

Abstract
Malignant infantile osteopetrosis (MIOP), an autosomal-recessive disorder, is extremely rare, presenting early in life with extreme sclerosis of the skeleton and reduced activity of osteoclasts. It was first described by Albers Schonberg in 1904. Disease manifestations include compensatory extramedullary haematopoiesis at sites such as the liver and spleen, hepatosplenomegaly, anaemia and thrombocytopaenia. Neurological manifestations can also occur due to narrowing of osseous foramina resulting in visual impairment, hearing loss, facial palsy and hydrocephalus. In addition, growth retardation and recurrent infections requiring long-term antibiotic use are common. The incidence of MIOP is 1/2 000 000 and if untreated, then it has a fatal outcome, with the majority of cases occurring within the first 5 years of life. At present, the only potentially curative option is a haematopoietic stem cell transplant. We present a 21-year-old woman, diagnosed with malignant infantile osteopetrosis, due to a mutation in the T-cell immune regulator 1 gene when aged 6 weeks, presenting with chronic osteomyelitis of her left mandible. As malignant infantile osteopetrosis has a high mortality in infancy, we felt it prudent to report this rare case in a patient surviving to adulthood.
Keywords: dentistry and oral medicine, infections
Background
Osteopetrosis is a rare genetic disorder and belongs to a highly heterogeneous group of conditions that share the hallmark of increased bone density on radiographs due to abnormalities in osteoclast differentiation or function. Four types have been reported. Our case discusses malignant infantile osteopetrosis, the autosomal recessively inherited form of the disease that generally appears in utero and presents at birth or within the first year of life. Phenotypic features such as macrocephaly, frontal bossing or hydrocephalus can develop. Delayed tooth eruption is also observed. As in our case, there is a predisposition to a short stature, fractures and a risk of developing osteomyelitis. Other subtypes include the adult benign autosomal-dominant form, intermediate autosomal-recessive osteopetrosis and a distinct form due to carbonic anhydrase 11 deficiency. Genetic testing can be used to confirm the diagnosis and differentiate between the different subtypes.
Case presentation
A 21-year-old woman presented to the emergency department with trismus and swelling of her left mandible. Clinical examination revealed a short stature, prominent frontal bossing, a depressed nasal bridge and a dysmorphic craniofacial appearance. She was feverish with a temperature of 37.9°C. On examination, there was a firm swelling over the left mandible with two sinuses in the submandibular region, one of them discharging pus (figure 1). Her interincisal opening was 1 cm. Intraoral examination was difficult due to her trismus, but only a lower left central incisor and canine were observed in the left mandible. She had multiple missing teeth. She was born at term by normal vaginal delivery without complications to non-consanguineous parents in the UK. She reported developing osteomyelitis of her mandible aged 12 years, requiring periodic debridement and protracted courses of clindamycin at Birmingham Children’s Hospital. A diagnosis of malignant infantile osteopetrosis (MIOP) was rendered at the age of 6 weeks following a chest infection with her chest radiograph demonstrating a homogeneous increased density of her ribs. She was blind in her left eye due to optic nerve compression. Her surgical history included optic nerve decompression surgery in 2001, which preserved the vision in her right eye. In addition, she underwent skull realignment surgery to reduce intracranial pressure in 2008.
Figure 1.
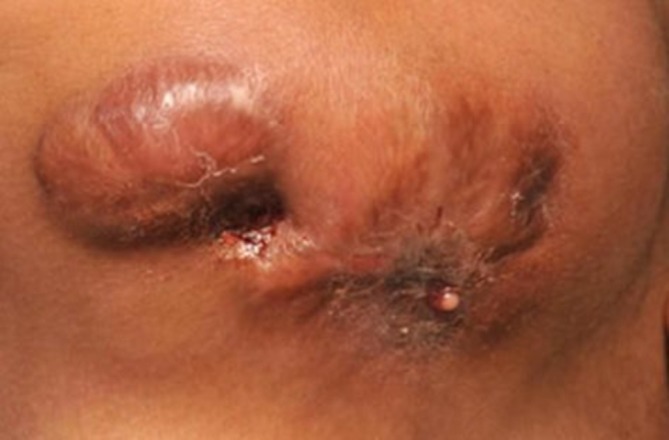
Two sinuses in the left submandibular region, one of them discharging pus.
