

Abstract
Macroautophagy is a complex degradative intracellular process by which long-lived proteins and damaged organelles are cleared. Common methods for the analysis of autophagy are bulk measurements which mask organelle heterogeneity and complicate the analysis of interorganelle association and trafficking. Thus, methods for individual organelle quantification are needed to address these deficiencies. Current techniques for quantifying individual autophagy organelles are either low through-put or are dimensionally limited. We make use of the multiparametric capability of mass cytometry to investigate phenotypic heterogeneity in autophagy-related organelle types that have been isolated from murine brain, liver, and skeletal muscle. Detection and phenotypic classification of individual organelles were accomplished through the use of a lanthanide-chelating membrane stain and organelle-specific antibodies. Posthoc sample matrix background correction and nonspecific antibody binding corrections provide measures of interorganelle associations and heterogeneity. This is the first demonstration of multiparametric individual organelle analysis via mass cytometry. The method described here illustrates the potential for further investigation of the inherently complex interorganelle associations, trafficking, and heterogeneity present in most eukaryotic biological systems.
Graphical Abstract
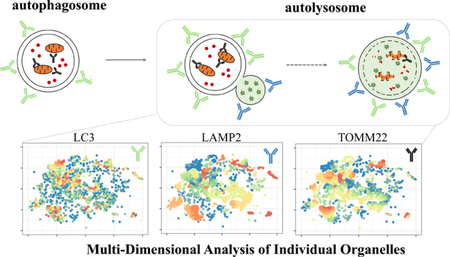
A growing body of evidence demonstrates that organelles of various types interact with each other to coordinate complex biological processes.1 For example, bulk intracellular recycling (macroautophagy) degrades long-lived proteins and damaged organelles releasing biomolecules that are used as biological building blocks. Macroautophagy is constitutively active and proceeds through at least four phenotypically unique organelle types whose interactions are dynamically coordinated to respond to intra-and extra-cellular stimuli. Dysfunctional macroautophagy has been implicated in age-related health decline, including neurodegenerative diseases, sarcopenia, and liver pathologies,2,3 where autophagy activity is generally decreased. Currently, the role of organelle heterogeneity in the development of these and other diseases hallmarked by dysfunctional organelle-related processes is unclear because bulk or whole-cell analyses (i.e., Western blots, flow cytometry) cannot distinguish different organelle types. Previously reported methods for individual organelle analysis include: transmission electron microscopy,4 capillary electrophoresis with laser-induced fluorescence detection,5,6 and flow cytometry.7,8 These techniques are either low-throughput, lack sensitivity, or are dimensionally limited.
Here, we report a multidimensional analysis of individual organelles detected via mass cytometry to reveal organelle population heterogeneity in macro-and mitophagy across multiple tissues. Mass cytometry has been used to conduct multidimensional analyses of individual cells,9 but has not been used for analyses of organelles. Mass cytometry relies on the use of antibodies, in which each unique antibody of the panel binds and identifies its specific molecular target. Each antibody is conjugated to a chelating polymer loaded with a different lanthanide isotope. Labeled particles are flowed through a nebulizer to produce single-particle droplets which are vaporized, atomized, and ionized via an inductively coupled plasma to produce ion clouds corresponding to individual particles. The temporally resolved ion clouds are analyzed via a time-of-flight mass spectrometer where the amount of lanthanide bound to each particle is quantified.10
Posthoc correction for sample matrix background signal and nonspecific antibody interactions were carried out on an individual organelle basis to reveal true signal for each reporter in each organelle. Through the analysis of filtered organelle suspensions, we confirm that mainly individual particles are detected rather than organelle aggregates, assessed by comparison of distribution of membrane stain intensity as a marker for aggregates. The corrected, individual organelle data are visualized using the Barnes-Hut implemented T-distributed stochastic neighbor embedding algorithm (t-SNE) to assess intrapopulation heterogeneity that is evident even within phenotypically similar organelle populations.
EXPERIMENTAL SECTION
Materials, Reagents, Buffers, and Solutions.
