

Abstract
A new levodopa‐carbidopa intestinal gel (LCIG) system featuring a higher levodopa/carbidopa (LD/CD) concentration and viscosity, LCIG‐HV, is being developed to reduce the intrajejunal volume of LD/CD that is administered as compared to the current commercial formulation, LCIG‐LV. This study characterizes the LCIG‐HV formulation and compares it to the LCIG‐LV formulation via dissolution testing and a clinical pharmacokinetic bioequivalence study. In vitro release profiles of LD/CD were determined using a USP Dissolution Apparatus 2 with 500 mL of phosphate buffer (pH 4.5) operating at 25 RPM. A single dose, open‐label study was conducted according to a two‐period, randomized, crossover design in 28 healthy subjects. The point estimate (PE) of the levodopa C max geometric mean for the LCIG‐HV formulation was 4% higher than that of the LCIG‐LV formulation. PEs of levodopa AUC t and AUC inf geometric means were comparable for both formulations. PEs of carbidopa C max, AUC t and AUC inf geometric means for the LCIG‐HV formulation were 3%‐5% higher than those of the LCIG‐LV formulation. For both formulations, the median T max for levodopa was 1.0 and 3.0 hours for carbidopa. The levodopa half‐life harmonic mean was 1.6 hour for both formulations. The carbidopa half‐life harmonic mean was 1.9 and 2.0 hour, respectively, for the LCIG‐HV and LCIG‐LV formulations. C max, AUC t and AUC inf of LD/CD carbidopa were comparable for both formulations. The current study demonstrates that the LCIG‐LV and LCIG‐HV formulations are clinically bioequivalent for LD/CD according to FDA guidance. However, the dissolution method was over discriminatory of formulation differences.
Keywords: bioequivalent, carbidopa, intestinal gel, levodopa
1. INTRODUCTION
Parkinson's disease (PD) is a chronic neurodegenerative disorder characterized by progressive loss of dopamine‐producing neurons and presents with motor symptoms including bradykinesia, rigidity, tremor, and postural instability.1, 2, 3 Levodopa (LD), a dopamine precursor, is considered the most effective therapy for treating patients with PD.4 LD is actively absorbed from the intestines and is rapidly metabolized to dopamine by L‐amino acid decarboxylase (L‐AAD). To block the peripheral activity of L‐AAD, LD is co‐administered with carbidopa (CD), an L‐AAD inhibitor that is unable to cross the blood‐brain barrier (BBB). The LD/CD combination increases central nervous system (CNS) penetration of exogenously administered LD across the BBB and decreases adverse effects associated with peripheral metabolism of LD to dopamine such as nausea, vomiting, cardiac arrhythmias, and orthostatic hypotension.5 However, high variability in LD exposure following oral LD/CD6 can result in suboptimal efficacy due to the rapid absorption and short half‐life of LD in the plasma, particularly for patients with advanced PD that have a narrow therapeutic window for LD.7 The large fluctuations in plasma LD exposure associated with oral administration of LD/CD may not adequately control the motor symptoms and dyskinesia of PD patients and may lead to increased “Off” time (period of significant PD symptoms), decreased “On” time (periods of adequate PD symptomatic control) and increased periods of dyskinesia.8
Levodopa‐carbidopa intestinal gel (LCIG) was developed by AbbVie and approved by the U.S. Food and Drug Administration (FDA) and European Medicines Agency (EMA) for treating PD patients by providing continuous delivery of LD and CD directly to the jejunum. Continuous delivery of LCIG into the jejunum via a percutaneous gastrojejunostomy (PEG‐J) tube has been previously shown overcome the limitations of oral LD/CD dosing and maintain stable LD and CD exposures for the majority of the dosing interval.9 In a double‐blind, double‐dummy, double‐titration Phase 3 trial conducted in advanced PD patients (the LCIG Horizon study), LCIG has been shown to significantly reduce the “Off” time and increase the “On” time without dyskinesia when compared to the LD/CD oral treatment.10
A new LCIG formulation is currently in development to facilitate reduced volume intrajejunal administration of levodopa and carbidopa. The new system features a new intestinal gel formulation with a higher LD/CD concentration (40/10 mg/mL LD/CD) and higher viscosity, LCIG‐HV compared to the current commercial intestinal gel formulation with lower LD/CD concentration (20/5 mg/mL LD/CD) and lower viscosity, LCIG‐LV. The new LCIG‐NG formulation reduces the volume of LCIG that must be administered daily and increases patient convenience and will allow the new system to have a smaller cartridge size. The current work characterizes the LCIG‐HV formulation and compares it to the LCIG‐LV commercial formulation via dissolution testing and a clinical pharmacokinetic (PK) bioequivalence (BE) study conducted in healthy volunteers and demonstrates that new LCIG‐HV formulation and the LCIG‐LV formulations are clinically bioequivalent for levodopa and carbidopa.