She had a history of multiple fractures since childhood and had pins placed in both legs to reduce bow deformity. Furthermore, she required regular blood transfusions from the age of 12 years, once every 8 weeks. She underwent splenectomy in 2009, following which her transfusion requirement markedly reduced (maintaining satisfactory haemoglobin between 11 and 12 g/dL) with the last transfusion undertaken in November 2010. She trialled gamma interferon injections subcutaneously three times weekly for a 2-year period, between the ages of 3 and 5 years in conjunction with high-dose oral calcitriol. This reduced the frequency of infections but was discontinued due to no significant change in bone turnover markers or bone density. During the Winter months, she received vitamin D supplements. She also required antibiotic prophylaxis (penicillin V 500 mg bd) postsplenectomy. There was a strong family history of osteopetrosis, with multiple affected children within the extended family, many of whom succumbed to their illness during childhood including her two brothers, with only a few reaching adolescence. The first brother died aged 8 years, following complications from a bone marrow transplant (BMT) and her other brother died aged 4 years following an unsuccessful BMT. The patient has not wished to pursue an attempt at BMT. The underlying aetiology of her osteopetrosis was clarified in 2004 following a bone biopsy, which confirmed the presence of numerous osteoclasts, but with defective bone resorption (figures 2 and 3). Genetic mutation studies confirmed that she had a mutation in exon 11 of the T-cell immune regulator 1 gene.
Figure 2.
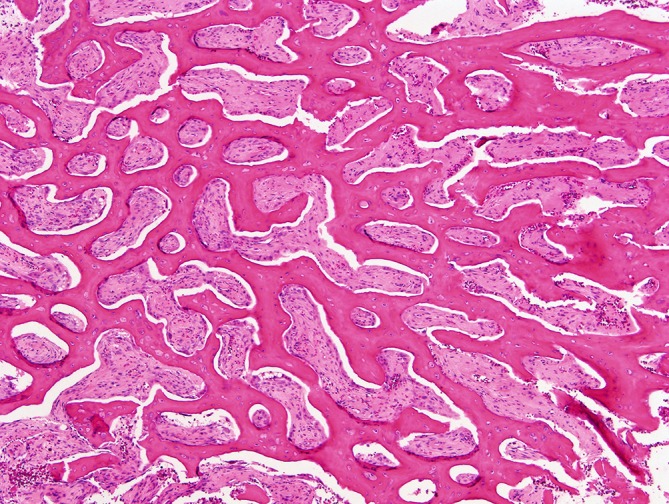
Low power view illustrating abnormally thick trabecular/lamellar bone with diffusely fibrotic medullary interstices and absence of haematopoiesis. There is widespread retraction cleft artefact separating bone from soft tissue due to differential shrinkage during tissue fixation/processing (H&E, original magnification ×4).
Figure 3.
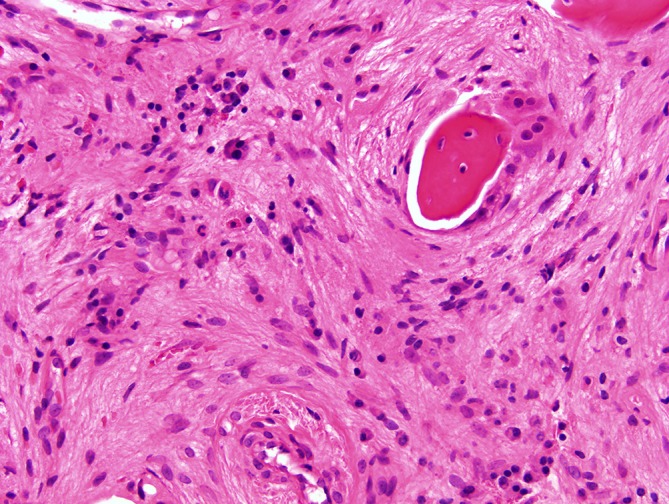
Medium power image demonstrating heavily fibrotic marrow spaces. There are no haematopoietic progenitors but the numerous plasma cells, lymphocytes and scattered polymorphonuclear leucocytes indicate ongoing osteomyelitis. Two multinucleate osteoclasts mantle the bone trabeculum without lacunar resorption (H&E, original magnification ×20).
Investigations
Baseline investigations including a full blood count, bone profile, parathyroid hormone level, vitamin D level, creatine kinase and acid phosphatase level were requested. Her haemoglobin was 11.7 g/dL, white cell count 27×109/L (neutrophils 5.3×109/L and lymphocytes 20 x109/L) and platelets 94×109/L. Further investigation with an orthopantomogram revealed several missing and impacted teeth, with sclerosis of the left body of her mandible (figure 4).
Figure 4.
