

Purpose:
The need for more representative tools to investigate the human tumor microenvironment (TME) has led to the development of humanized mouse models. However, the difficulty of immune system engraftment and minimal human immune cell infiltration into implanted xenografts are major challenges. We have developed an improved method for generating mismatched humanized mice (mHM), using a dual infusion of human hematopoietic stem and progenitor cells (HSPCs) and mesenchymal stem cells (MSCs), isolated from cord blood and expanded in vitro. Engraftment with both HSPCs and MSCs produces mice with almost twice the percentage of human immune cells in their bone marrow, compared to mice engrafted with HSPCs alone, and yields 9- to 38-fold higher levels of mature peripheral human immune cells. We identified a peripheral mHM blood human B cell threshold that predicts an optimal degree of mouse bone marrow humanization. When head and neck squamous cell carcinoma (HNSCC) tumors are implanted on the flanks of HSPC-MSC engrafted mice, human T cells, B cells, and macrophages infiltrate the stroma of these tumors at 2- to 8-fold higher ratios. In dually HSPC-MSC engrafted mice we also more frequently observed additional types of immune cells, including regulatory T cells, cytotoxic T cells, and MDSCs. Higher humanization was associated with in vivo response to immune-directed therapy. The complex immune environment arising in tumors from dually HSPC-MSC engrafted mice better resembles that of the originating patient’s tumor, suggesting an enhanced capability to accurately recapitulate a human TME.
Keywords: Humanized mouse model, cancer microenvironment, head and neck cancer, hematopoietic stem cell, patient-derived xenograft
INTRODUCTION
The tumor microenvironment (TME) is a critical interface in cancer biology and immunology, dictating not only tumorigenesis, but also tumor progression and susceptibility to conventional and immune-directed therapies. Immune-directed therapies have greatly advanced patient care but are limited by modest single-agent efficacy, lack of predictive biomarkers, and uncharacterized mechanisms of resistance limiting rational combinatorial strategies. In head and neck squamous cell carcinoma (HNSCC), programmed cell death 1 (PD-1) inhibitors are approved for recurrent/relapsed patients but have response rates of only ~25% (1). These barriers highlight the need for appropriate models to better understand the biologic basis of disease, identify and prioritize targets, and assist in devising more effective therapies.
To faithfully represent the human TME and develop therapies, a model must be capable of recapitulating the interactions between tumor and stromal cells observed in the patient. Cultured cell lines lack stromal cells and poorly predict patient response (2). Patient-derived xenografts (PDX) implanted on immunocompromised mice produce a more realistic TME, but the stroma and immune cells interacting with the growing tumor are of mouse origin, reconstitute only a partial TME while fostering xenogeneic tumor growth, and do not accurately represent a human environment (3,4). To overcome these obstacles, several humanized mouse (HM) models have been developed over the past 15 years (5–11). These models either infused immune cells directly or engrafted human hematopoietic precursors, so that ultimately stromal tissue interacts with implanted tumors to more accurately recreate the patient TME (11,12).
We previously reported the generation of a HM xenograft model (13). Purified human Myc and Bcl-2 proteins were used to drive in vitro expansion of umbilical cord blood-derived hematopoietic stem and progenitor cells (HSPCs) that were injected into previously irradiated NOD/SCID/IL2rg−/− (NSG) mice to produce animal cohorts with some human immune system components (14). Tumor tissue surgically resected from HNSCC patients was implanted onto the flanks of these HM and we observed that human T and B cells originating in the engrafted human bone marrow infiltrated these growing tumors. We also analyzed tumor mRNA expression and showed that the drift in gene expression typically observed in PDX was partially reversed (3,15). This model was limited by a low abundance of human immune cells within the mouse bone marrow and infiltrating the implanted xenografts. Some immune cell subtypes critical in immune biology and therapy, e.g., CD8, regulatory T (Treg) cells, and myeloid derived suppressor cells (MDSCs) were rare or undetectable.
Here we report a significantly improved HM xenograft model, constructed from patient- derived tumors implanted on allogeneic or mismatched humanized mice (mHM). To avoid manipulating the HSPCs and thus minimizing changes to the precursor environment, we focused on optimizing in vitro expansion and on identifying additional lineages for concurrent engraftment. We isolated HSPCs from donated cord blood, cultured them in cytokine-containing expansion media, and produced sufficient HSPCs to engraft large cohorts of mice. We also identified a mesenchymal stem cell (MSC)-like population within the expanding HSPCs which, when engrafted with the HSPCs, yields mHM with an incrementally greater proportion of human immune cells within their bone marrow. These produce exponentially more robust populations of lymphoid and myeloid cells in HM blood, consistent with the reported role of MSCs (16). To standardize model and cohort generation we sought to identify a peripheral mouse blood parameter that would accurately predict adequate humanization prior to tumor implantation. This is critical in reducing model variability, a consistent obstacle in HM platforms that limits their utility (11). A correlation of peripheral blood profiles of live animals with necropsy analysis of humanized bone marrow content allowed the identification of a threshold level of humanization at which mice can be considered experimentally useful. In these well-humanized mice, human monocytes, T cells, and B cells migrate into implanted HNSCC tissue creating a TME that more accurately resembles that of the originator patient.
METHODS
HSPC and MSC collection and expansion
Donated, de-identified cord blood was obtained from the University of Colorado cord blood bank (http://www.clinimmune.com/cordbloodbank/). Each cord was given an HM— numerical designation, and HSPCs were magnetically purified from the cord by CD34+ selection (Stemcell Technologies, Vancouver, BC; Cat#18086), suspended in expansion media supplemented with Flt3, SCF, IL-3, and IL-6 cytokines and human low-density lipoprotein (LDL; Stemcell Technologies; Cat#02698), and cultured at 37°C, 5% CO2 for 8–10 days. The initial HSPC population was calculated by flow cytometry, using human CD34 and CD45 (Biolegend, San Diego, CA; Cat#343608, 368510) antibodies. MSC-like cells were characterized by the presence of CD73 and CD166 antigens (Biolegend; Cat#344006, 343904).
HSC and MSC engraftment in mice
The University of Colorado Institutional Animal Care and Use Committee (IACUC) approved all experiments involving mice. On the final day of HSPC-MSC expansion, NOD/SCID/IL2rg−/− mice (NSG; Jackson Laboratories, Bar Harbor, ME) were prepared for engraftment by 1.5 Gy whole-body irradiation. Animals were placed in filter-topped mouse cages to reduce radiation scatter and irradiated in a in a RS-2000 irradiator (Rad Source Technologies, Suwanee, GA) using a 25mAmp 160KeV source at a dose rate of 115 cGy/min. After a recovery of several hours, each mouse was injected with 400,000 HPSCs and MSCs (mixed so that MCSs composed 5–10% of the total cells) suspended in 0.2 mL sterile PBS. A minimal volume of blood from each mouse was collected by submandibular puncture after 8 – 10 weeks to assess HSPC engraftment by flow cytometry, using human CD3, CD11b, CD19 and/or CD45 (Biolegend; Cat#300312, 301310, 392504, 304039) antibodies at a 1:10 concentration.
PDX generation and tissue collection
Patient-derived xenograft (PDX) generation has been previously reported (17). Briefly, a clinical pathologist examined patient tumors after resection and collected non-diagnostic, non-necrotic portions in DMEM supplemented with 10% fetal bovine serum (FBS), 200 units/mL penicillin, and 200 μg/mL streptomycin. Once in the lab, tumors were given a CUHN-numerical designation, then sections were cut into 3 mm3 pieces and implanted (F1 generation) on both flanks of several 6–10 week old female Fox1nnu nude mice (Envigo, Denver CO). Animals were anesthetized with isoflurane (induction at 5%, maintenance at 1–2%). In preparation for surgical implantation of PDX tumor tissue, and for 48h following the procedure, animals received buprenorphine injections (1 mg/kg) every 12h. Right and left hind flanks were sterilized, and small incisions made to create subcutaneous pockets. Prepared tumor pieces were dipped in Matrigel (Corning, Corning, NY; Cat#356234) and inserted into these pockets that were then closed with wound clips and tissue glue. Growing tumors were measured with calipers and their volume calculated using the formula (WxWxL)/2. When tumors reached 1,500mm3 they were passed either to nude mice or humanized and control NSG mice by the same process. When necessary, animals were euthanized by CO2 asphyxiation and cervical dislocation. Blood was collected by cardiac puncture in EDTA. Tumors and mouse tissues were kept in DMEM for cytometry, flash-frozen in liquid nitrogen, and/or preserved in formalin/paraffin.
