

Abstract
The compression of brain tissue by a tumour mass is believed to be a major cause of the clinical symptoms seen in patients with brain cancer. However, the biological consequences of these physical stresses on brain tissue are unknown. Here, via imaging studies in patients and by using mouse models of human brain tumours, we show that a subgroup of primary and metastatic brain tumours, classified as nodular on the basis of their growth pattern, exert solid stress on the surrounding brain tissue, causing a decrease in local vascular perfusion as well as neuronal death and impaired function. We demonstrate a causal link between solid stress and neurological dysfunction by applying and removing cerebral compression, which respectively mimic the mechanics of tumour growth and of surgical resection. We also show that, in mice, treatment with lithium reduces solid-stress-induced neuronal death and improves motor coordination. Our findings indicate that brain-tumour-generated solid stress impairs neurological function in patients, and that lithium as a therapeutic intervention could counter these effects.
Brain tissue compression and deformation by a tumour mass is believed to be a major cause of the life-threatening neurologic symptoms seen in patients with brain cancer. Brain tissue can be deformed and its function perturbed by two mechanical forces: tumour-associated edema and solid stress1. Solid stress refers to the compressive and tensile mechanical forces exerted by the solid components of the tissues, such as cells and extracellular matrix2–4. Solid stress, its origin, and its biological consequences are distinct from the fluid pressure that leads to edema, a well-studied mechanical abnormality in brain tumors5,6. Although the genesis and consequences of intratumor solid stress have been initially studied3,4,6, details of the mathematically predicted7 solid stresses have not been characterized in vivo, and they may have important consequences for the physiology of the normal brain surrounding the tumour. Moreover, no studies have been able to distinguish the mechanical effects from biological interactions between the tumour and normal brain tissue in vivo.
As a tumour grows in the cranium, it must displace or replace the surrounding tissue. The tumour growth-induced deformation of the brain – the so-called “mass effect” – can cause severe disability or death, and represents a negative prognostic factor8. Although the neurological consequences of the mass effect are empirically known from the clinical management of patients, the mechanical features, magnitudes, and biological consequences of the solid stresses from brain tumors remain unknown. The growth pattern of brain tumors (i.e. nodular versus infiltrative) has been implicated as one of the potential determinants of the intratumor solid stress4. It is unclear, however, how different tumour growth patterns affect the physiological functions of the surrounding tissue. As a consequence of this lack of mechanistic insight and characterization, effective therapeutic strategies aimed at neuroprotection and alleviation of neurological impairment caused by abnormal compressive forces in the brain are currently unknown.
In this study, we utilized multiple orthotopic mouse models of primary and metastatic brain tumors, a custom-designed in vivo compression apparatus to distinguish the mechanical effects from biochemical interactions between the tumour and normal brain tissue, mathematical modelling and intravital microscopy to establish a causal link between solid stress and neurological dysfunction in the surrounding brain. We show that solid stresses are higher around nodular tumors than around infiltrative tumors, and they deform the surrounding brain tissue and neuronal nuclei, thus reducing peritumoral vascular perfusion and inducing neuronal loss.
In concert with our preclinical findings, we also discovered that stratification of patients based on initial tumour nodularity and perfusion impairment assessed with magnetic resonance imaging (MRI) from patients with glioblastoma (GBM) or breast cancer (BC) brain metastases was able to predict functional status at the time of diagnosis. Finally, lithium – a neuroprotective drug – strongly attenuates compression-induced neuronal damage in mice administrated as either a preventive or intervention agent. This indicates that protecting brain tissue from the solid stress-induced dysfunction is pharmacologically possible, and suggests a rapidly translatable use of neuroprotective agents to improve quality of life in patients with nodular brain tumors.
Results
Measuring how nodular tumors mechanically affect the surrounding brain tissue
Little is known about how the tumour growth pattern observed in patients – nodular versus infiltrative – affects the magnitude, localization and consequences of tumour generated solid stress exerted on the surrounding brain tissue. Nodular tumors have well-defined margins, while infiltrative tumors invade into the surrounding tissue as individual cells (Fig. S1a). As a proof-of-concept study, we orthotopically implanted the nodular U87 cells and the highly infiltrative MGG8 GBM cells in nude mice. After 20 days of growth we measured the solid stress in the tumors and surrounding tissue using a planar-cut method4,9. Briefly, we removed the brain, and then released the residual solid stress by transversally cutting the brain and the tumour at the plane of interest (Fig. S1b). We measured the tissue deformation caused by the release of solid stress (Fig. 1a) as well as the Young’s modulus (stiffness) of the tumour and the brain tissue (Fig. S1c). The solid stress and stiffness measurement were performed on ex vivo tissues, which may differ from those in vivo setting due to presence of vascular and interstitial pressure (see Method Section for more detailed discussion). These measures of the tumour-brain construct were incorporated in a finite element model to estimate the radial (compression) and circumferential (tension) components of the solid stress exerted on the surrounding normal brain by the tumour (Fig. 1b-c, Movie 1 and Method section). Our results showed gradients of compression and tension in the surrounding brain tissue, more prominent around the nodular than the infiltrative tumors. At the tumour-brain interface, the circumferential solid stress was 0.110±0.005 kPa around nodular tumors – as opposed to 0.063±0.004 kPa for infiltrative tumors (Fig. 1b) – and the radial solid stress was 0.020±0.001 kPa around nodular tumors – as opposed to 0.014±0.001 kPa for infiltrative tumors (Fig. 1c). The force exerted by the tumors consistently distorted the micro-anatomy of the surrounding brain tissue. Consistent with the measured forces, the nodular U87 tumors locally deformed the surrounding tissue, as previously shown in other organs7, while the infiltrative MGG8 GBMs did not (Fig. 1d).
Fig. 1. Measuring how nodular tumors mechanically affect the surrounding brain.
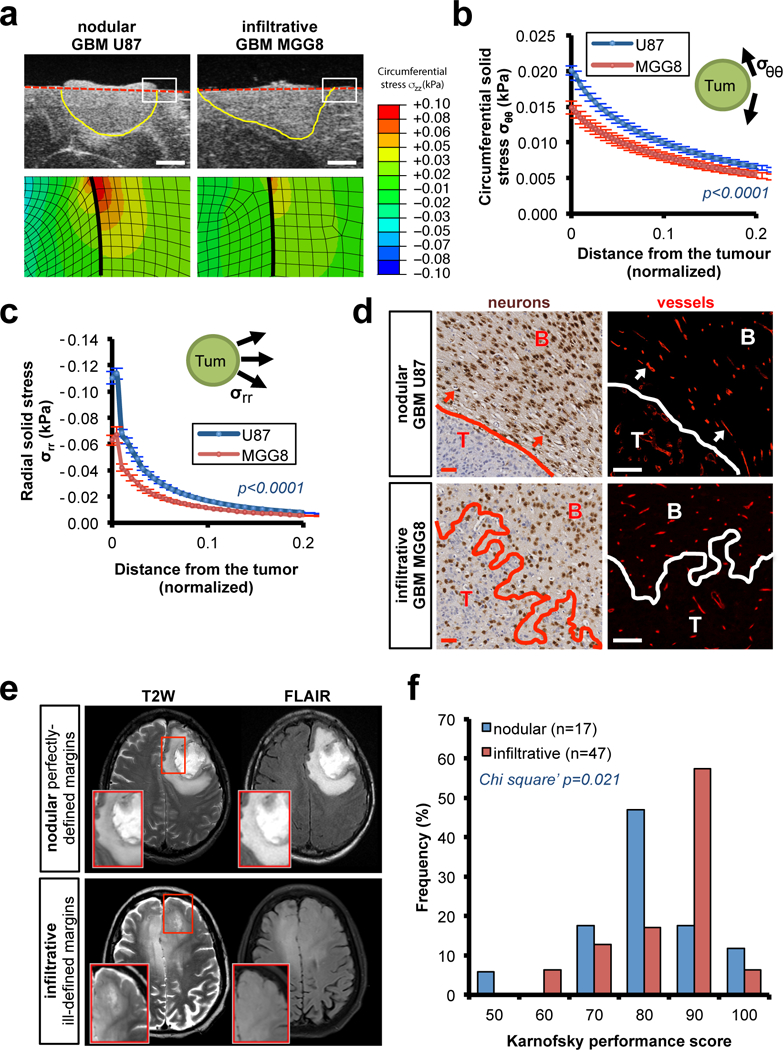
(a-c) Solid stress in mouse models of nodular (patient-derived U87 cell line) and infiltrative brain tumors (patient-derived MGG8 cell line) in nude mice. (a) High-resolution ultrasound imaging of the stress-induced deformation and representative stress profiles across the tumour diameter and the normal surrounding tissue. Scale bar: 1 mm. (b-c) Estimation of the circumferential (compression; σθθ) and radial (tension; σrr) stress in the surrounding tissue obtained from mathematical model (described in Figure S1b-d and Method section). Data are mean of three independent tumors ± s.e.m. (d) Micro-anatomic deformation of the brain tissue around nodular tumors. IHC of U87 and MGG8 tumors at the interface of the normal brain tissue. Neurons (NeuN staining) and vessels (Collagen IV staining). Arrows indicate the deformed region around the tumour, neurons are packed and vessels are displaced following the circumference of the tumour margin. Representative images from a cohort of 10 mice with tumour. Scale bar: 50 μm. (e) Representative T2W and FLAIR MRI of pre-surgery pre-treatment patients with perfectly-defined margins GBM (nodular) and ill-defined GBM (infiltrative). (f) Karnofsky performance score (KPS) histograms of patients with perfectly-defined margins (nodular) or ill-defined GBM (infiltrative). Cohort of 64 pre-surgery pre-treatment GBM patients.
Deformation of brains in patients phenocopies murine findings
The solid stress-mediated deformation of the surrounding tissue observed in preclinical models may reflect the “mass effect” macroscopically observed in radiological exams in brain tumour (i.e. tumor-induced deformation of the brain). We therefore investigated the deformation of the brain anatomy in patients. MRI allowed us to longitudinally observe the tumour growth in patients with GBM. Initial evidence in a post-surgery cohort suggested that nodular tumors had a larger mass effect than infiltrative ones (Fig. S2a and Movie 2-3). To investigate the association between mass effect and neurologic deficit, we correlated brain tumour growth patterns with pre-surgery Karnofsky performance scores (KPS), a clinical index used to estimate functional impairment. We used contrast-enhanced (CE)-T1 and T2/FLAIR images from MRI as well as the Visually Accessible Rembrandt Images (VASARI) criteria for characterization of tumor margins10. This allowed us to stratify a cohort of 64 pre-surgery, pre-treatment GBM patients using a scoring method of the definition of enhancing and non-enhancing margins (Fig. 1e). We discovered that patients with perfectly-defined margins of the tumour (i.e. nodular, 29.7% in our cohort) showed a lower pre-surgery KPS than patients with ill-defined margins (Fig. 1f). Stratification based on CE-T1 or T2-FLAIR showed comparable results (Fig. S2b). Importantly, the nodular and infiltrative cohorts did not differ in tumour (CE-T1) or edema (T2-FLAIR) volume (Fig. S2c), suggesting that the association between tumour growth pattern and KPS is independent from the well-known effect of peritumor edema. The significant difference in pre-operative KPS between nodular (median KPS=80) and infiltrative tumors (median KPS=90) was especially provocative, in light of previously reported differences between KPS measured pre- and post-surgical resection of the tumour, where we expect a complete removal of the solid stress caused by the tumor11.
These findings from mouse models and humans (measurements of solid stress, deformation of the surrounding brain, and the resulting functional impairments) suggest that nodular brain tumors – compared with those that are more infiltrative – exert significant chronic local compression and tension on the surrounding brain tissue and this may affect neurological function.
Nodular brain tumors impair vascular perfusion of adjacent normal tissue and neuronal function
To shed light on the consequences of the local tumour-generated solid stresses on the surrounding brain and to investigate the mechanopathological basis for the performance reduction in patients with nodular tumors, we assessed vascular perfusion in mice bearing GBM and BC brain metastasis. We previously demonstrated that solid stress compresses blood vessels within tumors3,6,12 and predicted this in non-CNS tissues7. Using Doppler optical coherence tomography (OCT13) we performed longitudinal high-resolution angiography in tumour-bearing live mice with transparent cranial windows. We observed that nodular brain tumors – U87 (GBM), GL261 (mouse GBM), and BT474 (BC) (Fig. S1a) – gradually displaced the surrounding vascular architecture and impaired vascular perfusion by reducing vascular volume fraction and vessel diameters (Fig. 2a-b, S3 and Movie 4-5). Moreover, we observed that the reduction in perfusion in the brain tissue strongly correlated with the tumour volume in nodular models (Fig. S4a). Notably, the infiltrative MGG8 GBMs did not cause vascular perfusion reduction in the surrounding brain tissue (Fig. 2b and S3a).
