

Abstract
Dysregulated activity of the protease matriptase is a key contributor to aggressive tumor growth, cancer metastasis, and osteoarthritis. Methods for the detection and quantification of matriptase activity and inhibition would be useful tools. To address this need, we developed a matriptase-sensitive protein biosensor based on a dimerization-dependent red fluorescent protein (ddRFP) reporter system. In this platform, two adjoining protein domains, connected by a protease-labile linker, produce fluorescence when assembled and are nonfluorescent when the linker is cleaved by matriptase. A panel of ddRFP-based matriptase biosensor designs was created that contained different linker lengths between the protein domains. These constructs were characterized for linker-specific cleavage, matriptase activity, and matriptase selectivity; a biosensor containing a RSKLRVGGH linker (termed B4) was expressed at high yields and displayed both high catalytic efficiency and matriptase specificity. This biosensor detects matriptase inhibition by soluble and yeast cell surface expressed inhibitor domains with up to a 5-fold dynamic range and also detects matriptase activity expressed by human cancer cell lines. In addition to matriptase, we highlight a strategy that can be used to create effective biosensors for quantifying activity and inhibition of other proteases of interest.
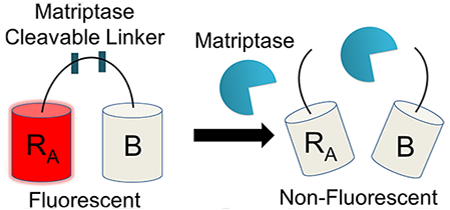
Molecular biosensors are tools that can be used to measure biological processes, such as protein–protein interactions, cell and molecular trafficking, or protease activity, through a signal readout.1,2 Proteases are increasingly discovered to play a critical role in pathological signal processes including inflammation and tumor metastasis.1,3 Thus, there is interest in creating novel biosensor designs that target specific proteases to detect their activity and inhibition. Since proteases naturally recognize and cleave peptide bonds, polypeptides can serve as natural protease substrates. A biosensor can be created by coupling the cleavage event to a signal readout. Recent biosensor designs have centered around detection signals resulting from fluorescent resonance energy transfer (FRET),4,5 luciferase,6,7 or fluorescent quenching.8,9 Through either acquisition or loss of a signal, biosensors have been designed to measure the activity of proteases critical to biological and pathological processes, including trypsin,5 caspase-3,10 matrix metalloproteinases (MMP),11 and cathepsin-B.12
Matriptase is a type II membrane anchored-serine protease expressed on the surface of epithelial cells that functions in developmental pathways as well as tissue regeneration.13,14 Overexpression and dysregulated activity of matriptase in the local tumor environment has been shown to correlate with poor patient prognosis in many human cancers, including breast, colorectal, pancreatic, cervical, and prostate cancers.13 The high turnover of key substrates, such as pro-hepatocyte growth factor (pro-HGF),15 urokinase plasminogen activator (uPA),15 pro-macrophage stimulating protein (pro-MSP),16 and platelet derived growth factor-D (PDGF-D)17 has been shown to contribute to cancer growth and metastasis. Furthermore, matriptase has been identified as a key driver of other disease systems, including osteoarthritis,18 activation of the H1N1 influenza virus,19 and human immunodeficiency virus (HIV).20
As matriptase expression does not always correlate with activity, tools that can distinguish active from inactive matriptase in disease settings would be useful.21–23 In addition, such reagents could serve as functional screening tools for isolating matriptase-based inhibitors. Small molecule and protein-based biosensors have been created that specifically detect activity by binding to the active site of matriptase.21–23 Although these strategies effectively allow for specific detection of active matriptase in vitro and in vivo, their inhibitor based designs result in matriptase inactivation and thus limit dual application for measuring continuous matriptase activity, which may be useful for inhibitor library functional screening applications. Polypeptide—small molecule compounds such as N-tert-butoxycarbonyl-Gln-Ala-Arg-7-amino-4-methylcoumarin (Boc-QAR-AMC) can also be used to measure matriptase activity and inhibition,24 but their spectral properties (ex/em: 380 nm/460 nm) require UV excitation, which restricts available instrumentation and can affect cell viability. Additionally, these organic compounds often require multistep synthesis which can be prohibitive in terms of cost, scale, and expertise needed.
To address these limitations, we created a matriptase-specific biosensor system based on a dimerization-dependent red fluorescent protein (ddRFP) system originally developed by Campbell and colleagues.10 The ddRFP system relies on interaction of two fluorescent protein domains: a quenched “A” copy and a nonfluorescent “B” copy (Figure 1a). When the A and B copies interact to form a heterodimer, a significant increase in fluorescence results. In order to facilitate this stabilizing interaction, a polypeptide linker can be used to genetically fuse the “A” and “B” copies together. The linker may also include a putative cleavage site that can be recognized by a protease of interest. Upon cleavage by a target protease, the “A” and “B” copies dissociate, resulting in a loss of the stabilizing interactions and return of the “A” copy to its quenched state. In this monomeric state, the “A” copy exhibits a predominately protonated (nonfluorescent) chromophore and a reduced quantum yield, rendering its fluorescence quenched. Therefore, a loss in fluorescence over time is indicative of protease activity, while retention of fluorescence indicates inactive protease (Figure 1b). Genetically encoded ddRFP biosensors have many potential benefits, such as a low cost of production, high dynamic range, robust activity under physiological and nonphysiological conditions, and ideal spectroscopic properties (ex/em: 535 nm/605 nm).
Figure 1.
