

SUMMARY
Cancers from sun-exposed skin accumulate “driver” mutations, causally implicated in oncogenesis. Since errors incorporated during translesion synthesis (TLS) opposite UV lesions would generate these mutations, TLS mechanisms are presumed to underlie cancer development. To address the role of TLS in skin cancer formation, we determined which DNA polymerase is responsible for generating UV mutations, analyzed the relative contributions of error-free TLS by Polη and error-prone TLS by Polθ to the replication of UV damaged DNA and to genome stability, and examined the incidence of UV induced skin cancers in Polθ−/−, Polη−/−, and Polθ−/− Polη−/− mice. Our findings that the incidence of skin cancers rises in Polθ−/− mice and is further exacerbated in Polθ−/− Polη−/− mice than in Polη−/− mice support the conclusion that error-prone TLS by Polθ provides a safeguard against tumorigenesis and suggest that cancer formation can ensue in the absence of somatic point mutations.
Keywords: UV signature mutations, error-prone translesion synthesis, DNA polymerase θ, DNA polymerase η, replication stress, genomic rearrangements, skin cancers
Graphical Abstract
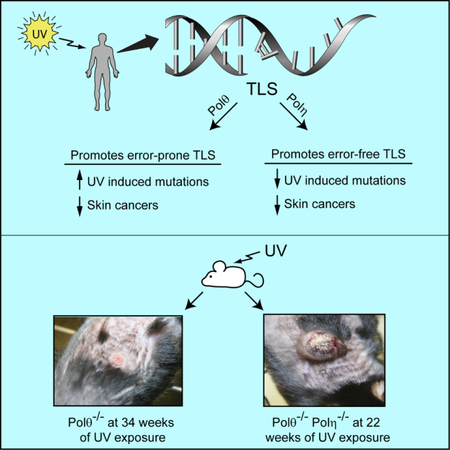
In Brief
Both error-free TLS by DNA polymerase η and error-prone TLS by DNA polymerase θ through UV lesions protect against replication stress induced chromosomal instability and prevent skin cancer formation.
INTRODUCTION
The UV component of sunlight is a major epidemiological risk factor for skin cancers that include basal cell carcinoma (BCC), squamous cell carcinoma (SCC), and melanoma (Glass and Hoover, 1989; Magnus, 1991; Oikarinen and Raitio, 2000). UV induces covalent links between two adjacent pyrimidines and causes the formation of two major types of photoproducts, cis-syn cyclobutane pyrimidine dimers (CPDs) and (6–4) pyrimidine-pyrimidone photoproducts [(6–4) PPs]. Because of their higher level of induction and slower rate of repair, CPDs constitute a much greater proportion of pro-mutagenic lesion than (6–4) PPs and it has been estimated that CPDs account for ~ 80% of UVB induced mutations in mammalian cells (Pfeifer, 1997; Yoon et al., 2000; You et al., 2001). The incidence of UV induced mutations is highly elevated in nucleotide excision repair (NER) defective xeroderma pigmentosum (XP) cells and DNA Polymerase (Pol) η defective XP variant (XPV) cells also exhibit high UV mutability (Friedberg et al., 2005). Since the incidence of UV induced mutations and UV induced skin cancers rises in the absence of NER, and also in the absence of Polη, which replicates through CPDs in a predominantly error-free manner in human cells (Yoon et al., 2009), the elevation in mutation frequency resulting from error-prone replication through UV lesions is thought to underlie skin cancers in XP and XPV patients. The evidence that “driver” mutations which confer selective clonal growth advantage accumulate in skin cancers (Durinck et al., 2011; Jayaraman et al., 2014; Martincorena et al., 2017; Pickering et al., 2014) is also compatible with a causal relationship between translesion synthesis (TLS) mechanisms which incorporate errors during replication and tumorigenesis.
To determine the contribution of error-prone TLS to skin cancer formation, we sought to identify the DNA polymerase responsible for generating UV-induced mutations. Here we show that Polθ is indispensable for mutagenic replication through CPDs and it is also essential for mutagenic TLS opposite (6–4) PPs. Since Polθ deficiency abolishes UV induced mutations, we generated Polθ−/− mice and examined their susceptibility to UV induced skin cancers. Contrary to the expectation of a reduction in tumorigenesis, we find that the susceptibility to UV induced skin cancers is elevated in Polθ−/− mice and it is further exacerbated in Polη−/− Polθ−/− mice; thus, error-prone TLS protects against skin cancer formation.
To evaluate how TLS mechanisms might prevent cancer formation, we analyzed the contributions of error-free TLS by Polη and error-prone TLS by Polθ to the replication of UV damaged DNA and to the prevention of genome instability. Our data indicate that by preventing the collapse of replication forks (RFs) stalled at DNA lesions, both error-free and error-prone TLS mechanisms act as potent inhibitors of DNA double strand break (DSB) formation and of the ensuing increase in genomic rearrangements; thereby, TLS mechanisms provide an effective barrier to chromosomal instability and tumorigenesis.
RESULTS
Requirement of Polθ for TLS Opposite UV Lesions in Human Cells
The SV40-based duplex plasmid system, in which bidirectional replication initiates from an origin of replication, provides a convenient and highly reliable means for analyzing the genetic control of TLS opposite a DNA lesion carried on the leading or the lagging strand DNA template. In this system, the frequency of blue colonies among the total Kan+ colonies provides a highly repeatable measure of TLS frequency (Yoon et al., 2009). Previously, we have provided evidence that TLS through a cis-syn TT dimer occurs via Polη-dependent pathway which acts in an error-free manner, and via two other pathways, which depend respectively upon Polκ and Polζ and act in an error-prone manner (Yoon et al., 2009) (Figure S1A). By contrast to Polη, which can proficiently incorporate a nt opposite the 3’T and 5’T of the cis-syn dimer (Biertumpfel et al., 2010; Johnson et al., 1999a; Johnson et al., 1999b; Masutani et al., 1999; Silverstein et al., 2010) Pols κ and ζ lack the ability to incorporate a nt opposite the 3’T of the cis-syn TT dimer, but they can insert a nt opposite the 5’T of the dimer and extend further synthesis (Johnson et al., 2000; Vasquez-Del Carpio et al., 2011; Washington et al., 2002). Hence, we had proposed that another Pol(s) would insert a nt opposite the 3’T of the cis-syn TT dimer from which Polκ or Polζ would extend synthesis (Yoon et al., 2009). Here we provide evidence that Polθ plays such a role for both Polκ and Polζ. As shown in Table 1, TLS opposite a cis-syn TT dimer present on the leading strand template in NER defective XPA cells treated with control siRNA, occurs with a frequency of ~41%, and it declines to ~18% in Polη depleted cells. Depletion of Polκ or Polζ catalytic subunit Rev3 reduces TLS to ~31%, and co-depletion of Polκ and Polζ reduces TLS to ~23%. Depletion of Polθ also reduced TLS frequency to ~23% and co-depletion of Polθ with Polκ or with Rev3 caused no further reduction in TLS. In striking contrast, co-depletion of Polθ with Polη resulted in a drastic reduction in TLS frequency to ~5%. These results indicating epistasis of Polθ over Polκ or Polζ and synergism of Polθ with Polη imply that Polθ functions together with Polκ or Polζ and that Polθ and Polη promote TLS opposite a cis-syn TT dimer via alternative pathways. Further, the observation that TLS in Polθ depleted XPV cells is reduced to ~3% adds to the evidence that Polη and Polθ function in alternate pathways for TLS opposite CPDs (Table S1 and Figure S1A). We also verified this conclusion for TLS opposite a cis-syn TT dimer present on the lagging strand template (Table 1 and Table S1).
Table 1.
The effects of siRNA knockdowns of Polθ and other TLS Pols on replicative bypass of a cis-syn TT dimer or a (6–4) TT photoproduct carried on the leading or lagging DNA strand template in XPA human fibroblasts
| Leading strand | Lagging strand | ||||||
|---|---|---|---|---|---|---|---|
| UV lesion | siRNA | Number of Kan+ colonies | Number of blue colonies among Kan+ | TLS (%) | Number of Kan+ colonies | Number of blue colonies among Kan+ | TLS (%) |
| cis-syn TT dimer | NC | 642 | 265 | 41.3 | 562 | 170 | 30.2 |
| Polη | 637 | 117 | 18.4 | 614 | 90 | 14.6 | |
| Polκ | 602 | 190 | 31.6 | 546 | 122 | 22.3 | |
| Rev3 | 587 | 181 | 30.8 | 483 | 94 | 19.5 | |
| Polκ + Rev3 | 508 | 115 | 22.6 | 424 | 64 | 15.1 | |
| Polθ | 762 | 174 | 22.8 | 623 | 102 | 16.4 | |
| Polθ + Polκ | 702 | 166 | 23.6 | - | - | - | |
| Polθ + Rev3 | 656 | 147 | 22.4 | - | - | - | |
| Polθ + Polη | 652 | 36 | 5.5 | 678 | 28 | 4.1 | |
| (6–4) TT PP | NC | 673 | 252 | 37.4 | 624 | 189 | 30.3 |
| Polη | 637 | 243 | 38.1 | 745 | 207 | 27.8 | |
| Polι | 582 | 232 | 37.8 | 723 | 190 | 26.3 | |
| Polη + Polι | 802 | 220 | 27.4 | 612 | 94 | 15.4 | |
| Pol θ | 623 | 167 | 26.8 | 685 | 100 | 14.6 | |
| Rev3 | 402 | 73 | 18.2 | 455 | 62 | 13.6 | |
| Polθ + Rev3 | 712 | 34 | 4.8 | 582 | 32 | 5.5 | |
See also Figures S1–S4 and Tables S1 and S3.
Previously, we have shown that TLS opposite a (6–4) TT PP occurs via two alternative error-prone pathways, dependent respectively upon Polη and Polι, and via an error-free Polζ-dependent pathway (Yoon et al., 2010b). Since Polη and Polι can insert a nt opposite the 3’T of the (6–4) TT PP but fail to extend synthesis (Johnson et al., 2001; Johnson et al., 2000), we had proposed that another TLS polymerase would act together with Polη and Polι to carry out such a role. Here we provide evidence that Polθ functions together with Polη and Polι in TLS opposite a (6–4) TT PP. As we showed previously (Yoon et al., 2010b), TLS opposite a (6–4) TT 5 PP present on the leading strand template of the plasmid occurs with a frequency of ~37% in XPA cells treated with control siRNA, and TLS is reduced to ~18% in Rev3 (Polζ)-depleted cells (Table 1). Even though TLS frequency is not affected in cells depleted for Polη or Polι, it declines to ~27% in cells co-depleted for Polη and Polι, consistent with their role in alternate TLS pathways. Importantly, TLS in Polθ-depleted XPA cells is reduced to the same level (~27%) as in cells co-depleted for Polη and Polι, and co-depletion of Polθ with Rev3 results in a drastic reduction in TLS to ~5%. These results (Table 1) and the TLS analyses in XPV cells (Table S1) provide evidence that Polθ functions together with Polη and Polι and that Polθ and Polζ provide alternative pathways for replication through a (6–4) TT PP (Figure S1B).
Polθ is Indispensable for UV Induced Mutations
The requirement of Polθ for Polκ and Polζ-dependent error-prone TLS opposite a cis-syn TT dimer (Figure S1A) and for Polη and Polι-dependent error-prone TLS opposite a (6–4) TT PP (Figure S1B) predicted that mutagenic TLS opposite both these UV lesions will be absent in Polθ-depleted cells. Accordingly, we did not find any evidence of mutations among TLS products generated from replication of plasmid carrying a cis-syn TT dimer or a (6–4) TT photoproduct on the leading or the lagging strand DNA template (Table S2).
Since mutagenic TLS opposite both the UV lesions occurs with a low frequency (~2%) (Yoon et al., 2009; Yoon et al., 2010b) (Table S2), and since these mutational analyses examine TLS only opposite a cis-syn TT dimer or a (6–4) TT PP, we examined the effects of Polθ depletion on mutations resulting from TLS opposite CPDs and (6–4) PPs formed at the various TT, TC, and CC dipyrimidine sites in the cII gene that has been integrated into the genome of big blue mouse embryonic fibroblasts (BBMEFs). In response to UV and other DNA damaging treatments, the cII gene exhibits mutational responses similar to those observed with endogenous chromosomal genes (Besaratinia and Pfeifer, 2006; You et al., 2001; You and Pfeifer, 2001). To examine UV mutations resulting from TLS opposite CPDs, the (6–4) PPs are selectively removed from the genome by expressing a (6–4) PP photolyase gene in the BBMEF cell line (You et al., 2001) and the effects of siRNA depletions analyzed. In unirradiated cells treated with control siRNA, spontaneous mutations in the cII gene occur at a frequency of ~18 × 10−5, and this frequency rises to ~45 × 10−5 in UV irradiated (5 J/m2) mouse cells exposed to photoreactivating light to activate (6–4) PP removal by (6–4) PP photolyase (Table 2). Thus, the additional increase in mutational frequency of ~27 × 10−5 CPDs formed at various dipyrimidine sites in the cII gene. results from mutagenic TLS opposite As expected from the requirement of Polθ for Polκ and Polζ-dependent error-prone TLS opposite CPDs, Polθ depletion reduced UV induced mutation frequency to a level similar to that in unirradiated cells; furthermore, the highly elevated UV induced mutation frequency in Polη depleted cells (~83 × 10−5) was reduced to ~16 × 10−5 in cells co-depleted for Polη and Polθ, a level similar to that in unirradiated cells (Table 2). Thus, Polθ is essential for the generation of UV induced mutations opposite CPDs.
Table 2.
UV induced mutation frequencies in the cII gene in BBMEF cells expressing a (6–4) PP photolyase,CPD photolyase, or no photolyase and treated with siRNA for Polθ or other TLS Pols
| Photolyase | siRNA | UVa | Photoreactivationb | Mutation frequencyc (x10−5) |
|---|---|---|---|---|
| (6–4) PP photolyase | NC | − | + | 17.6 ± 1.5 |
| Polθ | − | + | 16.8 ± 1.7 | |
| NC | + | + | 45.4 ± 2.2 | |
| Polη | + | + | 82.6 ± 2.4 | |
| Polθ | + | + | 17.8 ± 1.1 | |
| Polη + Polθ | + | + | 16.4 ± 1.4 | |
| CPD photolyase | NC | − | + | 17.2 ± 1.6 |
| Polθ | − | + | 17.0 ± 1.5 | |
| NC | + | + | 28.7 ± 1.6 | |
| Polθ | + | + | 18.1 ± 0.9 | |
| pNeo vector (no photolyase) | NC | − | + | 15.9 ± 1.4 |
| Polθ | − | + | 15.8 ± 1.2 | |
| NC | + | + | 55.1 ± 2.3 | |
| Polη | + | + | 97.1 ± 2.9 | |
| Polκ | + | + | 36.3 ± 1.7 | |
| Rev3 | + | + | 32.1 ± 1.1 | |
| Polθ | + | + | 17.4 ± 1.3 | |
| Polη + Polθ | + | + | 19.2 ± 1.6 | |
5 J/m2 of UVC (254 nm) light
Photoreactivation with UVA (360nm) light for 3h
Data are represented as mean ± SEM. Mean mutation frequencies and standard error of the mean were calculated for 4 independent experiments
See also Figures S1–S4 and Tables S2 and S3.