In vivo treatment studies
To study the effects of immune-directed therapy, mice received either a human IgG control (Gammagard; Shire, Lexington,MA; 10mg/kg) or nivolumab (Bristol-Myers Squibb; 10 mg/kg). Both were administered twice weekly by intraperitoneal injection for four weeks (8 doses). Two HM cohorts (HM034 and HM044) were subsequently implanted (together with contemporary equally sized cohorts of control, non-humanized NSG mice) with CUHN004 tumors, a case derived from an oral cavity HNSCC. After implantation tumor growth was monitored until the average cohort tumor size were ~100 mm3, at which time mice were allocated to treatment groups balanced by tumor size. Tumor growth was measured until tumors reached the size limits dictated by the animal welfare protocol, at which time they were euthanized. Effect was measured by the average normalized size of treated tumors divided by control tumors (T/C) ratio.
Fluorescence activated cell sorting (FACS) and flow cytometry
Collected tumor and mouse tissues were minced and dissociated in DMEM containing 1mg/ml collagenase IV (Worthington, Lakewood, NJ; Cat#LS004189) at 37°C for 1.5 hours. Bone marrow was isolated from femurs using a 27g syringe. Cells were filtered (40μm) and red blood cells lysed in ACK buffer (Life Technologies, Carlsbad, CA; Cat#A1049201) before resuspension in PBS containing 2% FBS for analysis. White blood cells were separated from whole blood by centrifugation and ACK lysis. Promiscuous antibody binding was blocked by incubation in Gammagard (5 mg/mL; Shire US, Inc., Lexington, MA) for 30 minutes. Cells were then washed and stained with the human antibodies CD3, CD19, CD11 b, and CD45 at a 1:10 concentration (Biolegend; Cat#300312, 301310, 392504, 304039). Cytometric analysis was carried out on a CyAn ADP (Beckman Coulter, Brea, CA) to determine the types and percentages of human immune cells present within these tissues.
Immunohistochemistry (IHC)
IHC analyses were performed as previously described (13). Briefly, slides were de-paraffinized and re-hydrated in graded concentrations of alcohol before antigen retrieval in citrate buffer, pH 6 (Dako, Carpinteria, CA; Cat#S1699) or EDTA buffer, pH 9 (Dako; Cat#2367) at 50°C for 10 minutes and rinsed in wash buffer (Dako; Cat#K8007). All staining was done in a Dako Autostainer. Slides were incubated in dual endogenous enzyme block (Dako; Cat#S2003) for 10 minutes, protein-free blocking solution (Dako; Cat#X0909) for 20 minutes, then in primary antibody for 60 minutes. Primary antibodies and dilutions: CD45 (Dako; Cat#M0701), 1:100; CD3 (Abcam; San Francisco, CA; Cat#ab5690), 1:100; CD20 (Abcam; Cat#ab9475), 1:100; CD56 (Bio-SB, Santa Barbara, CA; Cat#BSB5271), 1:50; CD8 (ThermoFisher, Waltham, MA; Cat#MS457-S1), 1:50; CD4 (Dako; Cat#M7310), 1:50; CD25 (Abcam; Cat#ab128955), 1:50; CD33 (Abcam; Cat#ab11032), 1:200; CD163 (BioCare, Pacheco, CA; Cat#CM353C), 1:100; and CD68 (Dako; Cat#M0876), 1:100; Granzyme B (Abcam; Cat#4059, 1:100). Staining was developed as follows: EnVision + Dual Link System HRP (Dako; Cat#K4061) for 30 minutes and substrate-chromogen (DAB+) Solution (Dako; Cat#K3468) for 5 minutes. Slides were counterstained with hematoxylin (Dako; Ct#S3301) for 10 minutes. When necessary, quantification of cells was performed by counting staining in at least three non-overlapping fields on a Zeiss Axio Imager A2 microscope.
Statistical analysis
In vitro and in vivo experiments were compared with a two-group t-test. Calculations were done using GraphPad Prism version 7.0 and SPSS version 11. Data are represented graphically as mean ± SEM. P-values < 0.05 were statistically significant. All statistical tests were two-sided. Significant changes in the mean of tumor infiltration as a function of human bone marrow percentage were assessed using the changepoint R package, data was ranked by human bone marrow percentage (Rank Index) then the Pruned Extract Linear Time (PELT) method with a Akaike’s Information Criterion (AIC) penalty was applied to the data to detect optimal changepoints, or the point at which the mean of the values changes (18).
RESULTS
HSPC isolation and expansion
We isolated HSPCs from donated cord blood by CD34+ selection, followed by culture in expansion media containing Flt3, stem cell factor (SCF), IL-3, IL-6, and LDL. We observed substantial variability in the percentage of the resident HSPC population in donated cords as well as in its potential for in vitro expansion. The percentage of HSPCs among the cord blood leukocytes, HSPC density (HSPCs/mL), and the total number of HSPCs purified from a cord did not independently predict HSPC expansion potential (P=0.90, P=0.22 and 0.22, respectively; Table 1), and neither did cord volume or the percentage of living cells, both of which had narrower ranges (T-test, P=0.78 and 0.87; data not shown). It takes 3–4 days in culture to be able to predict if a given cord will be responsive, and therefore capable of sufficient expansion for engraftment into a cohort of mice. By this point in the expansion, the cell population from responsive cords increased approximately 5-fold, from an initial average of 560,000 HSPCs to 2,750,00 HSPCs, while that of nonresponsive cords failed to even double, rising from an average of 370,000 HSPCs to 700,000 (P<0.01; Figure 1a). A combination of features (initial HSPC population and expansion over the first 3–4 days) predicted responsive cords according to one of two sets of conditions: (1) >500,000 HSPCs and ≥3-fold expansion, or (2) >200,000 HSPCs and at least ≥5-fold expansion. This rule correctly identified 62% and 85% responsive and non-responsive cords, respectively, yielding an overall 73% chance of a correct prediction.
Table 1.
A comparison of the characteristics of responsive and non-responsive cords prior to and during expansion. Data not shown for cord volume (P=0.78) and percentage of living cells (P=0.87).