Fig. 2. Reduced vessel perfusion in the brain tissue around nodular tumors.
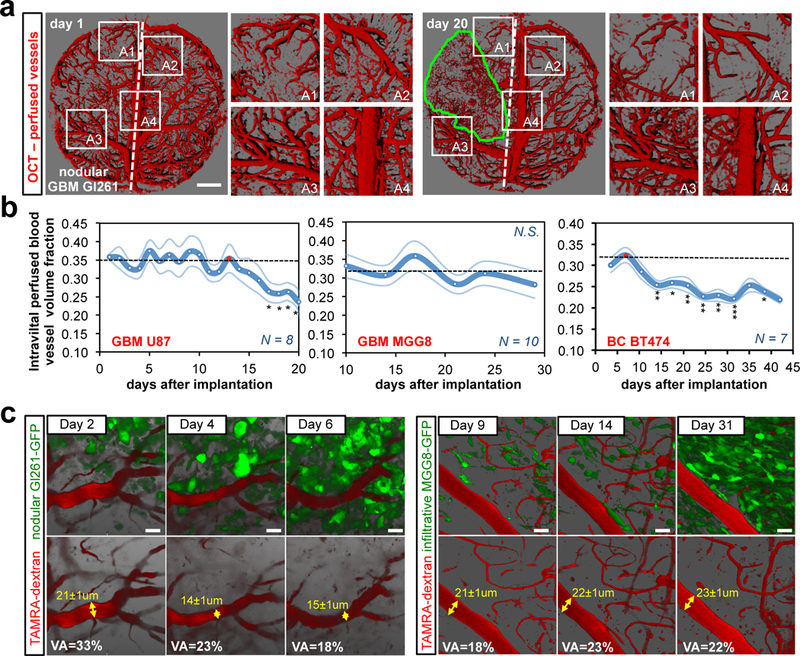
(a) Longitudinal OCT intravital angiography (perfused vessels) of the nodular Gl261 mouse model. U87 and BT474 are presented in Fig. S3B, D. Scale bar: 1 mm. (b) Longitudinal quantitative analysis of the vascular perfused volume fraction in the surrounding tissue. GBM U87: Day17 p=0.025; Day18 p=0.011; Day19 p=0.045; day20 p=0.037 vs the red-dotted time-point. BC BT474: Day14 p=0.001; Day18 p=0.024; Day21 p=0.003; day25 p=0.005; day28 p=0.001; day32 p<0.001; day38 p=0.035 vs the red-dotted time-point. Data are mean ± s.e.m. (c) Intravital longitudinal multi-photon imaging depicting changes in vessel diameter in the nodular Gl261-GFP and in the infiltrative MGG8-GFP GBM mouse models. GFP (green, tumour cells) and TAMRA-Dextran (red, 2MDa-dextran, blood flow). VA = vascular area. Scale bar: 20 μm.
To investigate the effect of mechanical stress on normal brain tissue at the cellular level, we imaged blood flow and tumour cells intravitally and longitudinally using multiphoton microscopy. We observed that the nodular GL261 tumors deformed vessels and reduced the vascular area fraction and vessel diameters in the observed region at the border of the tumour, while the infiltrative MGG8 cells penetrated into the brain tissue without perturbing vascular architecture and vessel perfusion (Fig. 2c).
We then investigated the effects of mechanical stress on neurons in the surrounding brain tissue. Using intravital multi-photon microscopy, we imaged EGFP-H2B mice (which have GFP-positive nuclei) bearing DsRed-GL261 tumors. Nuclei within the surrounding tissue were locally deformed (Fig. 3a), a potential consequence of compressive force. Solid stress compresses the cells, which transmit the compressive force to the nucleus, resulting in deformed nuclei. Since the intravital imaging of our reporter mouse model was not able to distinguish between neurons and the rest of the brain stromal cells, we performed immunohistochemistry (IHC) to stain the neuronal nuclei around nodular tumors. We observed the same nuclear deformation and a gradient of nuclear area reduction in neurons in the tissue surrounding the nodular tumors (Fig. 3b-c and S4b-c). Notably, no deformation or area reduction was detected in neuronal nuclei around infiltrative MGG8 tumors (Fig. 3b-c and S4b-c). Compression of neurons has been shown to cause neural cell death in vitro14,15. Similarly, we detected signs of apoptosis in the tissue surrounding nodular tumors (Fig. 3d and S4d).
Fig. 3. Neuronal deformation and death in the brain tissue around nodular tumors.
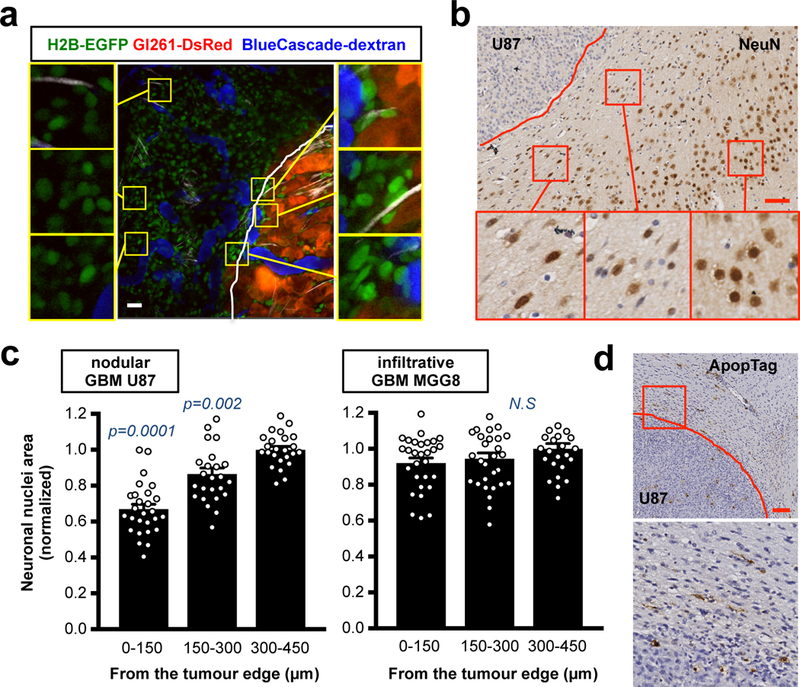
(a) Intravital multi-photon imaging of brain nuclei around the nodular mouse Gl261-DsRed GBM implanted in H2B-EGFP mouse (CAG::H2B-EGFP C57/BL6). GFP (green, nuclei within the brain tissue), DsRed (red, tumour cells) and BlueCascade (blue, 2MDa-dextran, blood flow). Insets are magnifications of the yellow squares. Vascular area (VA) is the quantification (%) of these exact images. Scale bar: 20 μm. (b) IHC of neuronal nuclei (NeuN) in the surrounding brain tissue of mice with the nodular U87 tumour. Representative image from a cohort of 10 mice with tumour. Insets are magnifications of the red squares. Scale bar: 50 μm. (c) Quantification of neuronal nuclear area in the brain tissue around nodular or infiltrative tumors. Data are mean ± s.e.m. p-values are vs “300–450 from the tumour edge”. (d) Apoptosis (ApopTag) at the interface between the nodular U87 tumour and the normal brain tissue. Representative image from a cohort of 10 mice with tumour. Inset is magnification of the red square. Scale bar: 100 μm.
Collectively, these results indicate that nodular tumors – in comparison to infiltrative ones – locally reduce perfusion by compressing blood vessels in the surrounding brain tissue as well as by compressing neuronal nuclei and inducing neuronal loss.
GBM and BC patients with nodular tumors present reduced vascular perfusion in the surrounding brain tissue and neurologic dysfunction
To test if our preclinical results on perfusion recapitulate those in patients, we investigated vascular perfusion in the surrounding normal-appearing tissues in a cohort of 64 pre-surgery GBM patients using MRI images. We assessed perfusion in the normal-appearing tissue, avoiding the regions of T2/FLAIR peritumoral edema and then normalizing with the whole brain perfusion levels (which are different in each individual; Fig. S5a). Our analysis showed that 53% of patients experienced a local gradient of reduced perfusion in the surrounding brain tissue (Fig. 4a-b). We also confirmed these perfusion results in an additional independent cohort of 14 post-surgery, but otherwise untreated, GBM patients (Fig. S5b-e).
Fig. 4. Reduced perfusion in the surrounding normal tissues in GBM and BC brain met patients.
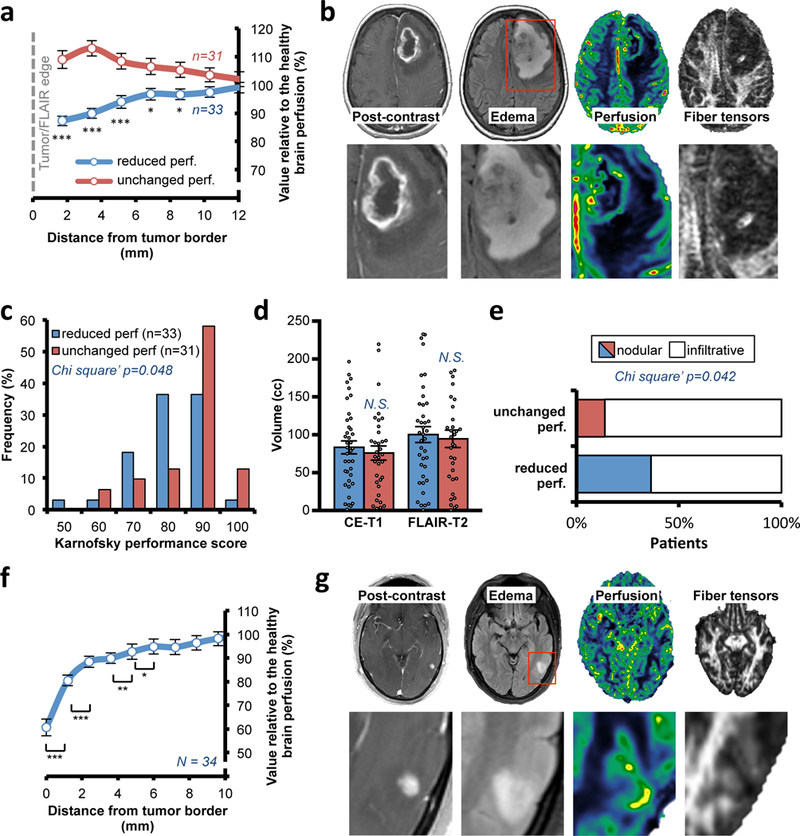
(a) Quantification of the local perfusion in surrounding normal brain tissue from immediately adjacent to the peri-edematous region of the GBM to the region 12 mm away from the tumor edge. The cohort of 64 pre-surgery GBM patients was divided in patients with a reduced perfusion in the surrounding tissue (“reduced perf”) and patients with no difference in perfusion (“unchanged perf”). Data are mean ± s.e.m. Point 1.72 p<0.001 vs “unchanged perf”; point 3.44 p<0.001; point 5.15 p=0.001; point 6.87 p=0.014; point 8.59 p=0.019. (b) Representative pre-surgery GBM patient from a cohort of 33 patients with local reduced perfusion and fiber tensor signal in the surrounding tissue as well as midline shift. (c) Histograms of KPS in “reduced perf” and “unchanged perf” GBM patients. (d) CE-T1 and FLAIR-T2 volumes in the two classes with “reduced perf.” or “unchanged perf.” in the surrounding brain tissue. N.S., not significant. Data are mean ± s.e.m (N = 64 patients). (e) Classification of the patients in the two perfusion subclasses in based on the edematous (FLAIR-T2) MRI margins (N = 64 patients). Comparable result with CE-T1 MRI, Fig. S6B. (f) Quantification of the local perfusion in the brain tissue immediately surrounding the BC brain mets. Cohort of 34 BC patients (26 HER2-positive; 8 HER2-negative). Data are mean ± s.e.m. Point 0 p<0.001 vs point 1.2; point 1.2 p<0.001 vs point 2.4; point 3.6 p=0.010 vs point 4.8; point 4.8 p=0.013 vs point 6. (g) Representative BC HER2-negative patient from a cohort of 34 patients with local reduction of perfusion and fiber tensor signal in the surrounding tissue.
