

Abstract
SIRT2 a member of the sirtuin family of protein lysine deacylases has been identified as a promising therapeutic target. In addition to catalyzing deacetylation, SIRT2 has recently been shown to remove fatty acyl groups from K-Ras4a and promote its transforming activity. Among the SIRT2 specific inhibitors, only the thiomyristoyl lysine compound, TM, can weakly inhibit the demyristoylation activity of SIRT2. Thus, more potent small molecule SIRT2 inhibitors are needed to further evaluate the therapeutic potential of SIRT2 inhibition, and to understand the function of protein lysine defatty-acylation. Here we report a SIRT2 inhibitor, JH-T4, which can increase K-Ras4a lysine fatty acylation. This is the first small molecule inhibitor that can modulate the lysine fatty acylation levels of K-Ras4a. JH-T4 also inhibits SIRT1 and SIRT3 in vitro. The increased potency of JH-T4 is likely due to the formation of hydrogen bonding between the hydroxyl group and SIRT1, SIRT2, and SIRT3. This is further supported by in vitro studies with another small molecule inhibitor, NH-TM. These studies provide useful insights for future SIRT2 inhibitor development.
Keywords: Sirtuins, SIRT2, Inhibitors, Cancer, Lysine Fatty Acylation
Graphical Abstract
A new SIRT2 inhibitor, JH-T4, can modulate both the deacetylation and defatty-acylation activities of SIRT2. JH-T4 is the first SIRT2 inhibitor that can control the lysine fatty acylation levels of the oncoprotein K-Ras4a in cells.
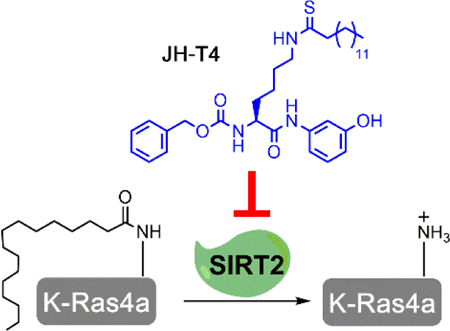
Sirtuins, the NAD-dependent class of the histone deacetylase family, have attracted a lot of research interest due to their connection to many biological processes such as transcription, metabolism, cancer, and aging.[1] Among the seven mammalian sirtuins, SIRT2 is the only sirtuin predominately localized in the cytosol.[1] Despite early studies suggesting a weak tumor suppressor role for SIRT2, many reports show that inhibiting SIRT2 has a broad anti-cancer effect.[2] Thus, SIRT2 is a promising anticancer target. SIRT2 was initially identified as a protein lysine deacetylase. However, it can also efficiently hydrolyze long chain fatty acyl groups from lysine residues.[3] Most of the current understanding of SIRT2 comes from its deacetylation activity. SIRT2 deacetylates various substrates ranging from metabolic enzymes, such as ENO1, to transcription factors such as HIF1-α.[4] Meanwhile, not much is known about the defatty-acylation activity of SIRT2. Recently, the first defatty-acylation substrate of SIRT2, K-Ras4a, was identified.[5] SIRT2-mediated lysine defatty-acylation promotes the transforming activity of K-Ras4a.[5] Since K-Ras4a is an important oncoprotein, the defatty-acylation activity of SIRT2 could be significant for the role of SIRT2 in cancer.
Several SIRT2 inhibitors have been developed. [2a, 6] Recent studies suggest that among these compounds, only TM can weakly inhibit the demyristoylation activity of SIRT2.[7] TM is a thiomyristoyl lysine compound, and is a mechanism-based SIRT2 inhibitor.[2a] TM has been shown to possess a broad anticancer effect.[2a, 6k, 8] In many cancer cell lines, TM inhibits SIRT2 and promotes c-Myc degradation.[2a] TM efficiently inhibits the deacetylation activity of SIRT2 in cells, while only weakly inhibits the defatty-acylation activity of SIRT2.[2a, 8] Due to the promising potential of SIRT2 as a therapeutic target, we are interested in developing more potent SIRT2 inhibitors that can inhibit both the deacetylation and the defatty-acylation activity of SIRT2. Here we report JH-T4, a TM analogue, which is also a potent SIRT2 inhibitor, but unlike TM has the ability to inhibit the defatty-acylation of K-Ras4a by SIRT2 both in vitro and in cells.
