

Abstract
Extracellular vesicles (EVs) and lipoproteins are abundant and co-exist in blood. Both have been proven to be valuable as diagnostic biomarkers and for therapeutics. However, EVs and lipoproteins are both on the submicron scale and overlap in size distributions. Conventional methods to separate EVs and lipoproteins are inefficient and time-consuming. Here we present an acoustofluidic-based separation technique that is based on the acoustic property differences of EVs and lipoproteins. By using the acoustofluidic technology, EVs and subgroups of lipoproteins are separated in a label-free, contact-free, and continuous manner. With its ability for simple, rapid, efficient, continuous-flow isolation, our acoustofluidic technology could be a valuable tool for health monitoring, disease diagnosis, and personalized medicine.
Introduction
Extracellular vesicles (EVs) and lipoproteins are both important biological particles in peripheral blood. EVs are small, membrane-bound phospholipid vesicles that are secreted by various types of cells.1 In the past decade, research on EVs has significantly intensified, because EVs have been identified as mediators of intercellular communications. Numerous reports have shown that EVs play important roles in both physiological and pathological processes, which makes them important targets for diagnostics and therapeutics.2–7 With these characteristics, EVs are considered promising biomarkers for cancer, Alzheimer’s disease,8 pregnancy monitoring,9 and hepatitis C infection.10 Lipoproteins are a lipid and protein complex whose major purpose is to transport triglycerides and cholesterol molecules among organs.11 Based on density and size, lipoproteins can be divided into five major subgroups: high density lipoprotein (HDL), low density lipoprotein (LDL), intermediate density lipoprotein (IDL), very low density lipoprotein (VLDL) and chylomicron. It has been repeatedly validated that lipoproteins play essential roles in the formation of fatty streaks in the wall of the artery.12,13 Therefore, levels of lipoproteins are used as a part of risk assessment for cardiovascular disease, coronary heart disease, heart attacks, peripheral vascular disease, aortic stenosis, thrombosis and strokes.14–20 The lipid profiles are taken into account when choosing specific therapies. Lipoproteins are also used as prognostic targets to evaluate therapeutic effectiveness and characterize disease pathogenesis, particularly in the context of atherosclerosis and cardiovascular heart disease.14,15,18 Among these classes, HDL is regarded as anti-atherogenic, while others are pro-atherogenic.21
The two important biological particles, EVs and lipoproteins are both abundant in blood and have similarities in terms of size and density. Plasma samples contain EVs from 40 nm to 2 μm in diameter (exosomes: 40–120 nm, microvesicles: 100 nm–1 μm, apoptotic bodies: 50 nm–2 μm);22 while lipoproteins subgroups HDL (5–12 nm), LDL (18–25 nm), IDL (25–35 nm), VLDL (30–80 nm), chylomicron remnants (30–80 nm), and chylomicrons (75–1200 nm) have similar size ranges as EVs.23 This size overlap makes it difficult to isolate each pair of particles by conventional methods such as size chromatography.24,25 Density gradient ultracentrifugation is another approach to isolate EV subgroups26 or lipoprotein subgroup;27 however, few protocols have been reported for separating EVs from lipoproteins. Moreover, density gradient ultracentrifugation is costly and timeconsuming (more than 48 h). In addition, some of the fractions of lipoproteins, e.g., HDL, have densities similar to EVs (EVs: 1.07–1.28 g ml−1; HDLs: 1.063–1.210 g ml−1).23 Therefore, despite the tremendous potential for medical diagnoses and therapeutics, the lack of effective isolation assays poses a significant hurdle in scientific studies and a barrier to implementing EVs or lipoprotein based analyses into clinical use.28 For example, Yuana et al. recently used cryo-electron microscopy to examine the morphological information of EVs. Surprisingly, they found that the majority of the particles are electron-dense and thus most likely represent lipoproteins rather than EVs, which challenged the prevailing opinion.29 This has also been verified by another recent study.30 In another study, Sódar et al. identified lipoproteins (predominantly LDL) which mimicked the characteristics of EVs and thus mixed up the downstream analysis process.31 Although specific antibodies and agents can be used to decrease the signal interference in some analytical methods, having a simple and rapid method that can separate EVs and lipoproteins in a label-free manner is beneficial in order to reduce time and cost.
In the past few years, the acoustofluidic (i.e., the fusion of acoustics and microfluidics) technique has been demonstrated to be rapid, efficient, and biocompatible.32–34 This approach has been successfully deployed for bioparticles ranging from cells35 and platelets36 to vesicles37,38 based primarily on the size differences of the objects. In addition to size differences, acoustofluidic separation technique can also separate particles based on physical properties such as particle density and compressibility. For example, Petersson et al. successfully separated polystyrene (PS) and poly(methacrylate) (PMMA) particles of the same size based on the density difference.39 Based on the combination of density and compressibility, the separation of lipid particles from erythrocytes has also been reported.40,41 These physical property-based separation approaches dealt with micro-objects with the size range of cells, and they may not be applicable for separating submicron objects (e.g., EVs and lipoproteins), because the acoustic radiation force is much smaller when acting on submicron objects than on micro-objects. In this regard, the frequency used in the acoustofluidic separation technique must be increased in order to separate submicron objects that have similar size and density.
In this work, we present an acoustofluidic method to separate EVs and lipoproteins based on their acoustic properties. To this end, we implement standing surface acoustic waves (SSAWs) which work at a frequency of 20 MHz, generating an acoustic radiation force that is strong enough to manipulate nanoscale objects. The acoustic pressure distributions within the fluid induced by SSAWs and the behavior of particles with different acoustic properties are investigated both numerically and experimentally. Upon identification of the proper working modes and conditions, the separation of EVs and lipoproteins from plasma samples can be achieved, and the separation performance is characterized. Our acoustofluidic separation technique, described here, is the first of its kind to separate EVs and lipoprotein contents in a label-free, continuous, and biocompatible manner.