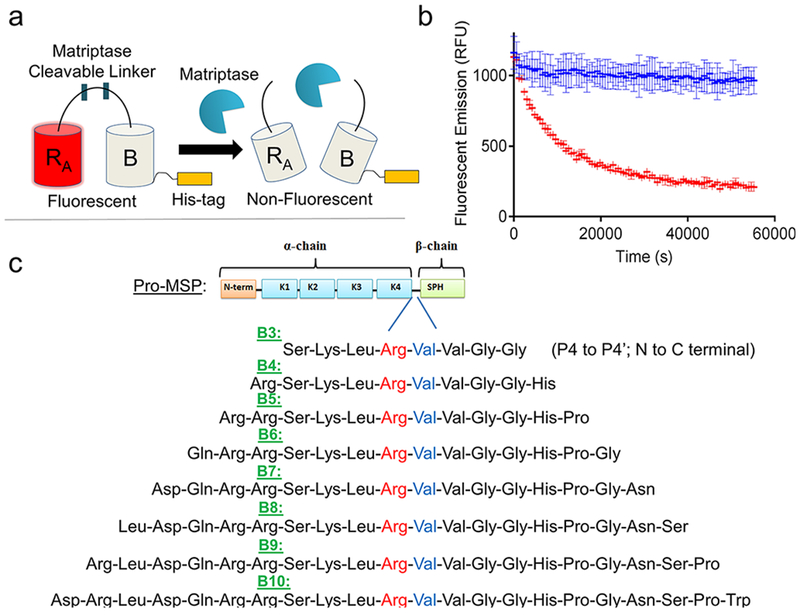
Matriptase biosensor design. (a) ddRFP-based biosensor schematic. Following proteolytic linker cleavage, the fluorescent “A” copy separates from the stabilizing “B” copy, resulting in loss of “A” copy fluorescence. (b) Example time course plot of biosensor mechanism; addition of matriptase cleaves the biosensor and reduces fluorescence over time (red), while absence of matriptase retains fluorescence over time (blue). (c) Matriptase cleavable linker designs and sequence information. Design name is derived from number of amino acids flanking the scissile bond. Scissile bond is highlighted in red (Arg) and blue (Val). Each sequence shown is derived from the natural pro-macrophage stimulating protein (Pro-MSP) sequence. N-term = N-terminus; K1–4 = Kringle domains 1–4; SPH = Serine Protease Homologue.
Previous work showed that the ddRFP system could be used to measure the activity of caspase-3 as a key participant in the apoptotic pathway.10 This study was limited to intracellular caspase-3 detection as a live cell optical imaging tool. Here, we developed the ddRFP system as a soluble protease-sensitive biosensor for measuring both matriptase activity and inhibition. For this goal, we built an optimized matriptase cleavable linker module that can be recognized by both soluble and cell associated forms of matriptase. This work is the first demonstration of this type of biosensor being applied outside the cell. Furthermore, we found that the length of the polypeptide linker in between the A and B domains of the biosensor was critical for conferring matriptase activity and selectivity. We showed that the optimal ddRFP biosensor could be used to measure both matriptase activity and inhibition, with up to a 5-fold dynamic range in a variety of experimental settings. This work also describes methods and design considerations that can be applied for developing similar biosensor reagents which target alternative proteases of interest.
RESULTS/DISCUSSION
To accomplish our goal of creating a ddRFP-based, matriptase sensitive biosensor, we first designed a series of amino acid linkers that could be recognized and cleaved by matriptase. A panel of linker designs was created based upon the sequence of a natural matriptase substrate, called pro-macrophage stimulating protein (pro-MSP). Pro-MSP is an inactive growth factor, that upon cleavage by matriptase, is converted into active MSP which binds its cognate receptor, RON, to stimulate downstream cellular events including growth and proliferation.16 The matriptase recognition site of pro-MSP has been well established and includes the sequence Ser-Lys-Leu-Arg-Val-Val-Gly-Gly (P4-P4’, N to C termini), with Arg483-Val484 representing the scissile bond.25 To develop a matriptase cleavable linker within the context of ddRFP, we first designed eight linkers of increasing lengths, each derived from the natural sequence of pro-MSP. The designs were named for the number of amino acids flanking each side of the scissile bond (i.e., B3 with three flanking residues, up to B10 with 10 flanking residues; Figure 1c), in which the entire sequence listed was derived from wild-type pro-MSP.
Following E. coli production and soluble purification, each biosensor construct was then characterized for its ability to detect matriptase activity. Figure 2a demonstrates a typical time course plot for each construct, in which fluorescent emission is measured over time in the presence or absence of matriptase. For biosensors exhibiting ideal behavior, fluorescence decreases over time in the presence of matriptase but is retained over time in the absence of matriptase. Interestingly, while constructs B4 through B9 portray these ideal fluorescent properties, B3 exhibits no change in fluorescence over time with matriptase, while B10 reveals low fluorescence. To further explore this trend, we next examined the change in each biosensor design’s initial velocity in response to increasing matriptase concentration to determine the effects of linker length on matriptase activity (Figure 2b). Once again, B3 shows low velocity of matriptase cleavage, while B10 exhibits essentially no change in fluorescence even with the addition of 10 nM matriptase. In contrast, the velocity of matriptase activity increases with increasing linker length for constructs B4 to B9. These results help support the presence of linker specific cleavage events by matriptase, as opposed to off target cleavage of the A or B fluorescent protein domains, in which all biosensor designs would show the same velocity profiles, including B3. Additionally, this trend was observed for conditions of both 10 nM and 1 nM matriptase, with a corresponding loss in velocity at lower concentration, suggesting a specific response to matriptase concentration and loss of fluorescence due to cleavage by matriptase.