2. METHODS
2.1. Dissolution study
Dissolution tests for both LD and CD were performed using a USP Apparatus 2 operating at a paddle speed of 25 RPM. The dissolution medium was 500 mL of pH 4.5 acetate buffer and the 5‐mL of gel sample (~equivalent to the morning dose) was delivered to the bottom of vessel for testing. An automatic sampler collected 1.5 mL of each of the sample at multiple time points (LCIG‐LV: 5, 10, 15, 20, 30, 45, and 60 minutes; LCIG‐HV: 15, 20, 30, 40, 50, 60, and 75 minutes) and the samples were then analyzed with high‐performance liquid chromatography (HPLC). In vitro drug release profiles were determined by plotting the cumulative percent dissolved in vitro at each time point until the full drug release was achieved.
2.2. Bioequivalence study design
This was an open‐label, randomized, single dose, 2 period cross‐over study in healthy subjects. All procedures performed in this study were in accordance with the ethical standards of the Helsinki Declaration and its later amendments or comparable ethical standards. The study protocol was approved by the local institutional review board and written informed consent was obtained from each subject before any study‐related procedures were performed.
Men and women (postmenopausal or surgically sterile) aged 18‐55 years with a body mass index between 18 and 30 kg/m2 and in general good health were eligible to enroll in the study. Subjects were excluded if they tested positive for human immunodeficiency virus (HIV) or hepatitis A, B, or C, or had alanine aminotransferase (ALT) or aspartate aminotransferase (AST) levels greater than 1.5 × the upper limit of normal or if they had contraindications to levodopa, (eg, narrow angle glaucoma, pheochromocytoma, Cushing's syndrome, malignant melanoma) or history of orthostatic hypotension, at the time of screening. Subjects must not have used or consumed any of the following before study drug administration: tobacco or nicotine products within 6 months; another investigational product within 6 weeks; a drug by injection within 30 days; over‐the‐counter or prescription medications, vitamins, or herbal supplements within 2 weeks; or grapefruit, grapefruit products, Seville oranges, star fruit, or alcohol within 72 h. Subjects were also excluded if placement of nasojejunal (NJ) tube was contraindicated or considered high risk for the NJ procedure (eg, severe mid‐face trauma, recent nasal surgery, esophageal stricture, esophageal varices, alkali ingestion, coagulation abnormalities or gastric bypass surgery).
The study had a standard two‐period crossover design in which the subjects were randomly assigned in a 1:1 ratio to the two sequences of formulations (Table 1). A total of 28 subjects were planned to be enrolled in this study (n = 14 per sequence of formulations). The expectation that there would be some subjects who would not provide data for both periods was taken into account. Power calculations were performed for levodopa C max and AUCinf for the test of the hypothesis of no difference between the formulations. The calculations were done for a difference defined by a ratio of central values of 0.80 or 1.25 (power is the same for these two ratios). For the cases of complete data for 24 subjects and 28 subjects with an equal number per sequence, the calculated power for levodopa C max was 85.9% and 91.0%, respectively. The power for AUCinf exceeded 99.0%.
Table 1.