| Responsive cords | Nonresponsive cords | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cord ID | HSPCs in cord blood (%) |
HSPC density (HSPCs/mL) |
HSPCs on day 1 |
HSPCs mid- expansion |
Fold change (Day 1-mid expans.) |
HSPCs Late- expansion |
Fold change (Day 1 - late expans.) |
Cord ID | HSPCs in cord blood (%) |
HSPC density (HSPCs/mL) |
HSPCs on day 1 |
HSPCs mid- expansion |
Fold change (Day 1-mid expans.) |
HSPCs late- expansion |
Fold change (Day 1 - late expans.) |
| HM013 | 0.45 | 2,158 | 204,400 | 1,509,240 | 7.38 | 16,712,640 | 81.76 | HM012 | 0.76 | 2,861 | 215,040 | 1,926,360 | 8.96 | 2,493,840 | 11.60 |
| HM016 | 0.51 | 1,907 | 278,880 | 1,411,440 | 5.06 | 24,030,840 | 86.17 | HM015 | 1 | 10,139 | 1,531,040 | 1,615,280 | 1.06 | 9,240 | 0.01 |
| HM025 | 0.36 | 4,020 | 419,240 | 3,400,200 | 8.11 | 13,582,800 | 32.40 | HM018 | ND | 542 | 62,520 | 846,480 | 13.54 | 1,469,880 | 23.51 |
| HM027 | 0.3 | 8,384 | 821,680 | 7,385,600 | 8.99 | 9,327,800 | 11.35 | HM023 | ND | 559 | 62,800 | 14,040 | 0.22 | 2,759,280 | 43.94 |
| HM028 | 0.2 | 5,003 | 503,840 | 1,574,640 | 3.13 | 6,051,240 | 12.01 | HM029 | 0.58 | 4,440 | 25,060 | 355,560 | 14.19 | 6,600 | 0.26 |
| HM031 | 1.25 | 1,332 | 158,960 | 3,474,600 | 21.86 | 36,103,032 | 227.12 | HM030 | 0.48 | 3,215 | 383,200 | 523,200 | 1.37 | 1,250,040 | 3.26 |
| HM034 | ND | 10,777 | 1,183,360 | 1,616,360 | 1.37 | 8,445,120 | 7.14 | HM032 | 0.27 | 16 | 2,000 | 120 | 0.06 | 0 | 0.00 |
| HM041 | 0.42 | 5,644 | 508,000 | 5,106,400 | 10.05 | 13,487,160 | 26.55 | HM040 | 0.14 | 4,699 | 256,180 | 2,002,400 | 7.82 | 2,337,360 | 9.12 |
| HM044 | 0.09 | 2,458 | 337,200 | 3,088,320 | 9.16 | 10,670,880 | 31.65 | HM050 | 0.15 | 7,217 | 792,480 | 41,640 | 0.05 | 2,828,160 | 3.57 |
| HM045 | 0.16 | 9,250 | 1,043,360 | 801,840 | 0.77 | 8,547,600 | 8.19 | HM052 | 0.26 | 4,466 | 465,120 | 243,960 | 0.52 | 2,084,880 | 4.48 |
| HM057 | 0.77 | 7,515 | 928,080 | 1,691,160 | 1.82 | 6,945,120 | 7.48 | HM056 | 0.25 | 2,091 | 223,200 | 1,064,200 | 4.77 | 34,200 | 0.15 |
| HM059 | 0.10 | 2,623 | 293,280 | 435,760 | 1.49 | 6,784,800 | 23.13 | HM066 | 0.05 | 752 | 74,480 | 318,480 | 4.28 | 70,800 | 0.95 |
| HM069 | 0.18 | 4,936 | 598,720 | 4,183,600 | 6.99 | 8,132,480 | 13.58 | HM067 | 0.26 | 5,567 | 705,920 | 115,920 | 0.16 | 1,116,840 | 1.58 |
| Avg. | 0.40 | 5,077 | 559,923 | 2,744,550 | 4.90 | 12,986,270 | 23.19 | Avg. | 0.38 | 3,581 | 369,157 | 697,511 | 1.89 | 1,266,240 | 3.43 |
| St. Dev. | 0.33 | 3,078 | 334,220 | 1,966,988 | 5.89 | 8,546,621 | 25.57 | St. Dev | 0.29 | 2,957 | 431,831 | 732,026 | 1.70 | 1,146,531 | 2.66 |
| P-value | 0.90 | 0.22 | 0.22 | <0.01 | <0.01 | ||||||||||
Figure 1. In vitro amplification of HSPCs and MSCs; pre-engraftment mouse irradiation.
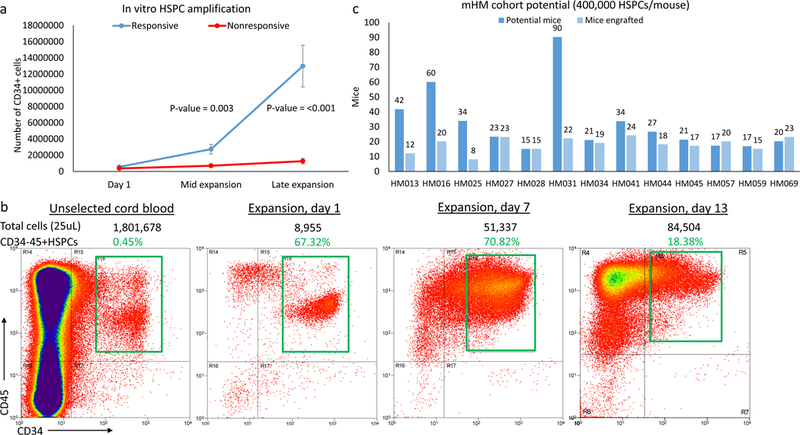
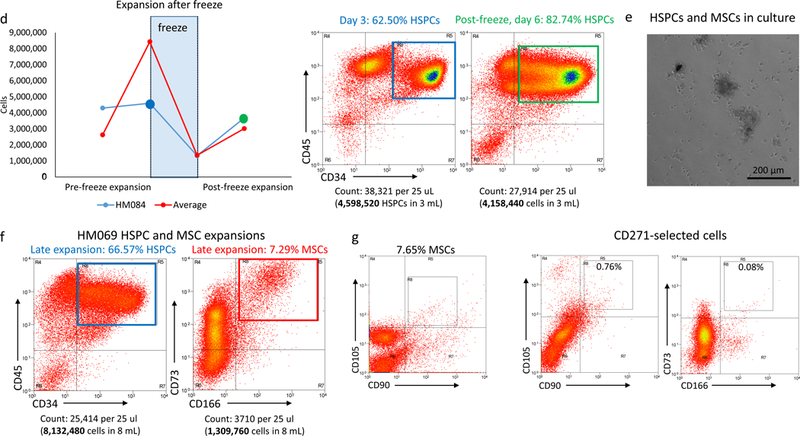
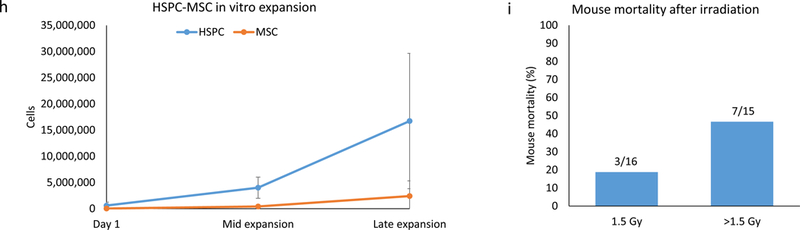
(a) Growth curves of HSPCs purified from responsive and non-responsive cords. Significant differences in proliferation can be seen after several days in culture (mid-expansion). (b) Flow cytometry depicting the amplification of HSPCs during in vitro expansion and, by day 13, the gradual loss of HSPCs due to increased immune cell differentiation. The total number of cells in the sample is recorded above each graph, along with the percentage of those cells (in green, corresponding to the green box superimposed on the graph) that are HSPCs. (c) Chart comparing potential and actual sizes of the mHM cohorts after HSPC expansion, based on an injection of 400,000 HSPCs per mouse. Cohorts HM057 and HM069 were generated using 300,000 cells/mouse, so their actual size is greater than their potential size. (d) HSPC loss due to freezing and subsequent re-expansion after removal of the cells from cryopreservation. Overall averages for all subsequently expanded cryopreserved HSPCs (red) and for the HM084 HSPCs (blue) are shown. For HM084, the cell numbers at the blue and green points on this graph are represented by cytometry, which shows pre-and post-freeze HM084 HSPCs in culture. (e) Picture of HSPCs (non-adherent circular cells) and MSCs (adherent spindle-like cells) in culture. Magnification is 10x and scale bar = 200μm. (f) Cytometry illustrating the relative abundance of HSPCs and MSCs at the end of in vitro expansion. (g) Cytometry indicates that many of the MSCs also express CD90 and CD105. (h) Relative expansion of HSPCs and MSCs from responsive cords in culture. (i) Comparison of radiation-induced mortality. Irradiation greater than 1.5 Gy resulted in 2.8 times as many deaths
Among responsive cords, the number of HSPCs increased rapidly for several days before plateauing around the ninth day of expansion, as increased differentiation overtook HSPC division (as depicted in the expansion of HM013, Figure 1b). The HSPC population in responsive cords overall increased by an average of 23-fold, to almost 13,000,000 cells, and the cell number among these cords ranged from 6,000,000 to 36,000,000. The number of HSPCs in non-responsive cords increased less than 4-fold, to only 1,250,000 cells on average (range zero to 2,750,000 HSPCs). In summary, the behavior after 3–4 days in culture but no baseline characteristic identified cords amenable to appropriate HSPC expansion.
Titration of HSPCs per mouse and mHM cohort size
Following this expansion protocol, we were able to generate HSPC populations of an average of ~13,000,000 cells, thus enabling the engraftment of large mHM cohorts using 400,000 HSPCs per mouse (Figure 1c). We have generated cohorts using as few as ~300,000 (HM057 and HM069) or as many as 1,000,000 HSPCs per mouse (HM025) and observed no significant variation in overall mouse humanization. On the other hand, when we engrafted a cohort (HM071) with only 150,000 HSPCs/mouse, the resulting mice showed poor humanization (with an average of only 7.1% human immune cells in their bone marrow).