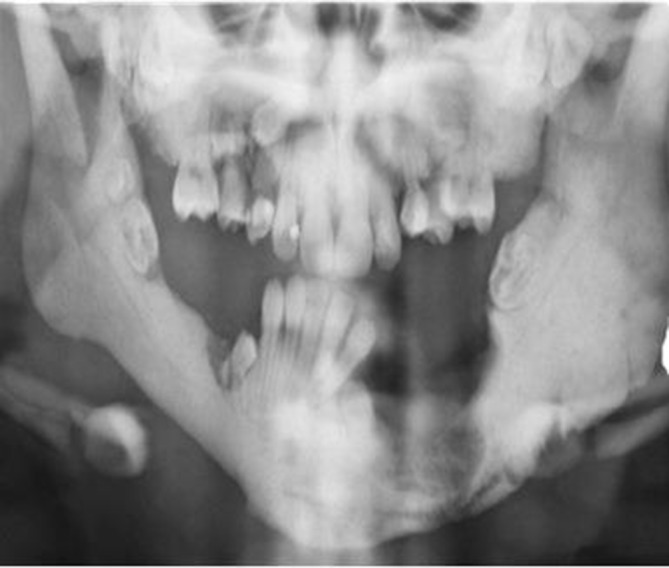
An OPG revealed several missing and impacted teeth, with sclerosis of the left body of her mandible. OPG, orthopantomogram.
Differential diagnosis
She was diagnosed with osteomyelitis of her mandible secondary to her malignant infantile osteopetrosis.
Treatment
She was admitted for intravenous antimicrobial therapy. A bone biopsy was performed under general anaesthesia and exposed bone was visible in the left mandible with pus draining from a fistula. The outer surface of the bone was debrided and three bone specimens were sent to microbiology. Postoperatively, following microbiology advise, she commenced treatment with meropenem 500 mg three times a day. Mixed skin organisms were cultured on her pus swab and her bone specimens were positive for Streptococcus mitis, S. oralis and S. constellatus. She commenced treatment with intravenous penicillin.
Outcome and follow-up
At present, her osteomyelitis is quiescent and she remains under review in the Department of Oral and Maxillofacial Surgery. She has also been referred for a prosthodontic assessment to try and improve her function. As already mentioned, the patient is aware that BMT is the only successful treatment available to her. However, infantile osteopetrosis remains an active research interest and potential treatment options may become available in the future.
Discussion
Osteopetrosis, otherwise known as Albers-Schonberg disease, is derived from the Greek ‘osteo’ meaning bone and ‘petros’ for stone. The German gynaecologist and radiologist, Henrich Albers-Schonberg, first described it in 1904, as a rare inherited metabolic bone disorder caused by impaired osteoclastic activity, resulting in impaired bone resorption.1 In 1926, Karshnen2 introduced the term osteopetrosis. This disease is also known as marble bone disease and is classified into three forms: infantile malignant autosomal-recessive osteopetrosis, intermediate osteopetrosis and autosomal-dominant osteopetrosis. 3 The incidence of osteopetrosis is estimated to be 1 case per 100 000–500 000 population, with the incidence of MIOP 1/2 000 000 and is usually fatal if untreated.4 Symptoms of significant haematological abnormalities with bone marrow failure, anaemia, hepatomegaly and life-threatening pancytopaenia commonly present in infancy. Delay in treatment will result in pneumonia, sepsis, haemorrhage and death.5 Other features include neurological abnormalities, frontal bossing, nystagmus, blindness, deafness and skeletal fractures. Osteomyelitis is a recognised complication and prevention of dental infection can be difficult. Due to impaired osteoclastic function, fractures with poor healing may occur. In addition, children with MIOP are at risk of developing hypocalcaemia, with associated tetanic seizures.6 Indeed, Kumar et al reported an extremely rare case of MIOP presenting with bronchopneumonia, anaemia and malaena in a 2-month-old child.4 The diagnosis is based on clinical and radiological features and biopsy should be avoided due to the infection risk. T-cell immune regulator 1 (TCIRG1) plays crucial roles in osteoclast function and its mutation causes autosomal-recessive osteopetrosis.7 The disease is fatal in infancy and is cured with haematopoietic stem cell transplantation, with a success rate of 50% and unsatisfactory rescue of growth and visual deterioration.8 In contrast, autosomal-dominant osteopetrosis, which has two types (I and II), is discovered later in life and presents with less serious manifestations. In type 1, which is caused by mutations in the lipoprotein receptor-related protein 5 gene, nerve compression is common, but fractures are rare. Fractures are frequent in type II and it is caused by mutations in the chloride channel protein 79.
The differential diagnosis includes myeloproliferative disease, leukaemia, sickle cell disease and congenital disorders such as hypoparathyroidism. Other sclerosing bone dysplasias, such as pyknodysostosis and craniometaphyseal dysplasia, should be included.9 In addition, secondary hyperparathyroidism caused by renal osteodystrophy may also produce a diffuse osteosclerosis. Chemical poisoning from lead, fluoride and beryllium should also be considered.
