protocol Subheading 3.1.2 to run the protein over the size-exclusion column. See Fig. 4 for examples of labeling specificity. Typical yields are about half of the yield for unlabeled complexes.
Fig. 4.
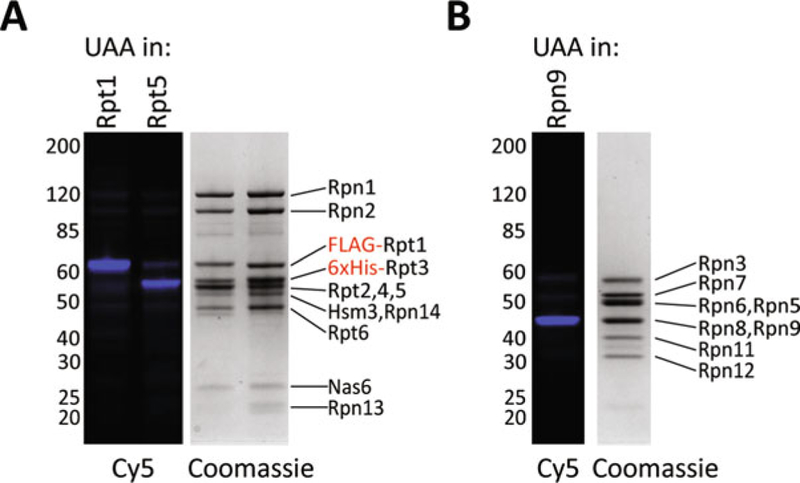
(a) Base with AzF incorporated at either Rpt1 or Rpt5 was labeled with Cy5-DBCO. Shown here are the Cy5 fluorescence channel and Coomassie staining of those samples run on an 8% NuPAGE gel. (b) Lid with AzF incorporated into Rpn9 was labeled with Cy5-DBCO. As in A, the Cy5 fluorescence channel and Coomassie staining of those samples run on an 8% NuPAGE gel are shown
4. Notes
It is important to use properly prepared electrocompetent cells. The protocol from Seidman et al. (2001) can be followed as a reference [29]. Chemically competent cells are not efficient at taking up the large plasmids used here. It is possible to make electrocompetent cells from BL21 Star (DE3) E. coli already transformed with pAM83. This is advantageous because the efficiency of a double transformation is much higher than that of the triple transformation. For protocol Subheading 3.3.1 it is necessary to make electrocompetent cells containing pAM87 or another plasmid because quadruple transformations are not feasible.
The wash step is important to remove any secreted β-lactamase in the media, which would diminish selection for the ampicillin resistance containing plasmid pAM81.
The minimum recommended volume to prep is three liters, though the protocol can be scaled up.
The proper sonication conditions will vary depending on the instrument used and should be optimized to maximize lysis without heating the sample. Other methods of cell lysis such as using a French press will also work.
The purification can also be performed using Ni-NTA agarose. Modify the procedure by batch binding 5 mL of Ni-NTA beads with the cleared lysate for 45 min and then transferring to a gravity drip column for the wash and elution.
The purification can be monitored using Bradford reagent. Aliquot 40 μL of Bradford reagent into the wells of a 96-well plate. Then take 5 μL of column eluate and mix thoroughly with the Bradford reagent. The presence of protein will be indicated by a color change from brown to blue. After 50 mL of washing, the eluate from the HisTrap may still react with Bradford, but you should nevertheless continue to the elution step. Excessive washing of the nickel column reduces yield of the final complex.
FLAG agarose must be regenerated before the first use by following the manufacturer’s recommended protocol. The resin can then be reused by regenerating after each purification.
The amount of FLAG agarose used is often the limiting factor for the final yield of base. Fresh FLAG agarose has higher binding capacity than used and regenerated resin. It is recommended to confirm good depletion of FLAG-Rptl. If FLAG- Rptl is still visible in the flow-through of the FLAG column, the FLAG binding, washing, and eluting steps (Subheading 3.1.2, steps 7–10) can be repeated with freshly regenerated FLAG agarose.
Be sure to use 3 × FLAG peptide (MDYKDHDGDYKDHDI- DYKDDDDK), as 1 × FLAG peptide does not effectively compete off the FLAG-Rptl.
