

Abstract
Nonaka myopathy is an autosomal recessive and slowly progressive distal myopathy. It is part of a rare group of myopathies predominantly affecting the distal limb musculature. Over 150 cases have been reported across the Middle East, Japan and Europe. We report the case of a 33-year-old woman presenting with symmetrical upper and lower limb weakness, most severely affecting the distal muscle groups. After extensive neurological investigation including neurophysiology, muscle biopsy and genetic analysis, she was finally diagnosed with Nonaka myopathy and treated conservatively with physiotherapy.
Keywords: neuromuscular disease, neuro genetics
Background
Myopathies are a heterogeneous group of disorders caused by primary dysfunction of the muscle fibres, often leading to weakness as a hallmark symptom. Classical teaching recognises a myopathic pattern of weakness as predominantly affecting proximal musculature, thus resulting in difficulty rising from chairs, climbing stairs or holding overhead items.1 While this is true for most myopathies, there is a distinct group of rarer genetic and acquired distal myopathies which result in mainly distal muscle weakness.2
Patients with distal weakness of the limbs are often first thought to have a peripheral neuropathic condition, however, awareness of distal myopathies and a high index of clinical suspicion are important in reaching the correct but rarer diagnosis of a distal myopathy.
Here, we will discuss a case of Nonaka myopathy and outline broad clinical and neurophysiological differences between distal myopathies and other mimicking conditions.
Case presentation
A 33-year-old Sudanese woman presented with a 5-year history of painless progressive weakness initially affecting her legs resulting in frequent falls, and later difficulty climbing up the stairs. She later also developed loss of power in her hands and was now struggling to open jars. She denied any sensory symptoms and there was no bulbar weakness or features of autonomic dysfunction.
Her medical history consisted of type 1 diabetes. She was otherwise healthy and had a normal childhood development. Her vaccination history was unknown, however, she denied infections. There was a history of diabetes in the family but no neurological illnesses or consanguinity. She never drank alcohol or smoked cigarettes.
On examination, she had a waddling gait with flaccid symmetrical weakness in the lower limbs which was more severe distally (grade 1/5 at ankles) than proximally (grade 3/5 weakness at hips). Power in the upper limbs was symmetrically reduced distally (grade 4/5) and normal more proximally. Tone was reduced in her lower limbs and there was generalised areflexia with downgoing plantar reflexes. There was no clinically delayed relaxation of muscles (ie, myotonia).
Sensation to touch, pain, vibration and joint position sense was globally normal and cranial nerve examination was unremarkable.
Investigations
Bloods tests were normal or negative for the following investigations:
Full blood count, urea and electrolytes, liver function tests, vitamin D, B12 and folate, creatine kinase (CK) (112 U/L, normal range 24–170), lactate dehydrogenase, viral serologies (for HIV, hepatitis B and C andcytomegalovirus (CMV)), antiganglioside antibodies, antinuclear antibodies, cardiolipin antibodies, antineutrophil cytoplasmic antibodies, immunoglobulin and protein electrophoresis.
Nerve conduction studies revealed normal peripheral sensory and motor nerve responses in her limbs. Needle electromyography revealed changes suggestive of an underlying myopathic process, that is, low-amplitude and short-duration polyphasic motor units.
MRI lumbosacral spine demonstrated very mild degenerative changes. Cerebrospinal fluid (CSF) examination was unremarkable with negative oligoclonal bands and viral PCR.
Differential diagnosis
Our patient presented with a clinical phenotype of symmetrical, pure motor and predominantly distal limb weakness with hypotonia and areflexia.
Prior to her investigations, the differential diagnosis included:
Peripheral neuropathy. Most peripheral neuropathies involve sensory fibres to a certain degree, thus leading to sensory symptoms which may either be negative (eg, numbness) or positive (eg, pain or paraesthesia). However, pure motor peripheral neuropathies, for example, due to lead toxicity, may present without sensory symptoms.3 Multifocal motor neuropathy with conduction block is another example, however, it generally presents in a more asymmetric fashion than our patient and typically has an upper limb predilection.4 Additionally despite pathological involvement of both sensory and motor fibres in some slowly progressive inherited peripheral neuropathies, such as Charcot-Marie-Tooth disease, patients may be unaware of any sensory symptoms, though neurophysiology typically reveals sensory involvement.5
Degenerative process affecting the motor anterior horn cell such as postpolio syndrome, lower motor neuron variants of motor neuron disease (progressive muscular atrophy) or distal spinal muscular atrophy (also known as distal hereditary motor neuropathy).6
A distal myopathy.
Her nerve conduction studies and needle Electromyogram (EMG) were important in differentiating between these possibilities. In general, diminished sensory nerve responses would immediately point to a peripheral neuropathy, while diminished motor nerve responses and needle EMG evidence of neurogenic abnormalities (large-amplitude and long-duration motor units) are expected to be seen in peripheral neuropathy or pathological processes affecting the motor anterior horn cell.