Working mechanism
Fig. 1 provides a schematic of the acoustofluidic lipoprotein separation device. When a radio frequency signal is applied to the interdigitated transducers (IDTs), two series of surface acoustic waves (SAW) are generated and propagate in opposite directions. The constructive interference of the two SAWs results in the formation of SSAW and generates parallel pressure nodes (regions of minimal pressure) and antinodes (regions of maximum pressure) within the microchannel. Particles in the SSAW field are subject to an acoustic radiation force (Fr), which can be expressed as42,43
| (1) |
where p0, Vp, βf, λ, ϕ, k, and x are pressure amplitude, particle volume, acoustic wavelength, acoustic contrast factor, wave vector, and distance from the pressure node, respectively. The direction of the acoustic radiation force is dependent on the acoustic contrast factor (ϕ), which can be expressed as
| (2) |
where ρp, ρf, βp, and βf are the densities of the particles and the fluid, and the compressibility of the particles and fluid, respectively. When the acoustic contrast is positive, the particles are pushed to the acoustic pressure nodes by the acoustic radiation force; whereas particles with negative acoustic contrast are pushed to pressure antinodes (Fig. 1b).
Fig. 1.
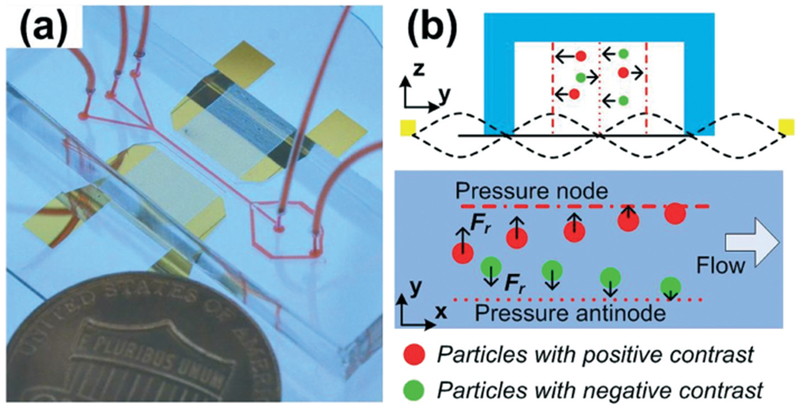
(a) A photo and (b) working mechanism of the acoustofluidic based EV/lipoprotein separation device. Our device separates particles based on acoustic properties. Particles with positive acoustic contrast are attracted to the acoustic pressure nodes, while particles have negative contrast are pushed to the antinodes by acoustic radiation force.
The parameters of EV and lipoprotein subclasses are listed in Tables 1 and 2. The fluid is plasma, with ρ = 1.025 g ml−1 and β = 4.23 × 10−10 Pa−1.37,45,46 As the smallest and densest subclass of lipoproteins, HDLs always appear to be positive in terms of their acoustic contrast factor. However, LDLs are intermediate, implying that due to the density and compressibility range, the calculated acoustic contrast factor can vary from −0.14 to 0.11. Because of the lack of measurement data for lipoproteins, no precise number of acoustic contrast factors is available for IDL, VLDL, and chylomicrons. Nevertheless, estimated values based on the density and lipid content percentage data are shown in Tables 1 and 2. The percentage of triglyceride is higher in IDL, VLDL, and chylomicrons than in HDL and LDL. Triglycerides have a compressibility of 5.34 × 10−10 Pa−1 and their acoustic contrast factor is −0.31,47 which means that a high percentage of triglycerides could lead to higher compressibility and negative acoustic contrast. On the other hand, the density of IDL, VLDL, or chylomicrons is lower than that of LDL. According to eqn (2), a particle of low density and high compressibility tends to exhibit a more-negative acoustic contrast. Therefore, IDL, VLDL, and chylomicrons must be negative in terms of acoustic contrast.
Table 1.
| Density (g ml−1) | Compressibility (10−10 Pa−1) | Acoustic contrast factor | |
|---|---|---|---|
| HDL | 1.063–1.210 | 3.39–4.03 | 0.21–0.23 |
| LDL | 1.019–1.063 | 3.93–4.79 | −0.14–0.11 |
| IDL | 1.006–1.019 | NA | Negative |
| VLDL | 0.930–1.006 | NA | Negative |
| Chylomicron | <0.930 | NA | Negative |
| EV | 1.130 | 3.5 | 0.27 |
Table 2.
| Lipoprotein subgroups | Triglyceride (%) |
|---|---|
| HDL | 8 |
| LDL | 4 |
| IDL | 31 |
| VLDL | 50 |
| Chylomicrons | 84 |
Owing to the difference in acoustic contrast factor, the acoustofluidic separation device is able to distinguish subgroups of lipoproteins. Specifically, IDL, VLDL, chylomicrons, and chylomicron remnants are focused to acoustic pressure antinodes, while HDL and EVs move to acoustic pressure nodes. By engineering the acoustic field pattern and channel dimension, the different groups can be directed to different outlets. Thus, within a single device, we can achieve the separation of subgroups of lipoproteins and EVs in a continuous manner.