Study design
| Sequence of formulations | N | Period 1 | Period 2 |
|---|---|---|---|
| 1 | 14 | LCIG‐LV Commercial 200/50 mg LD/CD (10.0 mL) | LCIG‐HV 200/50 mg LD/CD (5.0 mL) |
| 2 | 14 | LCIG‐HV 200/50 mg LD/CD (5.0 mL) | LCIG‐LV Commercial 200/50 mg LD/CD (10.0 mL) |
For these calculations, the error term variance for the analysis of the natural logarithm of levodopa C max was assumed to be 0.0592. This value was obtained from the results in a pilot study in healthy volunteers of LCIG 40/10 and LCIG 20/5 formulations. The study had a crossover design. The error term variance for the analysis of the natural logarithm of levodopa AUC was assumed to be 0.0232. This value was obtained as the average of the estimate of the variance (0.0090) obtained from the pilot study and the estimate (0.0373) from a crossover study in healthy volunteers11 for the comparison of tablet doses. Prior to the morning of dosing in each period, the subjects had an NJ tube placed by an interventional radiologist. A CADD‐Legacy® 1400 pump (Smiths Medical, Minneapolis, MN) was used for priming and dosing of LCIG. Prior to dosing in each period, the NJ tube was primed with 7.6 mL of LCIG according to the volume of the NJ tube described by the manufacturer. A dose of 200/50 mg LD/CD was delivered through the NJ tube over 30 minutes for both formulations. The volume of LCIG delivered to each subject was approximately 5.0 mL for the LCIG‐HV formulation and 10.0 mL for the LCIG‐LV commercial formulation. The drug cassette containing LCIG was weighed before and after dosing to calculate the actual dose of LCIG delivered for each administration. Following each dose, an abdominal X‐Ray was taken to confirm the placement of the NJ tube. No food or beverages, except for water to quench thirst, were allowed for 10 hours prior to dosing for each period.
2.3. Pharmacokinetics sample collection and bioanalysis
Blood samples for determination of LD and CD plasma concentrations were obtained by venipuncture prior to priming of NJ tube, prior to infusion, at 5, 10, 15, 30, 45 minutes and 1, 1.5, 2, 3, 4, 6, 8, 12, and 24 hours after the start of infusion for each dosing period. All samples were collected in evacuated tripotassium (K3) ethylenediaminetetraacetic acid (EDTA)‐containing collection tubes pre‐chilled in an ice bath. Processing of the pharmacokinetic samples and analysis of levodopa plasma concentrations using liquid chromatography with tandem mass spectrometric detection (LC‐MS/MS) are previously described in detail.12 The analytical method was validated over a concentration range of 10‐5000 ng/mL. For levodopa, the intra‐assay accuracy was between −4.74% and 7.59% and the intra‐assay precision was ≤14.2%, respectively. For levodopa, the inter‐assay accuracy was between 0.125% and 2.65% and the inter‐assay precision was ≤8.27%, respectively. For carbidopa, the intra‐assay accuracy was between −3.93% and 15.2% and the intra‐assay precision was ≤12.6%, respectively. For carbidopa, the inter‐assay accuracy was between −2.06% and 6.26% and the inter‐assay precision was ≤6.43%, respectively.
2.4. Pharmacokinetic statistical analysis and f 2 test
Non‐compartmental methods were used to determine values of the pharmacokinetic parameters. Pharmacokinetic parameters values that were determined include the maximum observed plasma concentration (C max), time to C max (peak time, T max), terminal phase elimination half‐ life (t 1/2), and the area under the plasma concentration‐time curve (AUC) from time 0 to the time of the last measurable concentration (AUCt) or to infinite time (AUCinf).
For statistical analysis, C max and AUC parameters were normalized to the actual dose delivered. The dose delivered was calculated as the difference of the weight of the LCIG cassette before and after dosing minus the weight of LCIG needed for priming.
A linear mixed effects model was used to perform the analysis on logarithmically transformed data. The model included fixed effects for period, formulation, and sequence. The subjects were viewed as a random sample. A 90% confidence interval for the ratio of formulation C max geometric means and the ratio of formulation AUC geometric means was provided by exponentiation of the confidence limits for the difference of the logarithm means within the framework of the linear mixed effects model. The 90% confidence intervals for these ratios were used to perform the 21‐sided tests procedure for an assessment on equivalence. A similarity factor f 2 test was used to determine differences between the in vitro dissolution profiles for the LCIG‐HV and LCIG‐LV commercial formulation. The methodology for the f 2 test has been previously described.13, 14
2.5. Safety and tolerability
Safety and tolerability were assessed throughout the study. All adverse events (AEs) reported from the time of NJ tube placement until 30 days following discontinuation of study drug administration had elapsed were collected. In addition, serious adverse events (SAEs) and protocol‐related non‐serious adverse events were collected from the time the subject signed the study‐specific informed consent. In addition to spontaneous reports by the subjects and observations by the investigator, adverse events were monitored by measurements on vital signs, physical examination, ECG, clinical laboratory test assessments, and Columbia‐Suicide Severity Rating Scale. Adverse events were coded using Medical Dictionary for Regulatory Activities (MedDRA). The number and percentage of subjects having treatment‐emergent adverse events were tabulated by primary System Organ Class (SOC) and preferred term and with a breakdown by formulation.