Immediate vs delayed utilization of cord blood precursors
We next assessed the feasibility of cryopreserving aliquots of partially expanded HSPCs from responsive cords, giving us the capability of performing experiments with cells from the same source at different timepoints. To this end, we purified and cryopreserved successfully expanding HSPCs and generated a bank of cells from responsive cords for future cohort generation. We successfully generated several cohorts (HM059, HM071, HM084, and HM060) from frozen HSPCs. Although on average only 20% of the cells tolerate the freezing process, the surviving HSPCs remained capable of undifferentiated division in culture for several days after thawing (shown for the average of all cryopreserved HSPCs and for HM084 in Figure 1d). Additionally, we observed no reduction in the degree of mouse humanization in the HM059 cohort (see Figure 2a, below).
Figure 2. Evidence of humanization in mHM cohorts.
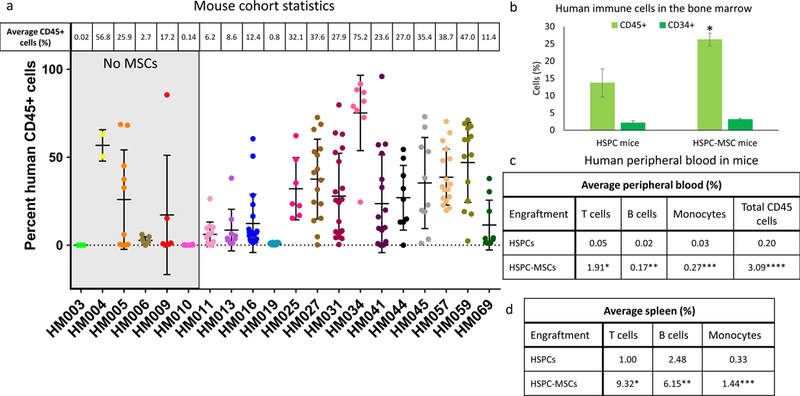
(a) Comparison of the average percentage of human CD45+ immune cells within the bone marrow of cohorts engrafted with HSPCs only (first six cohorts on the left, shaded in gray) and those engrafted with an HSPC- MSC mixture, along with the number of mice in each cohort. (b) Chart comparing the average CD45+ cell percentage (light green) and the average CD34-45+ HSPCs percentage (dark green) in mouse cohorts generated using only HSPCs and those created from HSPC-MSCs. *P=0.007. (c) Comparison of the percentage of human T cells, B cells, and monocytes found in the blood of mice engrafted with HSPCs and HSPC-MSCs. *P=<0.01, **P=0.01, ***P<0.01. Also compared are the percentages of total human CD45+ cells observed in these two mouse types. ****P<0.01. (d) Comparison of the percentage of human T cells, B cells, and monocytes found in the spleen of mice engrafted with HSPCs and MSPC-MSCs. *P<0.01, **P<0.01, ***P<0.01.
MSC isolation and expansion
Within 2–3 days of culture we observed adherent cells dividing among the non-adherent HSPCs. These cells were spindle-like in appearance (Figure 1e), and upon characterization they expressed the cell surface antigens CD73 and CD166, indicative of MSCs (Figure 1f). Further characterization showed that many of these cells also express the MSC markers CD90 and CD105 (Figure 1g). The proportion of these cells growing in cultures is variable, averaging 10% at mid-expansion, and 12% at the time of engraftment (Figure 1h).
Pre-engraftment irradiation
Whole-body irradiation is commonly used to help direct human precursor cells toward mouse bone marrow (19). To determine an ideal dose for optimal conditioning and minimal radiation-induced mouse mortality, we irradiated different groups of mice with 1.5, 1.8, 2.0, 2.2, or 2.4 Gy prior to the injection of 400,000 expanded HSPCs from the same cord, then compared mortality and humanization (Figure 1i). Although increased radiation doses led to small increases in humanization, mice receiving greater than 1.5 Gy were 2.8 times as likely to die as those receiving 1.5 Gy (3 of 16, or 19%, vs. 8 of 15, or 53%), encouraging the selection of a 1.5 Gy dose. To ensure all mice received an equivalent dose of radiation, we used containers designed to minimize mouse movement beneath the irradiation source.
mHM cohort generation
We have generated and analyzed mouse humanization on 20 mHM cohorts (Table 1, Figure 2a). We created the initial six cohorts using only non-adherent HSPCs and the later 14 cohorts using a mixture of 90–95% HSPCs and 5–10% MSCs. Overall, mice engrafted with the HSPC-MSC mixture were more thoroughly humanized. The average percentage of CD45+ human immune cells in mouse bone marrow rose from 13.7% in HSPC-engrafted mice to 26.2% in mice engrafted with HSPC-MSCs, a 91% increase (P=0.01; Figure 2b). The proportion of CD34+ precursors among these human immune cells remained constant between mice engrafted with HSPCs and those engrafted with HSPC-MSCs, at 15% and 12%, respectively (2.1% and 3.1% of all CD45+ cells; Figure 2b). Along with a ~2-fold increase in the bone marrow pool of human cells, blood of mice engrafted with HSPC-MSCs contained 38-, 9-, and 9-fold increases in peripheral T cells, B cells, and monocytes (Figure 2c; P<0.01 for T cells, P=0. 01 for B cells, and P<0.01 for monocytes), indicating that dual HSPC-MSC use leads to superior humanization. Spleens of mice engrafted with HSPC-MSCs also harbored 9-, 2-, and 4-fold larger populations of human T cells, B cells, and monocytes respectively (Figure 2d; P<0.01 for all comparisons).
Establishing a threshold to predict adequate mouse humanization
For effective generation of experimentally useful humanized xenograft cohorts, it is critical to have a means of predicting the degree of humanization of each individual mouse, preferably based on easily accessible peripheral blood. By comparing human cells in peripheral blood with a retrospective analysis of the relative human immune cell composition in the bone marrow of over 200 mice from 20 cohorts, we determined that 70% of all mice whose blood contained at least 0.05% human B cells also harbored bone marrow comprised of at least 20% human immune cells and are the most likely candidates to be well-humanized. Conversely, only 31% of the mice with less than a 0.05% human B cell population had bone marrow containing more than 20% human immune cells.
Adequate humanization led to enhanced human stroma invasion of HNSCC tissue implanted on mice flanks (Figure 3a, shown for both well- and poorly-humanized mice). An analysis of the relationship between human immune cells in mouse bone marrow and immune cells infiltrating tumors indicated that although there is not a linear relationship between human immune cells in bone marrow and tumor infiltration (Figure 3b, first panel), there is a significant increase in immune infiltration in mice in which >20% of bone marrow cells are of human origin (Figure 3b, second panel; P=0.01). This corresponds with results from peripheral blood, allowing us to use the percentage of B cells in peripheral blood as an early indicator of both the degree of humanization and subsequent tumor immune cell infiltration. Using these guidelines, well humanized mice (defined as all those whose bone marrow contains >20% human cells) averaged an almost 7-fold greater percentage of peripheral B cells (P=0.01) and an almost 4-fold greater percentage of immune cells infiltrating tumors (Figure 3c; P<0.01) than poorly humanized mice.
Figure 3. Defining the degree of humanization at which immune cells infiltrate into implanted tumors.
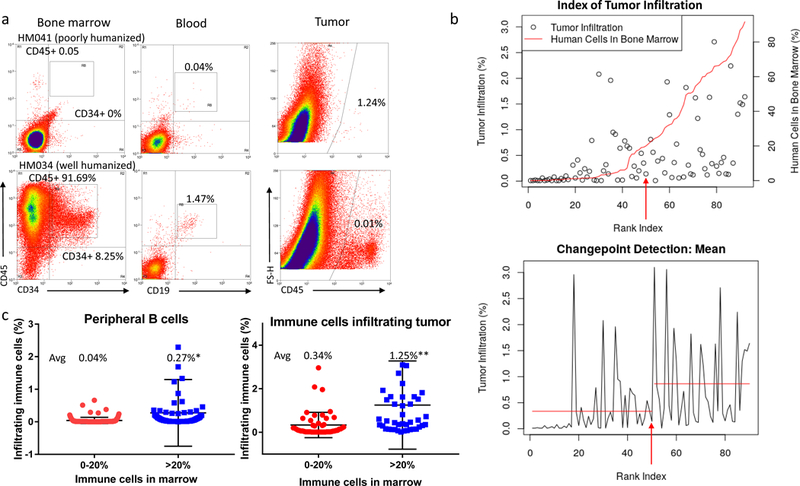
(a) Cytometry showing animals with a higher percentage of human immune cells in their bone marrow are also more likely to have human immune cells within their peripheral blood and infiltrating implanted tumors. (b) Panel 1: Graph showing the relationship between immune cells in the bone marrow (red line) and immune cell infiltration into tumors (circles), organized by mouse – ranked according to their percentage of human bone marrow – on the X axis. The arrow indicates the mouse identified as the changepoint between poorly- and well-humanized mice. Panel 2: Changepoint analysis showing the increase in the mean percentage of immune infiltration in mice whose bone marrow is composed of at least 20% human immune cells. The arrow indicates the changepoint mouse. (c) Plots depicting increased human B cell content in the peripheral blood of well-humanized mice (*P=0.01; left) and increased immune cells infiltration in tumors implanted on well-humanized mice (**P<0.01; right).