The above imaging method allowed us to stratify the patients from our pre-surgery cohort based on their level of perfusion in the surrounding tissue (i.e. “reduced perf” or “unchanged perf”). Patients with reduced perfusion in the surrounding tissue had significantly poorer KPS (Fig. 4c). Notably, these two patient cohorts showed similar levels of tumour volume and edema (Fig. 4d) and had equal pre-treatment with steroids (Fig. S6a). Moreover, diffusion tensor imaging (DTI) MRIs displayed local deformation of the white matter anatomy in the surrounding brain tissue (Fig. 4b), as previously shown16. We observed that the tumors with local reduction of perfusion (“reduced perf”) were more often classified as nodular (Fig. 4e and S6b), but no difference in the tumour shape was detected (Fig. S6c-d).
Because metastases in the brain are reported to be primarily nodular17, we then analysed a cohort of 34 patients with BC metastases in the brain with the same imaging approach as described above. Indeed, we observed markedly reduced perfusion in the surrounding tissue in proximity to the tumour (Fig. 4f-g and S6e). These results indicate that both primary and metastatic nodular tumors locally reduce vascular perfusion in the surrounding brain tissue and that this is correlated with neurological impairment.
In vivo solid stress application impairs vascular perfusion and induces neuronal dysfunction
Taken together, the above results in preclinical and clinical settings demonstrate a strong association between the gradient of solid stress and the reduction in local vessel and neural function in the surrounding brain tissue. To mechanistically confirm this link and distinguish the mechanical effects from biological interactions between the tumour and normal brain tissue (e.g., cytokines and inflammation associated with cancer cells that are cytotoxic to normal cells18), we directly and chronically applied mechanical compression to the brains in living mice with no tumour. This also avoided the confounding effect of the peritumoral edema. Our custom-designed in vivo apparatus was composed of a set-screw mounted on a modified cranial window and separated from the brain tissue by a biocompatible and deformable membrane. By turning the screw into the cranial window in nude mice without tumour, we can control the displacement of brain tissue so that it resembles that caused by nodular tumors (Fig. 5a and S7a). We expanded the compressing volume at around 1.3 mm3/day for 14 days and reached a maximum volume of approximately 19.0±0.7 mm3. The rate of compression was comparable to the rate of growth in the nodular U87 and BT474 intracranial models over the same time period, while the maximum volume was less than the tumors at endpoint (Fig. S7b). This method gradually and chronically compressed the brain and deformed the micro-anatomy of the cortex (Fig. 5b and S7c) mimicking the effects of a nodular tumour (Fig. 1d). We then performed longitudinal angiography on mice with controlled and gradual compression on the brain and discovered that vascular perfusion and vessel diameter were reduced (Fig. 5c and S7d), confirming our observation in nodular tumour-bearing brains (Fig. 2a-b).
Fig. 5. Direct chronic compression of brain reduces vessel perfusion and induces neuronal damage.
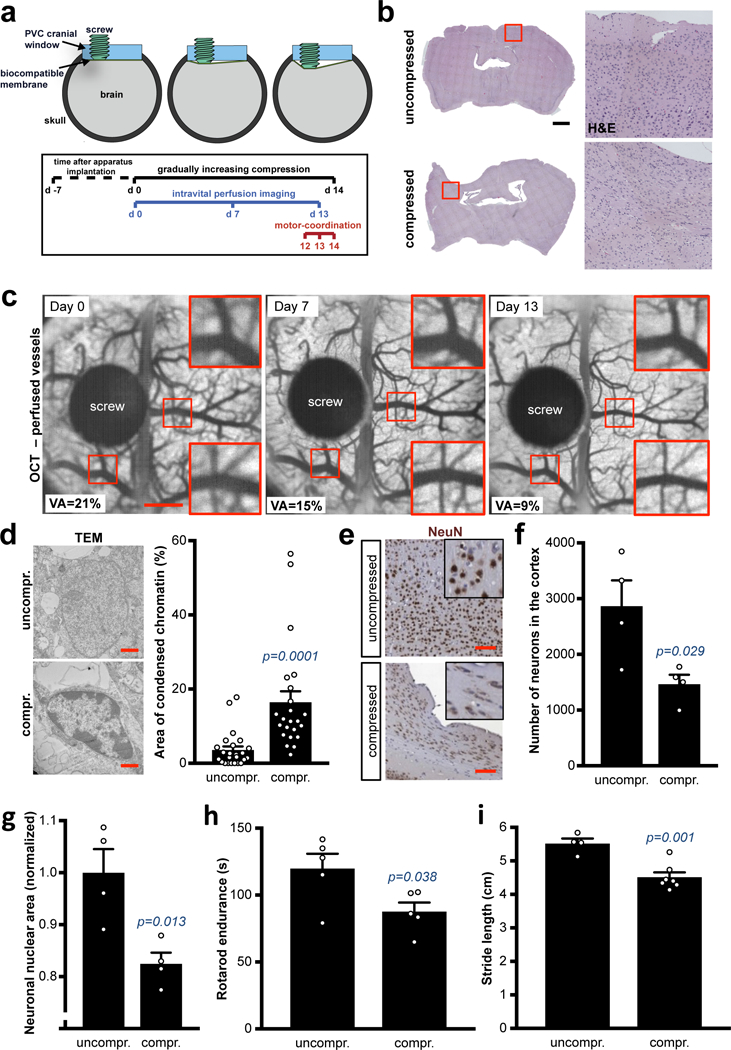
(a) Schematic of the intravital compression device procedure. (b) Deformation of the compressed tissue. Hematoxylin-eosin (H&E) of the whole brain gradually and chronically compressed for 14 days. Representative image from a cohort of 7 mice with compression device. Insets are magnifications of the white squares. Scale bar: 1 mm. (c) OCT longitudinal intravital angiography of the brain seen from the compression apparatus (average projection of the 3D imaging). Vascular area (VA) is the quantification (%) of the shown images. Scale bar: 1 mm. (d) Ultrastructural imaging of nuclei in uncompressed and compressed cortexes via electromicroscopy. Representative images from a cohort of 3 mice per experimental point. Scale bar: 1 μm. Quantification of the percentage of condensed chromatin area in n nuclei. Data are mean ± s.e.m. (e-f) NeuN (neuron nuclei) in the compressed cortex. Representative images from a cohort of 4 mice per experimental point and quantification of number of neurons (NeuN+ nuclei) per cortex exposed to the cranial window. Data are mean ± s.e.m. Scale bar: 100 μm. (g) Quantification of neuronal nuclear area in uncompressed or compressed cortexes. Normalized results. Data are mean ± s.e.m. (h) Rotarod endurance (index of motor coordination and balance) after 14 days of compression of the motor and somatosensory cortex. Data are mean of 3 consecutive days (1 measure/day) and 5 mice per group ± s.e.m. (i) Static gait test (index of locomotion); footprint analysis of the stride length. Footprint in Fig. S8E. Data are mean of 3 technical replicates and n mice per group ± s.e.m.
Next, we investigated the effects of compressive stresses on neurons and other cellular components in brain. First, ultrastructure analysis revealed signs of cell distress (condensed chromatin) in the compressed cortexes (Fig. 5d and S7e). In addition, we discovered that the NeuN-positive neuronal nuclear area was significantly reduced and deformed. The total number, area and circularity of neuronal nuclei were substantially reduced in the cortex after chronic compression (Fig. 5e-g and S8a). In the compressed cortexes we also detected apoptosis, signs of degenerative autophagy (LC3-II-positive vesicles), reduction of oligodendrocyte processes (CNPase+ filaments), and recruitment/activation of astrocytes (Fig. S8b) and signs of necrosis (TNFα and RIP1; Fig. S8c), all indicating neuroinflammation and neuronal damage.
Neural damage in the motor and somatosensor cortex—the regions compressed with our device—may consequently impair sensorimotor coordination and balance in mice, mimicking the neurological impairment that commonly occurs in brain tumour patients. By using Rotarod and static gait tests (Movie 6 and19,20), we found that motor-coordination and locomotion was significantly reduced in mice where the cortex was compressed (Fig. 5h-i and S8d-e). The mice undergoing brain compression maintained normal body weight (Fig. S8f).
These findings mechanistically explain the results shown in patients and mice with nodular tumors (Fig. 1 and 3) and the clinical evidence of the correlation of the mass effect events with performance in patients. The human brain has been shown to have a pronounced compression-tension asymmetry21 and heterogeneity in its stiffness22,23, as well as a broad spectrum of functions for each specific brain region. Consequently, we expect that the local compression of different areas of the brain might trigger different mechanical and neurological responses from the brain tissue. Therefore, further studies are needed to assess the regional effects of local solid stress from brain tumors.
In vivo removal of solid stress restores vascular perfusion and neurological function
Next, we asked whether the release of compression—simulating clinical surgical resection—leads to the rescue of the biological consequences of solid stress. First, we investigated a short-term decompression in brains compressed by a nodular tumour. After 20 days of nodular tumour growth in the brain of nude mice, we decompressed the brains by removing the cranial windows, mimicking craniotomy or surgical resection where consequent displacement has been previously shown24,25. Thus, instead of a full surgical resection that would damage the surrounding tissue, this approach allowed us to partially release the tumour compressive forces and investigate intravital brain vessel perfusion. We observed a strong increase in perfusion in the surrounding brain after alleviation of the tumour-induced compressive forces (Fig. 6a and S9a-c).
Fig. 6. Decompression restores vessel perfusion and partially rescues motor-coordination.
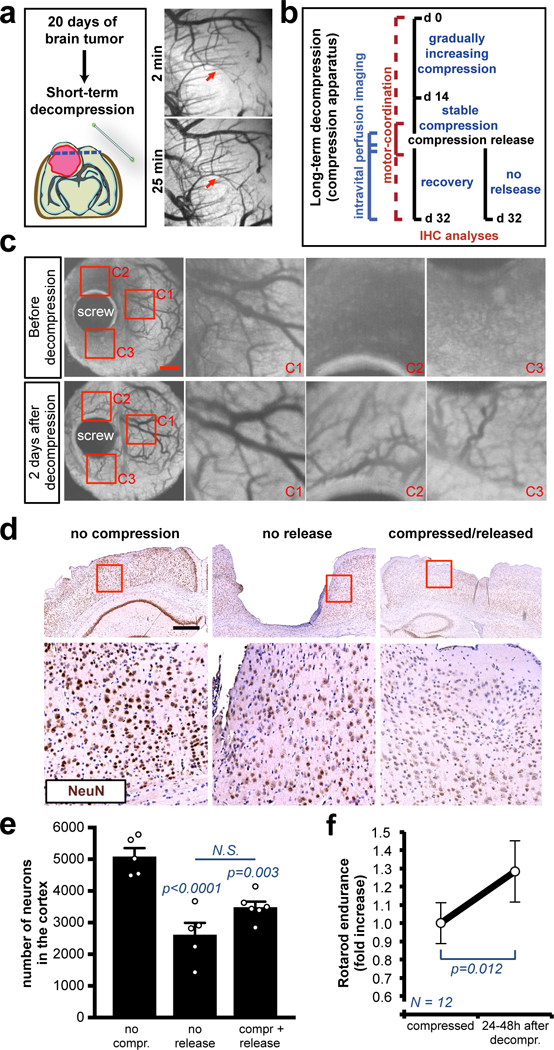
(a) OCT angiography after decompression of brains with nodular U87 tumors by removing cranial window (craniotomy). Complete time-course in Figure S9B-C. (b) Schematic of the long-term compression/decompression timeline via the in vivo chronic apparatus. (c) Longitudinal OCT angiography before and after decompression. Images of a representative mouse from a cohort of 3 mice. Insets are magnifications of the red squares. See Movie S7 for the complete time-lapse. Scale bar: 1 mm. (d) Representative cortexes stained with anti-NeuN from a cohort of 3 mice. The long-term compression induces tissue loss and the decompression partly restores the micro-anatomy of the cortex. Insets are magnifications of the red squares. Scale bar: 500 μm. (e) Quantification of the NeuN+ neurons in the cortexes, decompressed or compressed and released. Data are mean ± s.e.m. (f) Rotarod endurance (index of motor coordination and balance) 24–48 hours after decompression of the motor and somatosensory cortex. Data are mean of 2 consecutive days and 12 mice per group ± s.e.m (normalized with the value before decompression).
Using the in vivo compression device in mice with no tumour, we next investigated if the removal of solid stress—modelling a full tumour resection—leads to the restoration of vascular perfusion, neuronal damage and motor-coordination (Fig. 6b). Removing solid stress after long-term compression – achieved by turning the screw backwards toward its original set-point – restored vascular perfusion (Fig. 6c, S9d and Movie 7). Histological analysis of cortexes after 32 days of compression (long-term) showed massive brain tissue loss in the compressed area (Fig. 6d). Interestingly, 2 weeks after decompression, the brain showed a partial restoration of the micro-anatomy and number of neuronal cells (Fig. 6d-e and S10a). The release of the compression significantly improved motor-coordination at 2 days after decompression (Fig. 6f) – in line with previous data in post-surgery GBM patients11. Motor-coordination then dropped back again (Fig. S10b), indicating an incomplete recovery.