In our efforts to develop a more potent SIRT2 inhibitor, we decided to carry out structure activity relationship studies based on the structure of TM. We specifically were interested in modifying the aniline portion of the small molecule, to study what modifications would increase or decrease SIRT2 potency and selectivity. To this end, we found that adding a single hydroxyl group on the aniline moiety, leading to the compound JH-T4 (Figure 1), produces a sirtuin inhibitor with a very different inhibition profile. We measured the in vitro IC50 values (Table 1) of JH-T4 toward SIRT1, SIRT2, SIRT3, and SIRT6 under pre-incubation conditions (enzymes, NAD, and inhibitors were first incubated for 15 min before substrates were added to start the enzymatic reaction) and compared them to the IC50 values of TM. For SIRT2, we determined the IC50 values for both deacetylation and demyritoylation activities. For these assays the H3K9-Ac and H3K9-Myr peptides were used as substrates, as SIRT1,2,3 and 6 have efficient activity on these peptides, which are commonly used for Sirtuin studies.[2a, 3b, 7a, 9]
Figure 1.
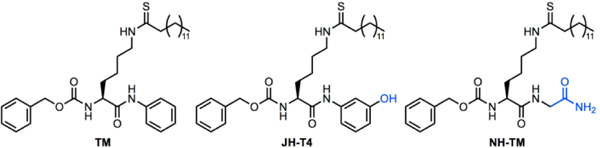
Chemical Structures of different Sirtuin Inhibitors
Table 1.
In vitro IC50 values (μM) of TM, JH-T4 and NH-TM for inhibiting sirtuin deacylation activity (ND = not determined). Values shown in brackets are from assays without pre-incubation.
| TM | JH-T4 | NH-TM | |
|---|---|---|---|
| SIRT1 Deacetylation | 25 ± 15 (59 ± 10) | 0.3 ± 0.2 (43 ± 3) | 0.4±0.1 |
| SIRT2 Deacetylation | 0.04 ± 0.02 (0.04 ± 0.01) | 0.03 ± 0.01 (0.03 ± 0.02 ) | 0.088±0.003 |
| SIRT2 Defatty-Acylation | 0.05±0.03 (>200) | 0.04 ± 0.03 (111 ± 10) | ND |
| SIRT3 Deacetylation | >50 (77 ± 6) | 15±5 (50 ± 8) | 2.42±0.01 |
| SIRT6 Defatty-Acylation | >200 | >100 | ND |
| Selectivity[a] | 650 | 10 | 4.8 |
(SIRT1 deacetylation IC50)/ (SIRT2 deacetylation IC50)
Despite its similarity in structure to TM, JH-T4 showed a different sirtuin inhibition profile. TM is a SIRT2 selective inhibitor, exhibiting a 650-fold selectivity for inhibition of SIRT2 over SIRT1 deacetylation activity.[8] In contrast, JH-T4 exhibited only a 10-fold selectivity for SIRT2 as it inhibits SIRT1, SIRT2, and SIRT3 in vitro with IC50 values of 15 μM or lower. Interestingly, under the pre-incubation assay condition, TM and JH-T4 inhibited both the deacetylation and defatty-acylation activity of SIRT2 in vitro comparably (IC50 values in the 30–50 nM range) (Table 1).