Materials and methods
Device fabrication
To fabricate the acoustofluidic separation device, we first fabricated IDTs on a lithium niobate (LiNbO3) substrate via photolithography, e-beam evaporation, and lift-off processes.35,42 A single-layer polydimethylsiloxane (PDMS) micro-channel of 50 μm height and 120 μm width was fabricated by soft-lithography methods. The PDMS microchannel and the LiNbO3 substrate were then carefully aligned and bonded together by plasma treatment.
Selection and synthesis of PDMS particles
PDMS has a negative acoustic contrast factor, so it was chosen in preliminary experiments to represent the IDL, VLDL, and chylomicrons. To prepare PDMS microspheres, PDMS base and curing agents (Dow Corning, Midland, USA) were first mixed at a weight ratio of 10 : 1, followed by degassing to remove air bubbles. The PDMS mixture was then added to a 0.5% PVA aqueous solution of 50 mL, and the mixed solution was stirred via a vortex mixer to form emulsion droplets. After that, the solution was placed at room temperature for 12 h, followed by baking at 65 °C for another 12 h to complete the curing reaction. Once cured, the prepared PDMS solution containing microspheres was centrifuged at 3000 rpm and rinsed with water. Then, the solution was filtered using a Falcon® cell strainer (Corning, USA) to remove large or aggregated particles and dust.
Experimental setup
Polystyrene particles, which have a positive acoustic contrast factor of 0.58 in water, were purchased from Bangs Laboratory, USA. The polystyrene particles’ positive contrast factor mimicked that of the HDLs and EVs in initial testing. Human plasma from healthy donors was purchased from Zen-Bio, Inc. USA. The plasma samples were frozen upon receipt at −20 °C until use. The acoustic separation device was placed on the stage of an inverted microscope (Eclipse Ti, Nikon, Japan). When processing plasma samples, the device was placed on an upright microscope (BX51WI, Olympus, Japan) with a Peltier cooling system (TEC1-12730, Hebei I.T., China) during experiments. The temperature of the Peltier cooling system was controlled via a variable DC power supply (TP1505D, Tek-power, USA). A CCD camera (Nikon DS-Qi2) recorded the separation process, and data was analyzed using Image J (NIH, USA). The fluid was controlled by syringe pumps (neMESYS, cetoni GmbH, Germany). When processing human plasma, samples were injected from two side inlets and PBS solution was injected from the central inlet. The flow rates for the three inlets were set to 0.5 μl min−1, 1 μl min−1, and 0.5 μl min−1, respectively. Separated samples were collected in microcentrifuge tubes. The SSAW was generated by applying a radio frequency signal to the IDTs on the LiNbO3 substrate. The radio frequency signal originated from a function generator (E4422B, Agilent, USA) and was amplified using an amplifier (Amplifier Research, USA). The input power was measured by an oscilloscope (DPO4104, Tektronix, USA). The working frequency was set at 19.573 MHz, and the voltage input to the device ranged from 20 to 40 Vpp.
Nanoparticle analysis
The size distribution and concentration of synthesized PDMS microspheres was tested with a Zetasizer Nano (ZEN0040, Malvern, UK). The plasma and processed samples were analyzed by nanoparticle tracking analysis (Nanosight LM10, Malvern, UK).
Lipid staining
To examine the lipoprotein particles, a fluorescent neutral lipid dye 4,4-difluoro-1,3,5,7,8-pentamethyl-4-bora-3a,4a-diaza-s-indacene (BODIPY 493/503, Thermo Fisher Scientific, USA) was used. The BODIPY was dissolved into PBS at 1 mg ml−1, followed by mixing vigorously to mechanically emulsify this solution. Then, 20 μl BODIPY solution was mixed with a 20 μl sample and incubated for 15 min in a dark environment. After that, the mixture was dropped onto Shandon™ Double Cytoslides™ (Thermal Fisher Scientific, USA) and incubated for 5 min. Next, the slide was washed 3 times with PBS. A cover slide (VWR, USA) was placed on the sample and observed under a fluorescent microscope.
Electron microscopy
The processed samples were mixed with paraformaldehyde solution at the final concentration of 4% w/v, and incubated at room temperature for 20 min. Then, a 300 mesh grid support film (Electron Microscopy Sciences, USA) was placed on the top of a drop of sample for 10 min. After that, the grid was transferred to a 100 μl drop of distilled water to rinse 3 times for 2 min each time. Then 100 μl uranyl-acetate solution was placed on the grid for 10 min for negative staining. Finally, the grid was rinsed with distilled water and then incubated for 10 min on a drop of methylcellulose uranyl. The samples were analyzed with an electron microscope.
ELISA
Lipoprotein contents were evaluated by ELISA. Human HDL, LDL, and VLDL ELISA kits were purchased from LifeSpan BioSciences, Inc. (Seattle, USA). ELISA tests were performed according to the user manual provided by the manufacturer. The results were read by a Synergy H1 microplate reader (Bio-Tek, Winooski, USA).
Western blot
Human plasma and the separated samples from both outlets were processed. All the samples were diluted 10 times for gel electrophoresis. Proteins were separated by SDS-PAGE electrophoresis and transferred to a polyvinylidene fluoride membrane (Bio-rad, USA). Then, membranes were incubated separately with three different antibodies: mouse anti-CD63 (sc-5275, 1 μg ml−1, Santa Cruz), rabbit anti-TSG101 (ab30871, 1 μg ml−1, Abcam), and mouse anti-HSP90 (ab13492, 1 μg ml−1, Abcam), followed by incubation with the appropriate HRP secondary antibody, including goat anti-mouse IgG (ab97040, 0.05 μg ml−1, Abcam) and goat anti-rabbit IgG (ab97080, 0.05 μg ml−1, Abcam). A Bio-Rad ChemiDoc XRS+ system was employed for quantification of protein expression levels.