To prevent aggregation at the bottom of the concentrator, mix the sample by inverting the concentrator every 5–10 min.
Occasionally not all of the protein solution will go through the spin filter. If the filter has clogged, it may be necessary to switch to a fresh filter. Pool the filtered protein before running on the size-exclusion column.
Using BSA as a standard may result in concentrations inaccurate by as much as twofold due to the different reactivity of BSA with the Bradford reagent compared to the base proteins. For a more accurate concentration, the sample can be submitted for amino acid analysis. The molecular weight of the complex is 652,000 kDa, including Rpt1–6, Rpn1, Rpn2, Rpn13, Nas6, Hsm3, and Rpn14. Nas2 does not stay bound.
It is recommended not to use a NanoDrop to measure the protein concentration as the high viscosity of the solutions interferes with accurate readings. Instead, measurements are typically taken in a 20 μL cuvette using a UV-Vis spectrophotometer.
The efficiency of incorporation seems to be dependent on the location of the amber codon within the protein, but this effect is difficult to predict [30]. Therefore, it is recommended to test out multiple locations within the protein to identify the ones that give the highest protein yields.
Another challenge is that some locations also yield truncated products, so it is also recommended to have a C-terminal tag or other means of selecting for full-length products. In the case of the base, truncated products do not incorporate well into the complex and are thus not purified with fully assembled base.
To ensure proper protein production, it is also necessary to replace any UAG codons that are being used as stop codons with either the ochre or opal stop codons.
The cells grow slowly under quadruple antibiotic selection, so it may be necessary to first inoculate a 5 mL culture, let that grow overnight, and then use it to inoculate the two 50 mL cultures at 1:500.
Concentrating the cells allows for a much higher cell density per liter of culture, allowing for more protein production for each gram of unnatural amino acid used.
This step gives the cells time to take up the AzF.
As AzF is sensitive to reducing agents, they should be avoided if possible until after the labeling step described in Subheading 3.3.2 step 3. It has been reported that the azide group is somewhat compatible with βME if reducing agent is required [31].
Enough DTNB should be added to be in excess of the estimated number of exposed cysteines in solution, which varies between protein complexes.
The optimal concentration of fluorophores varies between proteins due to differences in the accessibility of the AzF. It is recommended to optimize the concentration of DBCO- fluorophore. The efficiency of labeling can be calculated by the ratio of the concentration of fluorophore in the final protein solution (as measured by absorbance) to the concentration of protein. Specificity can be estimated by subjecting wild-type protein with no AzF incorporated to the same labeling conditions. The labeling was found to be significantly more specific at 4 °C than at room temperature.
Acknowledgments
We thank the members of the Martin Lab for helpful discussions and the Prof. P. Schulz Lab at the Scripps Research Institute as well as the Prof. F. Isaacs Lab at Yale University for providing plasmid constructs for unnatural amino acid incorporation. J.A.M.B. acknowledges support from the NSF Graduate Research Fellowship. This research was funded in part by the US National Institutes of Health (R01-GM094497 to A.M.), the US National Science Foundation CAREER Program (NSF-MCB-1150288 to A.M.), and the Howard Hughes Medical Institute (A.M.).