Characterization of the tumor infiltrating immune and stroma cells
In well-humanized mice generated from either HSPCs or HSPC-MSCs, we observed T cell, B cell, and macrophage infiltration within the stroma of implanted tumors (shown for CUHN013 in HM005 and CUHN004 in HM034; Figure 4a). In general, cytometry showed that tumor infiltration was greater in mice engrafted with HSPC-MSCs (P>0.01 for T cells, P=0.03 for B cells, and P=0.01 for macrophages; Figure 4b), similar to what was observed in the peripheral blood (Figure 2c). We also observed a population of uncharacterized immune cells invading the tumors (Figure 4c). In tumors grown on the early HM005 cohort, T cells, B cells, and macrophages collectively comprise 56% of the total immune cells infiltrating the tumor, whereas in the latter HM034 cohort these cells make up 74% of all CD45+ intra-tumoral immune cells.
Figure 4. Characteristics of immune cell infiltration in implanted tumors.
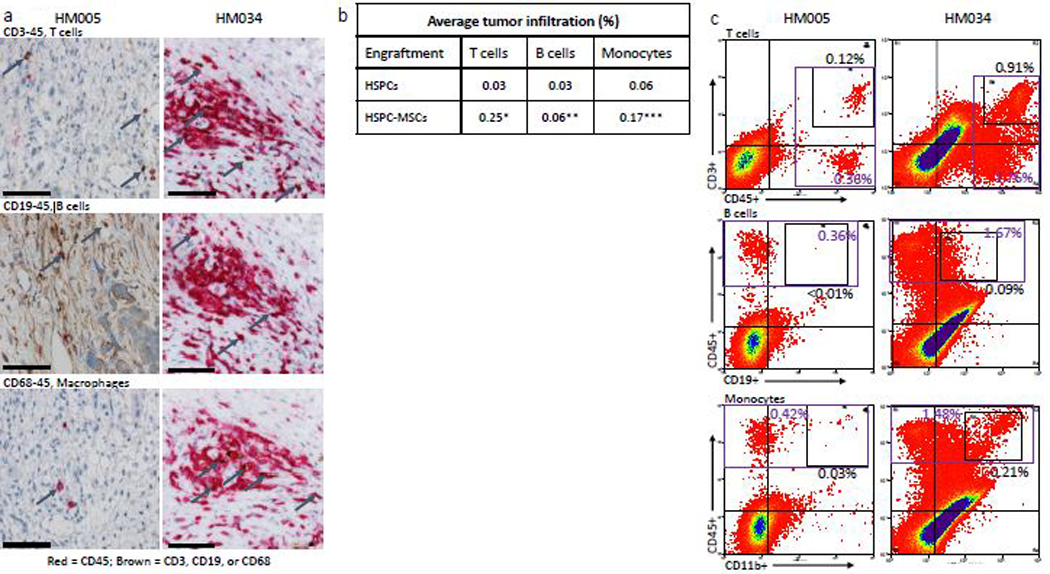
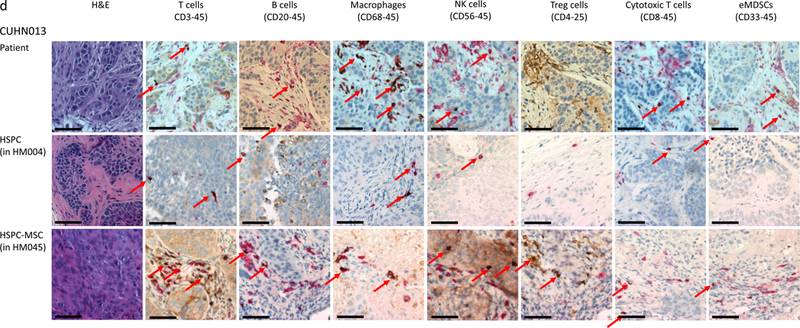
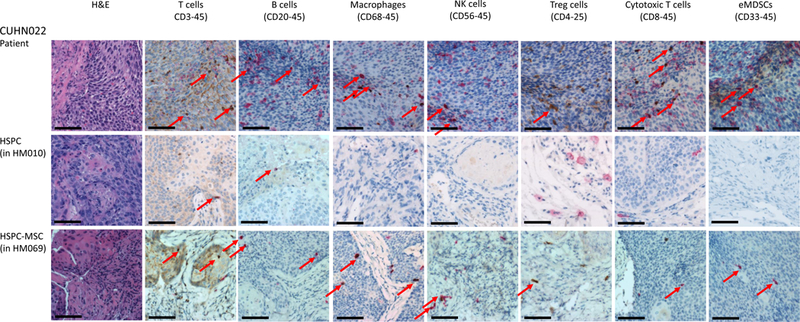
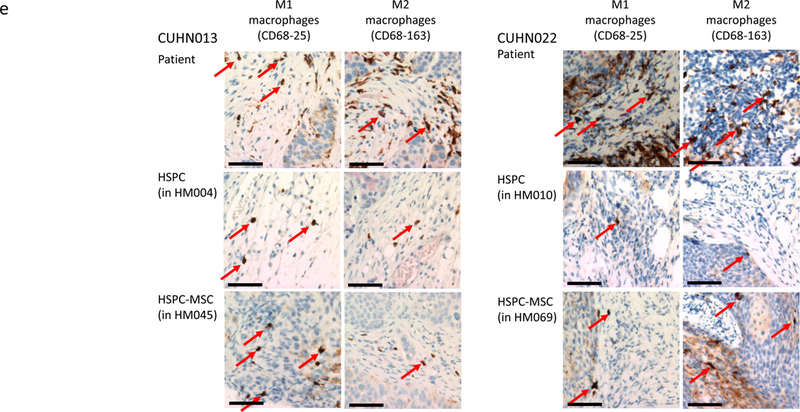
(a) IHC comparison of the relative presence of T cells (top panel), B cells (middle panel), and macrophages (bottom panel) within the tumors of well humanized mice engrafted either with HSPCs only (CUHN013 from HM005, whose bone marrow is composed of 32.74% human cells) or a HSPC-MSCs (CUHN004 from HM034, 91.69% human bone marrow). Magnification is 20x; scale bar = 100μm. (b) Comparison of the percentage of human T cells, B cells, and monocytes found in the tumors implanted on all tumor-bearing cohorts of mice engrafted with HSPCs and HSPC-MSCs. *P<0.01, **P=0.03, ***P=0.01. (c) Cytometry comparing the relative abundance of uncharacterized human CD45+ immune cells present in tumors implanted on mice engrafted with HSPCs (HM005) and HSPC-MSCs (HM034). In the HM005 mouse, immune cells make up 0.38% (average of the values in the colored boxes) of all cells in the tumor, yet T cells, B cells, and macrophages account for only 16% of all cells. In the HM034 mouse, immune cells make up 1.64% of all cells, while T cells, B cells and macrophages account for 1.21% of all cells in the tumor. (d) IHC staining for T cells, B cells, macrophages, NK cells, Treg cells, cytotoxic T cells, and eMDSCs within patient tumors and tumors implanted on mice engrafted with HSPCs or HSPC-MSCs. Top series = CUHN013 tumor from patient, from HM004 mouse, and from HM045 mouse. Bottom series = CUHN022 tumor from patient, from HM010 mouse, and from HM069 mouse. Magnification is 20x; scale bar = 100μm. (e) IHC staining comparing M1 and M2 macrophage differentiation within patient tumors and xenografts. Magnification is 20x; scale bar = 100μm.