Sucrose, hydroxyethyl piperazineethanesulfonic acid (HEPES), mannitol, ethylenediaminetetraacetic acid (EDTA), ethylene-bis-(oxyethylenenitrilo) tetraacetic acid (EGTA), 3-(N-morpholino) propanesulfonic acid (MOPS), Tris(hydroxymethyl)-aminomethane (Tris), Tris(hydroxymethyl)aminomethane hydrochloride (Tris-HCl), potassium chloride, sodium chloride, potassium phosphate monobasic (KH2PO4), sodium azide, methanol-free formaldehyde (16%), nitric acid, hydrochloric acid, sodium hydroxide, diethylenetriaminepentaacetic acid (DTPA), dodecan-1-amine, dimethylformamide, chloroform (CHCl3), trifluoroacetic acid (TFA), and α-cyano-4-hydroxycinnamic acid (αCHCA) were obtained from Sigma-Aldrich (St. Louis, MO). Tris(2-carboxyethyl) phosphine hydrochloride (TCEP), and terbium(III) chloride hexahydrate (TbCl3·6H2O) were obtained from Thermo Scientific (Waltham, MA). Phosphate buffered saline (PBS, 10× concentration, 1.37 M NaCl, 27 mM KCl, 80 mM Na2HPO4, and 20 mM KH2PO4, pH 7.4), and Tween-20 was obtained from Bio-Rad (Hercules, CA). Percoll density gradient media was obtained from GE Healthcare Life Sciences (Marlborough, MA). Maxpar X8 Multimetal Labeling Kit, Maxpar Fix & Perm Buffer, Cell-ID Intercalator, and 10× EQ Four Element Calibration Beads were obtained from Fluidigm (San Francisco, CA). Water was purified with a Millipore Synergy UV system (18.2mΩ/cm, Bedford, MA). Polytetrafluoroethylene (PTFE) syringe filters (0. Two μm pore size) were obtained from Pall Industries (Port Washington, NY). Polycarbonate membranes were obtained from EMD Millipore (Burlington, MA). MSEGTA buffer consisted of 225 mM mannitol, 75 mM sucrose, 5 mM HEPES, 1 mM EGTA, pH 7.4. When necessary additives were included such as 3 μM bovine serum albumin (MSEGTA-BSA), or Percoll density gradient media (12% or 24% Percoll-MSEGTA). Liver isolation buffer (IBL) consisted of 13 mM Tris, 14 mM MOPS, 10 mM EGTA, 200 mM sucrose, pH 7.4. Muscle isolation buffer 1 (IBm1) consisted of 67 mM sucrose, 50 mM Tris-HCl, 50 mM KCl, 10 mM EDTA, 3 μM BSA, pH 7.4. Muscle isolation buffer 2 (IBm2) consisted of 250 mM sucrose, 2 mM EGTA, 15 mM Tris-HCl, pH 7.4. Cell staining media (CSM) consisted of 20 mM KH2PO4, 150 mM NaCl, 7.5 μM bovine serum albumin, 800 μM NaN3, pH 7.2.
Antibody Panel Design and Characterization.
Selected antibodies were metal-labeled using a Maxpar metal-labeling kit, per manufacturer instructions. Briefly, isotopically pure lanthanide (Ln) ions were loaded into linear Ln-chelating polymer (similar structure to those published by Lou11). IgG antibodies were reduced using TCEP, and then incubated with Ln-chelating polymers for 90 min. Conjugates were rinsed to remove unbound Ln-chelated polymer and resuspended at 0.5 mg/mL. The average number of lanthanide atoms per antibody was calculated by quantifying the number of lanthanide ions and antibody molecules per unit volume. The number of lanthanide ions were quantified by comparison to a standard solution of lanthanide ions of known concentration. Briefly, metal-labeled antibodies were diluted 1:1000 from their stock concentrations into 2% HCl, followed by two subsequent 1:1000 dilutions into 2% HNO3 for a total dilution factor of 1:109. Lanthanide standard solutions were prepared in 2% HNO3 to a final concentration of 50pM. Solutions were sequentially flowed into the CyTOF2 mass cytometer, and data were acquired for all metal channels simultaneously in “solution mode” using the following parameters: step value = 1, settling time = 20 s, pushes/reading = 76 800. The steady signal resulting from the lanthanide standard was used to calculate the lanthanide atom concentration in the diluted antibody sample (Table S-1 of the Supporting Information, SI).