Figure 2.
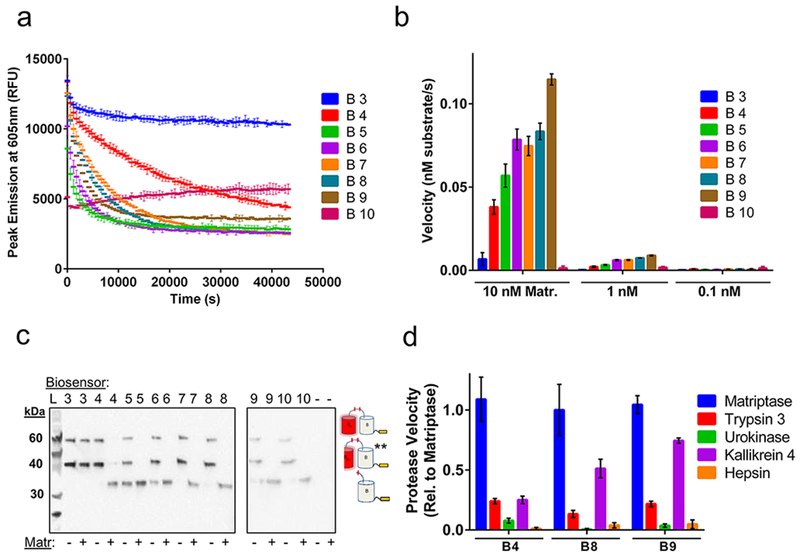
Biosensor kinetic measurements and characterization. (a) Time course trajectories of biosensors B3 to B10 with matriptase and (b) velocity graphs in the presence of varying concentrations of matriptase (Matr.). (c) Western blot bands correspond to three possible reaction products detected through the C-terminal hexahistidine-tag: uncleaved biosensor (top band, 59.4 kDa), **hydolytically cleaved biosensor (middle band, 40.3 kDa), and cleaved biosensor (bottom band, 32.5 kDa). Copy “A” product is not detected due to lack of a His-tag. Note, two gels were used for this experiment as indicated. (d) Velocity profile for B4, B8, and B9 in the presence of different serine protease family members, relative to matriptase activity. Experiments performed in triplicate and reports mean values with standard deviation. Data are normalized for background conditions of biosensor without matriptase.
To further investigate the effects of a linker-specific cleavage event by matriptase, we analyzed the reaction products of each biosensor design, incubated with or without 10 nM matriptase, using SDS-PAGE followed by Western blot. Western blot analysis enables detection of the hexahistidine tag of each biosensor and its matriptase cleaved products. These results reveal at most three bands for each construct (Figure 2c): the uncleaved biosensor substrate (59.4 kDa), the matriptase cleaved biosensor product (32.5 kDa), and the hydrolytically cleaved biosensor product (40.3 kDa), the latter of which is produced from boiling each protein for denaturation and has been a well-documented phenomenon.10,26 Western blot analysis reveals that no matriptase cleavage product is observed for the B3 linker design, confirming its lack of matriptase susceptibility and supporting the prior velocity data (Figure 2a,b). It is likely that the B3 linker is too short and thus sterically inaccessible for matriptase recognition and cleavage within the context of the ddRFP construct. In contrast, matriptase cleaved products are observed for B4 to B9, in agreement with the previous velocity data and confirming linker-specific cleavage by matriptase. Interestingly, B10 also reveals a cleavage product which indicates that the linker region is present and accessible in this construct. The diminished fluorescent signal of the intact B10 protein may be the result of misfolding or disruption of the A—B dimer interface, which would impair ddRFP stabilizing interactions and thus fluorescent output. Finally, we observed that the B5 and B6 constructs have cleavage products in lanes lacking matriptase (Figure 2c). These effects might also be present in time course data for the B5 and B6 constructs (Figure 2a) in which a precipitous loss in fluorescence is observed, and could indicate instability or degradation of these biosensor designs. Overall, the kinetic and Western blot results confirm linker-specific cleavage by matriptase and reveal poor functional characteristics of the B3, B5, B6, and B10 designs.
We next evaluated the effects of linker length on matriptase selectivity, testing biosensor designs B4, B8, and B9. Target selectivity is an important consideration in protease biosensor design for measuring protease activity in a heterogeneous environment. To test selectivity, a time course experiment was performed with a panel of serine proteases including trypsin 3,27 urokinase,28 kallikrein 4,29 and hepsin,25 in addition to matriptase. The resulting initial velocity values for each biosensor and protease tested were compared to matriptase activity (Figure 2d). Interestingly, minimal activity is observed for trypsin 3, urokinase, and hepsin, while the velocity of kallikrein 4 increases with increasing linker length, approaching the velocity of matriptase activity. These results indicate that a longer linker between the A and B domains allows for increased promiscuity and recognition by other serine proteases. Additionally, the observed lack of activity by hepsin, a known protease which recognizes wild-type pro-MSP,25 is likely attributed to the non-native amino acid residues that flank the cleavable linker region within the ddRFP context. For example, the B4 construct has the linker profile of GHGTGSTGNSGRSKLRVVGGHSEDNNMA with the region derived from wild-type pro-MSP underlined. Therefore, the unnatural flanking regions of the protease cleavable linker may confer further selectivity which favors matriptase and differs from the natural pro-MSP protease selectivity profile. In addition, for these selectivity studies, each protease was assumed to be 100% active. From these collective findings, we pursued further characterization of the B4 construct due to its superior stability, ideal kinetic properties, and matriptase selectivity. Additional studies revealed that B4 has a linear activity response to matriptase concentration and follows ideal Michaelis-Menten kinetics, with a quantified Km of 8.17 × 10−6 M and a kcat/Km of 2.09 × 107 M−1 s−1 (Supporting Information Figure 1). These parameters indicate high specificity and highly efficient turnover by matriptase.