Because much of the utility of the mHM xenograft model arises from its ability to recreate the TME of a patient tumor, we characterized the diverse immune cell populations in two of the originating patient tumors (CUHN013 and CUHN022) as well as in xenografts from mice engrafted with HSPCs and HSPC-MSCs. Using immunohistochemistry we identified T cells, B cells, both M1 and M2 macrophages, as well as Treg cells, cytotoxic T cells, NK cells, and immature (or early) myeloid derived suppressor cells (eMDSC) in both tumors (Figure 4d) (20). It appears that macrophage differentiation may mimic that frequently observed in patient tumors. M1 macrophages appear more predominantly in the CUHN013 patient tumor and xenografts; while M2 macrophages are more often observed in the CUHN022 patient tumor and xenografts (Figure 4e). Some cell types, such as macrophages, NK, Treg, and cytotoxic T cells, are much more common in the originating tumors and in tumors from mice engrafted with HSPC-MSCs than in mice engrafted only with HSPCs (Figure 4d), indicating that dual HSPC-MSC engraftment produces an mHM model that more faithfully replicates the immune environment of the patient, with a greater proportion of mature, identifiable immune cells suggesting it is an improved tool for recapitulating the tumor microenvironment.
Differential effect of immune-directed therapy
A key question is whether better humanization will lead to more effective antitumor effect in vivo. We prepared two different HM cohorts both bearing CUHN004 tumors, as well as non-HM NSG control cohorts. One cohort (HM034) was well humanized, with an average 82.4% and 69.8% human cells on the bone marrow and spleen; the second cohort (HM044) had 31.5% and 35.1% human cells on the bone marrow and spleen, respectively (both differences HM034 vs HM044 P<0.001). The programmed cell death receptor 1 (PD-1) inhibitor nivolumab induced tumor growth arrest with T/C of 0.70 and 0.63 at 28 and 32 days (P=0.15 and 0.046, respectively) in the HM034 cohort but had no effect (T/C 0.98 and 0.99; P>0.5) in the HM044 cohort. Treatment of non-humanized NSG controls with nivolumab did not have any significant anti-tumor effect (T/C=1.02; P>0.5). Although cytometry indicated that both groups of tumors were both well-invaded by T cells, nivolumab treatment increased invasion in HM034 tumors by an average of 2.1 times while treatment increased T cell invasion in the HM044 tumors by only a factor of 1.3. IHC analysis of T cell infiltration and T cell activity in these tumors showed a higher degree of pre- and post-treatment T cell invasion in HM034 vs HM044 (Figure 5a), as well as a corresponding increase of T cell effect as measured by granzyme B, a protein secreted by T cells to induce apoptosis of cancer cells and used as a surrogate for T cell activation (Figure 5b). In this initial assessment, the observed antitumor effect associated with a higher T cell infiltration and activation in the better humanized HM034, together with a lack of anti-tumor effect (similar to that of non-humanized NSG controls) in the less humanized HM044 suggests that higher humanization is advantageous for in vivo drug testing.
Figure 5. Mouse humanization and resultant T cell infiltration and activity within tumors dictate nivolumab response.
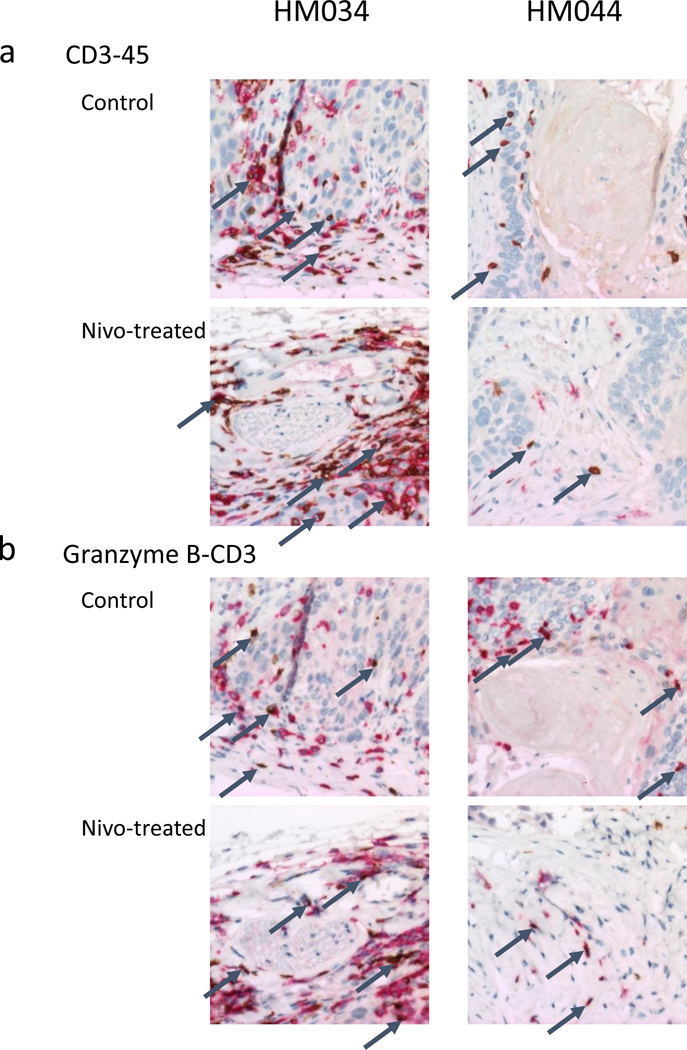
(a) Human immune cells, including T cells (identified by dual redbrown staining) are much more abundant in both control and nivolumab-treated CUHN004 tumors implanted on very well-humanized mice (HM034, average 82.4% human bone marrow) than in comparable tumors on a more moderately humanized cohort (HM044, average 31.5% human bone marrow.) (b) T-cell mediated cell killing (identified by granzyme B, stained brown) in the vicinity of CD3+ T cells (stained red) is a measure of the activity of tumor-infiltrating T cells in these cohorts before and after nivolumab treatment. The T/C of 0.70 in the HM034 cohort suggests that, when T cells are present in great enough numbers within a tumor, nivolumab can enhance their ability to destroy tumor cells.
DISCUSSION
The utility of a cancer model system lies in its ability to recreate conditions in the patient tumor and its microenvironment, ultimately enabling biologic discovery and therapy development. Although traditional mouse xenograft models can partially meet this need, they cannot recapitulate interactions between cancer and immune cells within the TME (21,22). Over the past decade, humanized mouse xenograft models have been developed to address this challenge (23). However, reconstitution of a human immune system within a mouse model depends on interrelated factors that must be coordinated to maximize the utility of this system.
There are several immunocompromised mouse strains that can be used in humanization studies (24). The NSG mouse is particularly suitable for this process, because it lacks T cells, B cells, NK cells, and mature macrophages, yet remains hardy enough to undergo pre- engraftment irradiation without undue mortality (25,26). Although some groups have reported that species-specific activity of some mouse cytokines prevents maturation of myeloid cells within the NSG mouse (27,28), it appears the presented dual HSPC-MSC engraftment process can, at least in part, overcome this obstacle; we can readily identify monocytes in the blood and macrophages infiltrating into tumors of well-humanized animals. Potential approaches for further improvements in myeloid differentiation include utilization of existing transgenic mouse strains containing human cytokines (29,30).
Several factors contribute to the substantial variability in the extent to which individual mice in a given mHM cohort are humanized. Although published humanization protocols recommend pre-engraftment mouse irradiation of up to 2.4 Gy (31,32), we have produced the humanized mice described in our results above using 1.5 Gy without excessive mouse (and subsequent cohort) attrition. Even at this lower dose, it is not atypical for 10% of the irradiated mice to die within a week of conditioning and engraftment. Although it is possible that higher doses produce greater consistency in humanization, mouse mortality can be as great as 50%, which has a substantial impact on cohort size and experimental utility. Humanization variability within individual cohorts could also be caused by movement of mice into and out of regions of increased radiation intensity. We have minimized this variation by irradiating the mice in smaller containers directly under the source.