Bones may be uniformly sclerotic, but alternating sclerotic and lucent bands may be noted in the iliac wings and near the ends of long bones.3 Radiographs may also show evidence of osteomyelitis or fractures, commonly reported in the femoral shaft and the posterior tibia. The radiological ‘bone within bone’ or ‘endobone’ appearance is characteristic and diagnostic and may be seen in radiographs of the hand, pelvis or scapula.4 According to Garcia, the dental pulp chamber is not clearly recognised due to increased bone density.10 Management of individuals with MIOP requires a multidisciplinary approach to deal with the haematological and metabolic abnormalities, recurrent infections such as osteomyelitis and neurological sequelae. Osteomyelitis associated with osteopetrosis tends to be refractory because of the reduced blood supply and accompanying anaemia. Antibiotic therapy combined with complete debridement of necrotic tissue, bacterial culture and sensitivity testing, followed by suturing of soft tissue is the main therapeutic approach.11 Indeed, Adachi et al suggest hyperbaric oxygen therapy in recalcitrant cases.12 However, improved oral hygiene and preventative care will minimise the infection risk. Early diagnosis and haematopoietic stem cell transplantation are the only curative options. Preconception genetics counselling is imperative, especially in those with a positive family history.
Learning points.
Osteopetrosis is a rare disease transmitted by autosomal-dominant or autosomal-recessive inheritance having variable penetrance. It is classified into three forms: malignant infantile autosomal-recessive osteopetrosis, intermediate osteopetrosis and autosomal-dominant osteopetrosis.
Malignant infantile osteopetrosis can present with fractures, osteomyelitis, short stature and frontal bossing. The abnormal expansion of bone interferes with medullary haematopoiesis resulting in life-threatening pancytopaenia and extramedullary haematopoiesis which may result in hepatosplenomegaly.
Haematopoietic stem cell transplantation is the only treatment that can offer potential cure. Interferon gamma 1b can be used in individuals unresponsive to a stem cell transplant or indeed, in those individuals awaiting a transplant.
Footnotes
Contributors: LD: wrote the case report. AW: histopathology report and the images. RW: edited the paper.
Funding: The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests: None declared.
Provenance and peer review: Not commissioned; externally peer reviewed.
Patient consent for publication: Obtained.
References
- 1. Albers-Schonberg HE. Rontgenbilder einer seltenem Knockererkrankung. Much Med Wochenschr 1904;51:365–8. [Google Scholar]
- 2. Karshner RG. Osteopetrosis. Am J Roentgenol 1926;16:405–19. [Google Scholar]
- 3. Baptista M, Vieira D, Abel C, et al. Type II osteopetrosis – Case report. Neuroradiology 2013. [Google Scholar]
- 4. Kumar KJ, Bandaru K, Prashanth SN, et al. Malignant infantile osteopetrosis. Indian J Hum Genet 2013;19:90–2. 10.4103/0971-6866.112911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Stark Z, Savarirayan R. Osteopetrosis. Orphanet J Rare Dis 2009;4:5–9. 10.1186/1750-1172-4-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Engiz O, Kara S, Bagrul D, et al. Infantile malignant osteopetrosis: a rare case of neonatal hypocalcaemia. Horm Res Paediatr 2012;78:1663–2818. [DOI] [PubMed] [Google Scholar]
- 7. Wada K, Harada D, Michigami T, et al. A case of autosomal dominant osteopetrosis type II with a novel TCIRG1 gene mutation. J Pediatr Endocrinol Metab 2013;26:575–7. 10.1515/jpem-2013-0007 [DOI] [PubMed] [Google Scholar]
- 8. Del Fattore A, Cappariello A, Teti A. Genetics, pathogenesis and complications of osteopetrosis. Bone 2008;42:19–29. 10.1016/j.bone.2007.08.029 [DOI] [PubMed] [Google Scholar]
- 9. Helfrich MH. Osteoclast diseases and dental abnormalities. Arch Oral Biol 2005;50:115–22. 10.1016/j.archoralbio.2004.11.016 [DOI] [PubMed] [Google Scholar]
- 10. Garcia CM, Garcia MAP, Garcia RG, et al. Osteomyelitis of the mandible in a patient with osteopetrosis. Case report and review of the literature. J Maxillofac Oral Surg 2011;3:120–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Barry CP, Ryan CD, Stassen LF. Osteomyelitis of the maxilla secondary to osteopetrosis: a report of 2 cases in sisters. J Oral Maxillofac Surg 2007;65:144–7. 10.1016/j.joms.2005.07.019 [DOI] [PubMed] [Google Scholar]
- 12. Adachi M, Iwai T, Watanuki K, et al. Osteomyelitis of the jaws associated with osteopetrosis: case report of two sisters. Oral Surg 2013;6:73–6. 10.1111/ors.12007 [DOI] [Google Scholar]