The B4 construct was next tested for its ability to detect the activity of matriptase expressed on tumor cells and inhibition in the context of a natural protein. We first characterized the expression profile of endogenous matriptase on three human cancer cell lines, PC3 (prostate cancer), A549 (lung cancer), and MDA-MB-231 (breast cancer). Matriptase expression and functional activity was tested using a human matriptase specific antibody and the commercially available matriptase substrate (Boc-QAR-AMC, R&D Systems), respectively (Supporting Information Figure 2a,b), and appeared to strongly correlate for each cell line tested. We next tested the ability of the B4 construct to detect matriptase activity expressed on human cancer cell lines (Figure 3a) and confirmed significant B4 processing in the presence of cancer cell lines compared with media only control. It is important to note that the heterogeneous environment of cancer cells may contribute additional proteases or other factors that may aid in B4 degradation. However, the distribution of matriptase activity measured by B4 for each human cancer cell line appears to correlate with the distribution measured by the matriptase specific antibody and commercial substrate, suggesting that B4 cleavage was the result of cell associated matriptase activity.
Figure 3.
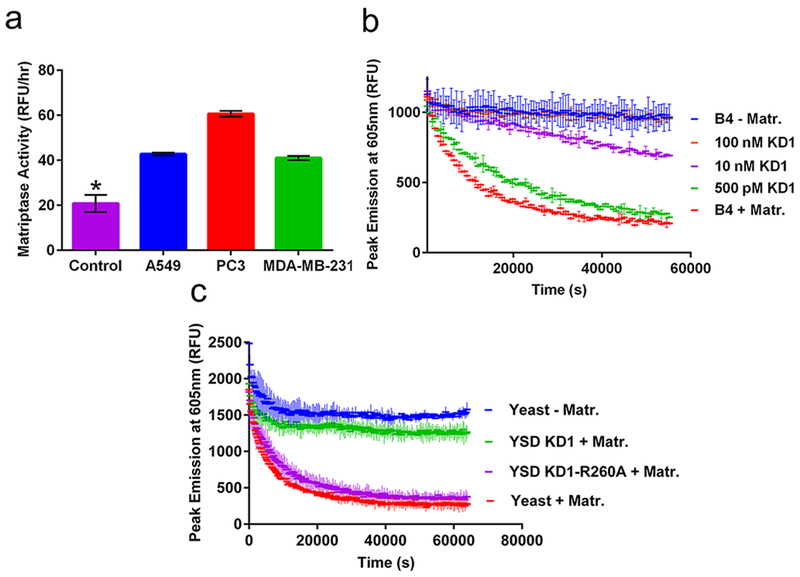
Application of B4 for measuring protease activity and inhibition. (a) B4 measurement of matriptase activity expressed on human A549 lung (blue), PC3 prostate (red), and MDA-MB-231 breast (green) cancer cell lines, compared with media alone (magenta); *p < 0.01 vs control. (b) B4 measurement of matriptase inhibition by soluble KD1 inhibitor. (c) B4 measurement of matriptase inhibition by yeast-displayed wild-type KD1 (green), KD1-R260A (magenta), or noninduced yeast controls with (red) or without (blue) matriptase.
To measure the inhibition of the matriptase-catalyzed cleavage of B4, we used an inhibitor domain derived from the matriptase hepatocyte growth factor activator inhibitor type-1 (HAI-1). HAI-1 naturally regulates matriptase activity in normal healthy tissue and is primarily expressed on the surface of epithelial cells to block the substrate-activating properties of matriptase. The multidomain structure of HAI-1 includes the extracellular Kunitz domain 1 (KD1; Supporting Information Figure 3a), which has been previously established to be the main inhibitory domain of HAI-1.30,31 To test inhibition of B4 activation, we first recombinantly expressed and purified KD1 as a soluble protein (Supporting Information Figure 3b). Following confirmation that soluble KD1 can inhibit matriptase using the Boc-QAR-AMC substrate as a positive control (Supporting Information Figure 3c), we then showed that KD1 inhibits matriptase cleavage of the B4 biosensor with a dose dependence on inhibitor concentration (Figure 3b). As expected, complete inhibition matched conditions without matriptase, with a 5-fold dynamic range for the assay.
We next showed that the B4 biosensor could detect matriptase inhibition by a yeast surface displayed KD1 inhibitor. Yeast surface display is an established protein engineering technology for characterizing and screening protein—based protease inhibitors32,33 However, yeast surface display inhibitor screens traditionally rely primarily on inhibitor—protease binding, rather than functional inhibition as a criterion for selection. By demonstrating that B4 can be used to detect matriptase inhibition by yeast displayed proteins, a future application could utilize B4 as a functional screening tool for identifying improved protease inhibitor candidates from a library of yeast displayed variants. KD1 was displayed on the surface of S. cerevisiae yeast cells as a fusion to the agglutinin mating protein Aga2p.33 We also displayed an inactive KD1 variant by introducing a point mutation at positon 260 (KD1-R260A) that disrupts matriptase binding (Supporting Information Figure 4a).31 This negative control protein will account for nonspecific interactions with the native yeast surface proteins. We showed that KD1 and KD1-R260A were both expressed on the surface of yeast, but only KD1 binds to matriptase, as expected (Supporting Information Figure 4b). We also confirmed that yeast-displayed KD1, but not KD1-R260A, inhibits activation of the commercial matriptase substrate (Boc-QAR-AMC; Supporting Information Figure 4c). Finally, using the B4 biosensor (Figure 3c) we observed distinct matriptase inhibition by yeast-displayed KD1 with a 5-fold dynamic range compared to KD1-R260A.