Results
Numerical studies of acoustic pressure and particle behavior
The SAW frequency was ~20 MHz, which indicated that the wavelength of SAW propagating on the LiNbO3 substrate was ~200 μm. Thus, the distance between pressure nodes of the SSAW on the substrate was ~100 μm. Therefore, the channel dimensions needed to be reduced to match the wavelength. The channel width was set as 120 μm in order to avoid the formation of multiple nodes and antinodes within the channel. The channel height was set as 50 μm, which was less than the wavelength of the acoustic wave in fluid (the acoustic wavelength in water is ~75 μm at 20 MHz).
Our acoustofluidic separation device was simulated using the finite element software package COMSOL Multiphysics® 4.3a. We previously reported our detailed modeling process.43,44 Fig. 2a shows the cross-section view of the simulated absolute acoustic pressure distribution within the rectangular microfluidic channel. When the center point of the channel surface has minimal vibration amplitude, a pressure antinode is generated in the middle of the channel. On the other hand, two pressure nodes are located at ~20 μm and ~100 μm in the y-direction. It is notable that besides the two channel pressure nodes, the area next to the side walls also had low acoustic pressure.
Fig. 2.
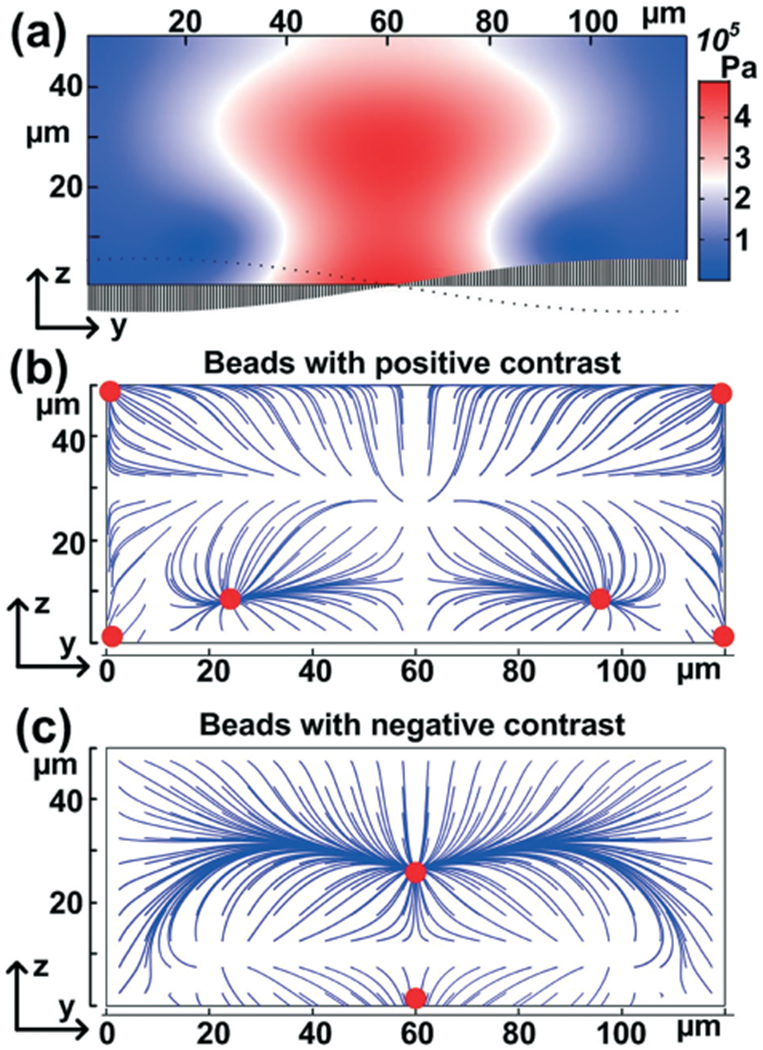
Numerical simulation of (a) acoustic pressure distribution induced by SSAW and (b and c) particle trajectories tracing. The plot is the cross-section plane of the channel.
Based on patterns of acoustic pressure, the behavior of particles under the acoustic field was also simulated. The trajectories and final focusing points when uniformly placing particles with positive or negative acoustic contrast are shown in Fig. 2b and c. The simulation results reveal that for particles with positive acoustic contrast, there were two primary focusing points located in between the center and two sidewalls and four minor focusing points located adjacent to the two sidewalls. On the other hand, the particles with negative acoustic contrast, as predicted, were focused in the center of the channel.
Acoustic manipulation of particles with different acoustic contrast factors
Based on our numerical simulation, we were able to find the acoustic pressure node and antinode patterns. The results (Fig. 2) suggested that distinct particle characteristics can be identified. Therefore, we explored the particle behavior experimentally using PS and synthesized PDMS beads, which have acoustic contrast factors of 0.58 and −1.16, respectively. Notably, there were two primary focusing points within the channel for 970 nm PS particles. Additionally, PS particles could also be pushed to the side walls (Fig. 3a). After being pushed away from the center of the channel, the PS particles were directed to two side outlets, which connected to one single outlet port for collection. We therefore referred to the side channel exit path as the positive contrast exit. We also tested 110 nm PS particles to mimic small bioparticles. Even though the 110 nm particles were not focused into a narrow line (because the acoustic force was less than the force experienced by larger particles), an obvious particle-free area was observed in the center of the channel (Fig. 3b), indicating that 110 nm PS particles were also pushed away from acoustic pressure antinode, and could be primarily directed to the side outlets.
Fig. 3.