These results allowed us to decouple the tumour mechanics from the biological interactions between the tumour and the surrounding normal tissue, thus mechanistically showing that both perfusion and neuronal function are impaired by solid stress and may be partially restored when the solid stress is removed, as occurs during surgical resection of the tumor25.
Systemic treatment with lithium protects neurons from solid stress-induced neuronal death and neurological dysfunction
To reduce the compression-induced neuronal damage and further improve neurological ability, we tested potential neuroprotective therapeutic strategies aimed at preserving neuronal function in chronically compressed brain tissues. The compression device allowed us to screen multiple drugs in a single animal study. We systemically treated mice with the following drugs during the chronic/gradual compression and the following two weeks after decompression (Fig. 7a): necrostatin-1 (Nec-1; 7 mg/kg/d i.p.; similar to 26), valproic acid (VPA; 400 mg/kg/d i.p.27), lithium chloride (LiCl; 50 mg/kg/d i.p.28) and dexamethasone (Dexa; a single dose at maximum compression as shown in Fig. S10d, 10 mg/kg i.p.29,30). These agents are known to cross the blood-brain barrier31–33, and have been shown to be neuroprotective during CNS ischemia or neurodegeneration26–28. No body weight loss was detectable in mice after 1 month of treatment (Fig. S10c), indicating no evident toxicity of the drugs. Our results identified lithium as a potential neuroprotective agent, while necrostatin-1, valproic acid and dexamethasone showed no evident neuroprotective improvement in our experimental setting (Fig. S10c-e). We replicated the compression/release experiment under lithium treatment (Fig. 7a), and found that lithium significantly reduced neuronal death, massively attenuated the reduction of cortical tissue area in the compressed brains (Fig. 7b-c). Moreover, ultrastructure analysis qualitatively showed reduced condensed chromatin (a sign of cell distress) in the lithium-treated cortexes (Fig. 7d). As a consequence, the Rotarod motor-coordination in lithium-treated mice was improved during both the compression and decompression phases with 34% higher endpoint endurance than in control mice (Fig. 7e and S10f). Similar results were obtained with the static gait test (Fig. 7f and S10g).
Fig. 7. Systemic treatment with lithium alleviates compression-mediated neuronal damage.
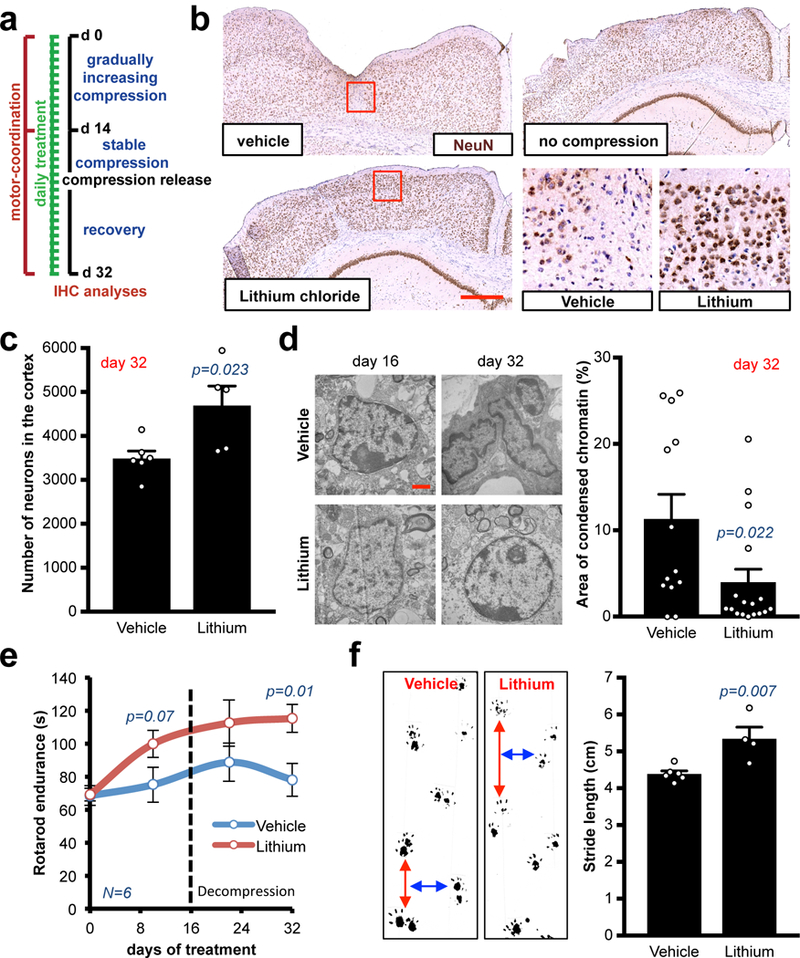
(a) Schematic of the treatments in long-term compression/decompression procedure. (b) Representative cortexes stained with anti-NeuN from a cohort of 5 mice per experimental point. Chronic lithium treatment preserves the cortex micro-anatomy after decompression. Insets are magnifications of the red squares. Scale bar: 500 μm. (c) Quantification of the NeuN+ neurons in the cortexes, decompressed or compressed and released. Data are mean ± s.e.m. (d) Ultrastructural imaging of nuclei in compressed cortexes treated with vehicle or lithium via electromicroscopy. Representative images from a cohort of 3 mice per experimental point. Scale bar: 1 μm. Quantification of the percentage of condensed chromatin area in n nuclei. Data are mean ± s.e.m. (e) Longitudinal analysis of the Rotarod endurance (index of motor coordination and balance). Data are mean of 2 consecutive days and 6 mice per group ± s.e.m. (f) Static gait test (index of locomotion); footprint analysis of the stride length at the time of highest compression (day 14). Data are mean of 3 technical replicates and n mice per group ± s.e.m. 115 steps measured for vehicle-treated mice and 62 for lithium-treated ones.
To gain mechanistic insight into the neuroprotective effect of lithium, we performed bulk RNA-sequencing (RNA Seq) on cortexes that were compressed, released and chronically treated with lithium or vehicle (Fig. 7a). With a sample size of 3 per group, a total of 21 genes were differentially expressed in lithium-treated cortexes versus vehicle control (Fig. 8a and Table S1–2; FDR<0.05). Of the 21 differentially expressed genes, 19 were up-regulated and 2 were down-regulated in the cortexes of lithium-treated mice (Fig. 8a, S11a and Table S1–2). The function of these genes in the CNS were linked to ion channels, mitochondrial function, and neuronal differentiation as well as protection from apoptosis, autophagy and ischemia (Table S1–2). To systematically analyse our RNA-Seq results, we performed gene-annotation enrichment analysis using the DAVID Bioinformatics Resource34. Significantly upregulated gene ontologies in the lithium-treated group included dendrite, postsynaptic density, neuron projection (cellular components), protein binding (molecular function), and protein localization to synapse (biological process) (Fig. S11b-e and Supp. Data 1a). Functional annotation clusters associated with neurons, synapses, microtubules, and calcium ion transport were also enriched in the upregulated genes in the lithium-treated group (Supp. Data 1b). Taken together, these results suggest that lithium treatment induces survival pathways in neurons (as demonstrated in Fig. 7b-c) and point out how lithium preserves neuronal function during the compression/release phase. Finally, we tested whether lithium is neuroprotective if given when the compression and resulting neurological symptoms are already present in mice. Notably, in this more clinically-relevant established compression model, lithium also efficiently increased the number of surviving neurons (Fig. 8b).
Fig. 8. Solid stress from nodular brain tumors compresses the surrounding brain tissue, thus causing neurological dysfunction.
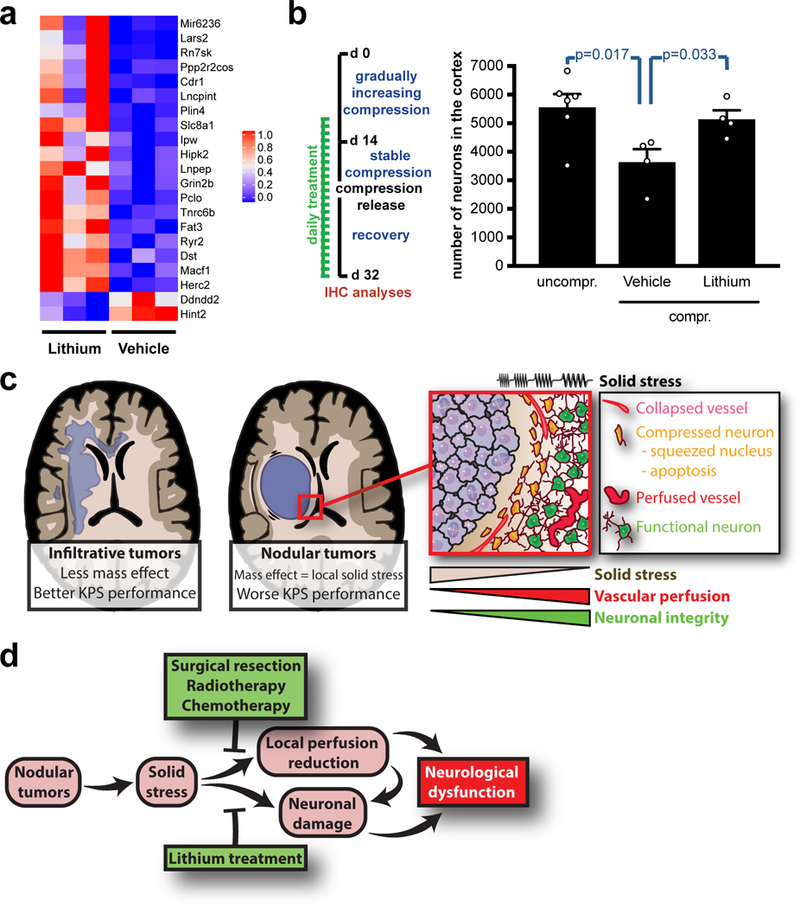
(a) Heatmap illustrating normalized gene expression of differentially expressed genes (FDR<0.05) from RNA-Seq analysis of compressed/released cortexes under chronic lithium treatment. (b) Quantification of the NeuN+ neurons in the cortexes, compressed and released lithium-treated with treatment start when compression and symptoms were already present. Data are mean ± s.e.m. (c) Schematic summarizing the effects of nodular tumors and their resulting solid stress on the surrounding brain tissue. The focus of the manuscript is on nodular tumors and the effects of solid stress on the surrounding tissue, while infiltrative tumors appeared to exert lower solid stress to the brain. (d) Schematic summarizing the causes, modifiers and consequences of solid stress from nodular tumors.
These results show the therapeutic potential of lithium in preserving neuronal and neurologic function in the CNS affected by brain tumor-generated solid stress.
Discussion
Brain tumors, particularly GBM and brain metastases, have poor prognosis and are associated with significant morbidity due to progressive neurologic dysfunction. Although huge efforts have been employed to inhibit or stop brain tumour growth, little is known about the reasons for neuronal loss in the surrounding brain tissue, which is the ultimate cause of significant neurological dysfunction, loss of quality of life and eventually death. One of the possible causes of neuronal loss is the tumour-generated solid stress exerted on the surrounding brain tissue. We previously predicted solid stress in and around non-CNS tumors7. However, the growth pattern of the tumour (infiltrative vs. nodular), the actual mechanical properties of the tissues and the impact on the brain function were not experimentally investigated previously. We therefore decided to examine the effects of growth pattern on the solid stress applied to the surrounding brain tissue. Our results showed higher solid stress in the tissue around nodular than infiltrative tumors and this prompted us to investigate whether this has consequences for neurological function. We found neuronal damage and reduced perfusion around nodular tumors with corresponding neurological dysfunction (Fig 8c). Importantly, these preclinical results allowed us to design a stratification method for GBM and BC patients with brain metastases using perfusion MRI, which can be used to predict which patients are likely to experience solid stress-mediated neurological impairment. We discovered an association between the growth pattern, reduced perfusion levels in the surrounding tissue and neurological impairment in a large set of patients across multiple cohorts. Notably, these associations were independent of peritumoral edema, widely recognized as an important determinant of neurological function in brain tumour patients, and treatment with steroids.