To examine UV mutagenesis resulting from TLS opposite (6–4) PPs formed at various dipyrimidine sites, the CPDs were selectively removed from the genome by expressing a CPD photolyase gene in BBMEF cells (You et al., 2001). In this cell line, spontaneous mutations occur at a frequency of ~17 × 10−5, and this mutation frequency rises to ~29 × 10−5 in UV irradiated (5 J/m2) cells exposed to photoreactivating light to activate CPD photolyase. In Polθ depleted cells, the elevation in mutation frequency resulting from mutagenic TLS opposite (6–4) PPs is reduced to a level similar to that in unirradiated cells (Table 2), thus indicating the indispensability of Polθ for UV induced mutations opposite (6–4) PPs.
The requirement of Polθ for mutagenic TLS opposite CPDs and (6–4) PPs implies that in the absence of any photolyase, Polθ depletion would inhibit the formation of UV induced mutations that would have resulted from error-prone TLS opposite CPDs and (6–4) PPs. To verify this, we analyzed mutation frequencies in the cII gene in a BBMEF cell line which expresses no photolyase. In this cell line, spontaneous mutations occur at a frequency of ~16 × 10−5, and mutation frequency rises to ~55 × 10−5 in UV irradiated cells. Polθ depletion reduced UV induced mutation frequency to ~17 × 10−5, and the highly elevated UV induced mutation frequency in Polη depleted cells was strongly inhibited in cells co-depleted for Polη and Polθ (Table 2).
UV induced C>T and CC>TT signature mutations resulting from TLS opposite CPDs and (6–4) PPs are clustered at particular hot spots in the cII gene. Mutagenic replication through CPDs generates mutational hot spots located at 11 dipyrimidine sequences, #1 to #11 (Yoon et al., 2009), whereas mutagenic TLS through (6–4) PPs generates mutational hot spots which occur primarily at the sequences labeled #3, #10, and #11 (Yoon et al., 2010b) (Figure S1C). The pattern of UV induced hot spot mutations in the cII gene in BBMEF cells treated with control siRNA and lacking any photolyase results from error-prone TLS opposite CPDs and (6–4) PPs, and as expected from the requirement of Polθ for mutagenic TLS opposite both the UV lesions, these mutational hot spots are absent in Polθ depleted cells (Figure S1C).
An earlier study reported a reduction in UV mutagenesis in Polθ depleted 293T cells (Ceccaldi et al., 2015). However, 293T cells differ strikingly from normal cells in that they exhibit cancer stem cell features (Debeb et al., 2010). In 293T cells, NER becomes highly deranged: it uses different combinations of proteins than are used in highly conserved NER, it occurs via alternative pathways, and rather than act in an error free manner, these NER pathways act in an error prone manner (J-H Yoon, L Prakash, S Prakash, unpublished data). One of these NER pathways requires XPA, Polθ, and additional proteins. UV induced mutation frequency is reduced to the same level in 293T cells depleted for XPA, Polθ, or for both XPA and Polθ (Figure S1D). The epistasis of Polθ with XPA is indicative of a role for Polθ in the aberrant XPA dependent NER pathway. This Polθ role in 293T cells has no biological relevance for the role of Polθ in error prone TLS we describe in this study and which occurs during DNA replication when RFs stall opposite UV lesions in normal cells.
The C-terminal Polymerase Domain is Sufficient for Polθ Function in TLS Opposite UV Lesions in Human Cells
Human Polθ is a 290 kDa protein comprised of an N-terminal ATPase/helicase domain, a large central domain, and a C-terminal polymerase domain (Figure S2A) that shares high similarity with A-family DNA Pols such as Escherichia coli pol1 (Seki et al., 2003). Previously, we showed that the C-terminal polymerase domain of Polθ comprised of residues 1708–2590 was sufficient for its role in TLS opposite the oxidative lesion thymine glycol (TG) in human cells (Yoon et al., 2014).
To determine whether the C-terminal polymerase domain is sufficient for TLS opposite UV lesions, we expressed Polθ (1708–2590) in normal human fibroblasts (HFs) (Figure S2A) which harbor the duplex plasmid containing a cis-syn TT dimer or a (6–4) TT PP on the leading strand template. In these cells treated with control siRNA, TLS opposite a cis-syn TT dimer occurs at a frequency of ~21%, irrespective of whether the cells carry just the vector plasmid with no Polθ or express the C-terminal Polθ polymerase domain (Table S3). However, in cells treated with Polθ siRNA and carrying the vector control, TLS is reduced to ~11% and TLS is restored to normal levels in cells expressing WT Polθ (1708–2590). Expression of WT Polθ (1708–2590) also restored normal TLS levels opposite (6–4) TT PP in cells treated with Polθ siRNA (Table S3). Thus, the N-terminal helicase and the central domains are dispensable and the C-terminal polymerase domain is sufficient for Polθ function in TLS opposite UV lesions.
To establish that Polθ DNA synthesis function was required for TLS opposite UV lesions, we examined TLS in human cells that express the Polθ (1708–2590) D2540A, E2541A mutant protein defective in DNA synthesis (Figure S2A). In human cells from which genomic Polθ has been depleted and that express catalytically inactive Polθ (1708–2590), TLS opposite both the UV lesions is reduced to the same level as in cells that carry the control vector (Table S3), thus indicating the requirement for Polθ DNA polymerase activity in the replication of UV damaged cells.
PCNA Ubiquitination Promotes the Assembly of Polθ (1708–2590) into Foci in UV Damaged Human Cells
The observation that Polθ (1708–2590) containing only the polymerase domain is sufficient for TLS opposite UV lesions in human cells implies that this Polθ protein is targeted to replication forks (RFs) stalled at UV lesions. To examine this, we expressed GFP-Polθ (1708–2590) in HFs and analyzed the frequency of GFP-Polθ (1708–2590) foci-containing cells. Whereas ~20% of unirradiated cells contain GFP-Polθ (1708–2590) foci, the frequency of cells containing these Polθ foci rises to ~50% in UV irradiated cells, and it rises to ~60% upon Polη depletion (Figures S2C,D). Similar to the requirement of Rad6-Rad18 mediated PCNA ubiquitination for the UV induced accumulation of Polη, Polκ, and Polζ into foci (Yoon et al., 2015), UV induced assembly of GFP-Polθ (1708–2590) into foci does not occur in Rad18 depleted cells (Figure S2D). Moreover, in chromatin fraction isolated from UV irradiated HFs, Polθ (1708–2590) co-immunoprecipitates with ub-PCNA and Rad18 (Figure S2B). Polθ (1708–2590) could bind to ub-PCNA via the PCNA binding PIP motif Q N L F Q T F I (conserved residues underlined) present between residues 1836–1843.
Biochemical Analysis of Polθ Role in TLS Opposite a cis-syn TT Dimer and a (6–4) TT PP
Previously, biochemical studies carried out with full length Polθ have indicated that it lacks the ability to insert a nt opposite the 3’T of the cis-syn TT dimer (Seki et al., 2004) but it could carry out extension of synthesis from an A opposite the 3’T of a (6–4) TT PP (Seki and Wood, 2007). Since the C-terminal polymerase domain is sufficient for the TLS function of Polθ in human cells (Table S3) and Polθ containing only this domain assembles into foci and co-immunoprecipitates with ub-PCNA and Rad18 (Figure S2), we examined purified Polθ (1708–2590) for its ability to insert dATP, dTTP, dGTP, or dCTP opposite the 3’T of a cis-syn TT dimer and to replicate DNA through the lesion in the presence of 4 dNTPS (Figure S3A). We find that Polθ inserts dATP or dGTP opposite the 3’T of the cis-syn TT dimer, and in the presence of 4 dNTPs, it fails to extend synthesis any further. On undamaged DNA, Polθ inserts dATP opposite the corresponding 3’T as well as opposite the next 3–4 template residues, and it also inserts dTTP or dGTP opposite the 3’T but to a lesser extent. Opposite (6–4) TT PP, Polθ (1708–2590) extends synthesis from the 3’T•A base pair (Figure S3B). Polθ exhibits a high error-proneness when inserting nts opposite the 5’T of (6–4) TT PP as well as opposite the corresponding 5’T in undamaged DNA.
Polθ Promotes Replication Fork (RF) Progression in UV Damaged Human Cells
To investigate the contribution of Polθ dependent mutagenic TLS to replication of UV damaged DNA, we monitored RF progression on single DNA fibers. siRNA treated HFs were pulse labeled with iododeoxyuridine (IdU) for 20 min, then irradiated with UV light (10 J/m2) followed by labeling with chlorodeoxyuridine (CldU) for 30 min. Compared to HFs treated with control siRNA, Polθ depletion conferred a reduction in CldU incorporation relative to IdU incorporation; Polη depletion, however, conferred a greater reduction in fork progression than Polθ depletion, and co-depletion of both Polη and Polθ resulted in a further additive reduction in fork progression in UV damaged cells (Figure 1A-C).
Figure 1. Requirement of Polθ for Replication through UV Lesions in Human Cells.
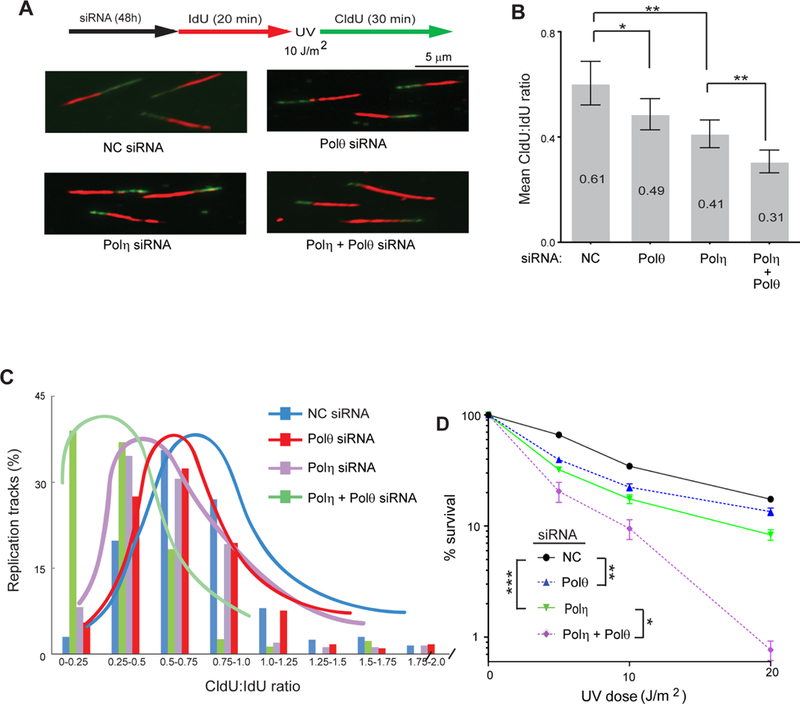
(A) Schematic of DNA fiber assay and representative images of stretched DNA fibers in UV damaged GM637 HFs treated with control (NC), Polη, Polθ, or Polη and Polθ siRNAs
(B) Quantitative analyses of RF progression through UV lesions (mean CldU:IdU ratio). The data represent ~400 DNA fibers from four independent experiments. Error bars indicate the standard deviation. Student’s two-tailed t-test p values, *, p<0.05; **, p<0.01.
(C) The % of replication tracts and the CldU:IdU ratios were measured in fibers from UV damaged GM637 HFs treated with NC, Polθ, Polη, or Polη and Polθ siRNAs. The data represent ~400 DNA fibers from four independent experiments.
(D) UV survival assay. GM637 HFs were treated with siRNAs for 48h and irradiated with UV light in PBS buffer. Cells were incubated for additional 48h after UV irradiation and UV survival was determined by the MTS assay. Error bars indicate the standard deviation of results of 4 independent experiments. Student’s two-tailed t-test p values, *, p<0.05; **, p<0.01; ***, p<0.001.
See also Figure S3.
As expected from the more significant role of Polη in replication of UV damaged DNA than Polθ, UV survival was reduced to a greater extent in HFs depleted for Polη than for Polθ and co-depletion of Polη with Polθ led to a further reduction in UV survival (Figure 1D).
Translesion Synthesis Opposite UV Lesions, Replication Fork Progression through UV Lesions, and UV Survival in Polθ−/−, Polη−/−, and Polη−/− Polθ−/− MEFs
Before analyzing the contribution of Polθ dependent mutagenic TLS to UV induced skin cancers, we confirmed that the results of siRNA depletions of Polθ and Polη in HFs were recapitulated in Polθ−/−, Polη−/−, and Polη−/− Polθ−/− MEFs (Figure 2). Similar to that in HFs, TLS opposite a cis-syn TT dimer and a (6–4) TT photoproduct was reduced by ~50% in Polθ−/− MEFs (Figure 2B). Compared to WT MEFs, RF progression through UV lesions was reduced by ~23% in Polθ−/−MEFs, ~36% in Polη−/−MEFs, and ~54% in Polη−/− Polθ−/− MEFs (Figure 2C). The rate of RF progression was not affected significantly in unirradiated Polθ−/−, Polη−/−, and Polη−/− Polθ−/− MEFs. (Figure 2D). Polθ−/− MEFs exhibited a very significant reduction in UV survival, but UV survival was reduced to a greater extent in Polη−/−MEFs than in Polθ−/− MEFs, and a further reduction in UV survival occurred in Polη−/− Polθ−/− MEFs over that in Polη−/− MEFs (Figure 2E).
Figure 2. Analysis of TLS through UV Lesions in Primary Polθ−/−, Polη−/−, and Polθ−/− Polη−/− MEFs.