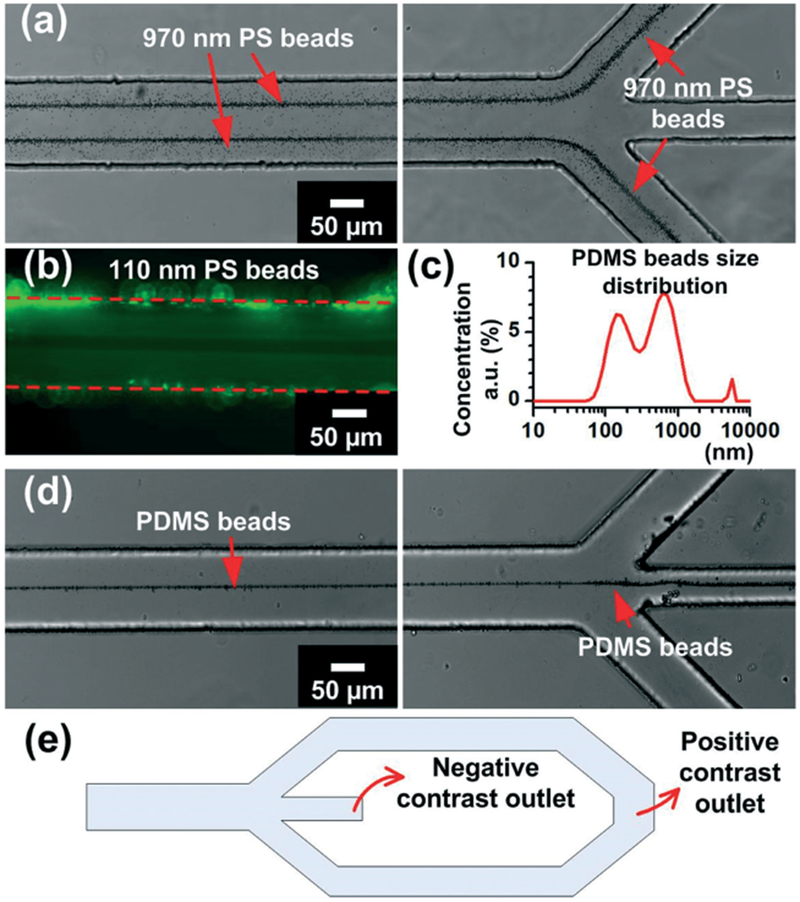
Acoustic manipulation of polystyrene (PS) and PDMS beads. (a) 970 nm PS beads are focused to acoustic pressure nodes and directed to side outlets. (b) 110 nm PS beads are driven away from the center of the channel. (c) Size distribution of synthesized PDMS beads. (d) PDMS beads are focused in the center of the channel at the acoustic pressure antinode. (e) Schematic of channel outlet design.
To contrast with the PS beads, we used synthesized PDMS beads to show the behavior of particles with negative acoustic contrast factors. As shown in Fig. 3c, the synthesized PDMS beads ranged from ~100 nm to ~2 μm in diameter, which is similar to the size range of lipoproteins. With the same experimental conditions, PDMS beads were focused in the center of the channel, and then exited through the central outlet (Fig. 3d). Because the particles with a negative acoustic contrast factor exited through the center channel, it was referred to as the negative contrast outlet. In summary, by using our acoustofluidic separation method, we successfully demonstrated the separation of PS and PDMS beads, relying on differences in acoustic contrast rather than particle size.
Acoustic separation of EVs and lipoproteins
Having identified the underlying mechanism and optimal working conditions of the acoustofluidic separation devices, we conducted the EV/lipoprotein separation. Human plasma samples were injected into the acoustofluidic device, and the output from the device was collected and characterized.
The size distribution of the bioparticles was first examined by nanoparticle tracking analysis, as shown in Fig. 4. It was predicted that samples collected from the negative contrast outlet (central outlet) contained particles with negative contrast factors, which likely refers to IDLs, VLDLs, and chylomicrons. The size of the particles collected from the central outlet ranged from ~20 nm to 600 nm, with very few particles smaller than ~20 nm. This is consistent with the size data for the lipoprotein subgroups of negative contrast factor, and it also suggests that smaller lipoproteins with positive contrast factors (i.e., HDLs) were not isolated. On the other hand, the sample from the positive contrast outlet (side outlet) showed a size range from several nanometers to >600 nm. This was consistent with the expected result because both HDLs and EV subgroups (i.e., exosomes and microvesicles) are collected through these outlets. Though a minor difference was noted, the size distribution curves for both samples were similar. This again indicated that our acoustofluidic separation, in this case, was not based on size.
Fig. 4.