Didodecyl-DTPA-Tb Synthesis and Metalation.
DDD-Tb was synthesized as reported by Leipold and co-workers, using 99.9% pure terbium chloride (aq) rather than the reported europium chloride (aq)12 The CyTOF2 at the University of Minnesota has higher transmission efficiency for 159Tb than any single isotope of Eu, resulting in improved sensitivity. Briefly, diethylenetriamine pentaacetic acid dianhydride (1.5 g, 4.2 mmol) was dissolved in DMF (75 mL) and maintained at 40 °C. Dodecan-1-amine (2.2 g, 11.7 mmol) was dissolved in CHCl3 (70 mL), added-dropwise to the reaction, and stirred overnight. The solid product was collected via filtration and resuspended in ultrapure H2O (100 mL) containing minimal NaOH (1 M, 10 mL) to solubilize the solid. The resulting solution was lyophilized, then resuspended in CHCl3 (70 mL) and filtered to remove excess NaOH. The solid was dried under reduced pressure to afford the trisodium didodecyl-DTPA salt (2.4 g, 301 μmol, 77% yield). Matrix-assisted laser desorption/ionization−mass spectrometry (MALDI-MS, positive reflectron mode, αCHCA matrix with 0.1% TFA) (m/z): [M + 3Na − H]+ calculated for C38H73N5O8Na3, 797.00; found 796.62. Electrospray ionization−mass spectrometry (ESI-MS, negative mode) (m/z): [M + H]− calculated for C38H70N5O8, 725.0; found 724.9. 1H NMR (400 MHz, D2O): δ 3.7−2.9 (m, br, 18 H, CH2), 1.22 (m, br, 44 H, CH2), 0.79 (t, 6 H, J = 5.6 Hz, CH3).
Didodecyl-DTPA-3Na ligand (200 mg, 252 μmol) was dissolved in 25 mM aqueous TbCl3 (141 mg, 378 μmol) and left at room temperature for 15 min. The solution was lyophilized and excess TbCl3 was removed using a CHCl3 extraction resulting in the metalated DDD-Tb ligand (200 mg, 227 μmol, 90% yield). DDD-Tb was dissolved to 0.2 mM in aqueous 50 mM NaCl and filtered through a 0.2 μm PTFE filter.
Organelle Isolation and Immuno-Labeling.
Female C57BL/6 mice were obtained from Jackson Laboratories or the National Institute on Aging. Mice were euthanized with sodium pentobarbital (200 mg/kg body mass, ip) followed by tissue dissection. All tissues were stored on ice or at 4 °C throughout organelle sample preparation unless otherwise noted. All protocols were approved by the Institutional Animal Care and Use Committee at the University of Minnesota (protocol # 1602–33497A).
Organelles were isolated from tissues of interest (brain, hind limb skeletal muscle, and liver) using procedures adapted from Chinopoulos or Frezza.13,14 Brain tissue was dissected and placed into MSEGTA-BSA buffer. The tissue was homogenized using a Potter-Elvehjem homogenizer (Wheaton, Millville, NJ) stroked ten times at 1600 rpm. The homogenate was transferred to clean microcentrifuge tubes and centrifuged for 5 min at 500g. The cloudy supernatant was transferred to clean microcentrifuge tubes as the postnuclear fraction and centrifuged at 14 000g for 10 min. The pelleted organelle fraction is resuspended in 12% Percoll–MSEGTA buffer, layered over 24% Percoll–MSEGTA, and centrifuged for 16 100g for 25 min. The resulting sample had two opaque layers sandwiching a clear band. The top two portions of the sample were removed, and the opaque bottom layer was washed twice with MSEGTA buffer producing a visible organelle pellet after the final wash.
Dissected liver was transferred to IBL. The tissue was rinsed with IBL until free of blood, then minced in a Petri dish with a flat razor blade. The minced tissue was homogenized using a Potter–Elvehjem homogenizer stroked four times at 1600 rpm. The homogenate was transferred to clean microcentrifuge tubes and centrifuged at 600g for 10 min. The cloudy supernatant was transferred to clean microcentrifuge tubes as the postnuclear fraction and centrifuged at 10 000g for 10 min. The resulting organelle pellet was washed once with IBL producing a final organelle pellet.