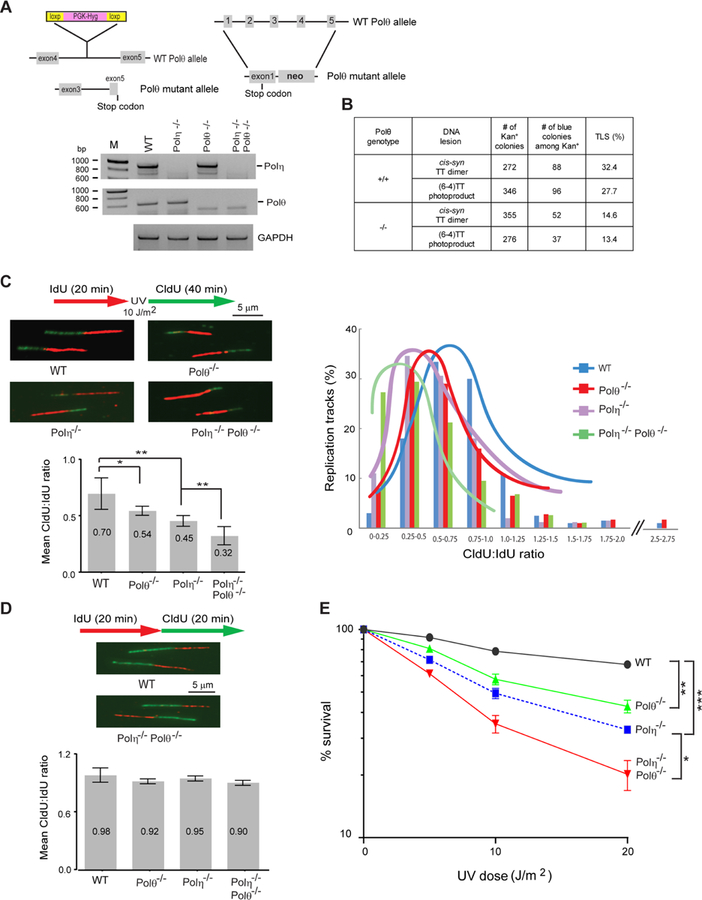
(A) Schematic for targeting the knock outs of Polη and Polθ genes and RT-PCR analyses of Polη−/−, Polθ−/− and Polη−/− Polθ−/− MEFs. GAPDH was used for a negative control.
(B) TLS opposite UV lesions in SV40 transformed Polθ−/− MEFs.
(C) Analyses of RF progression through UV lesions in primary WT, Polθ−/−, Polη−/−, and Polη−/− Polθ−/− MEFs. (Top left), schematic of DNA fiber assay and representative images of stretched DNA fibers. (Bottom left), quantitative analyses of RF progression through UV lesions (mean CldU:IdU ratio). The data represent ~400 DNA fibers from four independent experiments. Error bars indicate the standard deviation. Student’s two-tailed t-test p values, *, p<0.05; **, p<0.01. (Right), the % of replication tracts and the CldU:IdU ratios measured in fibers from UV damaged primary WT, Polθ−/−, Polη−/−, and Polη−/− Polθ−/− MEFs. The data represent ~400 DNA fibers from four independent experiments.
(D) Schematic of DNA fiber assay and images of stretched DNA fibers in unirradiated primary WT and Polη−/− Polθ−/− MEFs and quantitative analyses of RF progression (mean CldU:IdU ratio) in WT, Polθ−/−, Polη−/−, and Polη−/− Polθ−/− MEFs.
(E) UV survival of primary WT, Polθ−/−, Polη−/−, and Polη−/− Polθ−/− MEFs. Error bars indicate the standard deviation of results of four independent experiments. Student’s two-tailed t-test p values, *, p<0.05; **, p<0.01; ***, p<0.001.
DSB Formation Increases in Polθ−/−, Polη−/−, and Polη−/− Polθ−/−MEFs
Since impairment of RF progression through UV lesions would generate regions of unreplicated ssDNA, we carried out experiments to determine whether the extent of ssDNA accumulation corresponds to the RF progression defect engendered by inactivation of the respective Pol. The generation of ssDNA was detected by BrdU immunoassay carried out under non-denaturing conditions (Rubbi and Milner, 2001). The increase in the level of ssDNA in UV irradiated Polθ−/−, Polη−/−, and Polη−/− Polθ−/− MEFs (Figure 3A) corresponds to the level of RF progression defect in mutant MEFs (Figure 2C). A significant increase in ssDNA was also observed in non-UV irradiated Polη−/− Polθ−/− MEFs but not in Polθ−/− or Polη−/− MEFs (Figure 3A).
Figure 3. Generation of ssDNA and formation of DSBs in UV damaged primary Polθ−/−, Polη−/−, and Polη−/− Polθ−/− MEFs.
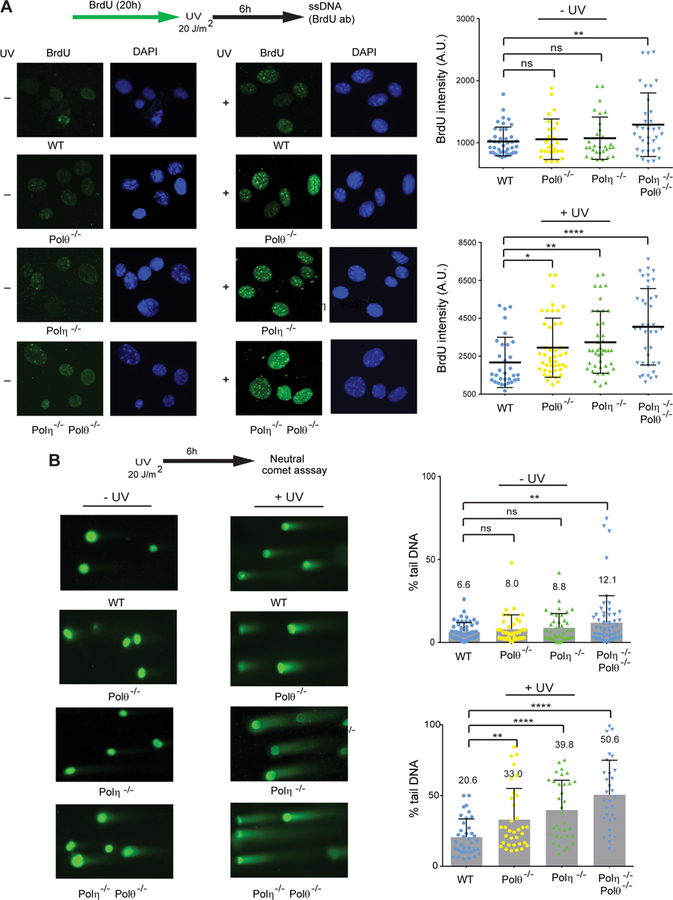
(A) BrdU immuno-assay for ssDNA detection in UV irradiated or unirradiated MEFs. Cells were treated with BrdU for 20h and irradiated with UV (20 J/m2) or not, followed by 6h incubation. Immuno-staining with BrdU was visualized by fluorescence microscopy. (Left) representative images of BrdU staining in unirradiated or UV irradiated primary WT, Polθ−/−, Polη−/−, and Polη−/− Polθ−/− MEFs; (Right) quantification of BrdU immuno-staining intensity in unirradiated and UV irradiated primary MEFs. The mean and standard deviation were analyzed from 4 independent experiments and are indicated by a horizontal and a vertical black bar, respectively. Student’s two-tailed t-test values, ns, not significant; *, p<0.05; **, p<0.01; ****, p<0.0001.
(B) Neutral comet assay for detection of DSBs in unirradiated or UV irradiated MEFs. Comet assay was performed on cells irradiated with UV (20J/m2) and incubated for 6h. (Left), representative images of neutral comet tails in DNA from unirradiated or UV irradiated primary WT, Polθ−/−, Polη−/−, and Polη−/− Polθ−/− MEFs. (Right), quantification of % of DNA in the comet tail in unirradiated and UV irradiated MEFs. The mean and standard deviation were analyzed from 4 independent experiments and are indicated by a numeral and a vertical black bar, respectively. Student’s two-tailed t-test p values, ns, not significant; **, p<0.01; ****, p<0.0001.
Since unreplicated ssDNA could become subject to endonucleolytic cleavage, we reasoned that the level of DSBs would rise in UV irradiated Polθ−/−, Polη−/−, and Polη−/− Polθ−/−MEFs. To assess this, we quantified DSB formation in UV irradiated MEFs by neutral comet assay (Figure 3B). As indicated by the percentage of DNA in tail, which is linearly related to break frequency (Collins et al., 2008; Gyori et al., 2014), Polη−/− MEFs exhibited a greater increase in DSBs than Polθ−/− MEFs, and the level of DSBs was higher in Polη−/− Polθ−/− MEFs than in Polη−/− MEFs. A significant increase in the level of DSBs was also observed in non-UV irradiated Polη−/− Polθ−/−MEFs (Figure 3B).
Sister Chromatid Exchanges (SCEs) and Chromosomal Aberrations are Elevated in Polθ−/−, Polη−/−, and Polη−/− Polθ−/− MEFs
Our observations indicating that impairment of TLS results in unreplicated regions of ssDNA and leads to DSB formation suggested that the one-ended DSBs thus generated could be repaired by homologous recombination (HR) when the opposing RF reaches the break site and the fully replicated unbroken sister chromatid is used for repairing the DSB by HR. Since SCEs are formed as cross-over products of HR, we examined the frequency of SCEs in UV irradiated (2 J/m2) Polθ−/−, Polη−/−, and Polη−/− Polθ−/− MEFs. Compared to that in WT MEFs, a highly significant increase in SCEs occurs in Polθ−/− MEFs; SCE frequency is elevated almost 2-fold in Polη−/− MEFs over that in Polθ−/− MEFs and a further rise in SCEs occurs in Polη−/− Polθ−/− MEFs over that in Polη−/− MEFs (Figure 4B).
Figure 4. Analysis of Sister Chromatid Exchanges (SCEs) and Chromosomal Aberrations in UV Damaged Primary Polθ−/−, Polη−/−, and Polη−/− Polθ−/− MEFs.
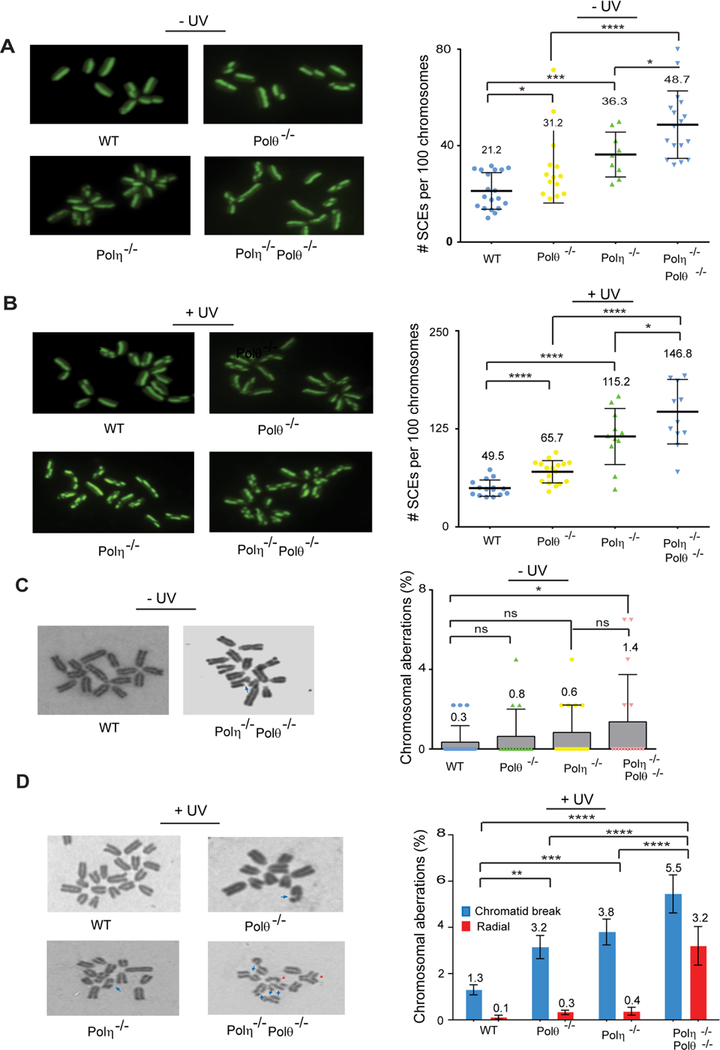
(A) SCEs in unirradiated MEFs. (Left), representative images of metaphases in unirradiated primary WT, Polθ−/−, Polη−/−, and Polη−/− Polθ−/− MEFs. (Right), quantification of SCE frequency in unirradiated primary WT, Polθ−/−, Polη−/− and Polη−/− Polθ−/− MEFs. Each datum point represents a single metaphase and ~1,000 metaphase chromosomes were analyzed. The mean and standard deviation were analyzed from 4 independent experiments and are indicated by a numeral and a vertical black bar, respectively. Student’s two-tailed t-test p values, *, p<0.05; ***, p<0.001; ****, p<0.0001.
(B) SCEs in UV irradiated MEFs. (Left), representative images of metaphases with UV (2 J/m2) induced SCEs in primary WT, Polθ−/−, Polη−/−, and Polη−/− Polθ−/− MEFs. (Right), quantification of scatterplot analysis of UV induced SCE frequency in primary WT, Polθ−/−, Polη−/−, and Polη−/− Polθ−/− MEFs. Each datum point represents a single metaphase and ~1,000 metaphase chromosomes were analyzed. The mean and standard deviation were analyzed from 4 independent experiments and are indicated by a numeral and a vertical black bar, respectively. Student’s two-tailed t-test p values, *, p<0.05; ****, p<0.0001.
(C) Chromosomal aberrations in unirradiated MEFs. (Left), chromatid breaks in Polη−/− Polθ−/− MEFs. (Right), quantification of chromosomal aberrations in unirradiated primary WT, Polθ−/−, Polη−/− and Polη−/− Polθ−/− MEFs. The data represent analyses of ~400 metaphases from four independent experiments. The mean and standard deviation were analyzed from 4 independent experiments and are indicated by a numeral and a vertical black bar, respectively. Student’s two-tailed t-test p values, ns, non-significant; *, p<0.05.
(D) Chromosome aberrations in UV irradiated MEFs. (Left), chromatid breaks (blue arrow) and radial structures (red arrow head) in UV irradiated primary WT, Polθ−/−, Polη−/−, and Polη−/− Polθ−/− MEFs. (Right), quantification of chromosomal aberrations in MEFs. The data represent analyses of ~400 metaphases from four independent experiments. The mean and standard deviation were analyzed from 4 independent experiments and are indicated by a numeral and a vertical black bar, respectively. Two way ANOVA p values, **, p<0.01; ***, p<0.001; ****, p<0.0001.
The very significant increase in ssDNA and DSBs observed in non-UV irradiated Polη−/− Polθ−/− MEFs (Figure 3) suggested a role for these Pols in the rescue of stalled RFs in undamaged cells. To further explore this possibility, we examined the frequency of SCEs in non-UV irradiated Polθ−/−, Polη−/−, and Polη−/− Polθ−/− MEFs. Our data show that a significant increase in SCEs occurs in Polθ−/− MEFs, SCEs occur at a higher frequency in Polη−/− MEFs than in Polθ−/− MEFs, and SCEs rise further in Polη−/− Polθ−/− MEFs over that in Polη−/− MEFs (Figure 4A). In cells not exposed to extraneous DNA damaging agents, these Pols may conduct replication through DNA lesions that derive from endogenous cellular reactions and they may play additional roles in the rescue of stalled RFs.
Next, we analyzed the frequency of chromosomal aberrations in UV damaged and undamaged Polθ−/−, Polη−/−, and Polη−/− Polθ−/− MEFs. In response to UV irradiation, chromosomal aberrations rise in Polθ−/− and Polη−/− MEFs and a more than additive increase in chromosomal aberrations occurs in Polη−/− Polθ−/− MEFs (Figure 4D). Chromatid breaks are the most prevalent lesion in Polθ−/− or Polη−/− MEFs, whereas the prevalence of both chromatid breaks and radial structures increases in Polη−/− Polθ−/− MEFs. The frequency of chromosomal aberrations also rises significantly in non-UV irradiated Polη−/− Polθ−/− MEFs and chromatid breaks are the predominant lesion (Figure 4C).