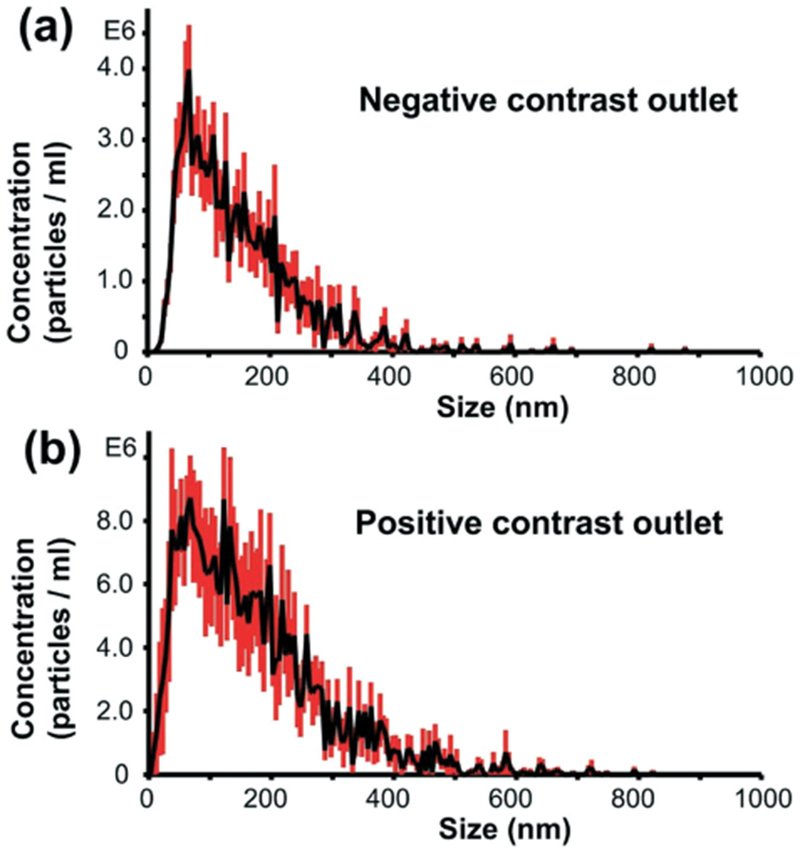
Particle size distributions of (a) samples collected from the negative contrast outlet and (b) samples emerging from the positive contrast outlet.
Characterization of separated lipoproteins and EVs
We collected samples from both negative contrast and positive contrast outlets after acoustofluidic separation. Then we characterized the contents to verify the presence of lipoprotein subgroups and EVs.
First, we performed lipid staining using a fluorescent neutral lipid dye. Lipoproteins are rich in lipids, especially VLDL, IDL, and chylomicrons, as indicated by Table 2. The results are shown in Fig. 5a and b. When comparing the left and right panels of Fig. 5a, all bright field visible particles were also positive for green fluorescence. Furthermore, the fluorescent image also showed submicron-sized particles that were barely visible under bright field. The results suggested that the particles collected from negative contrast outlet contain high proportions of lipid contents, which is a unique characteristic of lipoproteins. On the other hand, no fluorescence positive particles were observed in the sample collected from positive contrast outlet, as shown in Fig. 5b. The results showed that those visible particles in the positive contrast outlet were inadequate in terms of lipid contents. This indicated that the sample from positive contrast outlet contained few large subgroups of lipoproteins, e.g., chylomicrons or others.
Fig. 5.

Characterization of separated samples from (a and c) negative contrast outlet and (b and d) positive contrast outlet. (a and b): Bright field and fluorescent images of lipid staining. Particles with high lipid content level are stained by green fluorescence. TEM images reveal the presence of particles with different appearance. (c) Particles with a lipid core and monolayer membrane are enriched in the negative contrast outlet. (d) Bilayer membrane vesicle-like structures are observed in positive contrast outlet.
Noting that the optical microscope images had limited resolution to distinguish particles around or smaller than 100 nm, we examined the samples using a transmission electron microscope (TEM), as shown in Fig. 5c and d. As expected, particles in the sample collected from the negative contrast outlet presented distinct characteristics under the electron microscope that were referred to lipoproteins.30,48 The size of particles ranged from less than 100 nm to more than 500 nm, which also matched with the size of lipoproteins. In contrast, the particles in the positive contrast outlet were of a bilayer membrane structure, which is typical vesicular morphology.30,49 The sample contained large vesicles that exceeded 300 nm as well as exosomes which were ~100 nm in diameter, as shown in Fig. 5d.
To further verify the presence of EVs or lipoproteins, we used western blot and ELISA to probe the level of lipoprotein subclasses and EV protein markers in both outlets, as well as in the initial plasma sample (Fig. 6). Notably, the sample from negative contrast outlet contained high levels of VLDL, while there was a very small amount of VLDL present in the sample collected from the positive contrast outlets. This indicated that particles of negative acoustic contrast factor (e.g., VLDL) were directed to the central outlet by the acoustofluidic separation device. Fig. 6b shows that the two outlets contained equal amounts of LDLs. This was expected because the acoustic contrast factor of LDLs ranged from negative to positive across the known parameter values. In contrast, the HDL level in the side outlets was much higher than the central outlet, indicating that HDL particles were pushed to the side channels within the acoustofluidic separation device. We examined the expression level of EV markers in the samples collected from both outlets and dilute plasma sample by western blot, as shown in Fig. 6d. We analyzed EV endoplasmic reticulum protein HSP90, membrane-binding protein TSG101, and membrane tetraspanin CD63. The sample collected from the negative contrast outlet had a low expression for all three proteins, which indicated the absence of EVs. On the other hand, the sample collected from the positive contrast outlet expressed a high level of vesicular proteins, akin to that in plasma.
Fig. 6.
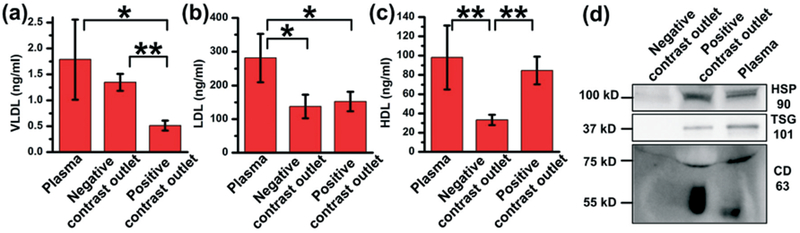
Characterization of lipoprotein contents separated by the acoustofluidic method. (a) VLDL, (b) LDL, and (c) HDL level in plasma sample, sample collected from negative contrast outlet, and sample from positive contrast outlet are tested. Data is obtained from three individual tests using the same plasma sample. *: p < 0.05, **: p < 0.01. (d) Western blot of samples collected from negative contrast outlet, positive contrast outlet and plasma. EV markers (HSP90, TSG101 and CD63) show high expression in positive contrast outlet and plasma, while no expression in negative contrast outlet.