3. RESULTS
3.1. Dissolution
Results for the dissolution profiles for LCIG‐HV and LCIG‐LV commercial formulations for LD and CD are shown in Figures 1 and 2. The f 2 values for levodopa and carbidopa were 23.9 and 25.0, respectively, comparing the LCIG‐HV (test) formulation to the LCIG‐LV commercial (reference) formulation.
Figure 1.
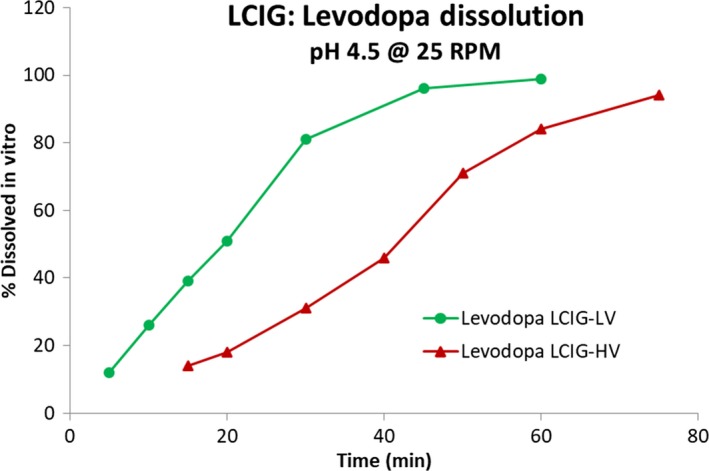
Levodopa dissolution profiles for LCIG‐LV commercial and LCIG‐HV formulations
Figure 2.
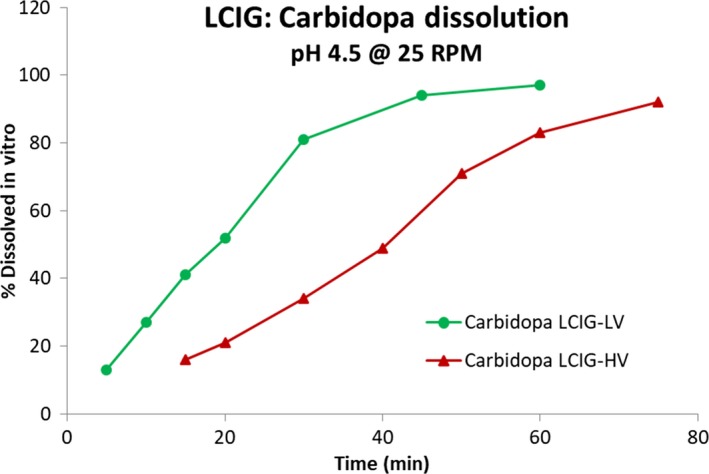
Carbidopa dissolution profiles for LCIG‐LV commercial and LCIG‐HV formulations
3.2. Participants in bioequivalence study
Twenty‐eight subjects were enrolled in this study. Demographic information for all subjects is presented in Table 2. Two subjects completed the first period of the study but not the second period. There was difficulty placing the NJ tube for one subject in Cohort 1 during Period 1 (LCIG‐LV commercial) and a decision was made by the primary investigator not to attempt placing the NJ tube for this subject during Period 2 (LCIG‐HV); therefore, the subject was prematurely discontinued. For an additional subject in Cohort 1 the NJ tube could not be successfully placed for Period 2 (LCIG‐HV) and the subject was prematurely discontinued. All available data were used in the PK analysis and BE assessment.
Table 2.