A protease biosensor created from dimerization-dependent fluorescent protein domains provides beneficial properties compared to small molecule-based strategies, including easy and low cost production, desirable spectral properties compatible with standard microscopes and flow cytometers, and robust activity in a range of assay conditions. In addition, the ddRFP system is modular and can potentially be adapted for measuring activity of alternative protease targets. Our efforts showed that the linker that joins the fluorescent protein domains was critical to its success as a biosensor component: a linker that is too short is not efficiently cleaved, and one that is too long exhibits nonspecific or promiscuous cleavage, or weak fluorescence. Biosensor 4, containing the linker sequence RSKLRVGGH, exhibited the highest matriptase selectivity, linker specific cleavage, stability, and follows ideal Michaelis—Menten kinetic behavior. Application of the B4 sensor enabled measurement of matriptase inhibition by both soluble and yeast-displayed inhibitor proteins, with up to a 5-fold dynamic range. Additionally, the B4 sensor was able to detect matriptase activity expressed by human cancer cell lines. Due to its ability to measure matriptase inhibition by soluble or cell surface proteins, the B4 biosensor can serve as a drug screening tool to identify matriptase inhibitors, as well as characterizing lead inhibitor candidates. In addition, detection of soluble matriptase and matriptase expressed on cancer cells also provides the opportunity to utilize B4 for diagnostic purposes.
METHODS
Media and Reagents.
YPD media: 20 g/L glucose, 20 g/L peptone, and 10 g/L yeast extract. SD-CAA media: 20 g/L dextrose, 6.7 g/L yeast nitrogen base lacking amino acids, 5.4 g/L Na2HPO4, 8.6 g/L NaH2PO4’H2O, and 5 g/L Bacto casamino acids. SG-CAA media: same as SD-CAA, but 20 g/L galactose is substituted for dextrose. BMMY, BMGY media, and RDB plates for P. pastoris strain GS115 were prepared exactly as described.34 Complete growth media: Dulbecco’s modified Eagle medium (Fisher Scientific), 10% fetal bovine serum (FBS; Fisher Scientific). Cell lines used include Human Embryonic Kidney (HEK) cells, PC3 (prostate cancer), MDA-MB-231 (breast cancer), and A549 (lung cancer; ATCC).
Biosensor Molecular Cloning.
The DNA sequence for each linker design was based upon the natural matriptase cleavage sequence found within the human pro-macrophage stimulating protein (Asp473 to Trp494; Genbank Accession ID: 4485). DNA encoding for each linker was genetically fused to the “A” domain using PCR and incorporated into the ddRFP construct with EcoRI and HindIII restriction enzyme sites into the pBAD vector.10 A C-terminal hexahistidine tag was included in each construct for downstream purification and Western blot detection.
Biosensor Production.
Following transformation into DH10B electrocompetent E. coli cells, each matriptase biosensor construct was expressed and purified. Briefly, transformed cells were expanded in Luria broth with 0.1 mg mL−1 ampicillin at 37 °C, and biosensor protein expression was induced by the addition of 0.2% (v/v) arabinose. Following overnight culture, each protein was extracted from E. coli inclusion bodies using Bacterial Protein Extraction Reagent (BPER-Thermo Fisher) and purified via Ni-NTA metal chelating chromatography. Absorbance values at 570 nm were measured to quantify concentration using a previously reported extinction coefficient (48 300 M−1 cm−1).10
Kinetic Protease Assays.
Matriptase Cleavage Screen.
First, 1 μM of each biosensor was added to 10 nM, 1 nM, or 0.1 nM recombinant human matriptase (R&D Systems) using manufacturer’s buffers in a 384-well plate at RT. Each biosensor without matriptase (buffer only) served as a negative background control. Fluorescent output (535 nm/605 nm) was monitored over time, and initial velocity values were quantified by first correcting for conditions without matriptase and then converting the initial reaction rate (RFU/h) to matriptase velocity (nM/s) using a determined standard curve.
Protease Selectivity Screen.
First, 1 μM of B4, B8, or B9 was separately added to 10 nM recombinant human trypsin 3, urokinase, kallikrein 4, and hepsin (all from R&D Systems) using manufacturer’s buffers in a 384-well plate at RT. Each biosensor without protease (buffer only) served as a negative background control. Fluorescent output measurement and initial velocity values were quantified for each condition as described above.
Michaelis—Menten Assay.
Varying concentrations of B4 (0 to 50 μM) were added to 10 nM recombinant human matriptase using manufacturer’s buffers in a 384-well plate at RT. B4 without matriptase (buffer only) served as a negative background control. Fluorescent output measurement and initial velocity values were quantified for each condition as described above. Final velocity values were plotted and fit to a Michaelis—Menten curve using Prism GraphPad software to quantify kinetic parameters, including: Km, Vmax, and kcat.
Western Blot Qualitative Cleavage Analysis.