To further compare the defatty-acylation inhibition by TM and JH-T4 we determined the IC50 value for inhibition of SIRT2 demyristoylation activity without pre-incubating the enzyme with NAD and inhibitor. Without preincubation, the in vitro IC50 value of TM was > 200 μM (42% inhibition at 200 μM), but the IC50 of JH-T4 was approximately 110 μM. This suggests that JH-T4 is more efficient at inhibiting the defatty-acylation activity of SIRT2 than TM is.
We also measured the IC50 values of TM and JH-T4 on the deacetylation activity of SIRT1, SIRT2, and SIRT3 without preincubation. Most IC50 value for inhibiting the deacetylation activity of SIRT1-3 without pre-incubation did not drastically change for TM and JH-T4 compared to that with pre-incubation (Table 1).[7a] However the IC50 value of JH-T4 on SIRT1 without pre-incubation increased dramatically JH-T4 (40 μM without pre-incubation, compared to 0.3 μM with pre-incubation).
We next wanted to compare the potency and selectivity of these compounds in cells. To evaluate the inhibition of SIRT1 deacetylation activity, we examined p53 acetylation levels, as Lys382 of p53 is a well-established SIRT1 substrate.[10] As expected, JH-T4, but not TM, increased Ac-p53 level on Lys382 in MCF-7 cells (Figure 2A). We further tested if these compounds could inhibit the deacetylation activity of SIRT2 in cells based on acetyl α-tubulin immunofluorescence, as acetyl α-tubulin is a widely used cellular readout of SIRT2 activity.[3a] Both the TM and JH-T4 treated samples showed a dramatic increase in acetyl α-tubulin levels compared to the sample treated with the vehicle control, ethanol. Thus, both compounds efficiently inhibit SIRT2 deacetylation activity in MCF-7 cells (Figure 2B).
Figure 2. In-Cell Sirtuin Inhibition by JH-T4.
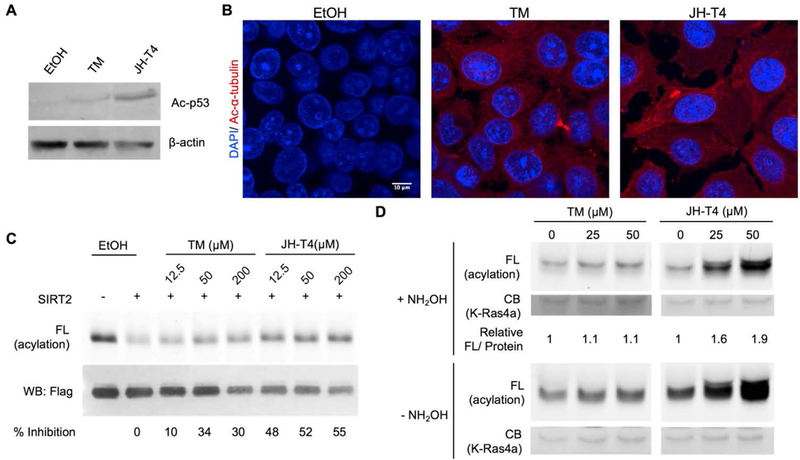
(A) Ac-p53 levels to evaluate the inhibition of SIRT1 in cells after 6 hr 25μM inhibitor and 200 nM trichostatin A (TSA) treatment in MCF-7 cells. (B) Ac-α-tubulin levels to detect inhibition of SIRT2 after 6 hr 25 μM inhibitor treatment in MCF-7 cells. (C) Inhibition of SIRT2 in vitro by TM and JH-T4 treatment by evaluating K-Ras4a lysine fatty acylation levels. (D) Detection of K-Ras4a lysine fatty acylation levels to evaluate in-cell inhibition of SIRT2 defatty-acylation activity. FL, fluorescence; CB, Coomassie blue staining.