Demographic summary for all subjects
| Characteristic | Mean ± SD (N = 28) | Min‐Max |
|---|---|---|
| Age (years) | 35.6 ± 11.2 | 19‐55 |
| Weight (kg) | 79.8 ± 8.04 | 63‐100 |
| Height (cm) | 177 ± 5.89 | 162‐187 |
| Sex | 28 Males (100%) | |
| Race | 16 White (57%), 9 Black (32%), 1 Asian (4%), 2 Multi‐race (7%) | |
Overall, the NJ tube placement and LCIG administration were well tolerated in 28 healthy volunteers. The most common AEs reported with reasonable possibility of being related to either LCIG formulation were nausea and vomiting. The nausea and vomiting AEs typically resolved within an hour of onset. The occurrence of AEs did not appear to be different between the 2 LCIG formulations. There were no SAEs in the study and no premature discontinuations due to AEs. A summary of all AEs from the current study can be found in Table 3.
Table 3.
Summary of all adverse events with reasonable possibility of being related to study drug
| System organ class | Dosing formulation | |
|---|---|---|
| LCIG‐HV | LCIG‐LV Commercial | |
| (N = 26) | (N = 28) | |
| n (%) | n (%) | |
| Any adverse event | 9 (34.6) | 7 (25.0) |
| Gastrointestinal disorders | ||
| Abdominal pain | 0 (0.0) | 1 (3.6) |
| Nausea | 5 (19.2) | 6 (21.4) |
| Vomiting | 5 (19.2) | 3 (10.7) |
| Nervous system disorders | ||
| Dizziness | 3 (11.5) | 0 (0.0) |
| General disorders | ||
| Fatigue | 0 (0.0) | 1 (3.6) |
3.3. Bioequivalence study
PK results are available for the 28 subjects who received the LCIG‐LV commercial formulation and 26 subjects who received the LCIG‐HV formulation. PK profiles confirm that both LCIG formulations were successfully delivered to all subjects through the NJ tube. Pharmacokinetic results from the study show that the levodopa and carbidopa PK profiles appear to be comparable for the LCIG‐LV commercial and LCIG‐HV formulations (Figure 3). Summary statistics for all of the PK parameters, normalized by dose delivered, are presented for both formulations in Table 4. The median T max was 1.0 hour for levodopa and 3.0 hours for carbidopa and showed no apparent difference between formulations. The harmonic mean t 1/2 was 1.6 and 2.0 hours for levodopa and carbidopa, respectively, with no apparent differences between formulations. The geometric means of C max, AUCt and AUCinf were very similar between the 2 formulations with the coefficient of variation percent ranging from 21 to 55%.
Figure 3.
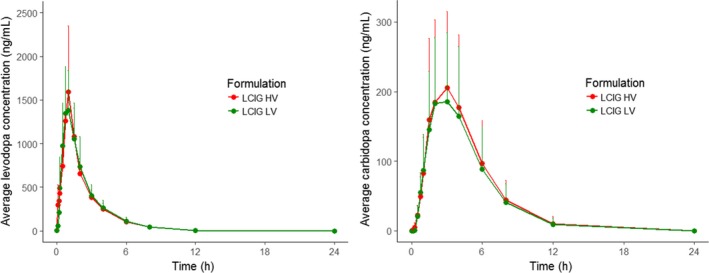
Comparison of levodopa and carbidopa pharmacokinetics between the LCIG‐HV formulation and the LCIG‐LV commercial formulation. PK profiles were normalized by dose delivered
Table 4.
Summary of PK parameters for LCIG‐HV and LCIG‐LV commercial formulations for levodopa and carbidopa
| Pharmacokinetic parameters (units) | Levodopa | Carbidopa | |||
|---|---|---|---|---|---|
| LCIG‐HV (N = 26) | LCIG‐LV commercial (N = 28) | LCIG‐HV (N = 26) | LCIG‐LV commercial (N = 28) | ||
| C max b | ng/mL | 1830 (1910, 32) | 1550 (1610, 25) | 225 (254, 49) | 187 (212, 51) |
| T max a | h | 1.0 (0.5‐1.5) | 1.0 (0.5‐2.0) | 3.0 (1.5‐4.0) | 3.0 (1.5‐4.0) |
| AUCt b | ng·h/mL | 3610 (3660, 16) | 3190 (3320, 25) | 1020 (1160, 50) | 837 (961, 53) |
| AUCinf b | ng·h/mL | 3680 (3720, 15) | 3260 (3380, 24) | 1090 (1220, 47) | 904 (1020, 50) |
| t 1/2 b | h | 1.60 (0.191) | 1.58 (0.155) | 1.94 (0.293) | 1.96 (0.408) |
| C max/Doseb | (ng/mL)/mg | 8.25 (8.81, 40) | 7.98 (8.25, 26) | 4.06 (4.66, 53) | 3.83 (4.35, 51) |
| AUCt/Doseb | (ng·h/mL)/mg | 16.3 (16.7, 21) | 16.4 (17.0, 26) | 18.4 (21.3, 55) | 17.2 (19.8, 55) |
| AUCinf/Doseb | (ng·h/mL)/mg | 16.6 (16.9, 21) | 16.7 (17.4, 25) | 19.6 (22.5, 52) | 18.6 (21.0, 51) |
Geometric Mean (Arithmetic Mean, %CV).