Dissected hind limb skeletal muscle was transferred to PBS containing 10 mM EDTA. Visible tendons, connective tissue and fat were trimmed, and tissue was minced using a flat razor blade on a Petri dish, then rinsed with PBS containing 10 mM EDTA. Minced muscle tissue was enzymatically digested (0.05% trypsin, 10 mM EDTA) for 30–45 min at 37 °C. Following digestion, tissue was centrifuged at 200g for 5 min and resuspended in IBm1. The tissue was homogenized using a Dounce homogenizer. Each sample was stroked 10 times with pestle A (clearance: 0.114 ± 0.025 mm), followed by 10 strokes with pestle B (clearance: 0.05 ± 0.025 mm). The homogenate was transferred to microcentrifuge tubes and centrifuged at 600g for 10 min. The resulting cloudy supernatant was transferred to clean microcentrifuge tubes as the postnuclear fraction and centrifuged at 10 000g for 10 min. The resulting organelle pellet was resuspended in IBm2, and centrifuged at 10 000g for 10 min, producing a final organelle pellet.
The organelle pellet for each tissue was resuspended in IBL to obtain samples of similar visual turbidity. Each sample was washed once with IBL, then stained with antibodies in CSM for 1 h at room temperature. Organelles were washed twice with CSM and resuspended in 4% formaldehyde for 30 min at room temperature. Fixed organelles were washed twice with PBS, then resuspended in 0.2 mg/mL DDD-Tb in 0.09% NaCl for 30 min at room temperature. Organelles were washed twice with PBS supplemented with 0.05% Tween-20 and resuspended in Maxpar Fix & Perm Buffer containing 63 nM Cell-ID Intercalator-Ir for 24 h. Prior to data acquisition, organelles were washed twice with CSM, and twice with ultrapure water.
Mass Cytometry Data Acquisition and Preprocessing.
Data was acquired on a CyTOF2 mass cytometer (Fluidigm, San Francisco, CA). Samples were diluted 1:106 in a solution of 1× EQ Four Element Calibration Beads in ultrapure water to attain an event detection rate of approximately 300 events/second. Organelle event data were acquired using the following instrument parameters: noise reduction, cell length = 10–150, lower convolution threshold = 200. To define a blank for each sample, the 1:106 diluted organelle suspension was filtered through a 0.2 μm polycarbonate filter to remove intact organelles. The resulting filtrate was analyzed in solution mode using the following parameters: step value = 1, settling time = 20 s, pushes/reading = 50. Intra-assay precision was assessed via technical replicates and showed detected event signal intensity was relatively stable in each monitored mass channel over 13 hours of data acquisition (Figure S-1).
Mass Cytometry Data Analysis.
Threshold values for the signal above sample matrix noise for each monitored mass channel were calculated for each sample as the mean signal in a given channel plus three times the standard deviation. Threshold values were used as the lower bound of gates when identifying phenotypically distinct organelle populations (Figure S-2, red bars).
Flow cytometry standard (.fcs) files were bead normalized (Helios software, version 6.7, Fluidigm), and then converted to plain text (.txt) files for further analysis in R. Using R, binary tags were added to each detected event with dual count (DC) signal intensity equal to or above the lower signal threshold for the organelle identification marker (i.e., DDD-Tb) and any combination of organelle phenotypic markers (i.e., anti-TOMM22-154Sm, anti-LAMP2-162Dy, anti-LC3-142Nd). Each data file was then subsetted using the binary tags to include only phenotypically identified organelles.
Quantification of the number of antibody tags per organelle was performed via a comparison of per organelle signal at each mass channel (i.e., particle data acquisition) to the transmission efficiency of each mass channel (i.e., solution data acquisition). Similar comparisons between particle and solution data has been used to quantify analytes in individual organelles via capillary electrophoresis with laser-induced fluorescence.15 The number of ions associated with each organelle event (# ionsAb) was calculated using the equation below, where DCAb represents the dual count signal of the antibody chelating a unique lanthanide isotope, defined as m. TEm is the empirically determined transmission efficiency of unique lanthanide isotope m for the CyTOF2 at the University of Minnesota.
| (1) |
Then, the number of each unique antibody present per organelle (#Ab per organelle) was calculated using a correction factor via the following equation.
| (2) |
The correction factor describes the number of lanthanide ions chelated to each antibody (listed in Table S-1). This calculation was carried out for all antibodies, regardless of specificity.