Next, we investigated whether either of the two compounds could inhibit the defatty-acylation activity of SIRT2 by evaluating the lysine fatty acylation level of K-Ras4a, the only reported SIRT2 defatty-acylation target. We made use of the biorthogonal palmitic acid analogue Alk14 following the same methods previously described.[5] First, we looked at the ability of the compounds to inhibit SIRT2 defatty-acylation on K-Ras4a in vitro. Alk14 labeled K-Ras4a was purified from HEK-293T cells, and subsequently treated with SIRT2, NAD, and either TM, JH-T4, or the vehicle control ethanol. Click chemistry was used to attach a fluorophore to quantify the fatty acylation level of K-Ras4a based on the in-gel fluorescence intensity. K-Ras4a samples treated with JH-T4 had higher lysine fatty-acylation levels (hydroxylamine resistant fatty acylation) than those treated with either the control, ethanol, or TM (Figure 2C, note that JH-T4 treated cells have lower protein loading but stronger fluorescence signal). This is supported by the quantification of the percent inhibition of the defatty-acylation activity (Figure 2C) and demonstrate that JH-T4 is more potent than TM at inhibiting the activity of SIRT2 on K-Ras4a lysine fatty acylation. We note that higher concentrations of TM or JH-T4 did not further increase K-Ras4a acylation levels, which is likely due to their limited solubility. At concentrations above 50 uM, the inhibitors may precipitate out, decreasing the actual concentrations.
Next, we evaluated whether JH-T4 was able to inhibit SIRT2-catalyzed K-Ras4a defatty-acylation in cells. Consistent with the in vitro inhibition of SIRT2 defatty-acylation of K-Ras4a, JH-T4 could also increase the fatty acylation level on K-Ras4a efficiently in cells (Figure 2D). In contrast, TM had very little effect on the lysine fatty acylation of K-Ras4a. We further quantified the increase in fatty acylation, by comparing the fluorescence signal to the protein output as determined by a Flag western blot, to confirm that JH-T4 had a more profound effect on the lysine fatty acylation level of K-Ras4a (Figure 2D). JH-T4 is therefore the first inhibitor to efficiently inhibit the defatty-acylation activity on K-Ras4a in cells.
The cellular effect of JH-T4 on K-Ras4a lysine fatty acylation was much stronger than TM, this is consistent with the fact that JH-T4 is better at inhibiting SIRT2 defatty-acylase activity in vitro. However, it is also possible that the inhibition of SIRT1 could contribute to this much stronger effect. We previously showed that SIRT1 can catalyze defatty-acylation of K-Ras4a in vitro, but knockdown of SIRT1 in cells did not affect K-Ras4a lysine fatty acylation, while knockdown of SIRT2 did.[5] It is possible that SIRT1 may partially rescue the loss of SIRT2 activity and thus inhibiting both SIRT1 and SIRT2 could be more effective at increasing K-Ras4a lysine fatty acylation.
Given that JH-T4 is a potent SIRT2 inhibitor that inhibits the defatty-acylation of K-Ras4a, we next evaluated the anti-cancer effect of these compounds in a wide range of cancer cells. We looked at their GI50 values in pancreatic cancer (ASPC1), colon cancer (HCT116), lung cancer (NCI-H23), and breast cancer (MDA-MB-231) cells, as well as in two normal mammary epithelial cells (HME1 and MCF10A). JH-T4 was more potent than TM in all the cell lines evaluated. However, unlike TM that exhibited cancer cell selectivity over normal cells, JH-T4 was also toxic to the normal cell lines HME1 and MCF10A.
The cytotoxicity of JH-T4 to normal cell lines may come from the fact that JH-T4 can inhibit several Sirtuins (Table 1). To evaluate if the anticancer effect of JH-T4 is through SIRT2 inhibition we evaluated the inhibition of anchorage independent growth in HCT116 cells overexpressing pCDH and Flag-SIRT2. While cells overexpressing SIRT2 were less sensitive to TM treatment, there was no difference in JH-T4 potency between the two cell lines (Table 3).[8] This suggests that the potency of JH-T4 is not primarily through SIRT2 inhibition. Thus, future SIRT2 inhibitor development should focus on regaining SIRT2 selectivity, while maintaining the potency of JH-T4.