Using a custom-designed in vivo compression apparatus, we simulated the physical forces from nodular tumors exerted on the brain and we demonstrated a causal link between solid stress and a reversible reduction of vascular perfusion and semi-irreversible neurological dysfunction. Importantly, the neurons died as a consequence of the slow and chronic compression of the compression apparatus. This preclinical model allowed us to experimentally isolate the mechanical forces on the brain from other potential reasons of neuronal death and neurological dysfunction caused by brain tumors, such as excitotoxicity, inflammation, tumour release of neurotoxic factors and physical contact between tumour cells and normal brain. Moreover, we demonstrated that part of the dysfunction caused by the compression of the normal tissue could be rescued by the removal/release of solid stress, as happens during surgical resection, chemotherapy and radiotherapy. As removal is not sufficient to completely restore the cortex tissue and the neurological function (semi-irreversible effect); we therefore proposed the use of neuroprotective drugs to counter these effects.
Our results suggest lithium treatment as a potent neuroprotective therapeutic strategy in patients with nodular brain tumors, characterized by low perfusion in the surrounding tissue and lower KPS. For decades, lithium has been in use for psychiatric disorders, such as mania, depression and bipolar disorder. Notably, in our experiments the lithium dose administrated here (50 mg/kg) was comparable to or lower than that given to mice in anti-depressant experiments (50–400 mg/kg35). In preclinical models and clinical studies, lithium has been proven to have neuroprotective effects in several neurodegenerative, brain-injury and ischemic models36–41 and this is in line with our RNA-Seq results. Notably, lithium also exerts anti-tumour effects by inhibiting tumour growth, chemosensitizing GBM preclinical models to temozolomide42,43 and reducing invasion of cultured GBM cells44, although its clinical efficacy remains to be determined in a clinical trial setting. These previous reports and our own findings support a potential therapeutic use of lithium in brain tumour patients.
Preservation of neurological function is increasingly taken into consideration in current clinical practice and neuro-oncologists now consider quality of life as a critical end-point45. Our results now implicate solid stress as one of the causes of brain tumor-induced loss of neurological functions. Thus, our preclinical findings provide rigorous evidence for immediate translation of the concept of neuroprotection in the specific subclass of patients with nodular brain tumors who experience neurological defects due to the compression of the surrounding tissue. These agents can be used in addition to treatments that alleviate tumour-generated mass effect to permit neuronal preservation while treatments to reduce tumour mass by surgery, radiation and chemotherapy are on-going.
In conclusion, we showed that the compression-induced neuronal damage may be attenuated by decompression, mimicking surgical resection, and chronic systemic treatment with lithium (Fig. 8d). These results indicate neuroprotective treatments as potential therapeutic strategies in patients with nodular “pushing” tumors to reduce the pathological effects of solid stress on the surrounding brain neurons, even in cases of inoperable or advanced brain cancer to improve quality of life for as long as possible.
Methods
Mice
All animal protocols were approved by and in accordance with the guidelines established by the Institutional Animal Care and Use Committee (IACUC) at Massachusetts General Hospital (MGH). The mouse colony (MGH Cox7) was maintained in accordance with National Institutes of Health and MGH guidelines. MGH is accredited with the International Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) and the NIH Office of Laboratory Animal Welfare (A3596–01). All experiments were performed in 8 to 10 week-old female nude mice. In the case of Figure 2D, we used CAG::H2B-EGFP (C57/BL6) mice from Jackson Laboratory.
Reagents and Cell culture
Lithium chloride (LiCl; L4408), valproic acid sodium salt (VPA; P4543) and necrostatin-1 (Nec-1; N9037) were from Sigma.
The human U87 and BT474 cells were from ATCC and the mouse Gl261 cells were from the National Cancer Institute Repository. MGG8, a cell line derived from a GBM patient, was previously established in the Department of Neurosurgery at Massachusetts General Hospital (MGH)46. MGG8 and Gl261 cells were grown in serum-free conditions using the NeuroCult NS-A proliferation kit (Stemcell Technologies)47–49. Both cell lines were stably transduced with GFP or Discosoma red fluorescent protein (dsRed) using a lentiviral construct. U87 cells were cultured in DMEM with 10% (vol/vol) FBS. Human HER2-amplified breast cancer BT474 cells were cultured in RPMI 1640 supplemented with 10% FBS (Atlanta Biologicals, Flowery Branch, GA, USA)50. All cell lines were repeatedly tested and were negative for mycoplasma using the Mycoalert Plus Mycoplasma Detection Kit (Lonza) and were authenticated before use by IDEXX Laboratories (IDEXX, Human 9 and 16 species-specific STR marker profile).
Planar-cut method - measurement of stress-induced deformation
We slightly modified previous methods4,9. The tissue deformation map was acquired via high-resolution ultrasound (Vevo 2100 system; FUJIFILM Visual Sonics) on the half of the tumour–brain that was immersed and relaxed in PBS. We used the probe MS550S for large samples (depth of view = 13 mm, frequency = 32–56 MHz) and the probe MS700 for smaller samples (depth of view = 9 mm, frequency = 30–70 MHz). The 3D images were exported from VisualSonics software to MATLAB (The MathWorks) for post-processing. The post-processing included smoothing, which was performed with moving averaging on the cut surface and bottom surfaces of the brain-tumour to remove any noise introduced by ultrasound imaging. The top and bottom surfaces were then exported to SolidWorks (Dassault System) to reconstruct a 3D object. This solid object was then exported to the compatible finite-element model ABAQUS (Dassault System), as described below.
Atomic force microscopy (AFM)-based measurement of the indentation modulus (stiffness)
The indentation moduli of tumors and normal brain were quantified using an Asylum MFP3D AFM (Asylum). We used polystyrene colloidal probe tips with an end diameter of ~25 μm (Polysciences) attached to tipless cantilevers with nominal spring constant k ~ 0.12 N/m (Bruker). The colloidal probes were attached to the cantilever by the lift-off process: a dot of low-viscosity glue (M-Bond 610, Structure Probes/SPI Supplies) was placed on a tipless cantilever by making quick contact between the cantilever and a thin layer of glue (1-μl-thick) spread over a 10×10 mm mica surface. We then made immediate contact between the tip of the cantilever and a colloid probe resting on a glass slide and waited for 1 min with the cantilever pushing against the colloid. This process was followed by heat curing for 2 h at 150°C. For each probe tip, the exact spring constants of the cantilevers were directly measured using the thermal calibration method. The relationship between the detected voltage and the applied force was calibrated by bringing the cantilever into contact with a glass slide and calculating the slope of the voltage–displacement curve. The displacement, d, was translated to force, F, using Hooke’s law (F = kd; k is the cantilever spring constant). The indentation was performed under a force control scheme (maximum force ~20 nN) limiting the indentation depths to 0.5–3 μm to stay in the linear regime. The tip displacement was obtained by subtracting the cantilever deflection from the vertical movement of the piezoelectric actuator. An indentation approach velocity of 2 μm/s ensured that the elastic modulus was probed at rates lower than the characteristic frequency of tumour as a poroelastic tissue51,52, to be close to equilibrium conditions. The effective indentation modulus Eind was computed using Hertzian contact mechanics models via least-squares linear regression of the experimental loading force–displacement curves. For the spherical colloidal probe tip with end radius R on the sample with thickness h » R (here, h ~5 mm, R ~12.5 μm)
where F is the indentation force and D is the indentation depth (we assumed υ = 0.1, as measured in other soft tissues53,54)..
Despite differences in absolute values, the relative differences between the stiffness of brain parenchyma and tumour tissue are consistent with previous publication where Magnetic Resonance Elastography (MRE) has been utilized for stiffness measurement 55. The discrepancies between our AFM measurement and MRE methods are potentially due to: (i) difference in methods of measurements: the Young’s modulus is estimated from the indentation modulus in AFM measurement in quasi-equilibrium indentation, while the MRE is based on measurement of shear wave speed when the tissue is vibrated at a certain frequency, e.g. 1000 Hz55, which has strong effect on the reported stiffness values56; (ii) in vivo vs. ex vivo settings: our AFM measurement is performed in vivo, while the MRE has been performed in vivo, when vascular and interstitial fluid pressures are present and may affect the stiffness values57, and (iii), strain-stiffening effect: our measurements have been performed on stress-relaxed samples, while the stressed tissue may exhibit higher stiffness value due to material nonlinearity58,59.
Mathematical model
By developing a mathematical model (2-D axisymmetric finite element model), the map of stress-induced deformation is converted to the strain tensor, , from which the stress tensor is estimated based on Hooke’s Law, written in index notation as60:
where E is the modulus of elasticity (stiffness), and ν is the Poisson’s ratio of the tissue based on linear isotropic assumption, and δij is the Kroenecker delta. We chose a linear elastic model to reduce model parameters, and avoid associated assumptions. However, the mathematical model can be improved in multiple directions1: (i) the use of hyperelastic models61 to account for nonlinearity in the material properties of the normal brain tissue and brain tumors, e.g. strain-stiffening58,59 in which stiffness values are altered in stressed tissues; (ii) the use of poroelastic model1 to account for the role of interstitial, vascular, and cerebrospinal fluid pressure, and (iii) triphasic model to account for electromechanics of brain and tumour tissue with fixed charge density62.
We developed our mathematical model using the general-purpose commercial finite element software ABAQUS (Version 6.9, SIMULIA, Providence, RI). The 3-D geometry of the tumour is first smoothed to reduce any noise in MATLAB (MathWorks). Since the geometries of the tumors of interest are mainly spherical, we used an axisymmetric model (Fig. S1D) to reduce the modelling and computational time. We used the 8-node axisymmetric element CAX8R for both the tumour and surrounding brain tissue with two elastic parameters: the Young’s modulus and the Poisson’s ratio, which are obtained as described above. We have previously shown that the solid stress relaxation time constant is on the order of 10 min or less4, following the linear poroelastic time constant predicted from the Young’s modulus and hydraulic permeability in poroelastic materials51,63. Since the time constant of the biological processes that give rise to solid stress in tumors, such as proliferation, ECM deposition and cell contraction, ranges from several hours to days, much longer than the stress relaxation time constant, the equilibrium mechanical properties that we are measuring reflect the appropriate properties in the generation and application of solid stress.
Since the tumors of interest are all surrounded by the brain tissue, free of solid stress at locations far from the tumour, the cut surface in the brain provides the reference plane that represents the original plane of the tumour tissue before stress relaxation. A displacement profile is assigned to the top deformed surface of the tumour to translate it back to the original reference plane (Movie S1). This assigned deformation is only in the direction perpendicular to the surface of the tumour (z-direction), and the tumour top surface is free to move parallel to the surface direction (x-y plane). This displacement assignment translates the tumour geometry from the relaxed state (deformed surface) to the original stresses state prior to the cut (flat surface). The stress component that is generated after the displacement assignment is reported as the circumferential solid stress in the intact brain-tumour. The boundary condition on the bottom surface of the surrounding brain tissue, far from the tumour, is defined as zero stress.
We are able to estimate the radial stress from, the stress component normal to the cut surface that is obtained from planar-cut method if the following assumptions hold: (i) the geometry and material properties of the tumor hold spherical symmetry around the center of the tumor, (ii) the incision from planar cut is going through the centre of the tumour, i.e., is equivalent to , and (iii) the stress field goes to zero at the outer edges of the surrounding brain far from the tumor. With the above assumption, the following equation holds between and 60:
which results in the following equation after integration:
Having from the planar-cut method for the range of interest, and assuming at , the outer edge of the brain far from the tumour, the radial stress is estimated (Fig. 1A-B).
Cranial window
To implant transparent cranial windows, a 6-mm circle was drawn over the frontal and parietal regions of the skull bilaterally. Using a high-speed air-turbine drill with a burr-tip 0.5 mm in diameter, a groove was made on the margin of the drawn circle, until the bone flap becomes loose. The bone flap was separated from the dura mater underneath. The dura and arachnoid membranes were cut completely from the surface of both hemispheres, avoiding any damage to the sagittal sinus. The window was sealed with a 7-mm cover glass, glued to the bone with histocompatible cyanoacrylate glue.
Tumor implantation
To generate orthotopic tumors, ten days after the cranial window implantation the coverslip was removed and 1 μl of 1–500.000 cells (MGG8, GL261, BT474) were stereotactically and slowly injected with a 28-gauge micro-syringe into the left striatum of 8- to 10-wk-old female nude mice 2 mm left of the sagittal suture, 0.1 mm rostral of the bregma, and at a depth of 2 mm from the brain surface. In case of U87 tumors, 1-mm chunks were implanted in the same region described above. Then a new cover glass was glued. Of note, the brain tumour models we used are characterized by different degrees of infiltration in the surrounding tissue. Regarding the GBM models, U87 is the most nodular, GL261 – although largely nodular – presents a low degree of infiltration and MGG8 is the most infiltrative. While U87 and MGG8 tumors rarely show necrosis when the tumors are not too large, GL261 tumors usually have multiple necrotic regions48,49. BT474 tumors are mostly nodular and present necrosis when tumour volumes are large50.