In reported descriptions of mouse humanization, the number of injected HSPCs needed for substantive engraftment varies from 100,000 to 700,000 per mouse (5,32). We have determined that injecting 400,000 HPSCs per mouse results in predictably stable humanization while still allowing for production of large mHM cohorts necessary for multi-arm treatment studies. Our experiments examining the range of HSPCs needed for adequate engraftment indicated that ~300,000 HSPCs can still humanize mice adequately, especially when using dual HSPC-MSCs (47% mouse bone marrow human HSPCs). However, when we lowered the HSPC number to 150,000, we observed a significant loss of humanization in the form of reduced human CD34 and CD45+ cell populations in the bone marrow (7.1%), fewer human peripheral blood cells, and less immune cell infiltration into implanted tumors. Conversely, the injection of up to 1,000,000 HSPCs per mouse did not significantly improve HM immune profiles.
Our results also indicate that inclusion of MSCs significantly increases the percentage of human bone marrow within engrafted mice and assists in immune cell differentiation and trafficking, consistent with the reported roles of MSCs (16,33–35). A doubling in the percentage of human bone marrow in mice engrafted with HSPC-MSCs compared to HSPC-only cohorts led to 15-fold higher number of human cells in the peripheral blood (3.09% vs. 0.20%). In HSPC-only mice T cells, B cells, and monocytes accounted for 50% of all human peripheral blood cells (0.10% of the 0.20%; see Figure 2c), while they constituted 76% (2.35% of the 3.09%) of human peripheral blood cells in mice created from HSPC-MSCs. This indicates the presence of MSCs is associated with an exponential enhancement in human bone marrow precursors that generate mature blood elements. It will be critical to identify bone marrow terminal differentiation markers to ascertain active versus inactive human precursors responsible for this significant increase in maturation. Additionally, treatment of two cohorts with varying degrees of humanization implanted with the same HNSCC patient-derived tumor gave a preliminary insight into mHM use for drug development; the PD-1 inhibitor nivolumab, approved for use in relapsed HNSCC patients, induced tumor arrest only in the better humanized cohort. These mice averaged 2.6 times the percentage of human cells in the bone marrow, and after treatment the number of T cells infiltrating the tumors increased by more than two-fold.
Although a high percentage of human bone marrow is necessary in the generation of useful HM, this may not be sufficient as there are additional potentially relevant factors (12). Myeloid blood cells depend on environmental cytokine cues for their maturation and trafficking, and mouse cytokines largely inhibit proper differentiation (30). Likewise, human progenitor T cells must migrate from the bone marrow to the thymus to mature, and a mouse thymus lacks the human major histocompatibility complexes to guide proper T cell education and selection, leading to a T cell population that may be largely anergic (9). Indeed, the in vivo nivolumab treatment study implied that a large population of T cell-producing HSCs might be necessary to produce a sufficient population of mature and functionally responsive T cells (as identified by granzyme B production) capable of invading tumors. Also, B cells may not mature properly without the presence of a human thymus (10). Further, when the human immune system in the mouse is derived from donated cord blood and the tumors implanted on these mice come from a different source, this allogeneic context may trigger unchecked immune responses unrelated to cancer progression or treatment (36). Graft-versus-host (GvH) disease, although uncommon, was evidenced by a hunched posture, lethargy, and weight loss in 9/213 mice. Analysis of GvH mouse tissues indicated that their blood and many of their organs were inundated with auto-reactive T cells, either inadvertently introduced along with the HSPCs during engraftment or mis-educated in the mouse thymus. The presence of MSCs can reduce aberrant activation of human T cells, so the rarity of this phenomenon in our mHM mice may be a consequence of the HSPC-MSCs used to engraft them (37).
There are many strategies being studied to overcome these challenges. Transgenic mouse strains expressing human cytokines can aid in immune cell development and trafficking. Mice can also be engineered to express human major histocompatibility complex molecules, such as the common HLA-A*02:01 variant, allowing them to undergo specific T cell responses to introduced tumor antigens (38). Engrafted human thymus and bone marrow can help guide T and B cell maturation (39). Attempts are also underway to harvest cancer patient HSPCs to create HM with an immune system autologous to the implanted tumor.
Despite formidable barriers to achieving a mouse system with appropriate and functional human immune components, these sophisticated mouse models can serve as platforms to advance our understanding of the TME and the development of immune-directed therapies and vaccines in cancer, as well as other areas of human disease. Indeed, much of the initial HM work was conducted to further autoimmunity and stem cell research, and today the increased relevance of the immune system and inflammation in cancer and degenerative human disease has made improved development of these models increasingly germane.
Translational Relevance.
In this report we describe in detail the evolving procedures to optimize humanized mouse cohort generation, including optimal conditioning, choice of lineage for engraftment, threshold for successful engraftment, HNSCC tumor implantation, and immune and stroma cell analyses. We developed a dual infusion protocol of human hematopoietic stem and progenitor cells (HSPCs) and mesenchymal stem cells (MSCs), leading to incremental human bone marrow engraftment, and exponential increase in mature peripheral human immune cells, and intratumor homing that includes a more complete lineage reconstitution. Additionally, we have identified practical rules to predict successful HSPC/MSC expansion, and a peripheral human cell threshold associated with bone marrow engraftment, both of which will optimize cohort generation and management. The tremendous advances in immune therapy in cancer have made the need for appropriate and standardized models more acute than ever, and therefore we anticipate that this manuscript will have an immediate impact in cancer-related research.
ACKNOWLEDGEMENTS
The authors are indebted to the patients who donated their tissue, blood and time, and to the clinical teams who facilitated patient informed consent, as well as sample and data acquisition. The authors wish to thank Karen Helm for helpful discussions and Pamela Garl for her manuscript review.
Support
This work was primarily supported by National Institutes of Health grants R01CA149456 (AJ), R01DE024371 (XJW, AJ), P30-CA046934 (University of Colorado Cancer Center Support Grant), Ruth L. Kirschstein National Research Service Award T32CA17468 (XJW, PNL trainee), Training in Otolaryngology Research T32DC012280 (CN trainee), the Daniel and Janet Mordecai Foundation (AJ), and the Peter and Rhonda Grant Foundation (AJ)
Footnotes
Competing financial interests
The authors declare that they have no potential conflicts of interest.