Table 3.
GI50 values (μM) of TM[7a] and JH-T4 for inhibiting anchorage independent growth in pCDH and SIRT2 overexpressing HCT116 cells. Average values from three independent experiments are presented.
| Inhibitor | pCDH Expression |
SIRT2 Expression |
Fold Change[a] |
|---|---|---|---|
| TM | 15 ± 5 | 27 ± 8 | 1.8 |
| JH-T4 | 12.1 ± 0.5 | 12.6 ± 0.3 | 1 |
GI50 for SIRT2 Expression / GI50 for pCDH Expression
To facilitate future inhibitor development efforts, we became interested in understanding how the introduction of the hydroxyl group on JH-T4 leads to decreased SIRT2 selectivity and increased inhibition of the defatty-acylation activity of SIRT2. Modeling of SIRT2 with TM and JH-T4 based on the previously reported crystal structure of Sirt2 in complex with a myristoyl peptide (4×3o), suggested that the introduction of a hydroxyl group on TM allowed the compound to form hydrogen bonds with the amino acid backbone of SIRT2, specifically with Gln265, Val266, and Asn267 (Figure 3).[11] Similar hydrogen bonds were also seen when a model was generated with JH-T4 and the established SIRT3 and H3K9 myristoyl peptide crystal structure, 5BWN (Figure S1).[9a] The ability to form hydrogen bonds makes JH-T4 a more efficient suicide substrate, which leads to the increased inhibition of SIRT1 and SIRT3, as well as better inhibition of the defatty-acylation activity of SIRT2.
Figure 3.
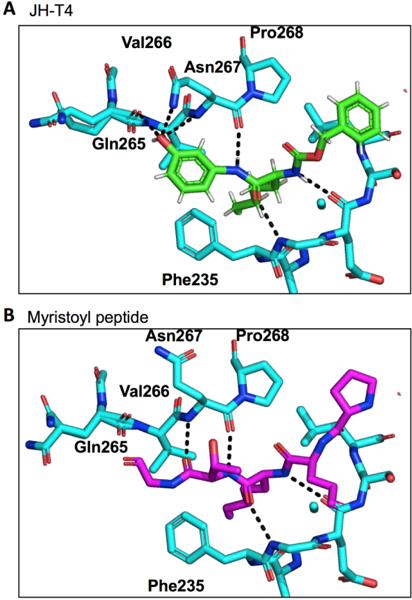
Modeling of (A) JH-T4 and (B) H3K9-Myr with SIRT2 using the 4×3o crystal structure to show the potential hydrogen bonding between the small molecule JH-T4 and the amino acid backbone of SIRT2.
To further test this hypothesis, we synthesized another TM analogue, NH-TM (Figure 1), where the aniline moiety was replaced with glycine. NH-TM, like JH-T4, should be able to form more hydrogen bonds than TM with SIRT1, SIRT2 and SIRT3, which was supported by modeling with the established SIRT2 and SIRT3 X-ray crystal structures (Figure S2).[9a, 11] If our hypothesis was correct, we expected NH-TM to behave similarly to JH-T4 at inhibiting SIRT1, SIRT2, and SIRT3 deacetylation activities. Indeed, NH-TM was potent against SIRT2, exhibiting an IC50 value of 0.088 μM (Table 1). NH-TM, like JH-T4, inhibits both SIRT1 and SIRT3 with IC50 values of 0.43 μM and 2.4 μM, respectively (Table 1).
Our study here not only provides the first SIRT2 inhibitor (JH-T4) that can inhibit the defatty-acylation of K-Ras4a in cells, but also highlights that a small change in structure can lead to a significant change in the potency and selectivity of a small molecule inhibitor. The introduction of a hydroxyl group on the selective SIRT2 inhibitor TM (JH-T4) decreased the selectivity, but increased the ability to inhibit the defatty-acylation activity of SIRT2. Our data suggest that this is due to the ability of the compound to form additional hydrogen bonds with SIRT1, SIRT2 and SIRT3.