Organelle suspensions were stained with host-matched, isotype control antibodies, each chelating a unique lanthanide isotope, to estimate the nonspecific binding properties of each specific marker antibody. The use of isotype control antibodies to assess nonspecific binding is often debated in the literature16,17 but is the only available method for simultaneous detection of specific and nonspecific antibody binding via mass cytometry, as other controls such as preincubation of clone-matched unlabeled antibodies would provide a population level assessment rather than at the individual particle level. To obtain specific marker signal (corrected # specific Ab), the number of isotype antibodies per organelle (# isotype Ab) was subtracted from the number of specific antibodies per organelle (# specific Ab), using the following expression.
| (3) |
Where appropriate, the corrected # of specific antibodies was normalized to the number of DDD-Tb molecules present in each organelle (corrected # specific Ab per organelle). This was done using the following equation, where DCDDD‑Tb is the dual count signal measured for the DDD-Tb mass channel and TE159Tb is the empirically derived transmission efficiency specific to the 159Tb mass channel.
| (4) |
t-SNE analysis was done using the Rtsne package in the R statistical programming environment. The following parameters were used: perplexity = 20, θ = 0.5, maximum iterations = 5000, and η = 10, resulting in a set of two-dimensional coordinates for each organelle used to plot clusters of individual organelles in two-dimensional space. Distributions for each relevant marker in a phenotypically identified population of organelles were binned into deciles. Coloring of the individual organelles in each t-SNE plot indicate the decile in which a given organelle falls.
RESULTS AND DISCUSSION
Individual Organelle Detection via Didodecyl-DTPA-Tb Ligand.
Mass cytometry analysis of whole cells requires a diagnostic signal that distinguishes intact particles from debris and triggers mass spectra integration. In the case of whole cells, this is accomplished through the use of pentamethylcyclopentadienyl-Ir(III)-dipyridophenazine, a nucleic acid stain.18 Because not all organelles contain nucleic acids, we sought to find a reagent to label lipid bilayers to identify organelles. The generic identification of organelles by mass cytometry makes use of a didodecyl-diethylenetriaminepentaacetic acid (DTPA) ligand chelating 159Tb3+ (DDD-Tb, Figure 1A). The long alkyl chains of DDD-Tb are incorporated into lipid bilayers,19 making it an ideal reagent to label organelle membranes for mass cytometry analysis. A variation of the reagent was first described for use in mass cytometry by Leipold and co-workers who used it in the analysis of individual bacteria.12 Incorporation of DDD-Tb into lipid bilayers was stable as noted by the lack of background drift of 159Tb signal as a function of time (data not shown).
Figure 1.
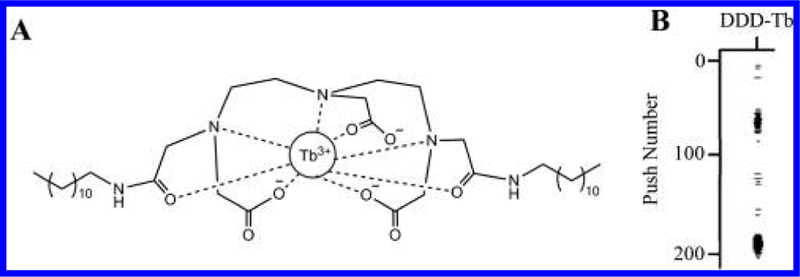
Structure and function of DDD-Tb ligand. (A) Structure of DDD-Tb, a general membrane stain used in this study to identify organelles. (B) Rain plot showing low background between two detected organelle events, n = 29 858 organelle events. The y-axis identifies the spectra (in sequence) over which detected organelle event signals are integrated. The x-axis shows the relevant m/z channel, 159Tb. Dark lines in the plot area indicate detected 159Tb3+ ions.