In summary, the acoustofluidic device successfully demonstrated the separation of lipoprotein subgroups based on their acoustic contrast factors. Subgroups with negative contrast factors were extracted from the whole population of lipoproteins and also separated from other plasma bioparticles (such as EVs). Thus, the samples collected from positive contrast outlet contained purified EVs.
Discussion and conclusion
Acoustofluidics is an efficient, biocompatible method for separating biological objects based mainly on size. In this work, we further expanded the capability of the acoustofluidic separation technology for separating submicron scale objects based primarily on acoustic properties. The acoustofluidic device presented here is the first on-chip approach that can separate EVs and subgroups of lipoproteins. Thus far we have demonstrated the separation of EVs and lipoprotein subgroups that exhibit negative contrast vs. positive contrast. Specifically, IDLs, VLDLs, and chylomicrons are separated from HDLs, exosomes and other EV subgroups. We demonstrated that VLDL was reduced by ~70% in the EV collections, and no expressions of EV markers were found in lipoprotein fractions. Efficiency could be improved by increasing the length of microfluidic channel. Compared to conventional isolation methods, the acoustofluidic separation system is label-free, fast, inexpensive, and biocompatible to vesicles and lipoproteins, making it an excellent tool for EV-related or lipoprotein-related studies and clinical applications.
Although this work aims to separate EVs and all the lipoprotein subgroups, some of the components, e.g., HDL and LDL, were not isolated. To further develop an approach that can purify all lipoprotein sub-classes, additional strategies should be deployed using the current acoustofluidic separation system. For example, the acoustic contrast factor of LDL, as mentioned, is not always negative in plasma. A possible solution may be in altering the medium properties. Increasing the density of plasma with additives will change the contrast factor of LDL to be totally negative. By doing so, LDL can be extracted along with IDL, VLDL and chylomicrons from those particles with positive acoustic contrast. This approach will also improve separation efficiency, since the absolute values of acoustic contrast factors will be larger, causing an increase of the acoustic radiation force. Furthermore, it is also possible to separate HDL from EVs, since HDL is the smallest subgroup of lipoproteins, and smaller than exosomes. In our recent work, we have demonstrated that by integrating two acoustofluidic separation modules into a single device, we can separate exosomes from other EVs.35 In forthcoming work, an integrated acoustofluidic separation system can be implemented to separate all subgroups of EVs and lipoproteins.
Acknowledgements
We gratefully acknowledge financial support from the National Institutes of Health (R01HD086325 and R01GM127714), United States Army Medical Research Acquisition Activity (W81XWH-18-1-0242), and National Science Foundation (ECCS-1807601).
Footnotes
Conflicts of interest
The authors declare no conflict of interest.
References
- 1.EL Andaloussi S, Mager I, Breakefield X and Wood M, Nat. Rev. Drug Discovery, 2013, 12, 347–357. [DOI] [PubMed] [Google Scholar]
- 2.Théry C, Ostrowski M and Segura E, Nat. Rev. Immunol, 2009, 9, 581–593. [DOI] [PubMed] [Google Scholar]
- 3.Vader P, Breakefield X and Wood M, Trends Mol. Med, 2014, 20, 385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cocucci E, Racchetti G and Meldolesi J, Trends Cell Biol, 2009, 19, 43–51. [DOI] [PubMed] [Google Scholar]
- 5.Shao H, Chung J, Balaj L, Charest A, Bigner D, Carter B, Hochberg F, Breakefield X, Weissleder R and Lee H, Nat. Med, 2012, 18, 1835–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ko J, Carpenter E and Issadore D, Analyst, 2016, 141, 450–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sunkara V, Woo H and Cho Y, Analyst, 2016, 141, 371–381. [DOI] [PubMed] [Google Scholar]
- 8.Rajendran L, Honsho M, Zahn T, Keller P, Geiger K, Verkade P and Simons K, Proc. Natl. Acad. Sci. U. S. A, 2006, 103, 11172–11177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sarker S, Scholz-Romero K, Perez A, Illanes S, Mitchell M, Rice G and Salomon C, J. Transl. Med, 2014, 12, 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ramakrishnaiah V, Thumann C, Fofana I, Habersetzer F, Pan Q, de Ruiter P, Willemsen R, Demmers J, Stalin Raj V, Jenster G, Kwekkeboom J, Tilanus H, Haagmans B, Baumert T and van der Laan L, Proc. Natl. Acad. Sci. U. S. A, 2013, 110, 13109–13113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moffatt R and Stamford B, Lipid metabolism and health, CRC/Taylor & Francis, Boca Raton, 2006. [Google Scholar]
- 12.Witztum J and Steinberg D, J. Clin. Invest, 1991, 88, 1785–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nicholls S and Nissen S, Curr. Opin. Lipidol, 2007, 18, 421–426. [DOI] [PubMed] [Google Scholar]