Protein lysine fatty acylation is emerging as an important protein post-translational modification. Recently, several Ras family of small GTPases have been reported to be regulated by lysine fatty acylation and sirtuin-catalyzed defatty-acylation.[5, 12] Small molecules that can inhibit the defatty-acylation activity of sirtuins would help to better understand the functions of protein lysine fatty acylation. Here, we identify JH-T4 as the first small molecule that can modulate the level of lysine fatty acylation on K-Ras4a. JH-T4 can be used as a tool to further study protein lysine fatty acylation, an understudied but important protein post-translational modification.
Interestingly, JH-T4 is more cytotoxic than TM in several human cell lines, likely due to inhibition of other sirtuins as SIRT2 overexpression did not protect the cells from the effect of JH-T4. However, compared to TM, JH-T4 loses the selective toxicity to cancer cells. At this point, it is not clear whether the lack of cancer cell selectivity is due to ability of JH-T4 to inhibit other sirtuins or due to inhibition of the defatty-acylation activity of SIRT2. To address this question, future SIRT2 inhibitor development should focus on obtaining compounds that can inhibit the defatty-acylation activity and are SIRT2-selective.
Experimental Section
All experimental details can be found in the supplemental information document.
Supplementary Material
Table 2.
GI50 values (μM) of TM and JH-T4 in several cancer and normal cells. The values are an average of three independent experiments, each done in duplicate. WT = wild type
| Cell Line | Cell Type | K-Ras Mutation |
TM | JH-T4 |
|---|---|---|---|---|
| ASPC1 | Pancreatic Cancer |
G12D | 24 ± 5 | 8.6 ± 0.9 |
| HCT116 | Colon Cancer |
G13D | 19 ± 1 | 6.2 ± 0.2 |
| NCI-H23 | Lung Cancer |
G12C | 29 ± 6 | 6.6 ± 0.3 |
| MDA-MB-231 | Breast Cancer |
G13D | 13 ± 4 | 6 ± 2 |
| HME1 | Normal Mammary Epithelial Cells |
WT | >50 | 11 ± 4 |
| MCF10A | Normal Mammary Epithelial Cells |
WT | >50 | 3 ± 1.2 |
Acknowledgements
We thank Dr. Pornpun Aramsangtienchai and Dr. Sushabhan Sadhukhan for providing the substrate peptides. This work is supported in part by a grant from NIH/NIDDK, DK107868. The work made use of the Cornell University NMR Facility, which is supported, in part, by the NSF through MRI award CHE-1531632. Imaging data was acquired through the Cornell University Biotechnology Resource Center, with NYSTEM (CO29155) and NIH (S10OD018516) funding for the shared Zeiss LSM880 confocal/multiphoton microscope. H.J. is a Howard Hughes Medical Institute International Student Research Fellow.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References:
