

If heart failure (HF) pathogenesis was a theatrical production, the script would typically place the cardiomyocyte on center stage. Indeed, significant drug development efforts in HF are focused on targeting cardiomyocyte pathobiology. However, sometimes a talented supporting cast can steal the show, especially when one of the supporting actors turns out to be the unexpected villain. This may very well be the case for a stress-activated cell type called the myofibroblast, a well-known player in wound healing responses 1. In this issue of Circulation Research, Meng et al., elegantly demonstrate that excessive activation of myofibroblasts fuels chronic HF progression in mice, even when the root-cause of the disease is a cardiomyocyte-intrinsic mutation in the sarcomere apparatus 2. These data add momentum to the emerging concept that dampening the activation of myofibroblasts may be a new therapeutic strategy in HF.
The adult heart contains a large number of resident cardiac fibroblasts, which are developmentally derived from the epicardium and play important roles in maintaining tissue architecture. In response to tissue injury, stress, or stimulation by morphogens (e.g., TGFβ), resident tissue fibroblasts undergo a dramatic cell state transition to become activated myofibroblasts, a highly synthetic cell type that rapidly induce the marker gene Periostin (Postn). Consistent with the role of myofibroblasts in wound healing, these cells acquire the capability to contract, migrate, proliferate, elaborate extracellular matrix, secrete cytokines, and recruit immune cells. While the cardiac myofibroblast has been long suspected to be a player in HF pathogenesis, defining a causal role for this cell type has been historically challenging due to lack of robust tools to incisively manipulate this cell population in vivo. In 2016, Jeff Molkentin’s laboratory knocked-in a Mer-Cre-Mer cassette into the endogenous mouse Postn locus, allowing for tamoxifen-inducible and myofibroblast restricted gene recombination in vivo 3. Using this powerful tool for lineage tracing in the adult mouse heart, this group definitively demonstrated that cardiac myofibroblasts derive from existing resident cardiac FBs. Furthermore, they provided early evidence that inhibition of myofibroblast activation could attenuate HF in a mouse models of acute pressure overload. However, it was not clear if these findings had broader applicability to other forms of HF, such as genetically-driven cardiomyopathies, particularly those in which the tempo of disease progression is slow and insidious.
Since the discovery of the first human mutation in the MYH7 locus that caused familial hypertrophic cardiomyopathy 4, a myriad of variants affecting sarcomeric proteins have been identified as principal molecular defects in heritable cardiomyopathy syndromes 5. Mutations of the cardiac myosin binding protein-C (cMyBP-C/MYBPC3) are among the most prevalent of these mutations 6. Genetic cardiomyopathy caused by mutations in MYBPC3 are not typically associated with a mutant full-length protein, but rather with truncated protein products that function as ‘poison’ peptides within the cardiomyocyte 7. Interestingly, cardiac stress occurring in the absence of a MYBPC3 mutation can trigger the production of a 40-kDa cMyBP-C ‘poison’ fragment that has been shown to accumulate in failing rodent and human hearts 8, 9. In contrast to several other ‘sarcomeropathies’, one of the most prominent features of MYBPC3-based hypertrophic cardiomyopathy is the extensive interstitial and perivascular fibrosis that takes root early in the course of disease. However, it has remained unclear until now whether fibrosis was simply a histological feature associated with disease progression or a bona-fide disease driver.
In this issue, Meng and colleagues use their published mouse model of human cMyBP-C cardiomyopathy in which they transgenically overexpress a 40kDa ‘poison’ fragment of MyBP-C (Mybpc340kDa) in a cardiomyocyte-specific and tetracycline-responsive manner 10. Previous characterization of this model has shown that it recapitulates key aspects of human cMyBP-C-based hypertrophic cardiomyopathy and HF 10. First, the authors provide strong evidence that their Mybpc340kDa transgenic model features robust activation of TGFβ-signaling and induction of pro-fibrotic markers, including Periostin, a marker of myofibroblasts. To evaluate whether specifically interfering with myofibroblast activation could attenuate disease severity in this sarcomere-driven cardiomyopathy, the authors use an elegant multi-allelic strategy to conditionally delete a key TGF-β receptor (Tgfbr2) in myofibroblasts using the PostnMerCreMer line 3. They found that myofibroblast-specific deletion of Tgfbr2 at a very early stage of disease pathogenesis (age 2 months; prior to the onset of gross structural remodeling of the heart) leads to reduced fibrosis and hypertrophy, improved left ventricular (LV) ejection fraction, and dramatically increased lifespan in Mybpc340kDa mice. Phosphorylation of SMAD3 in fibroblasts, an indicator of canonical TGF-β signaling, was upregulated in the Mybpc340kDa model and inhibited by myofibroblast-specific deletion of Tgfbr2. In addition, markers of myofibroblast transdifferentiation, such as Periostin and α-smooth muscle actin (α-SMA), were also decreased in these hearts. Importantly, the authors confirm that deletion of Tgfbr2 in myofibroblasts did not alter abundance of the 40kDa MyBP-C protein fragment, excluding the possibility that reduction in levels of the toxic peptide itself was an explanation for the observed improvements in cardiac function. These data demonstrate that early inactivation of myofibroblasts in this mouse model can have significant impact not only on fibrosis, but also on global LV remodeling, all in a manner that alters the natural history of this progressive and lethal genetic cardiomyopathy (Figure 1).