Immunohistochemistry
Whole head were harvested after intracardiac perfusion with 2% formaldehyde in PBS and then fixed with 2% formaldehyde in PBS for 24–72 hours. We next embedded the brains in paraffin, and 5-μm-thick paraffin sections were cut using microtome and mounted on glass slides. For IHC (immunohistochemistry), tissues were deparaffinized, and antigen retrieval was performed in a pH 9 solution (DAKO) at 96°C. To prevent nonspecific staining, sections were incubated with 5% normal donkey serum (Jackson ImmunoResearch) in PBS before incubation with the respective primary and secondary antibodies. The primary antibodies used are listed in Table S3. Apoptag (ApopTag Peroxidase In Situ Apoptosis Detection Kit, #S7100, Millipore) was used as a marker for apoptosis.
The number of neurons in the cortex was assessed by quantifying the number of NeuN+ nuclei in mosaics of the coronal sections of the region of the cortex exposed to the compression apparatus, i.e. the anterior cingulate area, the primary and secondary motor area and the first part of primary somatosensory area. The structures were identified using the mouse brain atlas map.
Intravital optical frequency domain imaging and perfused vessel analysis
In vivo imaging of perfused vessels and analysis of the vascular fraction and diameter was performed using the optical coherence tomography (OCT) as previously described13,64. OCT assessment was performed at indicated time until the mouse death/euthanasia. The construction, design and algorithms used to derive and quantify OCT images were previously described in detail13. In Figure 2A, we segmented and rendered vessels using a semi-automated algorithm (Bitplane Imaris Image Analysis software).
Multi-photon intravital microscopy
For intravital microscopy, the cranial window-bearing animals were anesthetized with isoflurane and fixed with a bilateral plastic holder. The MPLSM consisted of a MilleniaX pumped Tsunami Ti:sapphire laser (Spectra-Physics). MPLSM microscope consisted of an Olympus Fluoview FV300 system customized for multiphoton imaging. For TAMRA and GFP 810-nm excitation laser light, while for EGFP/DsRed/BlueCasade sequential imaging with 730- and 930-nm excitation laser lights were used. Power at the sample was estimated to be 1–3 mW. The used emission band pass filters were the following: 610±75 nm for TAMRA/DsRed, 535±43 nm for GFP/EGFP and 413±4 nm (CascadeBlue). We performed vessel angiography after retro-orbital injection of 0.1 ml of 10 mg/ml TAMRA- or CascadeBlue-tagged dextran (500 kDa; in-house conjugated). The images shown are 3D rendering of high-resolution z-stacks: 706×706 μm (xy-voxel of 0.69) and at least 50 μm of z-stack (z-steps 2 μm). We segmented and rendered vessels and tumour cells using a semi-automated algorithm (Bitplane Imaris Image Analysis software).
Ultrasound measurements of intracranial tumor volume
To assess tumour size in U87 and BT474 preclinical models, in vivo imaging was performed through plastic cranial windows using a small animal ultrasonography device (Vevo 2100, FujiFilm VisualSonics Inc., Toronto, Canada).
In vivo lectin perfusion
For lectin perfusion analyses, mice were slowly (~2 min) injected with 100 ml of 1 mg/ml biotin-conjugated lectin (Vector Labs) via retro-orbital injection 5 min before brain harvest, long enough for lectin to reach perfused vessels. The brains were then excised, fixed in 4% formaldehyde in PBS overnight and embedded in paraffin blocks4.
Patient eligibility
Study A:
Sixty-four previously untreated (“pre-surgical”) adult patients (mean age= 62 years; range 32–87; 29 females and 35 males) with a subsequent histologically confirmed diagnosis of a glioblastoma were retrospectively recruited from a cohort of patients examined at our institution65. All patients were referred for a conventional MRI examination, including structural and contrast-enhanced perfusion imaging. Evidence of contrast-enhancing tumor on structural MRI scans (>1cm in at least one dimension and/or > 10cc) was required. No forms of anti-cancer medicines or Gliadel wafers were allowed for study inclusion, except for the use of steroids as part of standard-of-care. Other inclusion criteria included a Karnofsky Performance Status (KPS) score of >60. Use of steroids and KPS scores were recorded at the time of the MRI exam.
Study B:
A second cohort of 14 adult patients (mean age 59 years; range 35–77; 8 females and 6 males) with histologically confirmed newly diagnosed glioblastoma (‘post-surgical’) at the time of the MRI exam was also retrospectively included66. The MRI exams were pulled from the baseline data (pre-therapy) part of an observational imaging study assessing the effects of chemo-radiation monotherapy (clinicalTrials.gov identifier NCT00756106). Except for the post-surgical status, the inclusion criteria matched that of the pre-surgical study described above. A minimum of >1cm residual tumour in at least one dimension was required and patients with gross total resections were excluded.
Study C:
Finally, baseline MRI data from a cohort of 34 patients (mean age 47 years; range 31–62; all females) with metastatic breast cancer to the brain were pulled from an open-label, non-randomized, phase II study on carboplatin and bevacizumab (clinicalTrials.gov identifier NCT01004172). Patients were either HER2-negative (8 patients) or HER2-positive (26 patients). Evidence of contrast-enhancing tumour on structural MRI scans (>1cm in at least one dimension) was required. Other inclusion criteria were no contraindication to MRI, an Eastern Cooperative Oncology Group performance status (ECOG PS) of 0–2, no increase in corticosteroid dose in the week prior to the baseline MR exam, left ventricular ejection fraction >50%, and adequate bone marrow, liver and renal function. Patients could have received prior carboplatin monotherapy. Prior bevacizumab was allowed if it had not been given since or within 6 months prior to the diagnosis of CNS metastases. Prior radiation therapy to the brain was allowed, conditioned by radiologic confirmation of subsequent brain lesions progression, or presence of one or more untreated residual lesions for analysis in our study. Patients who had not received radiotherapy were allowed if they did not require corticosteroids. There was no limit on the number of prior therapies. A two-week washout period was required from chemotherapy or any other investigational therapy. A one-week washout period was required for lapatinib. No washout period was required for trastuzumab.
All patient studies were approved by the Institutional Review Boards of MGH or Dana-Farber, including the use of informed consent.
MRI acquisition:
For all studies, each subject was scanned prior to the start of therapy. For study A65, patients were scanned on either a 1.5 Tesla MRI system (Signa; GE Healthcare) or a 3.0 Tesla MRI system (TimTrio, Siemens Healthcare). For studies B and C, all MRI exams were performed at 3.0 Tesla (TimTrio, Siemens Healthcare)66. The MRI scans specific for our study were typically as follows;
-
1.
FLAIR images:
Axial images with a repetition time (TR) 10000ms, echo time (TE) 70ms, 5mm slice thickness, 1mm inter-slice gap, 0.43mm in-plane resolution, 23 slices and a 512×512 matrix.
-
2.
T1 images:
Axial T1-weighted images acquired prior to – and after - injection of contrast agent (gadopentetate-dimeglumine, Gd-DTPA, Magnevist, Bayer Schering Pharma AG, Berlin, Germany). TR 600ms, TE 12ms, 5mm slice thickness, 1mm inter-slice gap, 0.43mm in-plane resolution, 23 slices and a 512×512 matrix.
-
3.
Diffusion images:
Axial images using a twice refocused echo-planar diffusion weighted sequence with TR ~8000ms, TE 80–85 ms, low and high b-values of 0s/mm2 and 700s/mm2 in 7 and 42 directions, respectively, for diffusion tensor reconstruction. Resolution was 2 mm isotropic, 64 slices and a 128 × 128 matrix.
-
4.
Perfusion images:
Axial dynamic susceptibility contrast (DSC) images using a gradient-echo echo-planar imaging sequence with TR 1400–1500ms, TE 30–35ms, flip angle 90°, 5mm slice thickness, 1.5mm inter-slice gap, 1.2mm in-plane resolution, 12 slices and a 160×160 matrix. A total of 100 volumes were acquired and 0.1mmol/kg of Gd-DTPA was injected at 5cc/s after approximately 81s of imaging. In addition, for studies B and C as part of a dynamic contrast enhanced image acquisition preceding the DSC imaging, patients also received a 0.1mmol/kg of Gd-DTPA acting as a pre-dose to minimize computational errors due to T1-shortening effects induced by contrast agent leakage in regions of blood–brain barrier breakdown or resection.
MRI post-processing
-
1.
Volumetrics: Using a previously described volumetric approach67, areas of signal intensity abnormality on FLAIR and CE-T1 images were outlined by an experienced neuroradiologist blinded to the order of the scans. Total tumour volume was estimated by summing all voxels in the enhancing tumour. All images, including the tumour outlines from the second visit, were realigned to the perfusion MR images at the first visit using normalized mutual information coregistration in Matlab (MathWorks, Natick, MA, USA).
-
2.
Diffusion: Apparent diffusion coefficient (ADC) maps estimating the relative water self-diffusion on a voxel-by-voxel basis were created from the low and high b-value images using standard Steskjal–Tanner diffusion approximation68 as well as measures of fractional anisotropy (FA)69.
-
3.
Perfusion: Cerebral blood flow (CBF) maps from the gradient-echo perfusion MRIs were calculated using established models in nordicICE (NordicNeuroLab, Bergen, Norway) and corrected for contrast-agent leakage70. Patient-specific variations were reduced by normalization of all brain voxel values to an average value from a normal-appearing gray- and white-matter tissue map excluding pathological areas as identified by the neuroradiologist on the MRIs71.
-
4.
Perfusion-gradient analysis: An image voxel-level, unsupervised morphological dilatation in Matlab was performed from the edge of the pathologic area (CE and FLAIR) and in steps equaling one image voxel growth in all outwards directions (Figure S5A). The algorithm was only allowed to grow in regions with voxels included in the normal-appearing gray- and white-matter tissue map, excluding large vessels, regions of necrosis and/or cysts71,72. A stable/increase (“unchanged perf”) or reduction (“reduced perf”) in the perfusion gradient in the peri-edematous region was defined as being over/below a 5% difference from the average perfusion value of normal-appearing tissue in regions outside a >10 pixels range (>12mm distance from the peri-edematous zone).
VASARI Scoring of MRI
Pre-surgical MRI images for the 64 treatment naive glioblastoma (Study A) were qualitatively scored for morphological features associated with nodular and infiltrative phenotypes by a board certified neuroradiologist reader with 10 years of clinical and research experience on brain tumors. The reader was blinded to clinical information (KPS, overall survival) and advanced MRI data (perfusion metrics) and classified tumors as “well defined” versus “poorly defined” according to features f12 and f13 of the VASARI criteria (reference 8 and https://wiki.cancerimagingarchive.net/display/Public/VASARI+Research+Project) utilizing CE-T1 and FLAIR images respectively. In addition, tumors were scored according to their shape on CE-T1 and FLAIR images utilizing an ordinal four-tier scheme: (1) sphere/oval, (2) sphere/oval + lobulations, (3) irregular and (4) non mass or diffuse/patchy. Nodular, well-defined tumors usually grow concentrically and are expected to have more spherical/oval shapes, while infiltrative, poorly marginated tumors are expected to have more irregular or diffuse/patchy appearances.
Compression apparatus model
The compression apparatus is comprised of a polyvinyl chloride (PVC) circular disk (2 mm thick, 7 mm in diameter) acting as a modified cranial window, a stainless steel set screw (M2×5mm) to apply the controlled chronic displacement on the brain tissue, a PVC membrane (0.01 mm thick) to separate the brain tissue from the set screw and other components, and a custom-made PDMS disk (1 mm thick, 3 mm in diameter) that surrounds the set screw from the top of the window to reduces potential backlash, backing off, or loosening of the set screw. All the components are purchased from McMaster-Carr. The PVC membrane is glued to the edge of the PVC disk by glue. The outer side of the PVC membrane, which is in touch with brain tissue, is clear of any glue to avoid any toxicity. The device is rinsed with ethanol prior to the surgical procedure.
The compression was performed under gas anaesthesia (isoflurane) every 2–3 days. Each turn of the screw was measured by OCT in order to be sure that equal amount of compressing volume was applied to every cortex. No tissue toxicity was detected in brain in contact with the membrane of the compression apparatus. No haemorrhages happened in any mouse as a consequence of compression or decompression of the cortex. In rare cases of rupture of the membrane of the compression apparatus, the apparatus inevitably felt off from the implantation caused of change in intracranial pressure; in those cases we immediately euthanized and excluded the mice from the study. Mice were daily checked for potential pain and distress. At endpoint each compression apparatus was checked to confirm that no breach of the membrane happened.