First, 1 μM of each biosensor was added to 10 nM recombinant human matriptase and kinetically monitored for fluorescent output overnight as described above. Each biosensor without matriptase (buffer only) served as a noncleaved control. The reaction products, along with matriptase alone or buffer alone controls, were first denatured by boiling at 95 °C for 10 min, then loaded onto a 12% polyacrylamide gel (GenScript) and subjected to electrophoresis for protein fragment separation. Following SDS-PAGE, proteins were transferred to a nitrocellulose membrane, washed once with Milli-Q water, and blocked with 5% bovine serum albumin (BSA) in 1xTBST (1 mL Tween 20:1 L Tris-buffer saline) buffer for 1 h at RT. Membranes were then transferred to 1xTBST containing 5% dry milk (BioRad) and a (1:10 000) dilution of primary anti-His tag antibody (GenScript) for 1 h at RT. After three 5 min wash steps in 1xTBST, membranes were transferred to 1xTBST containing 5% dry milk and a (1:10 000) dilution of anti-rabbit horseradish peroxidase (HRP) secondary antibody (Fisher Scientific) for 1 h at RT. Membranes were again washed as above, and protein bands were then detected using the SuperSignal West Femto HRP substrate (Fisher Scientific).
Soluble Inhibitor Production.
DNA encoding the open reading frame of the human KD1 domain of the HAI-1 protein (amino acids Cys250 to Val303; Genbank Accession ID: AY358969.1) was cloned into the pPIC9 yeast expression plasmid, amplified using DH10B electrocompetent E. coli cells, and confirmed by sequencing. Methods of pPIC9 cloning, transformation into P. pastoris, and protein expression were completed using reagents, media, and protocols exactly as previously described.34 The KD1 protein was expressed in P. pastoris and purified by Ni-NTA metal chelating chromatography and size exclusion chromatography. Purified protein was characterized using polyacrylamide gel electrophoresis (PAGE), and concentration was measured by UV-vis absorbance (280 nm) and an extinction coefficient of 11 835 cm−1 M−1. Purified protein was stored in 1xPBS at 4 °C (short-term storage) or flash frozen with 0.01% Tween 80 at −80 °C (long-term storage).
Yeast Display of Matriptase Inhibitor.
The human KD1-R260A variant was created using standard overlap PCR assembly site-directed mutagenesis techniques. Human KD1 gene products were cloned into the pTMY yeast display vector as previously described34 using Nhel and MluI restriction sites. Plasmids were transformed into EBY100 yeast cells using electroporation, expanded in SD-CAA media at 30 °C, and then induced for surface protein display using SG-CAA media at 20 °C. Antibodies to measure yeast surface expression (1:250 dilution of anti-HA mouse primary antibody (Fisher Scientific) and a 1:100 dilution of anti-HAI-1 rabbit primary antibody (Fisher Scientific)) were added and incubated with yeast for 30 min at RT followed by washing cells with manufacturer’s matriptase assay buffers (R&D Systems). Primary antibody binding was then detected by incubating yeast with a 1:100 dilution of anti-mouse phycoerythrin (PE; Invitrogen) and a 1:100 dilution of anti-rabbit fluorescein isothiocyanate (FITC; Abcam) for 15 min at 4 °C. Yeast were then washed with assay buffer, and fluorescent antibody binding was measured using flow cytometry (BD Accuri) and the data analyzed (BD CSampler software).
Matriptase binding to yeast-displayed KD1 proteins was carried out by first combining 105 yeast (OD = 107 yeast/mL) with 10 nM human matriptase using manufacturer’s buffer conditions, followed by incubation for 24 h at RT. Samples were then washed and stained for yeast expression (as above) and matriptase binding (1:100 dilution of anti-His FITC (Bethyl)). Samples were washed and maintained on ice until loading onto a flow cytometer for analysis as described above.
Matriptase Inhibition Assay.
First, 10 nM matriptase was added to purified KD1 soluble inhibitor (final concentrations ranging 0–750 nM) in a matriptase assay buffer. Immediately, 1 μM purified B4 was added to initiate the reaction. Yeast surface display matriptase inhibition was carried out using the same matriptase and substrate conditions as above, except induced yeast displaying inhibitor domains were first counted (10–108 yeast cells/sample) and incubated with matriptase prior to the addition of B4. Kinetic monitoring of fluorescent output from B4 was performed using methods and conditions described above. Conditions of B4 with matriptase alone or with buffer alone, with or without noninduced yeast, served as control samples. Separately, the same experiment was performed using commercial matriptase substrate, Boc-QAR-AMC (R&D), and fluorescent output (380 nm/460 nm) was measured over time.
Cancer Cell Matriptase Expression.
A total of 5 × 105 cancer cells were resuspended in cold 1xPBS with 1 mg mL−1 bovine serum albumin (0.1% BPBS) solution, containing a 1:100 dilution of mouse anti-human matriptase antibody (Fisher Scientific). Cell solutions were incubated at 4 °C for 2 h, washed with cold 0.01% BPBS, and stained for antibody binding using a 1:100 dilution of anti-mouse phycoerythrin (PE; Invitrogen). Following 15 min of incubation at 4 °C, cells were washed and analyzed for binding using flow cytometry (BD CSampler software). Mean cell binding (Mean-RFU) was quantified from at least 10 000 cell events and subtracted from fluorescent background labeling with secondary antibodies alone.
Cancer Cell Matriptase Activity Assay.