Figure 1.
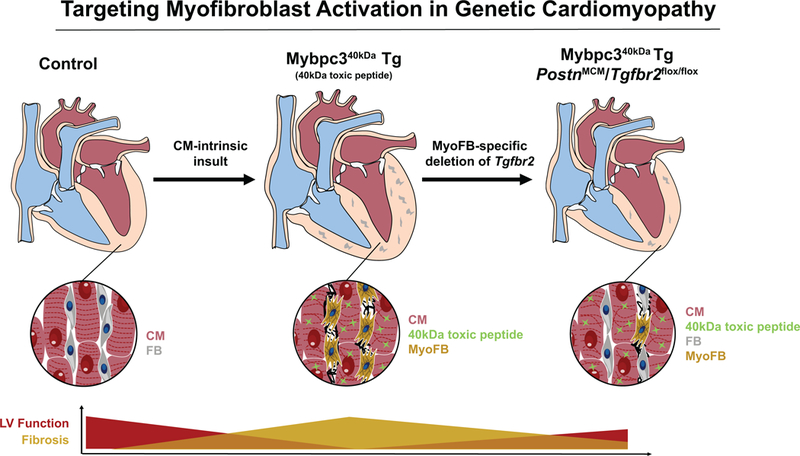
Transgenic (Tg) overexpression of a poison Mybpc340kDa peptide fragment in a cardiomyocyte (CM)-specific manner leads to hypertrophic cardiomyopathy and heart failure, characterized by severe fibrosis. Myofibroblast (MyoFB)-specific deletion of Tgfbr2 decreases fibrosis and improves cardiac function and survival, despite continued presence of the toxic peptide. FB=Fibroblasts. MCM=Mer-Cre-Mer.
Patients often present for medical attention due to pre-established HF, associated with chronic LV structural remodeling and cardiac fibrosis. Therefore, Meng and colleagues sought to determine whether late-stage deletion of myofibroblast-Tgfbr2, commenced by tamoxifen administration at 5 months of age in their Mybpc340kDa transgenic model, could also impact the disease phenotype. At the 5-month stage, the Mybpc340kDa transgenic mice feature significant cardiac fibrosis (~20% of LV), cardiac hypertrophy and systolic dysfunction. When assessed at 8 months age (i.e., 3 months after Tgfbr2 deletion), they observed significant improvements in cardiac fibrosis, LV systolic function, and survival, but not in cardiomyocyte hypertrophy or cardiac mass. Interestingly, comparison of their data to historic controls suggested that myofibroblast Tgfbr2-deletion could regress pre-established LV fibrosis, indicating that myofibroblast activity is important not only for initiation of fibrosis but also for its maintenance.