Median (minimum‐maximum).
Harmonic mean (pseudo‐standard deviation).
In the results of the statistical analysis for C max and AUC, the point estimate for the levodopa C max geometric mean of the LCIG‐HV formulation was 4% higher than the point estimate for the LCIG‐LV commercial formulation. The levodopa point estimates for AUCt and AUCinf parameters were nearly identical for the two formulations (Table 5). For carbidopa, the point estimates of the C max, AUCt and AUCinf geometric means for the LCIG‐HV formulation were 3%‐4% higher than the point estimates for the LCIG‐LV commercial formulation (Table 6). For all PK parameters for levodopa and carbidopa the 90% confidence interval for the ratio of the geometric mean of the LCIG‐HV formulation to geometric mean of the LCIG‐LV commercial formulation falls within the 0.8‐1.25 range.
Table 5.
Levodopa bioequivalence results LCIG‐HV vs LCIG‐LV Commercial (Test vs Reference)
| PK parameter | Geometric mean ratio | |
|---|---|---|
| Point estimate | 90% CI | |
| C max | 1.036 | 0.894‐1.201 |
| AUCt | 0.999 | 0.892‐1.117 |
| AUCinf | 0.995 | 0.891‐1.111 |
Table 6.
Carbidopa bioequivalence results LCIG‐HV vs LCIG‐LV commercial (Test vs Reference)
| PK parameter | Geometric mean ratio | 90% CI |
|---|---|---|
| C max | 1.031 | 0.906‐1.173 |
| AUCt | 1.041 | 0.917‐1.181 |
| AUCinf | 1.029 | 0.918‐1.153 |
4. DISCUSSION
The Levodopa‐Carbidopa Intestinal Gel system is intended for the long‐term treatment of motor fluctuations in patients with advanced idiopathic PD who are no longer adequately controlled by oral PD medications. The commercially available and next generation LCIG formulations are enteral suspensions in an aqueous gel that contains carmellose sodium (carboxymethylcellulose sodium) as the gelling agent. Levodopa is absorbed quickly and effectively from the small intestine through a high capacity transport system for amino acids.15, 16 Carbidopa, a decarboxylase inhibitor that is administered with levodopa to increase the bioavailability and decrease the clearance of levodopa, has slower and more variable absorption than levodopa and is believed to have a transport mechanism different from that of levodopa.17
The current study demonstrates that although the LCIG‐LV commercial and LCIG‐HV formulations are clinically bioequivalent for levodopa and carbidopa according to FDA guidance, these formulations have different dissolution profiles in vitro. The f 2 values for levodopa and carbidopa were 23.9 and 25.0, respectively, comparing the LCIG‐HV and LCIG‐LV formulations. This suggests that the dissolution profiles are not similar according to the FDA guidance, which specifies an f 2 value ≥50 is required.18, 19 The dissolution method used in this study was designed to provide sufficient discriminating capability against formulation and process attributes. A paddle speed of 25 RPM provided sufficient capability to discriminate the minor differences of the samples and dissolution at pH 4.5 provided the most relevant profiles since this pH is similar to the duodenum‐jejunal environment. In addition, CD is not stable at pH 6.8 and significant degradation was expected during testing at this pH. Since this would result in an inaccurate assessment of the dissolution rate, pH 4.5 is the most appropriate condition for dissolution assessment. However, this method was overly sensitive to the sample viscosity, a non‐critical characteristic to in vivo performance. Therefore, the in vitro dissolution results that were observed are not clinically relevant to the in vivo drug absorption. These in vitro and in vivo observations also suggest that USP apparatus 2‐based dissolution testing may not be appropriate for carmellose‐based aqueous gels used as enteral suspensions as it does not simulate a biorelevant condition. Additional dissolution methods are being considered which better represent the in‐vivo absorption of LCIG.