- 14.Miller G and Miller N, Lancet, 1975, 305, 16–19. [DOI] [PubMed] [Google Scholar]
- 15.Goldstein L and Brown S, Annu. Rev. Biochem, 1977, 46, 897–930. [DOI] [PubMed] [Google Scholar]
- 16.Gordon D, Probstfield J, Garrison R, Neaton J, Castelli W, Knoke J, Jacobs D, Bangdiwala S and Tyroler H, Circulation, 1989, 79, 8–15. [DOI] [PubMed] [Google Scholar]
- 17.Otvos J, Circulation, 2006, 113, 1556–1563. [DOI] [PubMed] [Google Scholar]
- 18.Grundy S, Circulation, 2002, 106, 2526–2529. [DOI] [PubMed] [Google Scholar]
- 19.Stockman J, Yearbook of Pediatrics, 2009, pp. 216–217. [Google Scholar]
- 20.Wilson P, D’Agostino R, Levy D, Belanger A, Silbershatz H and Kannel W, Circulation, 1998, 97, 1837–1847. [DOI] [PubMed] [Google Scholar]
- 21.Chroni A, Leondaritis G and Karlsson H, J. Lipids, 2011, 2011, 1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zaborowski M, Balaj L, Breakefield X and Lai C, BioScience, 2015, 65, 783–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schumaker V, Lipoproteins, apolipoproteins, and lipases, Academic Press, San Diego, 1994. [Google Scholar]
- 24.Böing A, van der Pol E, Grootemaat A, Coumans F, Sturk A and Nieuwland R, J. Extracell. Vesicles, 2014, 3, 23430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Welton J, Webber J, Botos L, Jones M and Clayton A, J. Extracell. Vesicles, 2015, 4, 27269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yuana Y, Levels J, Grootemaat A, Sturk A and Nieuwland R, J. Extracell. Vesicles, 2014, 3, 23262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chapman M, Goldstein S, Lagrange D and Laplaud P, J. Lipid Res, 1981, 22, 339–358. [PubMed] [Google Scholar]
- 28.Taylor D and Shah S, Methods, 2015, 87, 3–10. [DOI] [PubMed] [Google Scholar]
- 29.Yuana Y, Koning R, Kuil M, Rensen P, Koster A, Bertina R and Osanto S, J. Extracell. Vesicles, 2013, 2, 21494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang J, Zhang Y, Sze S, van de Weg S, Vernooij F, Schoneveld A, Tan S, Versteeg H, Timmers L, Lam C and de Kleijn D, Int. J. Mol. Sci, 2017, 19, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sódar B, Kittel Á, Pálóczi K, Vukman K, Osteikoetxea X, Szabó-Taylor K, Németh A, Sperlágh B, Baranyai T, Giricz Z, Wiener Z, Turiák L, Drahos L, Pállinger É, Vékey K, Ferdinandy P, Falus A and Buzás E, Sci. Rep, 2016, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ding X, Li P, Lin S, Stratton Z, Nama N, Guo F, Slotcavage D, Mao X, Shi J, Costanzo F and Huang T, Lab Chip, 2013, 13, 3626–3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen K, Wu M, Guo F, Li P, Chan C, Mao Z, Li S, Ren L, Zhang R and Huang T, Lab Chip, 2016, 16, 2636–2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang S, Lata J, Chen C, Mai J, Guo F, Tian Z, Ren L, Mao Z, Huang P, Li P, Yang S and Huang T, Nat. Commun, 2018, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li P, Mao Z, Peng Z, Zhou L, Chen Y, Huang P, Truica C, Drabick J, El-Deiry W, Dao M, Suresh S and Huang T, Proc. Natl. Acad. Sci. U. S. A, 2015, 112, 4970–4975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nam J, Lim H, Kim D and Shin S, Lab Chip, 2011, 11, 3361–3364. [DOI] [PubMed] [Google Scholar]
- 37.Lee K, Shao H, Weissleder R and Lee H, ACS Nano, 2015, 9, 2321–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu M, Ouyang Y, Wang Z, Zhang R, Huang P, Chen C, Li H, Li P, Quinn D, Dao M, Suresh S, Sadovsky Y and Huang T, Proc. Natl. Acad. Sci. U. S. A, 2017, 114, 10584–10589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Petersson F, Åberg L, Swärd-Nilsson A and Laurell T, Anal. Chem, 2007, 79, 5117–5123. [DOI] [PubMed] [Google Scholar]
- 40.Petersson F, Nilsson A, Holm C, Jönsson H and Laurell T, Analyst, 2004, 129, 938–943. [DOI] [PubMed] [Google Scholar]
- 41.Petersson F, Nilsson A, Holm C, Jönsson H and Laurell T, Lab Chip, 2005, 5, 20–22. [DOI] [PubMed] [Google Scholar]
- 42.Shi J, Huang H, Stratton Z, Huang Y and Huang T, Lab Chip, 2009, 9, 3354–3359. [DOI] [PubMed] [Google Scholar]
- 43.Mao Z, Xie Y, Guo F, Ren L, Huang P, Chen Y, Rufo J, Costanzo F and Huang T, Lab Chip, 2016, 16, 515–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen C, Zhang SP, Mao Z, Nama N, Gu Y, Huang P-H, Jing Y, Guo X, Costanzo F and Huang TJ, Lab Chip, 2018, 18, 3645–3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Feingold KR and Grunfeld C, Introduction to lipids and lipoproteins, In Endotext [Internet], MDText.com, Inc., 2018. [Google Scholar]
- 46.Hianik T, Rybár P, Kostner G and Hermetter A, Biophys. Chem, 1997, 67, 221–228. [DOI] [PubMed] [Google Scholar]
- 47.Garrett R and Grisham C, Biochemistry, Cengage Learning, Boston, 6th edn, 2016, p. 862. [Google Scholar]
- 48.Kondo A, Muranaka Y, Ohta I and Kann T, Clin. Chem, 1999, 45, 1974–1980. [PubMed] [Google Scholar]
- 49.Muller L, Mitsuhashi M, Simms P, Gooding W and Whiteside T, Sci. Rep, 2016, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]