A total of 1 × 105 cancer cells were seeded in 96-well plates in 2% fetal bovine serum-supplemented media. Plates were cultured for 24 h in a humidified tissue culture incubator at 37 °C in a 5% CO2 atmosphere. Following incubation, cells were washed twice with warm 1xPBS to remove residual serum, and serum free medium (DMEM alone) was added. Then, 1 μM purified B4 was immediately added to each condition and incubated at 37 °C in 5% CO2 for 1 h. B4 in serum free media alone served as a control condition. Plates were maintained at 37 °C in 5% CO2, and fluorescent output (535 nm/605 nm) was monitored every hour for at least 5 h. Rate of fluorescence change over time was quantified by fitting a linear curve using GraphPad software and averaging duplicate samples. Separately, the same experiment was performed using commercial matriptase substrate, Boc-QAR-AMC (R&D), and fluorescent output (380 nm/460 nm) was measured over time.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the Stanford Cancer Institute and the Stanford FACS facility for providing subsidized flow cytometry resources and expertise.
Funding
This work was funded by the National Institutes of Health (NIH) NIGMS Training Grant in Biotechnology (5T32GM008412) and NIH NCI R01 CA151706. A.C.M. is also grateful for support from a Stanford Agilent Fellowship, National Institutes of Standards and Technology (NIST) Fellowship, and Siebel Scholars Graduate Fellowship. S.A.H. kindly acknowledges support from a NSF Graduate Research Fellowship and a Stanford Graduate Fellowship.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.7b00715.
Kinetic characterization data of the biosensor 4 construct, matriptase expression and activity characterization on human cancer cell lines, production and inhibition data of the soluble KD1 monomer protein, and expression and inhibition data of the yeast displayed KD1 variants (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Liu Q, Wang J, and Boyd BJ (2015) Peptide-based biosensors. Talanta 136, 114–127. [DOI] [PubMed] [Google Scholar]
- (2).Tamura T, and Hamachi I (2014) Recent progress in design of protein-based fluorescent biosensors and their cellular applications. ACS Chem. Biol 9, 2708–2717. [DOI] [PubMed] [Google Scholar]
- (3).Turk B (2006) Targeting proteases: successes, failures and future prospects. Nat. Rev. Drug Discovery 5, 785–799. [DOI] [PubMed] [Google Scholar]
- (4).Zlobovskaya OA, Sergeeva TF, Shirmanova MV, Dudenkova VV, Sharonov GV, Zagaynova EV, and Lukyanov KA (2016) Genetically encoded far-red fluorescent sensors for caspase-3 activity. BioTechniques 60, 62–68. [DOI] [PubMed] [Google Scholar]
- (5).Grant SA, Weilbaecher C, and Lichlyter D (2007) Development of a protease biosensor utilizing silica nanobeads. Sens. Actuators, B 121, 482–489. [Google Scholar]
- (6).Hu K, Clément J-F, Abrahamyan L, Strebel K, Bouvier M, Kleiman L, and Mouland AJ (2005) A human immunodeficiency virus type 1 protease biosensor assay using bioluminescence resonance energy transfer. J. Virol. Methods 128, 93–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Zhou J, Wang D, Xi Y, Zhu X, Yang Y, Lv M, Luo C, Chen J, Ye X, Fang L, and Xiao S (2017) Assessing activity of Hepatitis A virus 3C protease using a cyclized luciferase-based biosensor. Biochem. Biophys. Res. Commun 488, 621–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Chen PT, Liao TY, Hu CJ, Wu ST, Wang SSS, and Chen RPY (2010) A highly sensitive peptide substrate for detecting two Aβ-degrading enzymes: Neprilysin and insulin-degrading enzyme. J. Neurosci. Methods 190, 57–62. [DOI] [PubMed] [Google Scholar]
- (9).Kwak SY., Yang JK, Jeon SJ., Kim HI., Yim J., Kang H., Kyeong S., Lee YS., and Kim JH. (2014) Luminescent graphene oxide with a peptide-quencher complex for optical detection of cell-secreted proteases by a turn-on response. Adv. Funct. Mater 24, 5119–5128. [Google Scholar]
- (10).Alford SC, Abdelfattah AS, Ding Y, and Campbell RE (2012) A fluorogenic red fluorescent protein heterodimer. Chem. Biol 19, 353–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Akers WJ, Xu B, Lee H, Sudlow GP, Fields GB, Achilefu S, and Edwards WB (2012) Detection of MMP-2 and MMP-9 activity in vivo with a triple-helical peptide optical probe. Bioconjugate Chem. 23, 656–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Wunder A, Tung C-H, Müller-Ladner U, Weissleder R, and Mahmood U (2004) In vivo imaging of protease activity in arthritis: A novel approach for monitoring treatment response. Arthritis Rheum. 50, 2459–2465. [DOI] [PubMed] [Google Scholar]
- (13).Uhland K (2006) Matriptase and its putative role in cancer. Cell. Mol. Life Sci 63, 2968–2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Lin CY, Tseng IC, Chou FP, Su SF, Chen YW, Johnson MD, and Dickson RB (2008) Zymogen activation, inhibition, and ectodomain shedding of matriptase. Front. Biosci., Landmark Ed. 13, 621–635. [DOI] [PubMed] [Google Scholar]
- (15).Lee SL, Dickson RB, and Lin CY (2000) Activation of hepatocyte growth factor and urokinase/plasminogen activator by matriptase, an epithelial membrane serine protease. J. Biol. Chem 275, 36720–36725. [DOI] [PubMed] [Google Scholar]