Taken together, this publication leverages genetically-engineered mouse strains to establish proof-of-concept that excessive myofibroblast activation is a major contributor to disease progression in a model of human heritable cardiomyopathy. Previous studies using the PostnMerCreMer mouse have focused on models of acute-onset cardiac stress (e.g., pressure overload by transverse aortic constriction or acute myocardial infarction). In these mouse models of acquired HF, multiple cells types are simultaneously and acutely stressed by a surgically imposed injury, making it difficult to assess whether myofibroblast activation is truly a primary versus secondary response. In contrast, the primary insult in the Mybpc340kDa transgenic model is entirely cardiomyocyte intrinsic, and therefore the activation of myofibroblast in this setting is unequivocally a secondary response to cardiomyocyte stress. Furthermore, unlike pressure overload or myocardial infarction models, the tempo of cardiac fibrosis in the Mybpc340kDa mouse is slow and insidiously progressive, leading to 50% mortality at approximately 9 months of age. As such, the current work definitively implicates that secondary activation of myofibroblasts can be a bona-fide disease driver. The observation that deletion of a critical TGF-β receptor in myofibroblasts confers protection in this setting supports a model in which the toxic peptide culminates in a cardiomyocyte stress response or altered biomechanics, ultimately eliciting endogenous TGF-β signaling in neighboring fibroblasts. The mechanism by which the diseased cardiomyocyte communicates with myofibroblasts may involve primary secretion of TGF-β by myocytes, paracrine stimulation of non-myocytes which then secrete TGF-β, or activation of pre-formed molecules of latent TGF- β (which can be triggered by mechanical stretch, proteolytic processing, reactive oxygen species, or allosteric interactions with proteins such as thrombospondins). Future studies that define these heterotypic signaling mechanisms may inform novel therapeutic approaches that can suppress excessive myofibroblast activation in the failing heart.
In addition to defining the most proximal signals that originate from the diseased cardiomyocyte, another major question remains: how does secondary activation of myofibroblasts fuel the vicious cycle of HF progression? We note that previous work has shown that myofibroblast-specific deletion of Tgfbr1/2 or Smad3 markedly attenuates pressure overload-induced cardiac pathology 11, suggesting that that interdicting canonical TGF-β signaling at different nodal points in this master regulatory pathway of myofibroblast identity can confer protection. Therefore, the salutary effects of myofibroblast-specific Tgfbr2 deletion in the Mybpc340kDa transgenic mice are likely related to wholesale suppression of this cell state and its pleiotropic consequences, including effects on matrix remodeling, cytokine elaboration, inflammation, altered ventricular mechanics and creation of a pro-arrthymic substrate. Although the deletion of Tgfbr2 in this study is myofibroblast specific, the phenotypic consequences may involve reciprocal and heterotypic interactions with cardiomyocytes, myeloid cells, smooth muscle cells and endothelial cells. In future work, it will be fascinating to perturb myofibroblast function and subsequently interrogate effects on each of these cell types to gain more insight into the mechanisms by which excessive myofibroblast activity contributes to HF. In addition to assessing the effects on wholesale suppression of the myofibroblast state (as is likely to occur with potent suppression of TGF-β signaling), the PostnMerCreMer mouse will also enable genetic manipulation of more restricted myofibroblast effectors functions (e.g. matrix production, cytokine secretion, proliferation, immune cell recruitment), thereby establishing the relative contributions of these individual processes to cardioprotection. Further investigation along these lines has the potential to elucidate new druggable targets in the myofibroblast.
This work has important therapeutic implications, particularly given the observation that myofibroblast inhibition that is commenced late in the course of disease retains beneficial effects, including an improvement in LV function and possible regression of fibrosis. Given the significant residual mortality in HF patients who are already using current guideline-directed medical therapy, identification of new therapeutic approaches is desperately needed. This paper adds to the emerging body of evidence that exuberant activation of cardiac myofibroblasts may represent a novel and potentially druggable axis in human HF. It will also be of great interest to explore whether targeting the myofibroblasts can have therapeutic effects in the highly prevalent syndrome of HF with preserved systolic function (HFpEF), for which there are no current therapies that improve mortality 12. For an anti-myofibroblast approach to be considered, the therapeutic index must be carefully considered. Kaniscak et. al. found that completely killing off the Periostin+ population using a toxigene-based genetic approach was poorly tolerated by the mouse heart, corroborating the role of myofibroblasts in basal cardiac homeostasis and early repair of cardiac injury 3. Thus, any approach that dampens myofibroblast activity must carefully navigate a therapeutic window that does not interfere with this cell’s role in tissue maintenance and organ repair throughout the body. Recent data suggest that there are temporally evolving and newly defined fibroblast cell states in the injured heart, suggesting more specialized roles for fibroblast subsets in acute injury responses versus chronic scar stabilization 13. A deeper understanding of the signal transduction pathways and epigenetic mechanisms underlying these cell state transitions may open new doors for targeting the fibroblast in HF 14, 15.
Studies such as the current one by Meng and colleagues add to the emerging body of exciting work that implicate the myofibroblast as a key driver of HF progression. If the current momentum in this field continues to grow, the next generation of theatrical productions in the HF genre may feature the fibroblast at the very top of the playbill.
Acknowledgements:
We thank Ana Caterina Silva from Gladstone Institutes for drawing the schematic figure.