Complementation of TLS and Associated Defects in Polθ−/− MEFs by the C-terminal Polθ Polymerase Domain
Since both the DNA helicase and DNA polymerase domains of Polθ have been deleted in Polθ−/− MEFs (Figure 2A), we verified that the TLS and TLS-associated defects in Polθ−/− MEFs accrue from the lack of polymerase function and not from the lack of helicase function. To this end, we expressed the C-terminal polymerase domain (1708–2590) of human Polθ in Polθ−/− MEFs (Figure S4A) and analyzed its effect on TLS and other associated defects. Polθ (1708–2590) complemented the TLS deficiency opposite both the UV lesions (Figure S4B), restored wild type level of RF progression in UV damaged cells (Figure S4C), reduced SCEs and chromosomal aberrations in UV damaged as well as undamaged MEFs (Figure S4D-F), and restored UV resistance (Figure S4G). Thus, all the TLS and TLS-associated defects in Polθ−/− MEFs derive from the lack of C-terminal polymerase domain and not from the lack of its N-terminal helicase domain. Since in addition to the N-terminal domain, most of the central domain is also missing in Polθ−/− MEFs carrying Polθ(1708–2590) (Figure S2A), both these domains are dispensable for Polθ function in TLS.
Using the DR-GFP assay, which consists of direct repeats of mutated GFP genes integrated close-by on the same chromosome and in which one of the repeats is targeted for DSB formation by the I-SceI endonuclease (Moynahan and Jasin, 2010), a role for Polθ N-terminal helicase domain in the inhibition of Rad51-ssDNA nucleofilament assembly and suppression of HR has been indicated (Ceccaldi et al., 2015). Our observation that the increase in SCEs in unirradiated or UV irradiated Polθ−/− MEFs is reduced to WT levels by the introduction of the Polθ (1708–2590) C-terminal polymerase domain (Figure S4D,E), however, indicates that the increased levels of SCEs in Polθ−/− MEFs result from the lack of Polθ C-terminal polymerase domain and not from the lack of its N-terminal helicase domain. Furthermore, our results that SCEs are reduced in Rad51 depleted undamaged or UV damaged Polθ−/− MEFs which either lack or express the Polθ C-terminal polymerase domain (Figure S4D,E) imply that SCEs in Polθ−/− MEFs arise from a Rad51-dependent pathway. Taken together, our data show that the increase in Rad51-dependent SCEs in Polθ−/− MEFs results from the lack of Polθ C-terminal polymerase domain.
The different effects of Polθ N-terminal helicase domain and C-terminal polymerase domain in HR as visualized by SCE formation in Polθ−/− MEFs or as determined by the analyses of a DSB generated by the I-SceI endonuclease can be reconciled by the fact that the I-SceI endonuclease initiated HR of DR-GFP would involve a two-ended DSB whereas SCEs arising from RF collapse in Polθ−/− MEFs would result from one-ended DSBs. Thus, while the Polθ N-terminal helicase domain functions in inhibiting Rad51-dependent HR of two-ended DSBs, it plays no such suppressive role in HR of one-ended DSBs, which would arise in cells under replication stress generated by UV lesions or by lesions in unirradiated cells. Since the rescue of stalled RFs by Polθ polymerase function prevents the formation of one-ended DSBs and the consequent generation of SCEs by HR, Polθ polymerase domain effects the suppression of HR generated by fork collapse in UV damaged or in undamaged cells.
Polθ−/− Mice are Prone to UV Induced Skin Cancers
To determine the impact of error-prone TLS on tumorigenesis, we UV irradiated (2 KJ/m2 UVB, 3 times/week) a cohort of wild type, Polθ+/−, and Polθ−/− mice and monitored them for skin tumor development on UV exposed dorsal skin. At ~ 45 weeks of UV exposure, 20% (4/20) of wild type mice had developed skin lesions that exhibited small focal epidermal hyperplasia, or non-invasive SCC with cellular atypia (Figure 5B,5E). Among Polθ+/− heterozygotes, by ~45 weeks of UV irradiation ~ 30% (6/21) of mice had developed skin lesions that included ulcerative dermatitis, hyperplasia, papilloma, or SCC (Figure 5B,5E). Polθ−/− mice were significantly more prone to skin cancers than Polθ+/− mice as ~75% of Polθ−/− mice had developed skin tumors by ~40 weeks and all of the Polθ−/− mice had skin tumors by ~45 weeks (Figure 5B). Almost all the tumors from Polθ−/− mice were SCCs and a majority of them were invasive deep into the muscular layer. Combinations of SCCs and carcinomas or carcinomas alone were infrequent in tumors from Polθ−/− mice (Figures 5E, S5, and S6). The evidence that the incidence of skin cancers rises in Polθ−/− mice shows that error-prone TLS by Polθ suppresses skin cancer formation and implies that skin cancers can form in the absence of UV induced driver mutations.
Figure 5. Skin Tumors Induced by Chronic Exposure to UVB irradiation in Polθ−/−, Polη−/−, and Polη−/− Polθ−/− Mice.
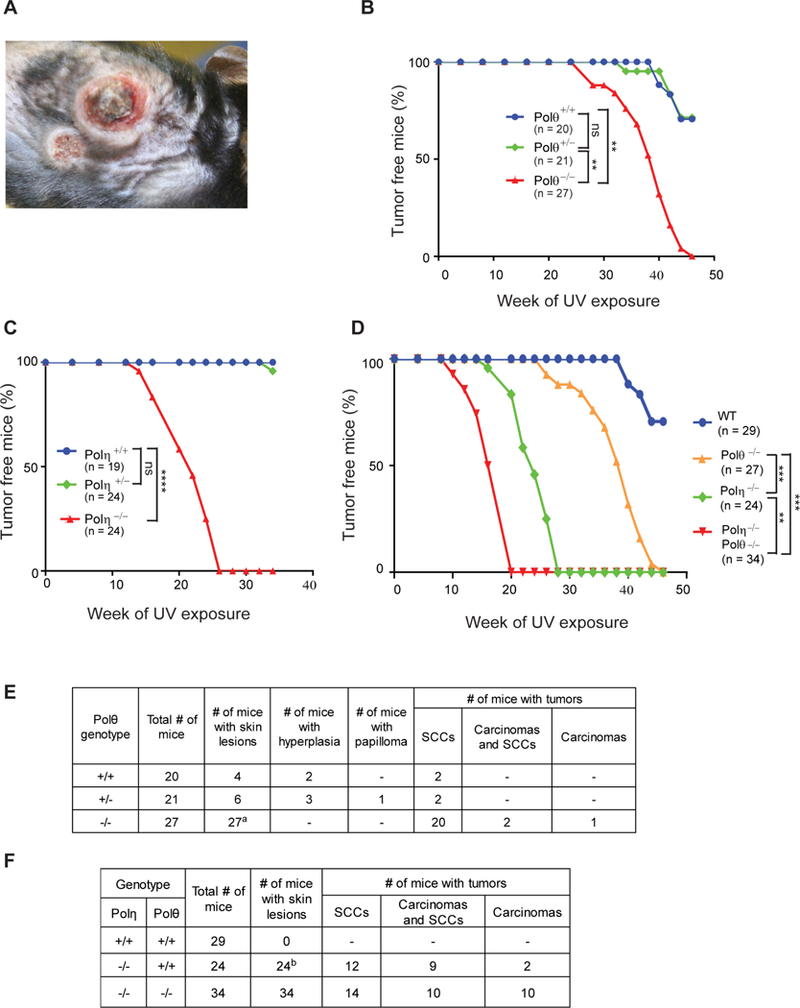
(A) UVB-induced skin tumors on the dorsal area of Polθ−/− mice at 42 weeks of UV exposure.
(B), (C), and (D) Kaplan-Meier curves of mice free of skin tumors after chronic UVB irradiation (2 KJ/m2, 3 times per week). (B) Polθ+/+, Polθ+/− and Polθ−/− mice. (C) Polη+/+, Polη+/− and Polη−/− mice. (D) WT, Polθ−/−, Polη−/− and Polη−/− Polθ−/− mice. Two way ANOVA p values, ns, non-significant; **, p<0.01; ***, p<0.001; ****, p<0.0001.
(E) Results of histopathological analyses of skin tumors from Polθ+/+, Polθ+/− and Polθ−/− mice. aTumors from 23 Polθ−/− mice were analyzed.
(F) Results of histopathological analyses of skin tumors from WT, Polη−/− and Polη−/− Polθ−/− mice. bTumors from 23 Polη−/− mice were analyzed.
See also Figures S5 and S6 and Table S4.
Polθ Deficiency Exacerbates the Susceptibility of Polη−/− Mice to UV Induced Skin Cancers
To examine the relative effectiveness of error-free and error-prone TLS in suppression of tumorigenesis, we first compared the susceptibility of wild type, Polη+/− and Polη−/− mice to UV induced skin cancers. None of the wild type or Polη+/− mice showed evidence of skin tumors by 35 weeks of UV exposure when the experiment was terminated. Tumors developed much earlier in Polη−/− mice than in Polθ−/− mice; 50% of Polη−/− mice had skin tumors by ~20 weeks of UV exposure and all of the Polη−/− experimental mice had skin tumors by ~28 weeks of UV exposure (Figure 5C). Among the tumors from Polη−/− experimental mice, we identified SCCs, a combination of carcinomas with SCCs, and carcinomas (Figure 5F, Figure S6). SCCs were the most frequent UV induced tumors identified in a previous study with Polη−/− mice (Lin et al., 2006).
To gain further understanding of Polθ’s role in cancer suppression, we examined the susceptibility of Polη−/− Polθ−/− mice to UV induced skin cancers. Polθ deficiency exacerbated the susceptibility of Polη−/− mice to UV induced skin cancers, as all the 34 experimental Polη−/− Polθ−/− mice had developed skin cancers by 20 weeks of UV exposure (Figure 5D). Tumors from Polη−/− Polθ−/− mice exhibited increased invasive tendency, were more poorly differentiated, and they presented multiple SCCs, carcinomas, or a combination of carcinomas and SCCs (Figures 5F and S6).
DISCUSSION
Polη Makes a More Significant Contribution to the Replication of UV Damaged DNA and is a More Effective Barrier to UV Induced Skin Cancers than Polθ
Polη is highly adapted for replicating through CPDs and our observation that UV induced mutations opposite CPDs are strongly inhibited in Polθ depleted MEFs (Table 2) documents the remarkable proficiency of Polη for error-free replication through this UV lesion. In addition, our evidence indicates that Polη plays a more significant role in the replication of UV damaged DNA than Polθ. Both in HFs and MEFs, Polη deficiency impedes replication of UV damaged DNA to a greater extent than Polθ deficiency, and UV survival is affected more adversely in Polη deficient cells than in Polθ deficient cells (Figures 1 and 2). In UV damaged MEFs, Polη mediated TLS protects RFs from collapse and the consequent formation of DSBs to a greater extent than Polθ dependent TLS (Figure 3B), and the much greater increase in SCEs in UV damaged Polη−/− MEFs than in Polθ−/− MEFS (Figure 4B) also points to a more prominent role of Polη in the replication of UV damaged DNA than Polθ. Furthermore, our data indicating that cancers arise much sooner in Polη−/− mice than in Polθ−/− mice (Figure 5D) provide strong evidence that Polη makes a much greater contribution to prevention of UV induced skin cancers than Polθ.
Error prone TLS by Polθ Protects Against UV Induced Skin Cancers
Even though by causing mutations in tumor suppressors and other cancer driver genes, error prone TLS by Polθ could contribute to tumorigenesis in sun exposed skin, our results show that rather than cause an increase in tumorigenesis, Polθ protects against it. This inference is supported by the increased incidence of skin cancers in Polθ−/− mice and the evidence that skin cancers arise much sooner in Polη−/− Polθ−/− mice than in Polη−/− mice (Figure 5D). The protective role of mutagenic TLS against skin cancers implies that DNA repair processes which come into play in the absence of TLS make a much more consequential contribution to tumorigenesis than would be conferred by error-prone TLS.
Cancer Driver Mutations in Normal Human Skin Cells
The protective role of error-prone TLS against cancer development suggests that cells can tolerate significant mutational burden before they become prone to transformation and invasion. In accord with this, clonal patches of keratinocytes carrying C>T UV signature mutations in the TP53 gene occur in normal sun exposed skin (Jonason et al., 1996; Klein et al., 2010; Ling et al., 2001; Nakazawa et al., 1994; Ziegler et al., 1994). Analyses of UV signature mutations in 74 cancer genes in sun-exposed epidermis have shown that normal skin cells harbor a high frequency of driver mutations in multiple cancer genes subject to strong selective pressure; moreover, the pattern of cancer driver mutations in normal skin cells is similar to mutations in skin SCCs (Martincorena et al., 2015). Yet normal skin cells exhibit no evidence of malignant transformation (Martincorena et al., 2015). Studies with TP53 mutations in UV exposed epidermis in mice have revealed that exponential growth of Tp53 mutant clones is slowed down relatively early in the expansion of the clones. This explains the limited range of the clone size and suggests that constraints on clonal growth provide a critical protection against progressive accumulation of driver mutations and malignant transformation (Martincorena et al., 2015). The observation that stem cell compartments act as physical barriers to the clonal expansion of Tp53 mutant keratinocytes in murine epidermis has led to the concept that such mutant clones become ‘imprisoned’, unable to escape the barrier presented by the stem cell compartment arrangement (Zhang et al., 2001).
The observation that about 25% of normal skin cells carry cancer driver mutations and that clones carrying 2 to 3 driver mutations show no evidence of malignant potential (Martincorena et al., 2015) suggests that mutations generated by error-prone TLS by Polθ would make relatively little contribution to the initiation of UV induced skin cancers. That raises the question of how TLS protects against tumorigenesis and the nature of genomic instability that may underlie tumorigenesis.
Role of TLS in Prevention of Genomic Rearrangements and Suppression of Tumorigenesis
Our observations that inactivation of error-free TLS by Polη or of error-prone TLS by Polθ results in unreplicated regions of ssDNA (Figure 3A), increased incidence of DSBs (Figure 3B), highly elevated SCEs (Figure 4B), and an increase in the frequency of chromosomal aberrations (Figure 4D) in UV damaged cells suggest that by promoting replication through DNA lesions, TLS mechanisms provide a key safeguard in preventing the collapse of RFs stalled at DNA lesions. In the absence of TLS, the collapse of RFs leaves unreplicated ssDNAs prone to nucleolytic attack, resulting in the formation of one-ended DSBs. Such DSBs could be repaired by Rad51-dependent HR or by non-homologous end-joining (NHEJ) when the opposing RF arrives at the break site. Consequently, SCEs and chromosomal aberrations increase in the absence of Polη or Polθ and they rise further in the absence of both Pols (Figure 4). While HR between homologous sequences will cause no genomic rearrangements, HR between paralogous repeat sequences or low copy repeats can result in genomic deletions and duplications (Liu et al., 2012). The joining of two distal one-ended DSBs on the same chromatid by NHEJ would result in loss of the intervening region, and joining of one-ended DSBs on different chromatids or chromosomes would lead to the formation of interchromatid or interchromosomal fusions, or to other types of aberrations. In addition to large chromosomal aberrations, additions or deletions of small genomic regions could result from the joining of the two one-ended DSBs by NHEJ when the opposing fork arrives at the break site.