Histology
Whole head were harvested after intracardiac perfusion with 2% formaldehyde in PBS and then fixed with 2% formaldehyde in PBS for 24–72 hours. We next embedded the brains in paraffin, and 5-μm-thick paraffin sections were cut using microtome and mounted on glass slides. Hematoxylin-eosin (H&E) and Cresyl violet (Nissl staining) stains are commonly used histological methods for the morphological assessments of the cells in brain tissue. Thus the paraffin slices were stained with H&E and Cresyl violet in the study for histologic evaluation.
Ultrastructural analysis by electromicroscopy
Whole head were harvested after intracardiac perfusion with 2% formaldehyde 2% glutaraldehyde in PBS and then fixed with 2% formaldehyde 2% glutaraldehyde in PBS for 24–72 hours. Vibratome sections (50 m) of mouse brain were post-fixed in 2.5% glutaraldehyde/PBS for 30 minutes, washed well in PBS, treated with 1% osmium tetraoxide/PBS for 1 hour, then 1% uranyl acetate/70% ethanol for 45 minutes. The tissue was then dehydrated in alcohol, embedded in EPON, thin-sectioned on an UltraCut E (Reichert-Jung), and examined using a JEOL JEM-1011 transmission electron microscope with AMTv601 software (Advanced Microscopy Techniques, Woburn, MA).
RNA isolation and quantitative Real Time PCR (qPCR)
Mouse brain tissues were homogenized and total RNA was isolated utilizing the RNeasy Mini kit (Qiagen, Valencia, CA) according to the manufacturer’s protocol. Genomic DNA was removed using RNase-Free DNase Set (Qiagen, Valencia, CA). 1 μg of RNA was used for the first strand cDNA synthesis (Bio-Rad, Hercules, CA). A no-reverse transcriptase control was included in the same PCR mixtures without reverse transcriptase to confirm the absence of DNA contamination in RNA samples. qPCR primers were synthesized at MGH DNA core facility (Table S4). 25-μl reactions were carried out in 96-well optical reaction plates using SYBR Green PCR Master Mix (Bio-Rad, Hercules, CA) with gene-specific primers, and the qPCR was run in the Stratagene Mx3000p Real-Time PCR System (San Diego, CA). Amplification of each sample was analysed by melting curve analysis, and relative differences in each PCR sample were corrected using GAPDH mRNA as an endogenous control and normalized to the level of control by using the 2-ΔΔCt method as previous described73,74.
Rotarod test
Neurological function of the mice was assessed by using an automated Rotarod (Rotamex 4/8 4-Lane Treadmill Shock Grid; Columbus Instruments, Livoniz, MI). To exclude the effect of motivational factors and to differentiate between motor learning and actual neurological function, mice were trained on the Rotarod every day for 3 days. Mice were randomized at the beginning of the study in order to have the same means in Rotarod endurance. The Rotarod test was performed always at similar period of the day and after 1 hour of acclimation of the mice in the room. We excluded mice that repeatedly fell off very early (<20 secs). Tests in two consecutive days were performed for each animal. Each test began with a 30 secs acclimation period at 4 rpm followed by acceleration by 4 rpm every 30 secs. The latency to fall was recorded as Rotarod endurance75.
Static Gait test
Gait characteristics (stride and track length) were analysed by applying ink to the animals’ hind paws and letting them walk on a strip of paper, inside a brightly lit alley (4.5 cm large, 40 cm long), towards a dark goal box. Mice were tested 3 consecutive times on the day of maximum compression of the cortex (day 12).
Short-term release of the solid stress
The release of the tumour-induced compression of the surrounding tissue was performed by removing the cranial window from anesthetized mice. This allowed us to obtain a map of the mechanical stress-induced deformation in a brain with or without the nodular U87 tumour. We then checked the dynamics of in vivo perfusion during the process of the release of the tumour-induced compression by OCT. Mice were then euthanized.
Therapeutic neuroprotection treatments
Mice were randomized at the beginning of the study by Rotarod endurance. We systemically treated mice with the following drugs during the chronic/gradual compression and the two following weeks after decompression: necrostatin-1 (Nec-1; 7 mg/kg/d i.p.; similar to26), valproic acid (VPA; 400 mg/kg/d i.p.27) and lithium chloride (LiCl; 50 mg/kg/d i.p.28). All these drugs are known to cross the blood-brain barrier31–33 and shown to be neuroprotective during CNS ischemia or neurodegeneration26–28. In the case of dexamethasone (Dexa), the steroid anti-edema drug was given once (10 mg/kg29,30) at the day of maximum compression (day 14). In the case of lithium treatment “intervention study” (i.e. with treatment start when compression was already applied to the brains), lithium (LiCl; 50 mg/kg/d i.p.28) was given from day 10 after the chronic compression initiation.
RNA-Sequencing
RNA-sequencing was performed by the Massachusetts Institute of Technology BioMicro Center. Quality of RNA samples was confirmed using a Fragment Analyzer (Advanced Analytical Technologies, Inc.) before Illumina libraries were prepared and indexed from ~250ng of total RNA using the Kapa Hyperprep kit following manufacturer’s recommendations (Roche). Libraries were confirmed using the Fragment Analyzer and quantified by qPCR prior to pooling and sequencing on an Illumina NextSeq500 using 40+40 paired end reads. Samples were demultiplexed using custom scripts allowing single mismatch to the index sequencing.
Analysis of RNA-Sequencing data
The raw FASTQ files were aligned by STAR76 to the Ensembl GRCm38 reference genome. Gene read counts were then generated using the Bioconductor R package GenomicAlignments77. The Bioconductor R package DESeq278 was then used to normalize and determine the differentially expressed genes. The DAVID Bioinformatics Resource34 was used for gene-annotation enrichment analysis on the differentially expressed genes.
Statistical Analysis
Unless otherwise specified, data are presented as mean ± s.e.m. Student’s two-tailed t tests based on Gaussian distributions were used to assess the significance. For comparisons among more than two groups, ANOVA or Kruskal-Wallis tests were followed by post hoc multiple comparisons. P<0.05 was considered significant. For comparison between categorical datasets, Fisher’s test was performed. Blinding during analysis was used for all the experiments and clinical analyses. Statistics were performed using Graphpad Prism or Microsoft Excel. The detailed list of the performed tests is provided in Table S5.
Supplementary Material
Acknowledgements
We thank Drs. A. Ivinson (UK Dementia Research Institute), M.A. Moskowitz and M.J. Whalen (MGH) for critical discussion and insightful suggestions; S. Roberge, M. Duquette, C. Smith and E. L. Jones (MGH) for the technical support, H. Wakimoto (MGH) for MGG8 cell line and O. Rapalino (MGH) for the help with the pre-operative clinical study. This work was supported by the National Cancer Institute (NCI; P01-CA080124, P50-CA165962, R01-CA129371, R01-CA208205, U01-CA 224348), NCI Outstanding Investigator Award (R35-CA197743), the Lustgarten Foundation, the Ludwig Center at Harvard, the National Foundation for Cancer Research and the Gates Foundation (RKJ), R01-HL128168 (to JWB, TPP and LLM), DP2OD008780 (TPP), R01CA214913 (TPP), P41EB015903 (Center for Biomedical OCT Research and Translation), NIH/NINDS P30NS045776 (EM facility core), P30-CA14051 from NCI (Koch Institute Genomics core). This work was also supported in part by the Susan G. Komen Foundation Fellowship PDF14301739, Fondation ARC pour la recherche sur le cancer and the INSERM-CNRS ATIP-Avenir grant (GS), NCI F32-CA216944–01 (HTN), the European Research Council (ERC) under the European Union’s Horizon 2020 (grant agreement No 758657), the South-Eastern Norway Regional Health Authority grants 2017073, 2016102 and 2013069, the Research Council of Norway grants 261984 and ES435705, the Norwegian Cancer Society grants 6817564 and 3434180 (KEE), F31HL126449 from the National Heart, Lung, and Blood Institute at the NIH (MD), SolidarImmun fellowship (JK), Feodor-Lynen Postdoctoral Fellowship from Alexander von Humboldt Foundation (MG), Deutsche Forschungsgemeinschaft AS422–2/1 (VA).
Footnotes
Competing interests: RKJ received honorarium from AMGEN, consultant fees from Pfizer, Ophthotech, Merck, SPARC, SynDevRx and XTuit. RKJ owns equity in Enlight, Ophthotech and SynDevRx, and serves on the Boards of Trustees of Tekla Healthcare Investors, Tekla Life Sciences Investors, the Tekla Healthcare Opportunities Fund and the Tekla World Healthcare Fund. No reagents or funding from these companies were used in these studies. KEE has intellectual properties with NordicNeuroLab AS, Bergen, Norway.
Data Availability
The authors declare that all data supporting the findings of this study are available within the paper and its supplementary information. Raw RNA-sequencing data from this study have been deposited in the NCBI Sequence Read Archive (SRA) under submission ID SUB4405185 and BioProject ID PRJNA486395.
Code availability.
The computer codes used in this study are available from the corresponding author upon reasonable request.