References
- 1.Finley D (2009) Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu Rev Biochem 78(1):477–513. 10.1146/annurev.biochem.78.081507.101607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goldberg AL (2007) Functions of the proteasome: from protein degradation and immune surveillance to cancer therapy. Biochem Soc Trans 35(Pt 1):12–17. 10.1042/BST0350012 [DOI] [PubMed] [Google Scholar]
- 3.Suraweera A, Munch C, Hanssum A, Bertolotti A (2O12) Failure of amino acid homeostasis causes cell death following proteasome inhibition. Mol Cell 48(2):242–253. 10.1016/j.molcel.2012.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Glickman MH, Rubin DM, Fried VA, Finley D (1998) The regulatory particle of the Saccharomyces cerevisiae proteasome. Mol Cell Biol 18(6):3149–3162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Groll M, Bajorek M, Kohler A, Moroder L, Rubin DM, Huber R, Glickman MH, Finley D (2000) A gated channel into the proteasome core particle. Nat Struct Biol 7 (11):1062–1067. 10.1038/80992 [DOI] [PubMed] [Google Scholar]
- 6.Rabl J, Smith DM, Yu Y, Chang SC, Goldberg AL, Cheng Y (2008) Mechanism of gate opening in the 20S proteasome by the proteasomal ATPases. Mol Cell 30(3):360–368. 10.1016/j.molcel.2008.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Glickman MH, Rubin DM, Coux O, Wefes I, Pfeifer G, Cjeka Z, Baumeister W, Fried VA, Finley D (1998) A subcomplex of the proteasome regulatory particle required for ubiquitin-conjugate degradation and related to the COP9-signalosome and eIF3. Cell 94 (5):615–623 [DOI] [PubMed] [Google Scholar]
- 8.Saeki Y, Tanaka K (2O12) Assembly and function of the proteasome. Methods Mol Biol 832:315–337. 10.1007/978-1-61779-474-2-22 [DOI] [PubMed] [Google Scholar]
- 9.Budenholzer L, Cheng CL, Li Y, Hochstrasser M (2017) Proteasome structure and assembly. J Mol Biol 429(22):3500–3524. 10.1016/j.jmb.2017.05.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi Y, Chen X, Elsasser S, Stocks BB, Tian G, Lee BH, Shi Y, Zhang N, de Poot SA, Tuebing F, Sun S, Vannoy J, Tarasov SG, Engen JR, Finley D, Walters KJ (2016) Rpn1 provides adjacent receptor sites for substrate binding and deubiquitination by the protea-some. Science 351(6275):aad9421 10.1126/science.aad9421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Husnjak K, Elsasser S, Zhang N, Chen X, Randles L, Shi Y, Hofmann K, Walters KJ, Finley D, Dikic I (2008) Proteasome subunit Rpn13 is a novel ubiquitin receptor. Nature 453(7194):481–488. https://doi.org/10.103S/nature06926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tomko RJ Jr, Funakoshi M, Schneider K, Wang J, Hochstrasser M (2010) Heterohexa-meric ring arrangement of the eukaryotic pro-teasomal ATPases: implications for proteasome structure and assembly. Mol Cell 3S (3):393–403. 10.1016/j.molcel.2010.02.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saeki Y, Toh EA, Kudo T, Kawamura H, Tanaka K (2009) Multiple proteasome-interacting proteins assist the assembly of the yeast 19S regulatory particle. Cell 137 (5):900–913. 10.1016/j.cell.2009.05.005 [DOI] [PubMed] [Google Scholar]
- 14.Funakoshi M, Tomko RJ Jr, Kobayashi H, Hochstrasser M (2009) Multiple assembly chaperones govern biogenesis of the protea-some regulatory particle base. Cell 137 (5):887–899. 10.1016/j.cell.2009.04.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roelofs J, Park S, Haas W, Tian G, McAllister FE, Huo Y, Lee BH, Zhang F, Shi Y, Gygi SP, Finley D (2009) Chaperone-mediated pathway of proteasome regulatory particle assembly. Nature 459(7248):861–865. 10.1038/nature08063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Park S, Li X, Kim HM, Singh CR, Tian G, Hoyt MA, Lovell S, Battaile KP, Zolkiewski M, Coffino P, Roelofs J, Cheng Y, Finley D (2O13) Reconfiguration of the protea-some during chaperone-mediated assembly. Nature 497(7450):512–516. 10.1038/nature12123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Verma R, Aravind L, Oania R, McDonald WH, Yates JR 3rd, Koonin EV, Deshaies RJ (2002) Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S protea- some. Science 298(5593):611–615. 10.1126/science.1075898 [DOI] [PubMed] [Google Scholar]