Genomic rearrangements can activate a gene to become an oncogene. The activated oncogenes would induce additional replication stress by deregulating the cell cycle, which would lead to further stalling and collapse of RFs, the formation of DSBs, and the generation of chromosomal rearrangements. Evidence of such a cycle of continuous formation of DSBs and oncogene activation in precancerous lesions and cancers has identified replication stress as an important driver of cancer (Halazonetis et al., 2008; Macheret and Halazonetis, 2015, 2018; Negrini et al., 2010).
Studies with cell lines derived from skin SCCs originating from sun-exposed site that represented stepwise progression of skin carcinogenesis and included primary tumor, two recurrences, and a metastatic lesion from the same patient have shown that genomic rearrangements occur early in cancer development being already present in the primary tumor (Popp et al., 2000). In pancreatic cancer evolution, preneoplastic cells acquire an extensive mutational burden, yet they remain non-invasive (Murphy et al., 2013); but changes in DNA copy number and acquisition of complex genomic rearrangements rapidly lead to invasion and metastasis (Notta et al., 2016). Topographic single-cell DNA sequencing of cells from ductal carcinoma in situ (DCIS) which is an early-stage breast cancer, and from invasive ductal carcinoma (IDC) in which tumor cells migrate to other areas of breast tissue has revealed that DCIS arises from a single tumor-initiating cell which acquires alterations in the number of copies of genes. IDC descends from DCIS either from a single tumor-initiating clone or from multiple clones which diverged from the initial founding clone by the acquisition of additional copy number aberrations (Casasent et al., 2018). The acquisition of genomic rearrangements early in tumorigenesis supports the notion that they play a causal role.
Polθ’s Role in Protection From Replication Stress Induced Genome Instability vs. Its Role in Alternative End-Joining
A role for Polθ in microhomology dependent alternative NHEJ has been deduced from endonuclease-mediated cleavage of reporter constructs. In this role, Polθ generates chromosome rearrangements; for example, induction of a site-specific DSB at loci in two different mouse chromosomes by the CRISPR/Cas9 system led to a large increase in translocation events in Polθ+/+ cells whereas the frequency of translocations was greatly reduced in Polθ defective cells (Mateos-Gomez et al., 2015; Mateos-Gomez et al., 2017). In striking contrast, Polθ’s role in TLS protects against chromosome rearrangements. In UV irradiated Polθ−/− or Polθ−/− Polη−/− MEFS, the frequency of chromosome aberrations rises (Figure 4D) instead of declines, as would occur from Polθ’s role in end-joining. By generating chromosome rearrangements, Polθ’s role in end-joining would be causal for cancers (Bunting and Nussenzweig, 2013), whereas Polθ’s role in TLS protects against skin cancers. These results, as well as the lack of any suppressive effect of Polθ N-terminal helicase domain on Rad51-dependent SCEs in UV damaged or undamaged Polθ−/− MEFs (Figure S4D,E) support the conclusion that the mechanisms for the repair of two-ended DSBs generated by endonuclease cleavage differ from those used for the repair of one-ended DSBs resulting from replication stress induced by TLS defects. .
Somatic Mutations and Cancers
The observation of a strong correlation between the number of stem cell divisions in the lifetime of a given tissue and the lifetime risk of cancer in that tissue has indicated that stochastic effects associated with DNA replication account for over 60% of the variation in cancer risk in a tissue (Tomasetti and Vogelstein, 2015) and the prevalence of driver point mutations in cancer cells has buttressed the notion that somatic mutations generated from replication errors play a causal role (Martincorena et al., 2017; Tomasetti and Vogelstein, 2015). Our findings, however, suggest that genomic rearrangements that result from fork collapse due to stochastic impairments in DNA replication would play an effective role in tumorigenesis and that cancer formation can ensue in the absence of somatic point mutations.
STAR * METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Satya Prakash (saprakas@utmb.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines and Cell Culture
Normal human fibroblast (GM00637), XPA deficient human fibroblast (GM04429), XPV deficient human fibroblast (GM03617) and mouse embryonic fibroblast cells were grown in DMEM medium (GenDEPOT) containing 10% fetal bovine serum (GenDEPOT) and 1% Antibiotic-Antimyocotic (GenDEPOT). Cells were grown on plastic culture dishes at 37 °C in a humidified incubator with 5% CO2. Normal (GM00637) and XPA (GM04429) cells are female and XPV (GM03617) is male.
Mice
All the animal manipulations and experiments described in this report have been approved by the UTMB Institutional Animal Care and Use Committee (IACUC protocol no. 0809061), and conducted according to the protocol. Our procedures complied with the policies and guidelines of UTMB Animal Resources Center and IACUC, and Health Research Extension Act of 1985 (Public Law 99–158). We followed the Public Health Service Policy on Humane Care and Use of Laboratory Animals (revised September 1986) and the NAS Guide for the Care and Use of Laboratory Animals (ISBN-13: 978-0-309-15400-0 revised 2010).
For UV irradiation experiment, we used 8–12-week-old males. The experimental animals were healthy and immunocompetent. They did not undergo any experimental manipulation or receive any drug before UV irradiation except for genotyping. We used C57BL/6J congenic single mutants: Polqtm1Jes/Polqtm1Jes, Polqtm1Jes/Polq+, Polhtm1Rak/Polhtm1Rak, and Polhtm1Rak/Polh+; C57BL/6J congenic double mutants: Polqtm1Jes/Polqtm1Jes Polhtm1Rak/Polhtm1Rak; and C57BL/6J congenic wild-type mice. The mice were specific-pathogen-free, maintained within the UTMB Animal Resources Center, and housed in individually ventilated cages with food and water ad libitum at a constant temperature and humidity on a 12 hr light-dark cycle (lights on 0600–1800 hrs).
METHOD DETAILS
DNA polymerase assays with human Polθ
The standard DNA polymerase reaction (5µL) contained 40 mM Tris-HCl (pH 7.5), 5 mM MgCl2, 1 mM DTT, 10% Glycerol, 100 µg/mL BSA and 100µM each of dGTP, dATP, dTTP and dCTP and 10nM of DNA substrate. The DNA substrate for TLS assays opposite a cis-syn TT dimer was generated by annealing a 75 nt template 5’-AGCAAGTCACCAATGTCTAAGAGTTCGTATTATGCCTACACTGGAGTACCGGAGCATCGTC GTGACTGGGAAAAC-3’, in which there was either an undamaged TT or a cis-syn TT dimer at the position indicated by TT, to a 5’ 32P labelled oligonucleotide primer N4309, 5’-GTTTTCCCAGTCACGACGATGCTCCGGTACT CCAGTGTAGGCAT-3’ (44 nt). For TLS assays opposite a (6–4) TT photoproduct, the above noted 75 nt template containing an undamaged TT or a (6–4) TT photoproduct was annealed to a 23 nt 5’ 32P labeled oligonucleotide primer 5’-TCCGGTACTCCAGTGTAGGCATA-3’. The reactions for both undamaged template and damaged template contained 0.5 nM DNA polymerase θ (1708–2590) and were incubated at 37°C for 5 min with undamaged DNA and for 20 min with DNA containing a cis-syn TT dimer or a (6–4) TT photoproduct. The reactions were stopped by the addition of loading buffer (25µL) containing EDTA (20nM), 95% formamide, 0.3% bromophenol blue and 0.3% xylene cyanol. The reaction products were resolved on a 12% polyacrylamide gel containing 8M urea and visualized with the Typhoon FLA 7000 Phosphoimager.
Construction of plasmid vectors containing a cis-syn TT dimer or a (6–4) TT photoproduct
The heteroduplex vectors containing a cis-syn TT dimer or a (6–4) TT photoproduct on the leading or lagging strand template were constructed as described previously (Yoon et al., 2010a; Yoon et al., 2009).
Translesion synthesis assays in human cells
For siRNA knock down of Polθ, HPLC purified duplex siRNA for human and mouse genes were purchased from Ambion. The sense sequence of siRNA target sequence is provided in Key Resources Table and the efficiency of Polθ knockdown was verified by western blot analysis (Figure S2). The siRNA knock down efficiency of other TLS Pols as well as the detailed methods for TLS assay have been described previously (Yoon et al., 2015).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Goat anti-rabbit AlexaFluor 488 | ThermoFisher Scientific | A-11034 |
| Goat anti-rabbit AlexaFluor 594 | ThermoFisher Scientific | A-11012 |
| Goat anti-rabbit AlexaFluor 488 | ThermoFisher Scientific | A-11034 |
| Goat anti-rat AlexaFluor 594 | ThermoFisher Scientific | A-11007 |
| Goat anti-mouse AlexaFluor 488 | ThermoFisher Scientific | A-11001 |
| Goat anti-rat IgG-HRP | Santa Cruz Biotechnology | sc-2006 |
| Goat anti-rabbit IgG-HRP | Santa Cruz Biotechnology | sc-2004 |
| Goat anti-mouse IgG-HRP | Santa Cruz Biotechnology | sc-2005 |
| Rabbit anti-goat IgG-HRP | Santa Cruz Biotechnology | sc-2768 |
| Mouse anti-BrdU | BD Biosciences | 347580 |
| Rat anti-BrdU | Abcam | Ab6326 |
| Mouse monoclonal anti-Flag | Sigma-Aldrich | F1804 |
| Rabbit polyclonal anti-GFP | ThermoFisher Scientific | A-11122 |
| Mouse monoclonal anti-β tubulin (D-10) | Santa Cruz Biotechnology | sc-5274 |
| Rabbit polyclonal anti-Rad51 (H-92) | Santa Cruz Biotechnology | sc-8349 |
| Mouse monoclonal anti-PCNA (PC10) | Abcam | ab29 |
| Rabbit monoclonal anti-Rad18 | Cell Signaling Technology | 9040 |
| Rabbit polyclonal anti-Polθ | Abcam | Ab80906 |
| Experimental Models: Cell Lines | ||
| Human: Normal fibroblast | Coriell Institute Cell Repository | GM00637 |
| Human: XPA deficient fibroblast | Coriell Institute Cell Repository | GM04429 |
| Human: XPV deficient fibroblast | Coriell Institute Cell Repository | GM03617, XP30RO |
| Mouse: big blue mouse embryonic fibroblast | Agilent | Cat# 726010 |
| Experimental Models: Organisms/Strains | ||
| Polη knock out mice | Jackson Laboratory | N/A |
| Polθ knock out mice | Jackson Laboratory | N/A |
| Chemicals | ||
| Polyacrylamide | Bio-Rad | 162–0177 |
| Acetic acid | EMD chemicals | AX0073–75 |
| Paraformaldehyde | Sigma-Aldrich | F1635 |
| Acridine Orange | ThermoFisher Scientific | A-3568 |
| BrdU | Sigma-Aldrich | B5002 |
| Chlorodeoxyuridine (CldU) | Sigma-Aldrich | C6891 |
| 5-Iodo-2’-deoxyuridine (IdU) | Sigma-Aldrich | I7125 |
| Colcemid | ThermoFisher Scientific | 15212012 |
| Geimsa stain solution | ThermoFisher Scientific | 10092013 |
| DAPI | ThermoFisher Scientific | D1306 |
| Sodium dodecyl sulfate | Bio-Rad | 161-0302 |
| Sodium deoxycholate | Sigma-Aldrich | D6750 |
| Sodium phosphate monobasic | Sigma-Aldrich | 71505 |
| Sodium phosphate dibasic | Sigma-Aldrich | S3264 |
| IGEPAL CA-630 | Sigma-Aldrich | I8896 |
| Isopropyl-beta-D-thiogalactopyranoside (IPTG) | GenDEPOT | I0355 |
| 5-bromo-4-chloro-indolyl-B-D- galactopyranoside (X-gal) | GenDEPOT | X0320 |
| Antibiotics | ||
| Antibiotic-Antimyocotic | GenDEPOT | CA002 |
| Zeocin | GenDEPOT | Z4500 |
| Hygromycin B | ThermoFisher Scientific | 10687010 |
| Kanamycin | MP Biochemicals | 0215002925 |
| Bacterial strain | ||
| XL1 blue super competent cells | Fisherscientific | 50–125-047 |
| MB7070 | N/A | |
| Media constituents, reagents, buffers | ||
| DMEM high glucose | GenDEPOT | CM002 |
| EMEM media | ThermoFisher Scientific | 11095080 |
| DPBS buffer | GenDEPOT | CA008 |
| HBSS buffer | GenDEPOT | CA507 |
| Fetal Bovine Serum opti Gold | GenDEPOT | F0900 |
| Prolong gold antifade mounting media | ThermoFisher Scientific | P-36930 |
| Trypsin-EDTA, 0.25% | GenDEPOT | CA014 |
| Opti-MEM media | ThermoFisher Scientific | 31985088 |
| Lipofectamine 2000 | ThermoFisher Scientific | 11668019 |
| IMfectin DNA transfection reagent | GenDEPOT | I7200 |
| West-Q Pico ECL solution | GenDEPOT | W3652 |
| West-Q Femto Clean ECL solution | GenDEPOT | W3680 |
| Constructs and Plasmids | ||
| p3xFLAG-CMV-7.1 | Sigma-Aldrich | E4026 |
| p3xFLAG-CMV-7.1/zeocin-human Polη | This study | Yoon et al, G&D 2015 |
| p3xFLAG-CMV-7.1/zeocin-human Rev1 | This study | Yoon et al, G&D 2015 |
| pEGFP-N1 vector | Clonetech | 6085–1 |
| pGFP-Polη | This study | Yoon et al, G&D 2015 |
| pcDNA3.1-myc-PolQ-flag | Addgene | Plasmid# 73132 |
| pBS vector/CPD | This study | Yoon et al, PNAS 2009 |
| pBS vector/(6–4)PP | This study | Yoon et al, G&D 2010 |
| Kits | ||
| Transpack packaging extract | Fisherscientific | 50–125-041 |
| QIAamp DNA mini kit | Qiagen | 51304 |
| QIAquick PCR purification kit | Qiagen | 28106 |
| FastLinky DNA ligation kit | GenDEPOT | F0661 |
| AmfiSure ultra fidelity PCR master mix | GenDEPOT | P0346 |
| Spin column for DNA purification | GenDEPOT | S1920 |
| MTS cell proliferation assay | Promega | G5421 |
| SV40 Large T Antigen Lentifect Lentiviral particles | GeneCopeia | LPP-SV40T- Lv105 |
| Comet assay kit | Trevigen | 4252–040 |
| Blood & cell culture DNA midi kit | Qiagen | 13343 |
| Restriction and other enzymes | ||
| MfeI | New England Biolab | R3589 |
| SpeI | New England Biolab | R0133 |
| DpnI | New England Biolab | R0176 |
| BamHI | New England Biolab | R3136 |
| SbfI | New England Biolab | R0642 |
| T4 DNA polymerase | New England Biolab | M0203 |
| T4 DNA ligase | New England Biolab | M0202 |
| Dam methyltransferase | New England Biolab | M0222 |
| RnaseA | ThermoFisher Scientific | EN0531 |
| Proteinase K | ThermoFisher Scientific | 25530015 |
| Softwares and Algorithms | ||
| Adobe illustrator | Adobe Systems | Version CS5 |
| GraphPad Prism | GraphPad software | Version6 |
| NIS-Elements AR | Nikon | Version4 |
| Oligonucleotides and siRNAs | ||
| human siPolθ 1 | ThermoFisher Scientific | siRNA |
| (GCCAAUGGUCUGAUCAAUC) | (Ambion) | ID#s122557 |
| human siPolθ 2 | ThermoFisher Scientific | siRNA |
| (CCGCUUUUGGAGUCAGUAA) | (Ambion) | ID#s21059 |
| human siRad51 | ThermoFisher Scientific | siRNA |
| (GGGAAUUAGUGAAGCCAAA) | (Ambion) | ID#s4467 |
| Mouse siPolθ | ThermoFisher Scientific | siRNA |
| (GCGAAGAGUUUCUGAUGAC) | (Ambion) | 4467ID#s174721 |
| mouse siRad51 | ThermoFisher Scientific | siRNA |
| (GCAGCAAAAUUGGUUCCAA) | (Ambion) | ID# s72671 |
Western blot analysis
48h after siRNA transfection, cells were washed with PBS buffer and lysed with RIPA buffer (1x PBS, 1% IP-40, 0.5% sodium deoxycholate, 0.1% SDS). After 15 min incubation on ice, cellular mixture was centrifuged and the supernatant was collected. Equivalent amounts (approximately 30µg) of prepared cellular extracts were separated on a 6% SDS-polyacrylamide gel and transferred to a PVDF membrane (Bio-rad). The membranes were probed with antibodies against human Polθ (monoclonal antibody in rabbit raised against polθ peptide by Abmart for us) or Flag (Sigma-Aldrich), followed by appropriate secondary antibodies conjugated with horseradish peroxidase. The signals were detected using ECL-Plus (GenDEPOT). For the loading control, anti-β-tubulin antibody (Santa Cruz Biotechnology) was used.