BIBLIOGRAPHY
- 1.Ferris RL, Blumenschein G Jr., Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. The New England journal of medicine 2016;375(19):1856–67 doi 10.1056/NEJMoa1602252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gillet JP, Varma S, Gottesman MM. The clinical relevance of cancer cell lines. J Natl Cancer Inst 2013;105(7):452–8 doi 10.1093/jnci/djt007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ben-David U, Ha G, Tseng YY, Greenwald NF, Oh C, Shih J, et al. Patient-derived xenografts undergo mouse-specific tumor evolution. Nat Genet 2017;49(11):1567–75 doi 10.1038/ng.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Day CP, Merlino G, Van Dyke T. Preclinical mouse cancer models: a maze of opportunities and challenges. Cell 2015;163(1):39–53 doi 10.1016/j.cell.2015.08.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen X, Chaleff S, et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. Journal of Immunology (Baltimore, Md: 1950) 2005;174:6477–89. [DOI] [PubMed] [Google Scholar]
- 6.Lan P, Tonomura N, Shimizu A, Wang S, Yang YG. Reconstitution of a functional human immune system in immunodeficient mice through combined human fetal thymus/liver and CD34+ cell transplantation. Blood 2006;108(2):487–92 doi 10.1182/blood-2005-11-4388. [DOI] [PubMed] [Google Scholar]
- 7.Wege AK, Ernst W, Eckl J, Frankenberger B, Vollmann-Zwerenz A, Mannel DN, et al. Humanized tumor mice--a new model to study and manipulate the immune response in advanced cancer therapy. International journal of cancer Journal international du cancer 2011;129(9):2194–206 doi 10.1002/ijc.26159. [DOI] [PubMed] [Google Scholar]
- 8.Rongvaux A, Willinger T, Martinek J, Strowig T, Gearty SV, Teichmann LL, et al. Development and function of human innate immune cells in a humanized mouse model. Nature biotechnology 2014. doi 10.1038/nbt.2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chung YS, Son JK, Choi B, Joo S-Y, Lee Y-S, Park JB, et al. Co-transplantation of human fetal thymus, bone and CD34(+) cells into young adult immunodeficient NOD/SCID IL2RY(null) mice optimizes humanized mice that mount adaptive antibody responses. Clinical Immunology (Orlando, Fla) 2015;157:156–65 doi 10.1016/j.clim.2015.02.005. [DOI] [PubMed] [Google Scholar]
- 10.Chung YS, Son JK, Choi B, Park JB, Chang J, Kim SJ. Transplantation of human spleen into immunodeficient NOD/SCID IL2RY(null) mice generates humanized mice that improve functional B cell development. Clinical Immunology (Orlando, Fla) 2015;161:308–15 doi 10.1016/j.clim.2015.09.001. [DOI] [PubMed] [Google Scholar]
- 11.Yong KSM, Her Z, Chen Q. Humanized Mice as Unique T ools for Human-Specific Studies. Arch Immunol Ther Exp (Warsz) 2018. doi 10.1007/s00005-018-0506-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morton JJ, Bird G, Refaeli Y, Jimeno A. Humanized Mouse Xenograft Models: Narrowing the Tumor-Microenvironment Gap. Cancer research 2016;76(21):6153–8 doi 10.1158/0008-5472.CAN-16-1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morton JJ, Bird G, Keysar SB, Astling DP, Lyons TR, Anderson RT, et al. XactMice: humanizing mouse bone marrow enables microenvironment reconstitution in a patient- derived xenograft model of head and neck cancer. Oncogene 2016;35(3):290–300 doi 10.1038/onc.2015.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bird GA, Polsky A, Estes P, Hanlon T, Hamilton H, Morton JJ, et al. Expansion of Human and Murine Hematopoietic Stem and Progenitor Cells Ex Vivo without Genetic Modification Using MYC and Bcl-2 Fusion Proteins. PloS one 2014;9(8):e105525 doi ARTN e105525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kresse SH, Meza-Zepeda LA, Machado I, Llombart-Bosch A, Myklebost O. Preclinical xenograft models of human sarcoma show nonrandom loss of aberrations. Cancer 2012;118(2):558–70 doi 10.1002/cncr.26276. [DOI] [PubMed] [Google Scholar]
- 16.Chen P, Huang Y, Womer KL. Effects of mesenchymal stromal cells on human myeloid dendritic cell differentiation and maturation in a humanized mouse model. Journal of Immunological Methods 2015;427:100–4 doi 10.1016/j.jim.2015.10.008. [DOI] [PubMed] [Google Scholar]
- 17.Keysar SB, Astling DP, Anderson RT, Vogler BW, Bowles DW, Morton JJ, et al. A patient tumor transplant model of squamous cell cancer identifies PI3K inhibitors as candidate therapeutics in defined molecular bins. Molecular oncology 2013;7(4):776–90 doi 10.1016/j.molonc.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Killick R, Eckley IA. changepoint: An R Package for Changepoint Analysis. J Stat Softw 2014;58(3):1–19. [Google Scholar]
- 19.Sugimoto K, Adachi Y, Moriyama K, Qiong W, Nakayama A, Hosono M, et al. Induction of the expression of SCF in mouse by lethal irradiation. Growth Factors 2001;19(4):219–31. [DOI] [PubMed] [Google Scholar]
- 20.Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun 2016;7:12150 doi 10.1038/ncomms12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Williams JA. Using PDX for Preclinical Cancer Drug Discovery: The Evolving Field. J Clin Med 2018;7(3) doi 10.3390/jcm7030041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McMillin DW, Negri JM, Mitsiades CS. The role of tumour-stromal interactions in modifying drug response: challenges and opportunities. Nat Rev Drug Discov 2013;12(3):217–28 doi 10.1038/nrd3870. [DOI] [PubMed] [Google Scholar]
- 23.Shultz LD, Ishikawa F, Greiner DL. Humanized mice in translational biomedical research. Nat Rev Immunol 2007;7(2):118–30 doi 10.1038/nri2017. [DOI] [PubMed] [Google Scholar]
- 24.Holzapfel BM, Wagner F, Thibaudeau L, Levesque J-P, Hutmacher DW. Concise review: humanized models of tumor immunology in the 21st century: convergence of cancer research and tissue engineering. Stem Cells (Dayton, Ohio) 2015;33:1696–704 doi 10.1002/stem.1978. [DOI] [PubMed] [Google Scholar]
- 25.Zhou Q, Facciponte J, Jin M, Shen Q, Lin Q. Humanized NOD-SCID I L2rg–/– mice as a preclinical model for cancer research and its potential use for individualized cancer therapies. Cancer letters 2014;344:13–9 doi 10.1016/j.canlet.2013.10.015. [DOI] [PubMed] [Google Scholar]
- 26.Knibbe-Hollinger JS, Fields NR, Chaudoin TR, Epstein AA, Makarov E, Akhter SP, et al. Influence of age, irradiation and humanization on NSG mouse phenotypes. Biology Open 2015;4:1243–52 doi 10.1242/bio.013201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen Q, Khoury M, Chen J. Expression of human cytokines dramatically improves reconstitution of specific human-blood lineage cells in humanized mice. Proc Natl Acad Sci U S A 2009; 106(51):21783–8 doi 10.1073/pnas.0912274106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ito R, Takahashi T, Katano I, Ito M. Current advances in humanized mouse models. Cell Mol Immunol 2012;9(3):208–14 doi 10.1038/cmi.2012.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wunderlich M, Chou FS, Link KA, Mizukawa B, Perry RL, Carroll M, et al. AML xenograft efficiency is significantly improved in NOD/SCID-IL2RG mice constitutively expressing human SCF, GM-CSF and IL-3. Leukemia 2010;24(10):1785–8 doi 10.1038/leu.2010.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rongvaux A, Willinger T, Martinek J, Strowig T, Gearty SV, Teichmann LL, et al. Development and function of human innate immune cells in a humanized mouse model. Nature Biotechnology 2014;32:364–72 doi 10.1038/nbt.2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ito M, Hiramatsu H, Kobayashi K, Suzue K, Kawahata M, Hioki K, et al. NOD/SCID/gamma(c)(null) mouse: an excellent recipient mouse model for engraftment of human cells. Blood 2002;100(9):3175–82 doi 10.1182/blood-2001-12-0207. [DOI] [PubMed] [Google Scholar]
- 32.Pearson T, Greiner DL, Shultz LD. Creation of “humanized” mice to study human immunity. Current Protocols in Immunology / Edited by John E Coligan [et al. ] 2008;Chapter 15:Unit 15.21 doi 10.1002/0471142735.im1521s81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Theocharides AP, Rongvaux A, Fritsch K, Flavell RA, Manz MG. Humanized hemato- lymphoid system mice. Haematologica 2016;101(1):5–19 doi 10.3324/haematol.2014.115212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Noort WA, Kruisselbrink AB, in’t Anker PS, Kruger M, van Bezooijen RL, de Paus RA, et al. Mesenchymal stem cells promote engraftment of human umbilical cord blood-derived CD34(+) cells in NOD/SCID mice. Exp Hematol 2002;30(8):870–8. [DOI] [PubMed] [Google Scholar]
- 35.Carrancio S, Romo C, Ramos T, Lopez-Holgado N, Muntion S, Prins HJ, et al. Effects of MSC coadministration and route of delivery on cord blood hematopoietic stem cell engraftment. Cell Transplant 2013;22(7):1171–83 doi 10.3727/096368912X657431. [DOI] [PubMed] [Google Scholar]
- 36.Patton J, Vuyyuru R, Siglin A, Root M, Manser T. Evaluation of the efficiency of human immune system reconstitution in NSG mice and NSG mice containing a human HLA.A2 transgene using hematopoietic stem cells purified from different sources. Journal of Immunological Methods 2015;422:13–21 doi 10.1016/j.jim.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roux C, Saviane G, Pini J, Belaid N, Dhib G, Voha C, et al. Immunosuppressive Mesenchymal Stromal Cells Derived from Human-Induced Pluripotent Stem Cells Induce Human Regulatory T Cells In Vitro and In Vivo. Front Immunol 2017;8:1991 doi 10.3389/fimmu.2017.01991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zitvogel L, Pitt JM, Daillere R, Smyth MJ, Kroemer G. Mouse models in oncoimmunology. Nature reviews Cancer 2016;16(12):759–73 doi 10.1038/nrc.2016.91. [DOI] [PubMed] [Google Scholar]
- 39.Brown ME, Zhou Y, McIntosh BE, Norman IG, Lou HE, Biermann M, et al. A Humanized Mouse Model Generated Using Surplus Neonatal Tissue. Stem Cell Reports 2018; 10(4): 1175–83 doi 10.1016/j.stemcr.2018.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]