- [1].Haigis MC, Sinclair DA, Annu Rev Pathol 2010, 5, 253–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a Jing H, Hu J, He B, Negron Abril YL Stupinski J, Weiser K, Carbonaro M, Chiang YL, Southard T, Giannakakou P, Weiss RS, Lin H, Cancer Cell 2016, 29, 767–768; [DOI] [PubMed] [Google Scholar]; b Zhao D, Zou SW, Liu Y, Zhou X, Mo Y, Wang P, Xu YH, Dong B, Xiong Y, Lei QY, Guan KL, Cancer Cell 2013, 23, 464–476; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Serrano L, Martinez-Redondo P, Marazuela-Duque A, Vazquez BN, Dooley SJ, Voigt P, Beck DB, Kane-Goldsmith N, Tong Q, Rabanal RM, Fondevila D, Munoz P, Kruger M, Tischfield JA, Vaquero A, Genes Dev 2013, 27, 639–653; [DOI] [PMC free article] [PubMed] [Google Scholar]; d Liu PY, Xu N, Malyukova A, Scarlett CJ, Sun YT, Zhang XD, Ling D, Su SP, Nelson C, Chang DK, Koach J, Tee AE, Haber M, Norris MD, Toon C, Rooman I, Xue C, Cheung BB, Kumar S, Marshall GM, Biankin AV, Liu T, Cell Death Differ 2013, 20, 503–514; [DOI] [PMC free article] [PubMed] [Google Scholar]; e Kim HS, Vassilopoulos A, Wang RH, Lahusen T, Xiao Z, Xu X, Li C, Veenstra TD, Li B, Yu H, Ji J, Wang XW, Park SH, Cha YI, Gius D, Deng CX, Cancer Cell 2011, 20, 487–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a North BJ, Marshall BL, Borra MT, Denu JM, Verdin E, Mol Cell 2003, 11, 437–444; [DOI] [PubMed] [Google Scholar]; b Teng YB, Jing H, Aramsangtienchai P, He B, Khan S, Hu J, Lin H, Hao Q, Sci Rep 2015, 5, 8529; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Jin J, He B, Zhang X, Lin H, Wang Y, J Am Chem Soc 2016, 138, 12304–12307; [DOI] [PMC free article] [PubMed] [Google Scholar]; d Cui Y, Li X, Lin J, Hao Q, Li XD, ACS Chem Biol 2017, 12, 47–51. [DOI] [PubMed] [Google Scholar]
- [4].a Cha Y, Han MJ, Cha HJ, Zoldan J, Burkart A, Jung JH, Jang Y, Kim CH, Jeong HC, Kim BG, Langer R, Kahn CR, Guarente L, Kim KS, Nat Cell Biol 2017, 19, 445–456; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Seo KS, Park JH, Heo JY, Jing K, Han J, Min KN, Kim C, Koh GY, Lim K, Kang GY, Lee J.Uee, Yim YH, Shong M, Kwak TH, Kweon GR, Oncogene 2015, 34, 1354–1362. [DOI] [PubMed] [Google Scholar]
- [5].Jing H, Zhang X, Wisner SA, Chen X, Spiegelman NA, Linder ME, Lin H, Elife 2017, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a Tiago Fleming Outeiro EK, Altmann Stephen M., Kufareva Irina, Strathearn Katherine E., Amore Allison M., Volk Catherine B., Maxwell Michele M., Rochet Jean-Christophe, McLean Pamela J., Young Anne B., Abagyan Ruben,, Feany B. T. H. Mel B., Kazantsev Aleksey G., Science 2007, 317, 516–519; [DOI] [PubMed] [Google Scholar]; b Rumpf T, Schiedel M, Karaman B, Roessler C, North BJ, Lehotzky A, Olah J, Ladwein KI, Schmidtkunz K, Gajer M, Pannek M, Steegborn C, Sinclair DA, Gerhardt S, Ovadi J, Schutkowski M, Sippl W, Einsle O, Jung M, Nat Commun 2015, 6, 6263; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Lain S, Hollick JJ, Campbell J, Staples OD, Higgins M, Aoubala M, McCarthy A, Appleyard V, Murray KE, Baker L, Thompson A, Mathers J, Holland SJ, Stark MJ, Pass G, Woods J, Lane DP, Westwood NJ, Cancer Cell 2008, 13, 454–463; [DOI] [PMC free article] [PubMed] [Google Scholar]; d Mellini P, Itoh Y, Tsumoto H, Li Y, Suzuki M, Tokuda N, Kakizawa T, Miura Y, Takeuchi