Stable expression of wild type and catalytic mutation of Polθ (1708–2590) in HF cells
Full length (7776 bp) human Polθ cDNA was obtained from Addgene plasmid Repository (plasmid #:73132). WT Polθ C-terminal domain (1708–2590) and its catalytic mutation were subcloned into pCMV7–3xFlag-zeo vector (Sigma). The vectors were transfected into GM637 HFs by Lipofectamine 2000 reagent (Invitrogen). After 24h incubation, 0.5 µg of Zeocin (GenDEPOT) were added to the culture media. After 3 days of incubation, cells were washed with PBS buffer and were continuously cultured with the media containing 250 ng of Zeocin for ~ 2 weeks. Protein expression and siRNA knock down efficiency were verified by western blot analysis (Figure S2A).
Co-immunoprecipitation of proteins in chromatin extracts
For chromatin bound nuclear extracts, GM637 HF cells were washed twice with ice-cold PBS buffer. Cells were lysed with CSK (Cytoskeleton) buffer (10mM Hepes pH 6.8, 100mM NaCl, 300mM sucrose, 3mM MgCl2, 1mM EGTA, 0.1% Triton X-100, e-complete protease inhibitors) and chromatin extracts were crosslinked with 1% formalin (Sigma-Aldrich) in PBS buffer for 10min at room temperature followed by 125mM glycine addition. Cell pellets were resuspended in micrococal nuclease (MNase) buffer containing 2,000 units/mL of MNase (NEB). Extracts were incubated at room temperature for 10min and then diluted with an equal volume of 2X immunoprecipitation buffer (300mM NaCl, 50mM Tris-HCl pH7.5, 2mM EDTA, 0.5% TritonX100, 10% glycerol, phosphatase inhibitor and protease inhibitors). The extracts were solubilized by sonication (4× 10sec with 30sec interval) and isolated by centrifugation at 17,000g for 15min at 4°C. 2mg of chromatin extracts were mixed with 40 µL of Flag agarose beads (Sigma-Aldrich) overnight at 4°C. Flag agarose beads were washed with IP buffer twice and bound proteins were eluted in 2x sample buffer (20% glycerol, 125mM Tris-HCl pH6.8, 5% β-mercaptoethanol, 50mM DTT, 0.05% bromophenol blue). PCNA, Rad18, Flag ab were used for western blot analysis.
UV survival assay
GM637 HF cells were transfected with siRNAs and 48h after siRNA transfection, cells were treated with UV. For primary MEFs, cells were seeded on duplicated 6 well plates and incubated overnight. For UV irradiation, cells were washed with PBS buffer and irradiated with various doses of UVC light in the presence of PBS buffer. After irradiation, fresh growth media were added into cells. Cells were incubated for additional 48h after UV irradiation. The UV cytotoxicity was determined by MTS assay (Promega). Briefly, 100 µL of MTS assay solutions were added to each well and incubated for 30 min. Cell viability was determined by measuring OD at 490nM, and four independent experiments were carried out.
Big blue transgenic mouse cell line and siRNA knockdown
The big blue transgenic mouse embryonic fibroblast (BBMEF) cells were grown in DMEM medium containing 10% FBS (GenDEPOT) and antibiotics. HPLC purified duplex siRNA for mouse Polθ was purchased from Ambion. The sense sequence of mPolθ siRNA is shown in Key Resosurces Table and the efficiency of its knockdown was verified by western blot analysis. For the cII mutation assay, cells were plated on 100 mm plates at 50% confluence (approximately 5 × 106 cells) and 500 pmoles of synthetic duplex siRNAs were transfected using 50 µl of Lipofectamine 2000 reagent (Invitrogen) following the manufacturer’s instructions.
UV irradiation, photoreactivation, and cII mutational assays in siRNA treated BBMEF cells
48h after siRNA knock down, cells were washed with HBSS buffer (Invitrogen) and irradiated at 5 J/m2 with UVC light, followed by photoreactivation for 3 h at room temperature as previously described (Yoon et al., 2009; Yoon et al., 2010b). Fresh growth medium was then added and cells were incubated for 24 h. After the 24h incubation period, the second siRNA transfection was carried out to maintain the siRNA knock down of the target gene(s). Cells were incubated for an additional 4 days to allow for mutation fixation. The mouse genomic DNA was isolated using the genomic DNA isolation kit (Qiagen). The LIZ shuttle vector was rescued from the genomic DNA by mixing DNA aliquots and transpack packaging extract (Stratagene), and the cII assay was carried out as previously described (Yoon et al., 2009; Yoon et al., 2010b). The mutation frequency was calculated by dividing the number of mutant plaques by the number of total plaques. For mutation analysis, the sequence of PCR products of the cII gene from the mutant plaques were analyzed as described previously (Yoon et al., 2009; Yoon et al., 2010b).
SupF UV mutation assay
293T cells were transfected with siRNAs (100 pmole) and 48 h after siRNA transfection, cells were cotransfected with siRNA (50 pmole) and with UV irradiated (UVC 500 J/m2) pSP189 shuttle vector. Plasmid DNA was rescued after 48 h incubation and treated with DpnI to remove the unreplicated plasmid DNA. The rescued plasmids were transformed into MB7070 bacterial cells which carry a lacZ gene harboring an amber mutation. The transformed bacterial cells were grown on LB plates containing ampicillin, IPTG, and X-gal, and mutation frequency was determined by the ratio of white (mutant) colonies to total (blue and white) colonies.
Foci formation assay
For Polθ (1708–2590) foci analysis, cells stably expressing GFP Polθ(1708–2590) and pcDNA3-zeo-GFP vector were treated with siRNA and cultured on a coverslip with 50% confluence. After 48h, cells were treated with UVC (30J/m2). After UV irradiation, fresh growth media were added and cells were incubated for 6h. After washing with PBS buffer, cells were pre-extracted in 0.2% Triton X-100 for 2min and fixed with 4% paraformaldehyde for 20min. Nuclear staining was performed with DAPI (Molecular probe) in PBS buffer for 20min. The fluorescent images were visualized and captured by fluorescence microscope (Nikon Eclipse 80i).
DNA fiber assay
GM637 HF cells were transfected with siRNAs (100 pmole) and 48h after siRNA transfection, cells were pulse-labelled with 25µM IdU (sigma) for 20 min. Cells were then washed with PBS buffer twice and irradiated with UVC (10 J/m2). After UV irradiation, cells were labelled with 250µM CldU for 30 min. DNA fibers were spread on glass slides, and slides incubated in 2.5M HCl for 90min and then washed with PBS buffer. The slides were incubated in blocking buffer, 5% BSA in PBS for 2h. Primary antibodies, rat anti-BrdU antibody (Abcam) and mouse anti-BrdU antibody (BD bioscience) were diluted in blocking buffer and incubated for 1h followed by extensive washing with PBS buffer. Secondary antibodies, goat anti-rat Alexa 594 and goat anti-mouse Alexa 488 were applied for 30min and slides were mounted with antifade gold mounting media (Invitrogen). Fibers were analyzed by Nikon Eclipse fluorescence microscope.
Generation of Polθ−/−, Polη−/−, and Polη−/− Polθ−/− mice
For Polθ−/− mutant mice, C57BL/6J congenic strain, B6.Cg-Polqtm1Jes/J was purchased from the Jackson Laboratory (Shima et al., 2004). Mutant mice were backcrossed to C57BL/6J for nine generations prior to arrival at the UTMB. We maintained the mutants by backcrossing them to C57BL/6J and generated experimental mice by heterozygous intercrosses. We genotyped 253 offspring generated by heterozygous intercrosses, and found 57 Polθ−/−, 123 Polθ+/−, and 73 Polθ+/+. The average number of viable pups was 6.4/litter (313/49 litters).
For Polη−/− mutant mice, B6;129-Polhtm1Rak (Lin et al., 2006) was backcrossed to C57BL/6J under the speed congenic program at the Jackson Laboratory. Experimental mice, heterozygous and homozygous males, were produced at the Jackson Laboratory and at the UTMB after a line >98% derived from C57BL/6 was established. Polη−/− mice were overtly normal and fertile. The average number of viable pups from homozygous incross breeders was 7.3/litter (86/12 litters).
For Polθ−/− Polη−/− mutant mice, we first crossed Polhtm1Rak/Polhtm1Rak females and Polqtm1Jes/Polq+ males to get double heterozygotes, and then intercrossed double heterozygotes to get Polθ−/− Polη−/− mice. From the Polθ+/− Polη+/− x Polθ+/− Polη+/− intercrosses, we recovered Polθ−/−Polη−/− and other genotypes (Table S4). We incrossed Polθ−/− Polη−/− for 2–3 generations to produce experimental Polθ−/− Polη−/− males, and backcrossed Polθ−/− Polη−/− mice to C57BL/6J to produce experimental Polθ+/− Polη+/− males. Polθ−/− Polη−/− mice were overtly normal and fertile, but they were poor breeders. The neonatal litter size of Polθ−/− Polη−/− incrosses appeared to be smaller, and pups were often lost before weaning. The average number of viable pups was 2.9/litter (176/60 litters). C57BL/6J females paired with Polθ−/− Polη−/− males produced larger litters and successfully raised them, i.e., the average number of viable pups was 7.8/litter (109/14 litters}. These results suggest that the poor reproductive performance of double homozygous breeders may be due to the dams, and/or that the lack of both Polθ and Polη may cause embryonic/perinatal lethality with incomplete penetrance or decreased survival rate in the mouse.
To identify the Polη knock out, tail DNA genotyping was performed using the following primers; for wild type allele (370bp PCR products), XPV-com primer: 5’– AAGGGACAAGCGAACAGAGA, and XPV-wt primer: 5’– TCACTTCAACACTAGCTTCCC, and for mutant allele (500bp PCR products), XPV-com primer: 5’–AAGGGACAAGCGAACAGAGA, and XPV-mut: 5’– AGCAATATCACAGGCCCAAC. All primers were purchased from Sigma-Aldrich. To identify the Polθ knock out, tail DNA genotyping was performed using the following primers; for wild type allele (300bp PCR products), QWT forward primer: 5’– TGCAGTGTACAGATGTTACTTTT, and QWT-reverse primer: 5’– TGGAGGTAGCATTTCTTCTC, and for mutant allele (190bp PCR products), Qmut-forward primer: 5’–TCACTAGGTTGGGGTTCT, and Qmut-reverse primer: 5’– CATCAGAAGCTGACTCTAGAG.
Isolation of Polθ−/− Polη−/−, and Polθ−/− Polη−/−MEFs
Primary MEFs were isolated from embryos derived from intercrossing of Polη+/− and Polθ+/− mice as described previously (Tommasi et al., 2005; Yoon et al., 2015). In brief, mouse embryos harvested in utero at 13.5 days of gestation were roughly minced and incubated with trypsin for 20 min at room temperature. Homogenous cell suspensions were then added to 25 ml of Dulbecco Modified Eagle’s Medium (DMEM, Genedepot), supplemented with 10% fetal calf serum. Early passage (P<5) mouse embryonic fibroblasts (MEFs) were used for all experiments. For verifying the Polθ−/− and Polη−/− genotypes, RT-PCR was carried out. Total RNA was extracted using Qiagen RNasy extraction kit (Qiagen) and 100 ng of total RNA were used for RT-PCR analysis. RT-PCR was performed with Qiagen one step RT-PCR kit following the manufacturer’s protocols. For mouse GAPDH, the amplification was carried out at 95 °C for 30 sec, 55 °C for 45 sec, and 72 °C for 50 sec for 24 cycles. Twenty six cycles for mouse Polθ and Polη were applied for the amplification at 95 °C for 30 sec, 55 °C for 45 sec, and 72 °C for 1 min. RT-PCR products were analyzed on 1.5% agarose. The primers used for RT-PCR analyses are given in Table S5. Assays for DNA synthesis in UV irradiated MEFs and for UV survival were performed as described for HFs.