In our case, however, normal peripheral sensory and motor nerve responses and needle EMG findings of low-amplitude and short-duration motor units favoured a distal myopathic process and the patient proceeded to a left deltoid muscle biopsy. This revealed features consistent with a vacuolar myopathy without any inflammatory cell infiltrate (figure 1).
Figure 1.
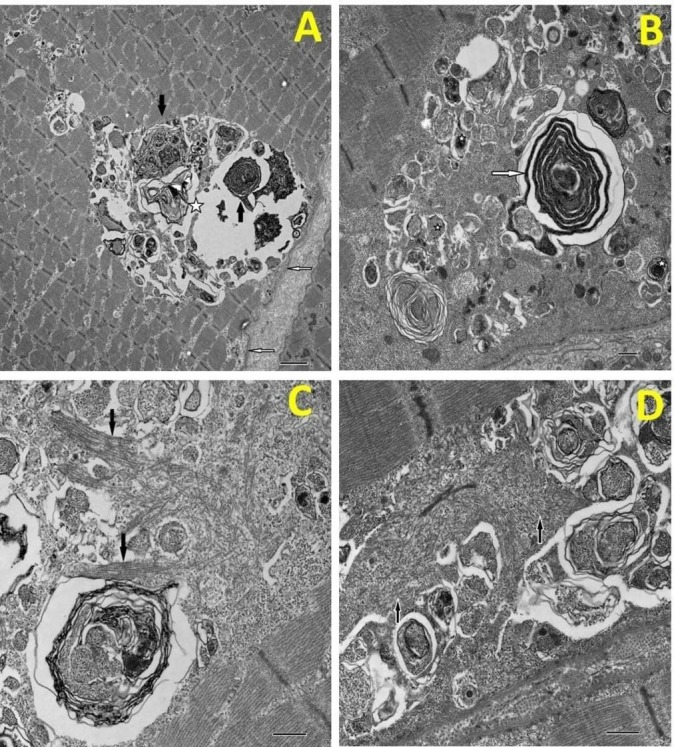
(A) Low magnification image of a rimmed vacuole (asterisk) located beneath the sarcolemma (open arrows). The vacuole contains darkly staining myelin figures (closed arrows). (B) a similar rimmed vacuole demonstrates a darkly staining membrane figure (arrow) and a number of smaller lysosomes (asterisks). (C) High magnification view of the characteristic thick filaments within the sarcoplasm. They occur both randomly orientated and in small bundles (arrows). (D) Masses of randomly orientated thick filaments (arrows). Bar A=2µm, B, C & D=500nm.
Subsequent genetic analysis of the muscle specimen revealed a pathogenic missense mutation in the GNE gene (c.878A>G), supporting the diagnosis of Nonaka myopathy, an autosomal recessive hereditary inclusion body distal myopathy.7
Treatment
A conservative treatment approach was decided on, involving a multidisciplinary team approach and emphasising physiotherapy. No specific medications are currently available, but as GNE mutations are thought to cause an impairment in sialic acid pathway (deficiency of end product),7 studies are underway to test whether replacing sialic acid or its precursors may help prevent or reverse muscle degeneration.8
Outcome and follow-up
The patient is under regular multidisciplinary neuromuscular follow-up with supportive therapy in place.
Discussion
This case illustrates the importance of considering distal myopathies as potential differential diagnoses of predominantly distal limb weakness with no sensory symptoms.
There are many different causes of myopathies which may be genetic or acquired including metabolic, endocrine, toxic or inflammatory aetiologies.1 Weakness is usually a cardinal feature of myopathy and it may present:
Proximally. The ‘classical’ well-recognised pattern affecting proximal limb girdle musculature. This is most commonly seen in clinical practice.1
Mainly proximally but occasionally with some distal involvement. An example of this is facioscapulohumeral dystrophy which predominantly involves facial, periscapular and proximal upper limb muscles but can additionally feature early distal lower limb weakness leading to foot drop.9
Mainly distally or with a significant distal component, that is, the distal myopathies.2
Distal myopathies are rarer and most are genetic although some are acquired (eg, inclusion body myositis). Examples of distal myopathies and some typical features are noted in table 1.2 10
Table 1.