The half‐life of levodopa and carbidopa in this study is similar to what has previously been reported in healthy volunteers following oral dosing11 although short infusions (30 minutes) of LCIG have not previously been administered to healthy volunteers. Both pharmacokinetic parameters and bioequivalence assessment in this study were completed based on the use of concentration time points normalized based on the actual dose delivered. This dose normalization was pre‐specified for the study with the objective being to compare the LCIG‐LV commercial and LCIG‐HV formulations without concern in regard to inconsistencies in delivery of the dose that may arise due to the NJ tube priming step and/or pump performance.
Overall, administration of the study drug was well tolerated in the healthy subjects that participated in this study. The most common safety findings were nausea and vomiting all of which have been previously observed with LCIG administration.10, 12 For the current study an NJ tube was used to deliver LCIG rather than a PEG‐J tube which is used by Parkinson's patients who receive LCIG. NJ tube placement is known to be associated with nausea and emesis so it is unclear if the nausea and emesis findings from this study were due to the NJ tube or LCIG administration although many of these AEs were reported near the LD T max suggesting the AEs may be related to study drug administration. Healthy volunteers are not dopamine deficient like Parkinson's disease patients and it is thought that levodopa is not as well tolerated in healthy volunteers compared to Parkinson's disease patients. The nausea and vomiting AEs in this study occurred in approximately 11%‐20% of the healthy volunteers. A previous study of Parkinson's disease patients treated with oral immediate release levodopa and carbidopa reported that nausea and vomiting occurred in only 2% of patients.20 Furthermore, a separate study in which oral immediate release levodopa and carbidopa was administered to PD patients reported levodopa values well above the 2000 ng/mL level suggesting that Parkinson's disease patients can tolerate higher levodopa exposure than healthy volunteers.9
5. CONCLUSIONS
The results from this study demonstrate that the LCIG‐LV and LCIG‐HV formulations are clinically bioequivalent for levodopa and carbidopa. However, the dissolution method was overly discriminating on the formulation differences.
DISCLOSURE
All authors are employees of AbbVie and may hold AbbVie stock or stock options.
DATA SHARING STATEMENT
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual and trial‐level data (analysis data sets), as well as other information (eg, protocols and Clinical Study Reports), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications.
This clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research, and will be provided following review and approval of a research proposal and Statistical Analysis Plan (SAP) and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time and the data will be accessible for 12 months, with possible extensions considered. For more information on the process, or to submit a request, visit the following link: https://www.abbvie.com/our-science/clinical-trials/clinical-trials-data-and-information-sharing/data-and-information-sharing-with-qualified-researchers.html.
ACKNOWLEDGEMENTS
The study was funded by AbbVie. AbbVie contributed to the study design, research, and interpretation of data, and the writing, review, and approval of the publication. Medical writing support was provided by Wesley Wayman, an employee of AbbVie. The authors thank the AbbVie Clinical Pharmacology Research Unit staff for conducting this study.
Rosebraugh M, Kalluri HV, Liu W, et al. Levodopa‐carbidopa intestinal gel high concentration formulation is clinically bioequivalent to commercial formulation. Pharmacol Res Perspect. 2019;e00473 10.1002/prp2.473
The authors confirm that the principal investigator for this manuscript is Dilraj Sidhu and that he had direct clinical responsibility for subjects.
REFERENCES
- 1. Kempster PA, O'Sullivan SS, Holton JL, Revesz T, Lees AJ. Relationships between age and late progression of Parkinson's disease: a clinico‐pathological study. Brain. 2010;133(Pt 6):1755‐1762. [DOI] [PubMed] [Google Scholar]
- 2. Kulisevsky J, Luquin MR, Arbelo JM, et al. Advanced Parkinson's disease: clinical characteristics and treatment. Part II.. Neurologia. 2013;28:558‐583. [DOI] [PubMed] [Google Scholar]
- 3. Poewe W, Mahlknecht P. The clinical progression of Parkinson's disease. Parkinsonism Relat Disord. 2009;15(Suppl 4):S28‐S32. [DOI] [PubMed] [Google Scholar]
- 4. Nutt JG. Pharmacokinetics and pharmacodynamics of levodopa. Mov Disord. 2008;23(Suppl 3):S580‐S584. [DOI] [PubMed] [Google Scholar]
- 5. DiPiro JT. Pharmacotherapy– a pathophysiologic approach. 2017.