Stable expression of full length Polθ and C-terminal (1708–2590) polymerase domain of Polθ in Polθ−/− MEFs
Polθ−/− MEFs were immortalized by lentivirus expressing SV40 large T antigen transfection (Genecopoeia). Transformed MEFs were transfected with plasmids carrying Myc-full length (pCDNA3.1-Myc-Polθ) or GFP-C-terminal (1708–2590) Polθ. Transformed Polθ−/− MEFs stably expressing Myc-full length or GFP-C-terminal (1708–2590) Polθ were confirmed by western blot analysis.
ssDNA detection assay (non-denaturing BrdU staining)
Primary MEFs were cultured with 20µM BrdU for 20h and irradiated with UVC (20J/m2). Cells were incubated in growth media for 6h to induce ssDNA accumulation. Cells were then harvested and treated with 75mM KCl for 20min at 37 °C, fixed with 3:1 methanol: acetic acid mix and spread on glass slides. Primary mouse anti-BrdU antibody (BD bioscience) were diluted in blocking buffer (PBS with 5% BSA) and incubated for 1h followed by extensive washing with PBS buffer. Secondary antibodies, goat anti-mouse Alexa 488 were applied for 30min and DAPI staining done for 10min. Slides were mounted with antifade gold mounting media (Invitrogen). ssDNA accumulation and DAPI staining were analyzed with a Nikon Eclipse 80i fluorescence microscope.
Neutral comet assay
To analyze UV induced DSBs in primary MEFs, neutral comet assays were performed. Cells were irradiated with UVC (20J/m2) and incubated in growth media for 6h. Cells were then harvested and mixed with agar and spread on glass slides. Neutral comet assay was done with Comet Assay kit (Trevigen) as described (Collins et al., 2008). Comet tail DNA was visualized and captured by fluorescence microscope (Nikon Eclipse 80i).
Sister chromatid exchange assay
Wild type and mutant primary MEFs were irradiated with UVC (2J/m2) and labeled with BrdU (100µM) in growth media for 48h followed by colcemid (0.2µg/ml, Invitrogen) treatment for 3 h. Cells were then harvested and treated with 75mM KCl for 20 min at 37 °C, fixed with 3:1 methanol: acetic acid mix and spread on glass slides. After drying for one day, slides were stained with Acridine Orange (Invitrogen, 0.1 mg/ml) for 5 min and incubated in Sorensen’s buffer (0.1M NaH2PO4 + 0.1M Na2HPO4, pH6.8). Metaphases were visualized and analyzed by Nikon Eclipse fluorescence microscope.
Chromosomal aberration assay
WT and mutant primary MEFs were irradiated with UVC (2J/m2) and incubated for 48h followed by colcemid (0.2µg/ml, Invitrogen) treatment for 3 h. Cells were then harvested and treated with 75mM KCl for 20min at 37 °C, fixed with 3:1 methanol: acetic acid mix and spread on glass slides. After drying for one day, slides were stained with Giemsa staining solution (4%, Invitrogen) Metaphases were visualized and analyzed by Nikon Eclipse microscope.
Analysis of UVB induced skin tumors in WT, Polθ−/−, Polη−/−, and Polη−/− Polθ−/− mice
8 week old male mice were treated with UVB (2KJ/m2 for 3 times per week) after removing hair on dorsal area of mice. For the Polθ−/− cohorts, mice were irradiated for 50 weeks and for Polη−/− and Polη−/− Polθ−/− cohorts, mice were irradiated for up to 30 weeks. All mice were carefully and extensively checked once per week for skin tumor development on the dorsal skin. For histopathological analyses, skin lesions (larger than 3mm diameter) were removed from the dorsal skin and fixed with 10% neural-buffered formalin. Samples were embedded in paraffin and sections were prepared.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical details of individual experiments, including number of observations and number of experiments done, mean values and standard deviations, and p values of two-tailed t-tests are described in the figure legends and indicated in the figures. Information on quantification of data is also included in Tables and described in Table legends. Statistical analyses were done using GraphPad software Prism6.
Supplementary Material
(A) Pathways for replicating through CPDs. Polη replicates through CPDs in an error-free manner. Polθ functions in inserting nucleotides opposite the 3’T or 3’C of a CPD in the error-prone TLS pathways dependent upon Polκ or Polζ for extension of synthesis.
(B) Pathways for replicating through (6–4) photoproducts. Polθ extends synthesis from the nucleotides inserted by Polη or Polι opposite the 3’T or 3’C of the (6–4) photoproduct and it promotes error-prone TLS. Polζ functions in an alternate error-free TLS pathway.
(C) UV induced (5 J/m2) mutational spectra in the cII gene in BBMEF cells. UV induced mutations that occur in control siRNA (NC) treated cells are shown above the sequence and the lack of UV induced hot spot mutations in cells treated with Polθ siRNA is shown below the sequence. The positions of UV induced hot spots #1–11 are indicated. A frequently occurring spontaneous hot spot at position 223 is indicated by lower case ‘g’.
(D) UV induced (500 J/m2) mutation frequencies in the supF gene in 293T cells depleted for Polθ, XPA, or both. Mutation frequencies represent the average of 3 different experiments; error bars represent ± SD. Students two-tailed t-test p values, ***, p<0.001; ****, p<0.0001.
(A) Map of human Polθ and constructs of C-terminal domain of WT and catalytic mutant of Polθ. Expression of 3xFlag-Polθ (1708–2590) or 3xFlag-Polθ (1708–2590) containing D2540A, E2541A (DE→AA) mutations in GM637 HFs. siRNA targeted to site 1 was used for depletion of genomic Polθ and siRNA targeted to site 2 was used for depletion of Polθ (1708–2590). The efficiency of siRNA depletion of genomic Polθ was verified by western blot with Polθ ab and the efficiency of depletion of Polθ (1708–2590) was verified by Flag ab. β-tubulin was used as the loading control.
(B) Co-immunoprecipitation of 3xFlag Polθ (1708–2590) with ub-PCNA in chromatin bound fractions. GM637 cells expressing 3xFlag-Polθ (1708–2590) were UV irradiated and incubated for 6h. Chromatin extracts were immunoprecipitated with Flag M2-agarose. Co-immunoprecipitation of Flag-Polθ (1708–2590) with PCNA and Rad18 was determined by western blot analysis.
(C) Accumulation of Polθ (1708–2590) into foci. GM637 cells expressing GFP-Polθ (1708–2590) were treated with control siRNA. After 48 h of siRNA treatment, cells were UV irradiated. GFP-Polθ foci were visualized by fluorescence microscopy. Representative images of GFP-Polθ foci in unirradiated or UV irradiated cells.
(D) Quantification of GFP-Polθ(1708–2590) foci containing cells treated with control, Polη, or Rad18 siRNA. The mean and standard deviation were analyzed from 4 independent experiments. Student’s two-tailed t-test p values, *, p<0.05; ****, p<0.0001.
(A) Incorporation of dNTPs opposite a cis-syn TT dimer by Polθ. 0.5 nM Polθ (1708–2590) was incubated with the DNA substrate (10 nM) in the presence of 100 μM of a single dNTP or each of four dNTPs (N) for 5 min at 37°C with undamaged DNA (lanes 2–6) or for 20 min with DNA containing a cis-syn TT dimer (lanes 8–12).
(B) Extension of synthesis from the nucleotide opposite the 3’T of a (6–4) TT photoproduct by Polθ. Assay conditions were the same as in (A), and the DNA substrates used were undamaged DNA (lanes 2–6) and (6–4) TT photoproduct containing DNA (lanes 8–12).
(A) Western blot analysis of expression of human GFP-Polθ (1708–2590) and of siRNA depletion of mouse Rad51 in SV40 transformed Polθ−/− MEFs. β-tubulin was used as the loading control.
(B) Complementation of the TLS deficiency in Polθ−/− MEFs by Polθ (1708–2590).
(C) Schematic of DNA fiber assay and images of stretched DNA fibers in UV irradiated Polθ−/− primary MEFs carrying vector control or expressing Polθ (1708–2590) are shown on the left and quantitative analyses of fork progression (mean CldU:IdU ratio) are shown on the right. The data represent ~400 DNA fibers from 4 independent experiments. Error bars indicate the standard deviation. Students two-tailed t-test p values, p< 0.05.
(D) Quantification of scatter plot analyses of SCE frequencies in non-UV irradiated Polθ−/− MEFs carrying the GFP vector, expressing Myc-full length Polθ, or GFP-Polθ (1708–2590), and untreated or treated with Rad51 siRNA. Each datum point represents a single metaphase and ~1,000 metaphase chromosomes were analyzed. The mean and standard deviation were analyzed from 4 independent experiments. Student’s two-tailed t-test values, *, p<0.05; **, p<0.01
(E) Quantification of scatter plot analysis of UV induced SCE frequency in Polθ−/− MEFs carrying GFP vector, expressing Myc-full length Polθ, or GFP-Polθ (1708–2590) and treated or not treated with Rad51 siRNA. Each datum point represents a single metaphase and ~1,000 metaphase chromosomes were analyzed. The mean and standard deviation were analyzed from 4 independent experiments. Student’s two-tailed t-test p values, ***, p<0.001; ****, p<0.0001.
(F) Quantification of chromosomal aberrations in unirradiated or UV irradiated Polθ−/− MEFs carrying vector or expressing Polθ (1708–2590). The data represent analyses of ~400 metaphases from 4 independent experiments. The mean and standard deviation were analyzed from 4 independent experiments. Student’s two-tailed t-test values, ns, not significant; **, p<0.01.
(G) UV survival assay in Polθ−/− MEFs carrying vector or expressing Polθ (1708–2590). Error bars indicate the standard deviation of results of 4 independent experiments. Student’s two-tailed t-test values, ****, p<0.0001.
(A) Normal skin from Polη−/− Polθ−/− mice.
(B) An example of well-differentiated SCC from Polθ−/− mice which shows flask-shaped lesions with prominent atypia and mitotic activity.
(C) An example from Polθ−/− mice of carcinoma in situ which were frequently present in multiples. Carcinomas show less differentiation than SCCs; unlike SCCs, they lack intercellular spinous processes and keratin pearls.
(A) Polθ−/− mice.
(B) Polη−/− mice.
(C) Polη−/− Polθ−/− mice.
SCCs show varying degrees of differentiation from well differentiated in (A,B) to less differentiated in (C).
Highlights.
DNA polymerase θ is essential for mutagenic replication through UV lesions
UV induced skin cancer formation rises in the absence of Polθ
Translesion synthesis by Polη or Polθ prevents replication fork collapse
TLS protects against chromosomal instability and tumorigenesis
ACKNOWLEDGMENTS
We thank Mariano Garcia-Blanco for discussions and helpful suggestions. This work was supported by NIH grants ES020833 and ES022948..
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Besaratinia A, and Pfeifer GP (2006). Investigating human cancer etiology by DNA lesion footprinting and mutagenicity analysis. Carcinogenesis 27, 1526–1537. [DOI] [PubMed] [Google Scholar]
- Biertumpfel C, Zhao Y, Kondo Y, Ramon-Maiques S, Gregory M, Lee JY, Masutani C, Lehmann AR, Hanaoka F, and Yang W (2010). Structure and mechanism of human DNA polymerase η. Nature 465, 1044–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunting SF, and Nussenzweig A (2013). End-joining, translocations and cancer. Nat. Rev. Cancer 13, 443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casasent AK, Schalck A, Gao R, Sei E, Long A, Pangburn W, Casasent T, Meric-Bernstam F, Edgerton ME, and Navin NE (2018). Multiclonal invasion in breast tumors identified by topographic single cell sequencing. Cell 172, 205–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccaldi R, Liu JC, Amunugama R, Hajdu I, Primack B, Petalcorin MI, O’Connor KW, Konstantinopoulos PA, Elledge SJ, Boulton SJ, et al. (2015). Homologous-recombination-deficient tumours are dependent on Poltheta-mediated repair. Nature 518, 258–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins AR, Oscoz AA, Brunborg G, Gaivao I, Giovannelli L, Kruszewski M, Smith CC, and Stetina R (2008). The comet assay: topical issues. Mutagenesis 23, 143–151. [DOI] [PubMed] [Google Scholar]
- Debeb BG, Zhang X, Krishnamurthy S, Gao H, Cohen E, Li L, Rodriguez AA, Landis MD, Lucci A, Ueno NT, et al. (2010). Characterizing cancer cells with cancer stem cell-like features in 293T human embryonic kidney cells. Mol. Cancer 9, 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durinck S, Ho C, Wang NJ, Liao W, Jakkula LR, Collisson EA, Pons J, Chan SW, Lam ET, Chu C, et al. (2011). Temporal dissection of tumorigenesis in primary cancers. Cancer Discov 1, 137–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedberg EC, Walker GC, Siede W, and Wood RD (2005). DNA Repair and Mutagenesis (American Society for Microbiology Press; ). [Google Scholar]
- Glass AG, and Hoover RN (1989). The emerging epidemic of melanoma and squamous cell skin cancer. J. Am. Med. Assoc 262, 2097–2100. [PubMed] [Google Scholar]
- Gyori BM, Venkatachalam G, Thiagarajan PS, Hsu D, and Clement MV (2014). OpenComet: an automated tool for comet assay image analysis. Redox Biol 2, 457–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halazonetis TD, Gorgoulis VG, and Bartek J (2008). An oncogene-induced DNA damage model for cancer development. Science 319, 1352–1355. [DOI] [PubMed] [Google Scholar]
- Jayaraman SS, Rayhan DJ, Hazany S, and Kolodney MS (2014). Mutational landscape of basal cell carcinomas by whole-exome sequencing. J. Invest. Dermatol 134, 213–220. [DOI] [PubMed] [Google Scholar]
- Johnson RE, Haracska L, Prakash S, and Prakash L (2001). Role of DNA polymerase η in the bypass of a (6–4) TT photoproduct. Mol. Cell. Biol 21, 3558–3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RE, Kondratick CM, Prakash S, and Prakash L (1999a). hRAD30 mutations in the variant form of xeroderma pigmentosum. Science 285, 263–265. [DOI] [PubMed] [Google Scholar]
- Johnson RE, Prakash S, and Prakash L (1999b). Efficient bypass of a thymine-thymine dimer by yeast DNA polymerase, Polη. Science 283, 1001–1004. [DOI] [PubMed] [Google Scholar]
- Johnson RE, Washington MT, Haracska L, Prakash S, and Prakash L (2000). Eukaryotic polymerases ι and ζ act sequentially to bypass DNA lesions. Nature 406, 1015–1019. [DOI] [PubMed] [Google Scholar]
- Jonason AS, Kunala S, Price GJ, Restifo RJ, Spinelli HM, Persing JA, Leffell DJ, Tarone RE, and Brash DE (1996). Frequent clones of p53-mutated keratinocytes in normal human skin. Proc. Natl. Acad. Sci. U. S. A 93, 14025–14029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein AM, Brash DE, Jones PH, and Simons BD (2010). Stochastic fate of p53-mutant epidermal progenitor cells is tilted toward proliferation by UV B during preneoplasia. Proc. Natl. Acad. Sci. U. S. A 107, 270–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Q, Clark AB, McCulloch SD, Yuan T, Bronson RT, Kunkel TA, and Kucherlapati R (2006). Increased susceptibility to UV-induced skin carcinogenesis in polymerase eta-deficient mice. Cancer Res 66, 87–94. [DOI] [PubMed] [Google Scholar]