Sources of funding:
MA was supported by the Swiss National Science Foundation (project P2LAP3_178056). SMH was supported by NIH R01 HL127240.
Footnotes
Disclosures:
SMH is an executive and shareholder of Amgen, Inc. and is a shareholder of Tenaya Therapeutics.
The authors of this article declare no conflict of interest related to the contents of this article.
References
- 1.Prabhu SD, Frangogiannis NG. The biological basis for cardiac repair after myocardial infarction: From inflammation to fibrosis. Circ Res. 2016;119:91–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meng Q, BB, MSB, JJ, HO, IV-A, KS-W, JG, JDM, Blaxall BC JR. Myofibroblast specific tgfβ receptor ii signaling in the fibrotic response to cardiac myosin binding protein c-induced cardiomyopathy. Circ Res (this issue). 2018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kanisicak O, Khalil H, Ivey MJ, Karch J, Maliken BD, Correll RN, Brody MJ, SC JL, Aronow BJ, Tallquist MD, Molkentin JD. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat Commun. 2016;7:12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Geisterfer-Lowrance AA, Kass S, Tanigawa G, Vosberg HP, McKenna W, Seidman CE, Seidman JG. A molecular basis for familial hypertrophic cardiomyopathy: A beta cardiac myosin heavy chain gene missense mutation. Cell. 1990;62:999–1006 [DOI] [PubMed] [Google Scholar]
- 5.Konno T, Chang S, Seidman JG, Seidman CE. Genetics of hypertrophic cardiomyopathy. Curr Opin Cardiol. 2010;25:205–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaski JP, Syrris P, Esteban MT, Jenkins S, Pantazis A, Deanfield JE, McKenna WJ, Elliott PM. Prevalence of sarcomere protein gene mutations in preadolescent children with hypertrophic cardiomyopathy. Circ Cardiovasc Genet. 2009;2:436–441 [DOI] [PubMed] [Google Scholar]
- 7.Barefield D, Sadayappan S. Phosphorylation and function of cardiac myosin binding protein-c in health and disease. J Mol Cell Cardiol. 2010;48:866–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sadayappan S, Osinska H, Klevitsky R, Lorenz JN, Sargent M, Molkentin JD, Seidman CE, Seidman JG, Robbins J. Cardiac myosin binding protein c phosphorylation is cardioprotective. Proc Natl Acad Sci U S A. 2006;103:16918–16923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sadayappan S, Gulick J, Klevitsky R, Lorenz JN, Sargent M, Molkentin JD, Robbins J. Cardiac myosin binding protein-c phosphorylation in a {beta}-myosin heavy chain background. Circulation. 2009;119:1253–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Razzaque MA, Gupta M, Osinska H, Gulick J, Blaxall BC, Robbins J. An endogenously produced fragment of cardiac myosin-binding protein c is pathogenic and can lead to heart failure. Circ Res. 2013;113:553–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khalil H, Kanisicak O, Prasad V, Correll RN, Fu X, Schips T, Vagnozzi RJ, Liu R, Huynh T, Lee SJ, Karch J, Molkentin JD. Fibroblast-specific tgf-beta-smad2/3 signaling underlies cardiac fibrosis. J Clin Invest. 2017;127:3770–3783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shah SJ, Kitzman DW, Borlaug BA, van Heerebeek L, Zile MR, Kass DA, Paulus WJ. Phenotype-specific treatment of heart failure with preserved ejection fraction: A multiorgan roadmap. Circulation. 2016;134:73–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fu X, Khalil H, Kanisicak O, Boyer JG, Vagnozzi RJ, Maliken BD, Sargent MA, Prasad V, Valiente-Alandi I, Blaxall BC, Molkentin JD. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J Clin Invest. 2018;128:2127–2143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duan Q, McMahon S, Anand P, Shah H, Thomas S, Salunga HT, Huang Y, Zhang R, Sahadevan A, Lemieux ME, Brown JD, Srivastava D, Bradner JE, McKinsey TA, Haldar SM. Bet bromodomain inhibition suppresses innate inflammatory and profibrotic transcriptional networks in heart failure. Sci Transl Med. 2017;9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Micheletti R, Plaisance I, Abraham BJ, Sarre A, Ting CC, Alexanian M, Maric D, Maison D, Nemir M, Young RA, Schroen B, Gonzalez A, Ounzain S, Pedrazzini T. The long noncoding rna wisper controls cardiac fibrosis and remodeling. Sci Transl Med. 2017;9 [DOI] [PMC free article] [PubMed] [Google Scholar]