- (16).Bhatt AS, Welm A, Farady CJ, Vásquez M, Wilson K, and Craik CS (2007) Coordinate expression and functional profiling identify an extracellular proteolytic signaling pathway. Proc. Natl. Acad. Sci. U. S. A 104, 5771–5776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Ustach CV, Huang W, Conley-LaComb MK, Lin CY, Che M, Abrams J, and Kim HR (2010) A novel signaling axis of matriptase/PDGF-D/β-PDGFR in human prostate cancer. Cancer Res. 70, 9631–9640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Milner JM, Patel A, Davidson RK, Swingler TE, Desilets A, Young DA, Kelso EB, Donell ST, Cawston TE, Clark IM, Ferrell WR, Plevin R, Lockhart JC, Leduc R, and Rowan AD (2010) Matriptase is a novel initiator of cartilage matrix degradation in osteoarthritis. Arthritis Rheum. 62, 1955–1966. [DOI] [PubMed] [Google Scholar]
- (19).Beaulieu A, Gravel É, Cloutier A, Marois I, Colombo É, Désilets A, Verreault C, Leduc R, Marsault E, and Richter MV (2013) Matriptase Proteolytically Activates Influenza Virus and Promotes Multicycle Replication in the Human Airway Epithelium. J. Virol 87, 4237–4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Wood MP, Cole AL, Eade CR, Chen LM, Chai KX, and Cole AM (2014) The HIV-1 gp41 ectodomain is cleaved by matriptase to produce a chemotactic peptide that acts through FPR2. Immunology 142, 474–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Häußler D, Schulz-Fincke A-C, Beckmann A-M, Keils A, Gilberg E, Mangold M, Bajorath J, Stirnberg M, Steinmetzer T, and Gutschow M (2017) A Fluorescent-Labeled Phosphono Bisbenzguanidine As an Activity-Based Probe for Matriptase. Chem. - Eur. J 23, 5205–5209. [DOI] [PubMed] [Google Scholar]
- (22).Godiksen S, Soendergaard C, Friis S, Jensen JK, Bornholdt J, Sales KU, Huang M, Bugge TH, and Vogel LK (2013) Detection of Active Matriptase Using a Biotinylated Chloromethyl Ketone Peptide. PLoS One 8, 2–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Darragh MR, Schneider EL, Lou J, Phojanakong PJ, Farady CJ, Marks JD, Hann BC, and Craik CS (2010) Tumor detection by imaging proteolytic activity. Cancer Res. 70, 1505–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Li P, Jiang S, Lee SL, Lin CY, Johnson MD, Dickson RB, Michejda CJ, and Roller PP (2007) Design and synthesis of novel and potent inhibitors of the type II transmembrane serine protease, matriptase, based upon the sunflower trypsin inhibitor-1. J. Med. Chem 50, 5976–5983. [DOI] [PubMed] [Google Scholar]
- (25).Ganesan R, Kolumam GA, Lin SJ, Xie M-H, Santell L, Wu TD, Lazarus RA, Chaudhuri A, and Kirchhofer D (2011) Proteolytic Activation of Pro-Macrophage-Stimulating Protein by Hepsin. Mol. Cancer Res 9, 1175–1186. [DOI] [PubMed] [Google Scholar]
- (26).Gross LA, Baird GS, Hoffman RC, Baldridge KK, and Tsien R Y. (2000) The structure of the chromophore within DsRed, a red fluorescent protein from coral. Proc. Natl. Acad. Sci. U. S. A 97, 11990–11995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Rolland-Fourcade C, Denadai-Souza A, Cirillo C, Lopez C, Jaramillo JO, Desormeaux C, Cenac N, Motta J-P, Larauche M, Taché Y, Berghe P, Neunlist M, Coron E, Kirzin S, Portier G, Bonnet D, Alric L, Vanner S, Deraison C, and Vergnolle N (2017) Epithelial expression and function of trypsin-3 in irritable bowel syndrome. Gut 66, 1767–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Smith HW, and Marshall CJ (2010) Regulation of cell signalling by uPAR. Nat. Rev. Mol. Cell Biol 11, 23–36. [DOI] [PubMed] [Google Scholar]
- (29).Klokk TI, Kilander A, Xi Z, Whre H, Risberg B, Danielsen HE, and Saatcioglu F (2007) Kallikrein 4 is a proliferative factor that is overexpressed in prostate cancer. Cancer Res. 67, 5221–5230. [DOI] [PubMed] [Google Scholar]
- (30).Zhao B, Yuan C, Li R, Qu D, Huang M, and Ngo JCK (2013) Crystal structures of matriptase in complex with its inhibitor hepatocyte growth factor activator inhibitor-1. J. Biol. Chem 288, 11155–11164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Kojima K, Tsuzuki S, Fushiki T, and Inouye K (2008) Roles of functional and structural domains of hepatocyte growth factor activator inhibitor type 1 in the inhibition of matriptase. J. Biol. Chem 283, 2478–2487. [DOI] [PubMed] [Google Scholar]
- (32).Glotzbach B, Reinwarth M, Weber N, Fabritz S, Tomaszowski M, Fittler H, Christmann A, Avrutina O, and Kolmar H (2013) Combinatorial Optimization of Cystine-Knot Peptides towards High-Affinity Inhibitors of Human Matriptase-1. PLoS One 8, 18–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Boder ET, and Wittrup DK (1997) Yeast Surface Display for Screening Combinatorial Polypeptide Libraries. Nat. Biotechnol 15, 553–557. [DOI] [PubMed] [Google Scholar]
- (34).Jones DS, Tsai PC, and Cochran JR (2011) Engineering hepatocyte growth factor fragments with high stability and activity as Met receptor agonists and antagonists. Proc. Natl. Acad. Sci. U. S. A 108, 13035–13040. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.