- 18.Yao T, Cohen RE (2002) A cryptic protease couples deubiquitination and degradation by the proteasome. Nature 419(6905):403–407. 10.1038/nature01071 [DOI] [PubMed] [Google Scholar]
- 19.Tomko RJ Jr, Hochstrasser M (2O11) Incorporation of the Rpn12 subunit couples completion of proteasome regulatory particle lid assembly to lid-base joining. Mol Cell 44 (6):907–917. 10.1016/j.molcel.2011.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Estrin E, Lopez-Blanco JR, Chacon P, Martin A (2013) Formation of an intricate helical bundle dictates the assembly ofthe 26S proteasome lid. Structure 21(9):1624–1635. 10.1016/j.str.2013.06.023 [DOI] [PubMed] [Google Scholar]
- 21.Chin JW, Santoro SW, Martin AB, King DS, Wang L, Schultz PG (2002) Addition of p-azido-L-phenylalanine to the genetic code of Escherichia coli. J Am Chem Soc 124 (31):9026–9027 [DOI] [PubMed] [Google Scholar]
- 22.Chin JW, Martin AB, King DS, Wang L, Schultz PG (2002) Addition of a photocrosslinking amino acid to the genetic code of Escherichiacoli. Proc Natl Acad Sci U S A 99 (17):11020–11024. 10.1073/pnas.172226299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen PR, Groff D, Guo J, Ou W, Cellitti S, Geierstanger BH, Schultz Pg (2009) A facile system for encoding unnatural amino acids in mammalian cells. Angew Chem 48 (22):4052–4055. 10.1002/anie.200900683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Amiram M, Haimovich AD, Fan C, Wang YS, Aerni HR, Ntai I, Moonan DW, Ma NJ, Rovner AJ, Hong SH, Kelleher NL, Goodman AL, Jewett MC, Soll D, Rinehart J, Isaacs FJ (2015) Evolution of translation machinery in recoded bacteria enables multi-site incorporation of nonstandard amino acids. Nat Biotechnol 33(12):1272–1279. 10.1038/nbt.3372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chatterjee A, Sun SB, Furman JL, Xiao H, Schultz PG (2013) A versatile platform for single- and multiple-unnatural amino acid mutagenesis in Escherichia coli. Biochemistry 52(10):1828–1837. 10.1021/bi4000244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Agard NJ, Prescher JA, Bertozzi CR (2004) A strain-promoted [3 + 2] azide-alkyne cycloaddition for covalent modification of biomolecules in living systems. J Am Chem Soc 126 (46):15046–5047. 10.1021/ja044996f [DOI] [PubMed] [Google Scholar]
- 27.Ning X, Guo J, Wolfert MA, Boons GJ (2008) Visualizing metabolically labeled glycoconjugates of living cells by copper-free and fast huis-gen cycloadditions. Angew Chem 47 (12):2253–2255. 10.1002/anie.200705456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van Geel R, Pruijn GJ, van Delft FL, Boelens WC (2012) Preventing thiol-yne addition improves the specificity of strain-promoted azide-alkyne cycloaddition. Bioconjug Chem 23(3):392–398. 10.1021/bc200365k [DOI] [PubMed] [Google Scholar]
- 29.Seidman CE, Struhl K, Sheen J, Jessen T (2001) Introduction of plasmid DNA into cells. Curr Protoc Mol Biol Chapter 1:Unit1 8 10.1002/0471142727.mb0108s37 [DOI] [PubMed] [Google Scholar]
- 30.Schinn SM, Bradley W, Groesbeck A, Wu JC, Broadbent A, Bundy bC (2017) Rapid in vitro screening for the location-dependent effects of unnatural amino acids on protein expression and activity. Biotechnol Bioeng 114 (10):2412–2417. 10.1002/bit.26305 [DOI] [PubMed] [Google Scholar]
- 31.Tian H, Sakmar TP, Huber T (2016) A simple method for enhancing the bioorthogonality of cyclooctyne reagent. Chem Commun 52 (31):5451–5454. 10.1039/c6cc01321j [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beckwith R, Estrin E, Worden EJ, Martin A (2013) Reconstitution of the 26S proteasome reveals functional asymmetries in its AAA+ unfoldase. Nat Struct Mol Biol 20(10):1164–1172. 10.1038/nsmb.2659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lander GC, Estrin E, Matyskiela ME, Bashore C, Nogales E, Martin A (2012) Complete subunit architecture of the proteasome regulatory particle. Nature 482 (7384):186–191. 10.1038/nature10774 [DOI] [PMC free article] [PubMed] [Google Scholar]