- 6. Othman AA, Dutta S. Population pharmacokinetics of levodopa in subjects with advanced Parkinson's disease: levodopa‐carbidopa intestinal gel infusion vs. oral tablets. Br J Clin Pharmacol. 2014;78:94‐105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shoulson I, Glaubiger GA, Chase TN. On‐off response. Clinical and biochemical correlations during oral and intravenous levodopa administration in parkinsonian patients. Neurology. 1975;25:1144‐1148. [DOI] [PubMed] [Google Scholar]
- 8. Fahn S. How do you treat motor complications in Parkinson's disease: medicine, surgery, or both? Ann Neurol. 2008;64(Suppl 2):S56‐S64. [DOI] [PubMed] [Google Scholar]
- 9. Othman AA, Rosebraugh M, Chatamra K, Locke C, Dutta S. Levodopa‐carbidopa intestinal gel pharmacokinetics: lower variability than oral levodopa‐carbidopa. J Parkinsons Dis. 2017;7:275‐278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Olanow CW, Kieburtz K, Odin P, et al. Continuous intrajejunal infusion of levodopa‐carbidopa intestinal gel for patients with advanced Parkinson's disease: a randomised, controlled, double‐blind, double‐dummy study. Lancet Neurol. 2014;13:141‐149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yeh KC, August TF, Bush DF, et al. Pharmacokinetics and bioavailability of Sinemet CR: a summary of human studies. Neurology. 1989;39(11 Suppl 2):25‐38. [PubMed] [Google Scholar]
- 12. Nyholm D, Odin P, Johansson A, et al. Pharmacokinetics of levodopa, carbidopa, and 3‐O‐methyldopa following 16‐hour jejunal infusion of levodopa‐carbidopa intestinal gel in advanced Parkinson's disease patients. AAPS J. 2013;15:316‐323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Diaz DA, Colgan ST, Langer CS, Bandi NT, Likar MD, Van Alstine L. Dissolution similarity requirements: how similar or dissimilar are the global regulatory expectations? AAPS J. 2016;18:15‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. O'Hara T, Dunne A, Kinahan A, Cunningham S, Stark P, Devane J. Review of methodologies for the comparison of dissolution profile data. Adv Exp Med Biol. 1997;423:167‐171. [DOI] [PubMed] [Google Scholar]
- 15. Lennernas H, Nilsson D, Aquilonius SM, Ahrenstedt O, Knutson L, Paalzow LK. The effect of L‐leucine on the absorption of levodopa, studied by regional jejunal perfusion in man. Br J Clin Pharmacol. 1993;35:243‐250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wade DN, Mearrick PT, Morris JL. Active transport of L‐dopa in the intestine. Nature. 1973;242:463‐465. [DOI] [PubMed] [Google Scholar]
- 17. Durso R, Evans JE, Josephs E, et al. Variable absorption of carbidopa affects both peripheral and central levodopa metabolism. J Clin Pharmacol. 2000;40:854‐860. [DOI] [PubMed] [Google Scholar]
- 18. Freitag G. Guidelines on dissolution profile comparison. Drug Inform J. 2001;35:865‐874. [Google Scholar]
- 19. FDA . Guidance for Industry. SUPAC‐IR. Immediate release solid oral dosage forms. Scale‐up and post approval changes. Chemistry, manufacturing and controls. In vitro dissolution testing and in vivo bioequivalence documentation. Available from: https://www.fda.gov/downloads/drugs/guidances/ucm070636.pdf. Accessed 18 September 2018. 1995.
- 20. Hauser RA, Hsu A, Kell S, et al. Extended‐release carbidopa‐levodopa (IPX066) compared with immediate‐release carbidopa‐levodopa in patients with Parkinson's disease and motor fluctuations: a phase 3 randomised, double‐blind trial. Lancet Neurol. 2013;12:346‐356. [DOI] [PubMed] [Google Scholar]