Clinical features of distal myopathies
| Distal myopathy | Clinical features |
| Nonaka myopathy | Autosomal recessive. See text. |
| Welander distal myopathy | Autosomal dominant. Age of onset >40 years. Predominant symmetrical weakness of finger extension. |
| Tibial muscular dystrophy (Udd myopathy) | Autosomal dominant. Age of onset >35 years. Predominant weakness of anterior leg compartment muscles. Upper limbs rarely affected. May be asymmetric. |
| Miyoshi myopathy | Autosomal dominant. Age of onset >15 years. Asymmetric posterior calf weakness. |
| Myofibrillar myopathies | Autosomal dominant. Variable age onset and distribution of weakness depending on subtype. May be associated with cardiomyopathy and respiratory failure. |
| Myotonic dystrophy | Autosomal dominant. Variable age of onset depending on severity. Symmetrical weakness of long finger flexors, ankle dorsiflexors, facial weakness and ptosis with clinical myotonia. Multisystem disturbance including endocrine and cardiac involvement. |
| Inclusion body myositis | Acquired inflammatory myopathy. Age of onset >30–50 years. Typically asymmetric insidious involvement of distal long finger flexors and quadriceps. |
Nonaka myopathy is an autosomal recessive neuromuscular disorder which is more common in the Middle East and Japan where it has an estimated prevalence of 1 in a million.11 There are over 150 GNE gene variants associated with GNE myopathy.12 It is characterised by early-adult onset weakness of distal lower limb muscles, and a normal or mildly elevated serum CK level.7 8 Later on, proximal lower limb muscles such as the glutei may become clinically affected, though the quadriceps are typically spared, and later distal hand muscle involvement is also recognised.7 8 Muscle biopsies show evidence of rimmed vacuoles in affected muscle fibres without inflammation,7 as illustrated in our case (see figure 1). As there is currently no specific treatment, a multidisciplinary supportive approach including physiotherapy is crucial. This will likely be supplemented in future by targeted treatments currently being investigated such as sialic acid replacement.8
Learning points.
Most myopathies manifest proximally, however, some rarer genetic and acquired myopathies can present with predominantly distal weakness.
A high index of clinical suspicion is required to diagnose a distal myopathy in a patient with gradual-onset distal weakness and no sensory disturbance.
The most important differential diagnoses include peripheral neuropathies and anterior horn cell disorders.
Nerve conduction studies and electromyography are helpful in excluding the more common peripheral neurogenic or anterior horn cell mimics.
Nonaka myopathy is an autosomal recessive slowly progressive disorder presenting initially with symmetrical distal lower limb weakness and later progressing more proximally but sparing the quadriceps. Mild distal upper limb weakness may be a feature.
Footnotes
Contributors: YN and AA wrote the initial draft. EC provided the microscopic pictures. SJ was responsible for patient care, making the diagnosis and took overall responsibility of the project, reviewed and edited manuscript/figures before final submission.
Funding: The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests: None declared.
Provenance and peer review: Not commissioned; externally peer reviewed.
Patient consent for publication: Obtained.
References
- 1. Suresh E, Wimalaratna S. Proximal myopathy: diagnostic approach and initial management. Postgrad Med J 2013;89:470–7. 10.1136/postgradmedj-2013-131752 [DOI] [PubMed] [Google Scholar]
- 2. Dimachkie MM, Barohn RJ. Distal myopathies. Neurol Clin 2014;32:817–42. 10.1016/j.ncl.2014.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hanewinckel R, Ikram MA, Van Doorn PA, et al. Peripheral neuropathies. Handb Clin Neurol 2016;138:263–82. 10.1016/B978-0-12-802973-2.00015-X [DOI] [PubMed] [Google Scholar]
- 4. Vlam L, van der Pol WL, Cats EA, et al. Multifocal motor neuropathy: diagnosis, pathogenesis and treatment strategies. Nat Rev Neurol 2011;8:48–58. 10.1038/nrneurol.2011.175 [DOI] [PubMed] [Google Scholar]
- 5. Rossor AM, Evans MR, Reilly MM. A practical approach to the genetic neuropathies. Pract Neurol 2015;15:187–98. 10.1136/practneurol-2015-001095 [DOI] [PubMed] [Google Scholar]
- 6. Rossor AM, Kalmar B, Greensmith L, et al. The distal hereditary motor neuropathies. J Neurol Neurosurg Psychiatry 2012;83:6–14. 10.1136/jnnp-2011-300952 [DOI] [PubMed] [Google Scholar]
- 7. Nishino I, Carrillo-Carrasco N, Argov Z. GNE myopathy: current update and future therapy. J Neurol Neurosurg Psychiatry 2015;86:385–92. 10.1136/jnnp-2013-307051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pogoryelova O, González Coraspe JA, Nikolenko N, et al. GNE myopathy: from clinics and genetics to pathology and research strategies. Orphanet J Rare Dis 2018;13:70 10.1186/s13023-018-0802-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pastorello E, Cao M, Trevisan CP. Atypical onset in a series of 122 cases with FacioScapuloHumeral Muscular Dystrophy. Clin Neurol Neurosurg 2012;114:230–4. 10.1016/j.clineuro.2011.10.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Udd B. Distal myopathies. Curr Neurol Neurosci Rep 2014;14:434 10.1007/s11910-013-0434-4 [DOI] [PubMed] [Google Scholar]
- 11. Kayashima T, Matsuo H, Satoh A, et al. Nonaka myopathy is caused by mutations in the UDP-N-acetylglucosamine-2-epimerase/N-acetylmannosamine kinase gene (GNE). J Hum Genet 2002;47:0077–9. 10.1007/s100380200004 [DOI] [PubMed] [Google Scholar]
- 12. Celeste FV, Vilboux T, Ciccone C, et al. Mutation update for GNE gene variants associated with GNE myopathy. Hum Mutat 2014;35:915–26. 10.1002/humu.22583 [DOI] [PMC free article] [PubMed] [Google Scholar]