References
- 1.Goriely A et al. Mechanics of the brain: perspectives, challenges, and opportunities. Biomech Model Mechanobiol 14, 931–965, doi: 10.1007/s10237-015-0662-4 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Helmlinger G, Netti PA, Lichtenbeld HC, Melder RJ & Jain RK Solid stress inhibits the growth of multicellular tumor spheroids. Nature Biotechnology 15, 778–783, doi: 10.1038/nbt0897-778 (1997). [DOI] [PubMed] [Google Scholar]
- 3.Stylianopoulos T et al. Causes, consequences, and remedies for growth-induced solid stress in murine and human tumors. Proc Natl Acad Sci U S A 109, 15101–15108, doi: 10.1073/pnas.1213353109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nia HT et al. Solid stress and elastic energy as measures of tumour mechanopathology. Nat Biomed Eng 1, doi: 10.1038/s41551-016-0004 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jain RK, Martin JD & Stylianopoulos T The role of mechanical forces in tumor growth and therapy. Annual Review of Biomedical Engineering 16, 321 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chauhan VP et al. Compression of pancreatic tumor blood vessels by hyaluronan is caused by solid stress and not interstitial fluid pressure. Cancer Cell 26, 14–15, doi: 10.1016/j.ccr.2014.06.003 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stylianopoulos T et al. Coevolution of solid stress and interstitial fluid pressure in tumors during progression: implications for vascular collapse. Cancer Res 73, 3833–3841, doi: 10.1158/0008-5472.CAN-12-4521 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gamburg ES et al. The prognostic significance of midline shift at presentation on survival in patients with glioblastoma multiforme. Int J Radiat Oncol Biol Phys 48, 1359–1362 (2000). [DOI] [PubMed] [Google Scholar]
- 9.Nia HT et al. Quantifying solid stress and elastic energy from excised or in situ tumors. Nat Protoc 13, 1091–1105, doi: 10.1038/nprot.2018.020 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mazurowski MA, Desjardins A & Malof JM Imaging descriptors improve the predictive power of survival models for glioblastoma patients. Neuro Oncol 15, 1389–1394, doi: 10.1093/neuonc/nos335 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chambless LB et al. The relative value of postoperative versus preoperative Karnofsky Performance Scale scores as a predictor of survival after surgical resection of glioblastoma multiforme. J Neurooncol 121, 359–364, doi: 10.1007/s11060-014-1640-x (2015). [DOI] [PubMed] [Google Scholar]
- 12.Chauhan VP et al. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat Commun 4, 2516, doi: 10.1038/ncomms3516 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vakoc BJ et al. Three-dimensional microscopy of the tumor microenvironment in vivo using optical frequency domain imaging. Nat Med 15, 1219–1223, doi: 10.1038/nm.1971 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bar-Kochba E, Scimone MT, Estrada JB & Franck C Strain and rate-dependent neuronal injury in a 3D in vitro compression model of traumatic brain injury. Sci Rep 6, 30550, doi: 10.1038/srep30550 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arundine M, Aarts M, Lau A & Tymianski M Vulnerability of central neurons to secondary insults after in vitro mechanical stretch. J Neurosci 24, 8106–8123, doi: 10.1523/JNEUROSCI.1362-04.2004 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Field AS et al. Diffusion tensor eigenvector directional color imaging patterns in the evaluation of cerebral white matter tracts altered by tumor. J Magn Reson Imaging 20, 555–562, doi: 10.1002/jmri.20169 (2004). [DOI] [PubMed] [Google Scholar]
- 17.Blanchet L et al. Discrimination between metastasis and glioblastoma multiforme based on morphometric analysis of MR images. AJNR Am J Neuroradiol 32, 67–73, doi: 10.3174/ajnr.A2269 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takano T et al. Glutamate release promotes growth of malignant gliomas. Nat Med 7, 1010–1015, doi: 10.1038/nm0901-1010 (2001). [DOI] [PubMed] [Google Scholar]
- 19.Balkaya M, Krober JM, Rex A & Endres M Assessing post-stroke behavior in mouse models of focal ischemia. J Cereb Blood Flow Metab 33, 330–338, doi: 10.1038/jcbfm.2012.185 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roth L et al. Impaired gait pattern as a sensitive tool to assess hypoxic brain damage in a novel mouse model of atherosclerotic plaque rupture. Physiol Behav 139, 397–402, doi: 10.1016/j.physbeh.2014.11.047 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Budday S et al. Mechanical characterization of human brain tissue. Acta Biomater 48, 319–340, doi: 10.1016/j.actbio.2016.10.036 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Budday S et al. Mechanical properties of gray and white matter brain tissue by indentation. J Mech Behav Biomed Mater 46, 318–330, doi: 10.1016/j.jmbbm.2015.02.024 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnson CL et al. Local mechanical properties of white matter structures in the human brain. Neuroimage 79, 145–152, doi: 10.1016/j.neuroimage.2013.04.089 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ohue S et al. Evaluation of intraoperative brain shift using an ultrasound-linked navigation system for brain tumor surgery. Neurol Med Chir (Tokyo) 50, 291–300 (2010). [DOI] [PubMed] [Google Scholar]
- 25.Paul DA et al. White matter changes linked to visual recovery after nerve decompression. Sci Transl Med 6, 266ra173, doi: 10.1126/scitranslmed.3010798 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang SH et al. Nec-1 alleviates cognitive impairment with reduction of Abeta and tau abnormalities in APP/PS1 mice. EMBO Mol Med 9, 61–77, doi: 10.15252/emmm.201606566 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dash PK et al. Valproate administered after traumatic brain injury provides neuroprotection and improves cognitive function in rats. PLoS One 5, e11383, doi: 10.1371/journal.pone.0011383 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Makoukji J et al. Lithium enhances remyelination of peripheral nerves. Proc Natl Acad Sci U S A 109, 3973–3978, doi: 10.1073/pnas.1121367109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rocksen D, Lilliehook B, Larsson R, Johansson T & Bucht A Differential anti-inflammatory and anti-oxidative effects of dexamethasone and N-acetylcysteine in endotoxin-induced lung inflammation. Clin Exp Immunol 122, 249–256 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Capasso A, Di Giannuario A, Loizzo A, Pieretti S & Sorrentino L Dexamethasone pretreatment reduces the psychomotor stimulant effects induced by cocaine and amphetamine in mice. Prog Neuropsychopharmacol Biol Psychiatry 19, 1063–1079 (1995). [DOI] [PubMed] [Google Scholar]
- 31.Forester BP et al. Brain lithium levels and effects on cognition and mood in geriatric bipolar disorder: a lithium-7 magnetic resonance spectroscopy study. Am J Geriatr Psychiatry 17, 13–23, doi: 10.1097/JGP.0b013e318172b3d0 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Phiel CJ et al. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem 276, 36734–36741, doi: 10.1074/jbc.M101287200 (2001). [DOI] [PubMed] [Google Scholar]
- 33.Jagtap PG et al. Structure-activity relationship study of tricyclic necroptosis inhibitors. J Med Chem 50, 1886–1895, doi: 10.1021/jm061016o (2007). [DOI] [PubMed] [Google Scholar]
- 34.Huang da W, Sherman BT & Lempicki RA Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4, 44–57, doi: 10.1038/nprot.2008.211 (2009). [DOI] [PubMed] [Google Scholar]
- 35.Can A et al. Antidepressant-like responses to lithium in genetically diverse mouse strains. Genes Brain Behav 10, 434–443, doi: 10.1111/j.1601-183X.2011.00682.x (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moore GJ, Bebchuk JM, Wilds IB, Chen G & Manji HK Lithium-induced increase in human brain grey matter. Lancet 356, 1241–1242 (2000). [DOI] [PubMed] [Google Scholar]
- 37.Foland LC et al. Increased volume of the amygdala and hippocampus in bipolar patients treated with lithium. Neuroreport 19, 221–224, doi: 10.1097/WNR.0b013e3282f48108 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rowe MK & Chuang DM Lithium neuroprotection: molecular mechanisms and clinical implications. Expert Rev Mol Med 6, 1–18, doi: 10.1017/S1462399404008385 (2004). [DOI] [PubMed] [Google Scholar]
- 39.Yazlovitskaya EM et al. Lithium treatment prevents neurocognitive deficit resulting from cranial irradiation. Cancer Res 66, 11179–11186, doi: 10.1158/0008-5472.CAN-06-2740 (2006). [DOI] [PubMed] [Google Scholar]
- 40.Yu F et al. Lithium ameliorates neurodegeneration, suppresses neuroinflammation, and improves behavioral performance in a mouse model of traumatic brain injury. J Neurotrauma 29, 362–374, doi: 10.1089/neu.2011.1942 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li Q et al. Lithium reduces apoptosis and autophagy after neonatal hypoxia-ischemia. Cell Death Dis 1, e56, doi: 10.1038/cddis.2010.33 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Han S et al. Lithium enhances the antitumour effect of temozolomide against TP53 wild-type glioblastoma cells via NFAT1/FasL signalling. Br J Cancer 116, 1302–1311, doi: 10.1038/bjc.2017.89 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Korur S et al. GSK3beta regulates differentiation and growth arrest in glioblastoma. PLoS One 4, e7443, doi: 10.1371/journal.pone.0007443 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nowicki MO et al. Lithium inhibits invasion of glioma cells; possible involvement of glycogen synthase kinase-3. Neuro Oncol 10, 690–699, doi: 10.1215/15228517-2008-041 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meyers CA & Brown PD Role and relevance of neurocognitive assessment in clinical trials of patients with CNS tumors. J Clin Oncol 24, 1305–1309, doi: 10.1200/JCO.2005.04.6086 (2006). [DOI] [PubMed] [Google Scholar]
- 46.Wakimoto H et al. Maintenance of primary tumor phenotype and genotype in glioblastoma stem cells. Neuro Oncol 14, 132–144, doi: 10.1093/neuonc/nor195 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Griveau A et al. A Glial Signature and Wnt7 Signaling Regulate Glioma-Vascular Interactions and Tumor Microenvironment. Cancer Cell 33, 874–889 e877, doi: 10.1016/j.ccell.2018.03.020 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kloepper J et al. Ang-2/VEGF bispecific antibody reprograms macrophages and resident microglia to anti-tumor phenotype and prolongs glioblastoma survival. Proc Natl Acad Sci U S A 113, 4476–4481, doi: 10.1073/pnas.1525360113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Peterson TE et al. Dual inhibition of Ang-2 and VEGF receptors normalizes tumor vasculature and prolongs survival in glioblastoma by altering macrophages. Proc Natl Acad Sci U S A 113, 4470–4475, doi: 10.1073/pnas.1525349113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Askoxylakis V et al. Preclinical Efficacy of Ado-trastuzumab Emtansine in the Brain Microenvironment. J Natl Cancer Inst 108, doi: 10.1093/jnci/djv313 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nia HT, Han L, Li Y, Ortiz C & Grodzinsky A Poroelasticity of Cartilage at the Nanoscale. Biophys J 101, 2304–2313, doi:DOI 10.1016/j.bpj.2011.09.011 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Netti PA, Berk DA, Swartz MA, Grodzinsky AJ & Jain RK Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer research 60, 2497–2503 (2000). [PubMed] [Google Scholar]
- 53.Kiviranta P et al. Collagen network primarily controls Poisson’s ratio of bovine articular cartilage in compression. J Orthop Res 24, 690–699, doi: 10.1002/jor.20107 (2006). [DOI] [PubMed] [Google Scholar]
- 54.Buschmann MD et al. Stimulation of aggrecan synthesis in cartilage explants by cyclic loading is localized to regions of high interstitial fluid flow. Arch Biochem Biophys 366, 1–7 (1999). [DOI] [PubMed] [Google Scholar]
- 55.Jamin Y et al. Exploring the biomechanical properties of brain malignancies and their pathologic determinants in vivo with magnetic resonance elastography. Cancer Res 75, 1216–1224, doi: 10.1158/0008-5472.CAN-14-1997 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Arani A et al. Acute pressure changes in the brain are correlated with MR elastography stiffness measurements: initial feasibility in an in vivo large animal model. Magnetic resonance in medicine 79, 1043–1051 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Weickenmeier J et al. Brain stiffens post mortem. Journal of the mechanical behavior of biomedical materials 84, 88–98 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pogoda K et al. Compression stiffening of brain and its effect on mechanosensing by glioma cells. New journal of physics 16, 075002 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Storm C, Pastore JJ, MacKintosh FC, Lubensky TC & Janmey PA Nonlinear elasticity in biological gels. Nature 435, 191 (2005). [DOI] [PubMed] [Google Scholar]
- 60.Timoshenko S & Goodier J Theory of elasticity. 1951. New York 412, 108. [Google Scholar]
- 61.Mihai LA, Chin L, Janmey PA & Goriely A A comparison of hyperelastic constitutive models applicable to brain and fat tissues. Journal of The Royal Society Interface 12, 20150486 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lang GE, Stewart PS, Vella D, Waters SL & Goriely A Is the Donnan effect sufficient to explain swelling in brain tissue slices? Journal of The Royal Society Interface 11, 20140123 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Grodzinsky AJ Fields, Forces, and Flows in Biological Systems. (Garland Science, 2011). [Google Scholar]
- 64.Ager EI et al. Blockade of MMP14 activity in murine breast carcinomas: implications for macrophages, vessels, and radiotherapy. J Natl Cancer Inst 107, doi: 10.1093/jnci/djv017 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Emblem KE et al. A generic support vector machine model for preoperative glioma survival associations. Radiology 275, 228–234, doi: 10.1148/radiol.14140770 (2015). [DOI] [PubMed] [Google Scholar]
- 66.Pinho MC et al. Low incidence of pseudoprogression by imaging in newly diagnosed glioblastoma patients treated with cediranib in combination with chemoradiation. Oncologist 19, 75–81, doi: 10.1634/theoncologist.2013-0101 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sorensen AG et al. Comparison of diameter and perimeter methods for tumor volume calculation. J Clin Oncol 19, 551–557, doi: 10.1200/JCO.2001.19.2.551 (2001). [DOI] [PubMed] [Google Scholar]
- 68.Oh J et al. Quantitative apparent diffusion coefficients and T2 relaxation times in characterizing contrast enhancing brain tumors and regions of peritumoral edema. J Magn Reson Imaging 21, 701–708, doi: 10.1002/jmri.20335 (2005). [DOI] [PubMed] [Google Scholar]
- 69.Basser PJ & Pierpaoli C Microstructural and physiological features of tissues elucidated by quantitative-diffusion-tensor MRI. J Magn Reson B 111, 209–219 (1996). [DOI] [PubMed] [Google Scholar]
- 70.Bjornerud A & Emblem KE A fully automated method for quantitative cerebral hemodynamic analysis using DSC-MRI. J Cereb Blood Flow Metab 30, 1066–1078, doi: 10.1038/jcbfm.2010.4 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Emblem KE & Bjornerud A An automatic procedure for normalization of cerebral blood volume maps in dynamic susceptibility contrast-based glioma imaging. AJNR Am J Neuroradiol 30, 1929–1932, doi: 10.3174/ajnr.A1680 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Emblem KE, Due-Tonnessen P, Hald JK & Bjornerud A Automatic vessel removal in gliomas from dynamic susceptibility contrast imaging. Magn Reson Med 61, 1210–1217, doi: 10.1002/mrm.21944 (2009). [DOI] [PubMed] [Google Scholar]
- 73.Ren J et al. Protein kinase C-delta (PKCdelta) regulates proinflammatory chemokine expression through cytosolic interaction with the NF-kappaB subunit p65 in vascular smooth muscle cells. J Biol Chem 289, 9013–9026, doi: 10.1074/jbc.M113.515957 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vitner EB et al. RIPK3 as a potential therapeutic target for Gaucher’s disease. Nat Med 20, 204–208, doi: 10.1038/nm.3449 (2014). [DOI] [PubMed] [Google Scholar]
- 75.Gao X et al. Anti-VEGF treatment improves neurological function and augments radiation response in NF2 schwannoma model. Proc Natl Acad Sci U S A 112, 14676–14681, doi: 10.1073/pnas.1512570112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21, doi: 10.1093/bioinformatics/bts635 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lawrence M et al. Software for computing and annotating genomic ranges. PLoS Comput Biol 9, e1003118, doi: 10.1371/journal.pcbi.1003118 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550, doi: 10.1186/s13059-014-0550-8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.