J, Lahtela-Kakkonen M, Suzuki T, Chem Sci 2017, 8, 6400–6408; [DOI] [PMC free article] [PubMed] [Google Scholar]; e Schiedel M, Rumpf T, Karaman B, Lehotzky A, Olah J, Gerhardt S, Ovadi J, Sippl W, Einsle O, Jung M, J Med Chem 2016, 59, 1599–1612; [DOI] [PubMed] [Google Scholar]; f Yang LL, Wang HL, Zhong L, Yuan C, Liu SY, Yu ZJ, Liu S, Yan YH, Wu C, Wang Y, Wang Z, Yu Y, Chen Q, Li GB, Eur J Med Chem 2018, 155, 806–823; [DOI] [PubMed] [Google Scholar]; g Wang C, Wang F, Chen X, Zou Y, Zhu H, Zhao Q, Shen J, Li Y, Li Y, He B, Bioorg Med Chem Lett 2018, 28, 2375–2378; [DOI] [PubMed] [Google Scholar]; h Yang L, Ma X, Yuan C, He Y, Li L, Fang S, Xia W, He T, Qian S, Xu Z, Li G, Wang Z, Eur J Med Chem 2017, 134, 230–241; [DOI] [PubMed] [Google Scholar]; i Tatum PR, Sawada H, Ota Y, Itoh Y, Zhan P, Ieda N, Nakagawa H, Miyata N, Suzuki T, Bioorg Med Chem Lett 2014, 24, 1871–1874; [DOI] [PubMed] [Google Scholar]; j Villalba JM, Alcain FJ, Biofactors 2012, 38, 349–359; [DOI] [PMC free article] [PubMed] [Google Scholar]; k Hu J, Jing H, Lin H, Future Med Chem 2014, 6, 945–966; [DOI] [PMC free article] [PubMed] [Google Scholar]; l Yoon YK, Oon CE, Anticancer Agents Med Chem 2016, 16, 1003–1016; [DOI] [PubMed] [Google Scholar]; m Rotili D, Tarantino D, Nebbioso A, Paolini C, Huidobro C, Lara E, Mellini P, Lenoci A, Pezzi R, Botta G, Lahtela-Kakkonen M, Poso A, Steinkuhler C, Gallinari P, De Maria R, Fraga M, Esteller M, Altucci L, Mai A, J Med Chem 2012, 55, 10937–10947. [DOI] [PubMed] [Google Scholar]
- [7].a Spiegelman NA, Price IR, Jing H, Wang M, Yang M, Cao J, Hong JY, Zhang X, Aramsangtienchai P, Sadhukhan S, Lin H, ChemMedChem 2018, 13, 1890–1894; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kudo N, Ito A, Arata M, Nakata A, Yoshida M, Philos Trans R Soc Lond B Biol Sci 2018, 373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Spiegelman NA, Price IR, Jing H, Wang M, Yang M, Cao J, Hong JY, Zhang X, Aramsangtienchai P, Sadhukhan S, Lin H, ChemMedChem 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a Gai W, Li H, Jiang H, Long Y, Liu D, FEBS Lett 2016, 590, 3019–3028; [DOI] [PubMed] [Google Scholar]; b Jiang H, Khan S, Wang Y, Charron G, He B, Sebastian C, Du J, Kim R, Ge E, Mostoslavsky R, Hang HC, Hao Q, Lin H, Nature 2013, 496, 110–113; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Feldman JL, Baeza J, Denu JM, J Biol Chem 2013, 288, 31350–31356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA, Cell 2001, 107, 149–159. [DOI] [PubMed] [Google Scholar]
- [11].Wang Y, Fung YME, Zhang W, He B, Chung MWH, Jin J, Hu J, Lin H, Hao Q, Cell Chem Biol 2017, 24, 339–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].a Zhang X, Spiegelman NA, Nelson OD, Jing H, Lin H, Elife 2017, 6; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhou Y, Huang C, Yin L, Wan M, Wang X, Li L, Liu Y, Wang Z, Fu P, Zhang N, Chen S, Liu X, Shao F, Zhu Y, Science 2017, 358, 528–531. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.