- Ling G, Persson A, Berne B, Uhlen M, Lundeberg J, and Ponten F (2001). Persistent p53 mutations in single cells from normal human skin. Am. J. Pathol 159, 1247–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Carvalho CM, Hastings PJ, and Lupski JR (2012). Mechanisms for recurrent and complex human genomic rearrangements. Curr. Opin. Genet. Dev 22, 211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macheret M, and Halazonetis TD (2015). DNA replication stress as a hallmark of cancer. Annu. Rev. Pathol 10, 425–448. [DOI] [PubMed] [Google Scholar]
- Macheret M, and Halazonetis TD (2018). Intragenic origins due to short G1 phases underlie oncogene-induced DNA replication stress. Nature 555, 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnus K (1991). The Nordic profile of skin cancer incidence. A comparative epidemiological study of the three main types of skin cancer. Int. J. Cancer 47, 12–19. [DOI] [PubMed] [Google Scholar]
- Martincorena I, Raine KM, Gerstung M, Dawson KJ, Haase K, Van Loo P, Davies H, Stratton MR, and Campbell PJ (2017). Universal Patterns of Selection in Cancer and Somatic Tissues. Cell 171, 1029–1041 e1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martincorena I, Roshan A, Gerstung M, Ellis P, Van Loo P, McLaren S, Wedge DC, Fullam A, Alexandrov LB, Tubio JM, et al. (2015). Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 348, 880–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masutani C, Kusumoto R, Yamada A, Dohmae N, Yokoi M, Yuasa M, Araki M, Iwai S, Takio K, and Hanaoka F (1999). The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase η. Nature 399, 700–704. [DOI] [PubMed] [Google Scholar]
- Mateos-Gomez PA, Gong F, Nair N, Miller KM, Lazzerini-Denchi E, and Sfeir A (2015). Mammalian polymerase theta promotes alternative NHEJ and suppresses recombination. Nature 518, 254–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateos-Gomez PA, Kent T, Deng SK, McDevitt S, Kashkina E, Hoang TM, Pomerantz RT, and Sfeir A (2017). The helicase domain of Poltheta counteracts RPA to promote alt-NHEJ. Nat. Struct. Mol. Biol 24, 1116–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moynahan ME, and Jasin M (2010). Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat. Rev. Mol. Cell Biol 11, 196–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy SJ, Hart SN, Lima JF, Kipp BR, Klebig M, Winters JL, Szabo C, Zhang L, Eckloff BW, Petersen GM, et al. (2013). Genetic alterations associated with progression from pancreatic intraepithelial neoplasia to invasive pancreatic tumor. Gastroenterology 145, 1098–1109 e1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa H, English D, Randell PL, Nakazawa K, Martel N, Armstrong BK, and Yamasaki H (1994). UV and skin cancer: specific p53 gene mutation in normal skin as a biologically relevant exposure measurement. Proc. Natl. Acad. Sci. U. S. A 91, 360–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negrini S, Gorgoulis VG, and Halazonetis TD (2010). Genomic instability--an evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol 11, 220–228. [DOI] [PubMed] [Google Scholar]
- Notta F, Chan-Seng-Yue M, Lemire M, Li Y, Wilson GW, Connor AA, Denroche RE, Liang SB, Brown AM, Kim JC, et al. (2016). A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature 538, 378–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oikarinen A, and Raitio A (2000). Melanoma and other skin cancers in circumpolar areas. Int. J. Circumpolar Health 59, 52–56. [PubMed] [Google Scholar]
- Pfeifer GP (1997). Formation and processing of UV photoproducts: effects of DNA sequence and chromatin envirnoment. Photochem. Photobiol 65, 270–283. [DOI] [PubMed] [Google Scholar]
- Pickering CR, Zhou JH, Lee JJ, Drummond JA, Peng SA, Saade RE, Tsai KY, Curry JL, Tetzlaff MT, Lai SY, et al. (2014). Mutational landscape of aggressive cutaneous squamous cell carcinoma. Clin. Cancer Res 20, 6582–6592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp S, Waltering S, Holtgreve-Grez H, Jauch A, Proby C, Leigh IM, and Boukamp P (2000). Genetic characterization of a human skin carcinoma progression model: from primary tumor to metastasis. J. Invest. Dermatol 115, 1095–1103. [DOI] [PubMed] [Google Scholar]
- Rubbi CP, and Milner J (2001). Analysis of nucleotide excision repair by detection of single-stranded DNA transients. Carcinogenesis 22, 1789–1796. [DOI] [PubMed] [Google Scholar]
- Seki M, Marini F, and Wood RD (2003). PolQ (Pol θ), a DNA polymerase and DNA-dependent ATPase in human cells. Nucleic Acids Res 31, 6117–6126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki M, Masutani C, Yang LW, Schuffert A, Shigenori I, Bahar I, and Wood RD (2004). High-efficiency bypass of DNA damage by human DNA polymerase Q. EMBO J 23, 4484–4494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki M, and Wood RD (2007). DNA polymerase θ (POLQ) can extend from mismatches and from bases opposite a (6–4) photoproduct. DNA Repair 7, 119–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shima N, Munroe RJ, and Schimenti JC (2004). The mouse genomic instability mutation chaos1 is an allele of Polq that exhibits genetic interaction with Atm. Mol. Cell. Biol 24, 10381–10389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverstein TD, Johnson RE, Jain R, Prakash L, Prakash S, and Aggarwal AK (2010). Structural basis for the suppression of skin cancers by DNA polymerase eta. Nature 465, 1039–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasetti C, and Vogelstein B (2015). Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 347, 78–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tommasi S, Dammann R, Zhang Z, Wang Y, Liu L, Tsark WM, Wilczynski SP, Li J, You M, and Pfeifer GP (2005). Tumor susceptibility of Rassf1a knockout mice. Cancer Res 65, 92–98. [PubMed] [Google Scholar]
- Vasquez-Del Carpio R, Silverstein TD, Lone S, Johnson RE, Prakash L, Prakash S, and Aggarwal AK (2011). Role of human DNA polymerase kappa in extension opposite from a cis-syn thymine dimer. J. Mol. Biol 408, 252–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washington MT, Johnson RE, Prakash L, and Prakash S (2002). Human DINB1-encoded DNA polymerase κ is a promiscuous extender of mispaired primer termini. Proc. Natl. Acad. Sci. U. S. A 99, 1910–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon J-H, Bhatia G, Prakash S, and Prakash L (2010a). Error-free replicative bypass of thymine glycol by the combined action of DNA polymerases κ and ζ in human cells. Proc. Natl. Acad. Sci. U. S. A 107, 14116–14122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon J-H, Lee CS, O’Connor TR, Yasui A, and Pfeifer GP (2000). The DNA damage spectrum produced by simulated sunlight. J. Mol. Biol 299, 681–693. [DOI] [PubMed] [Google Scholar]
- Yoon J-H, Prakash L, and Prakash S (2009). Highly error-free role of DNA polymerase η in the replicative bypass of UV induced pyrimidine dimers in mouse and human cells. Proc. Natl. Acad. Sci. U. S. A 106, 18219–18224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon J-H, Prakash L, and Prakash S (2010b). Error-free replicative bypass of (6–4) photoproducts by DNA polymerase ζ in mouse and human cells. Genes Dev 24, 123–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon JH, Park J, Conde J, Wakamiya M, Prakash L, and Prakash S (2015). Rev1 promotes replication through UV lesions in conjunction with DNA polymerases η, ι, and κ but not DNA polymerase ζ. Genes Dev 29, 2588–2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon JH, Roy Choudhury J, Park J, Prakash S, and Prakash L (2014). A role for DNA polymerase theta in promoting replication through oxidative DNA lesion, thymine glycol, in human cells. J. Biol. Chem 289, 13177–13185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Y-H, Lee D-H, Yoon J-H, Nakajima S, Yasui A, and Pfeifer GP (2001). Cyclobutane pyrimidine dimers are responsible for the vast majority of mutations induced by UVB irradiation in mammalian cells. J. Biol. Chem 276, 44688–44694. [DOI] [PubMed] [Google Scholar]
- You Y-H, and Pfeifer GP (2001). Similarities in sunlight-induced mutational spectra of CpG-methylated transgenes and the p53 gene in skin cancer point to an important role of 5-methylcytosine rsidues in solar UV mutagenesis. J. Mol. Biol 305, 389–399. [DOI] [PubMed] [Google Scholar]
- Zhang W, Remenyik E, Zelterman D, Brash DE, and Wikonkal NM (2001). Escaping the stem cell compartment: sustained UVB exposure allows p53-mutant keratinocytes to colonize adjacent epidermal proliferating units without incurring additional mutations. Proc. Natl. Acad. Sci. U. S. A 98, 13948–13953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler A, Jonason AS, Leffell DJ, Simon JA, Sharma HW, Kimmelman J, Remington L, Jacks T, and Brash DE (1994). Sunburn and p53 in the onset of skin cancer. Nature 372, 773–776. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Pathways for replicating through CPDs. Polη replicates through CPDs in an error-free manner. Polθ functions in inserting nucleotides opposite the 3’T or 3’C of a CPD in the error-prone TLS pathways dependent upon Polκ or Polζ for extension of synthesis.
(B) Pathways for replicating through (6–4) photoproducts. Polθ extends synthesis from the nucleotides inserted by Polη or Polι opposite the 3’T or 3’C of the (6–4) photoproduct and it promotes error-prone TLS. Polζ functions in an alternate error-free TLS pathway.
(C) UV induced (5 J/m2) mutational spectra in the cII gene in BBMEF cells. UV induced mutations that occur in control siRNA (NC) treated cells are shown above the sequence and the lack of UV induced hot spot mutations in cells treated with Polθ siRNA is shown below the sequence. The positions of UV induced hot spots #1–11 are indicated. A frequently occurring spontaneous hot spot at position 223 is indicated by lower case ‘g’.
(D) UV induced (500 J/m2) mutation frequencies in the supF gene in 293T cells depleted for Polθ, XPA, or both. Mutation frequencies represent the average of 3 different experiments; error bars represent ± SD. Students two-tailed t-test p values, ***, p<0.001; ****, p<0.0001.
(A) Map of human Polθ and constructs of C-terminal domain of WT and catalytic mutant of Polθ. Expression of 3xFlag-Polθ (1708–2590) or 3xFlag-Polθ (1708–2590) containing D2540A, E2541A (DE→AA) mutations in GM637 HFs. siRNA targeted to site 1 was used for depletion of genomic Polθ and siRNA targeted to site 2 was used for depletion of Polθ (1708–2590). The efficiency of siRNA depletion of genomic Polθ was verified by western blot with Polθ ab and the efficiency of depletion of Polθ (1708–2590) was verified by Flag ab. β-tubulin was used as the loading control.
(B) Co-immunoprecipitation of 3xFlag Polθ (1708–2590) with ub-PCNA in chromatin bound fractions. GM637 cells expressing 3xFlag-Polθ (1708–2590) were UV irradiated and incubated for 6h. Chromatin extracts were immunoprecipitated with Flag M2-agarose. Co-immunoprecipitation of Flag-Polθ (1708–2590) with PCNA and Rad18 was determined by western blot analysis.
(C) Accumulation of Polθ (1708–2590) into foci. GM637 cells expressing GFP-Polθ (1708–2590) were treated with control siRNA. After 48 h of siRNA treatment, cells were UV irradiated. GFP-Polθ foci were visualized by fluorescence microscopy. Representative images of GFP-Polθ foci in unirradiated or UV irradiated cells.
(D) Quantification of GFP-Polθ(1708–2590) foci containing cells treated with control, Polη, or Rad18 siRNA. The mean and standard deviation were analyzed from 4 independent experiments. Student’s two-tailed t-test p values, *, p<0.05; ****, p<0.0001.
(A) Incorporation of dNTPs opposite a cis-syn TT dimer by Polθ. 0.5 nM Polθ (1708–2590) was incubated with the DNA substrate (10 nM) in the presence of 100 μM of a single dNTP or each of four dNTPs (N) for 5 min at 37°C with undamaged DNA (lanes 2–6) or for 20 min with DNA containing a cis-syn TT dimer (lanes 8–12).
(B) Extension of synthesis from the nucleotide opposite the 3’T of a (6–4) TT photoproduct by Polθ. Assay conditions were the same as in (A), and the DNA substrates used were undamaged DNA (lanes 2–6) and (6–4) TT photoproduct containing DNA (lanes 8–12).
(A) Western blot analysis of expression of human GFP-Polθ (1708–2590) and of siRNA depletion of mouse Rad51 in SV40 transformed Polθ−/− MEFs. β-tubulin was used as the loading control.
(B) Complementation of the TLS deficiency in Polθ−/− MEFs by Polθ (1708–2590).
(C) Schematic of DNA fiber assay and images of stretched DNA fibers in UV irradiated Polθ−/− primary MEFs carrying vector control or expressing Polθ (1708–2590) are shown on the left and quantitative analyses of fork progression (mean CldU:IdU ratio) are shown on the right. The data represent ~400 DNA fibers from 4 independent experiments. Error bars indicate the standard deviation. Students two-tailed t-test p values, p< 0.05.
(D) Quantification of scatter plot analyses of SCE frequencies in non-UV irradiated Polθ−/− MEFs carrying the GFP vector, expressing Myc-full length Polθ, or GFP-Polθ (1708–2590), and untreated or treated with Rad51 siRNA. Each datum point represents a single metaphase and ~1,000 metaphase chromosomes were analyzed. The mean and standard deviation were analyzed from 4 independent experiments. Student’s two-tailed t-test values, *, p<0.05; **, p<0.01
(E) Quantification of scatter plot analysis of UV induced SCE frequency in Polθ−/− MEFs carrying GFP vector, expressing Myc-full length Polθ, or GFP-Polθ (1708–2590) and treated or not treated with Rad51 siRNA. Each datum point represents a single metaphase and ~1,000 metaphase chromosomes were analyzed. The mean and standard deviation were analyzed from 4 independent experiments. Student’s two-tailed t-test p values, ***, p<0.001; ****, p<0.0001.
(F) Quantification of chromosomal aberrations in unirradiated or UV irradiated Polθ−/− MEFs carrying vector or expressing Polθ (1708–2590). The data represent analyses of ~400 metaphases from 4 independent experiments. The mean and standard deviation were analyzed from 4 independent experiments. Student’s two-tailed t-test values, ns, not significant; **, p<0.01.
(G) UV survival assay in Polθ−/− MEFs carrying vector or expressing Polθ (1708–2590). Error bars indicate the standard deviation of results of 4 independent experiments. Student’s two-tailed t-test values, ****, p<0.0001.
(A) Normal skin from Polη−/− Polθ−/− mice.
(B) An example of well-differentiated SCC from Polθ−/− mice which shows flask-shaped lesions with prominent atypia and mitotic activity.
(C) An example from Polθ−/− mice of carcinoma in situ which were frequently present in multiples. Carcinomas show less differentiation than SCCs; unlike SCCs, they lack intercellular spinous processes and keratin pearls.
(A) Polθ−/− mice.
(B) Polη−/− mice.
(C) Polη−/− Polθ−/− mice.
SCCs show varying degrees of differentiation from well differentiated in (A,B) to less differentiated in (C).