To define signals of organelle events, background signal was obtained by mass spectrometric analysis of filtrates resulting from filtering organelle suspensions through a 0.2-μm polycarbonate membrane (Figure S-3). The diameters of lysosomes, autophagosomes, or mitochondria vary from 0.15 to 5 μm.20 Thus, the filtrate of a 0.2-μm polycarbonate filter is devoid of most organelles and contains unbound reporters and debris. The background signal of the filtrate plus three times the standard deviation was used as an organelle event identification threshold (Table S-2). We also sought to understand how organelle aggregates, detected as single events, would affect the results. Mass spectrometric analysis of filtrates of an organelle suspension filtered through 8.0-or 1.2-μm polycarbonate membranes, did not affect the statistical modes of the DDD-Tb signal distributions (Figure 2A). Also, a QQ-plot comparison of the 8.0-μm filtered and unfiltered distribution show almost no variation (Figure 2B), indicating that detected organelle events in the unfiltered sample are <8.0-μm, and mostly represent the detection of individual organelles. Autophagy-related organelles regularly fuse during the autophagy process, thus absolute autophagy-related organelle sizes will vary and likely be larger than that of the individual organelle alone. The comparison of 0.2-and 1.2-μm filtrate distributions to either the 8.0-μm filtered or unfiltered show that distributions show a similar trend of variation. A 0.2-μm pore size was used in subsequent experiments as the more stringent filtration cutoff for assessing signal from the matrix background.
Figure 2.
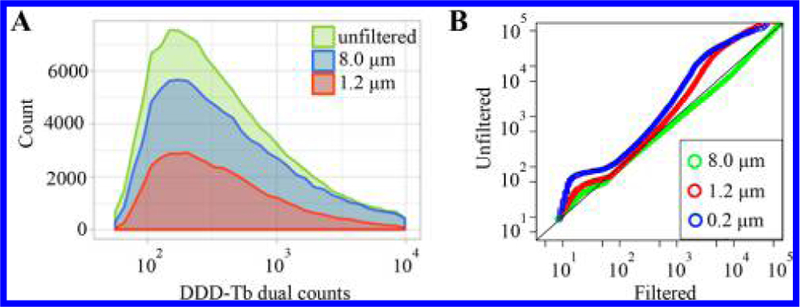
Individual organelle detection via mass cytometry. (A) Distributions of organelle frequency vs DDD-Tb dual count (DC) signal obtained after filtration. The similarity in the mode of DC signal (unfiltered: 294 ± 2.5 dual counts, 8.0 μm: 323 ± 4 dual counts, 1.2 μm: 287 ± 4 dual counts), indicates that detected events represent are likely individual organelles rather than aggregates. (B) QQ-plot comparing the DDD-Tb distributions of filtered or unfiltered samples. The adherence of the 8.0-μm trace to a y = x line suggests the majority of detected events are individual organelles.
Phenotypic Identification of Organelle types via Antibodies in Multiple Tissues.
Autophagy progression involves various organelle types. Here, these organelle types were identified by the unique pattern of antibodies bound to each organelle. Notably, autophagosomes and phagophores were detected through the binding of anti-LC3 antibodies and lysosomes were detected through the binding of anti-LAMP2 antibodies (Figure 3). These organelle types made up 6% and 46% of the total number of phenotyped organelle events from murine brain (Figure 3A). That is, their relative number abundance was ~1:8. The only surrogate for comparisons of relative abundance of organelles found in the literature were cross sections in TEM images of murine brain, which showed autophagosomal vacuoles and lysosomes, occupying 0.8 and 3.3% of the cytoplasmic cross-sectional area, respectively (relative abundance, ~ 1:4).21,22 While the relative abundance in this source and our data show the same trend, the remarkable feature of the mass cytometric results is the ability to deepen the classification of organelles detected, including autolysosomes (6%) and autolysosomes with mitochondrial association (4%), (Figure 3A). Association between autolysosomes with their cargo (e.g., mitochondria) is critical to assess selective forms of autophagy (e.g., mitophagy) and to uncover their roles in biological systems.
Figure 3.

Characterization of phenotypically unique organelle populations. (A) Phenotypically identified organelles are assigned to the unique organelles which make up the autophagy process. This identification suggests the percentage of each organelle type participating in either macroautophagy or mitophagy in brain tissue. See Figure S-4 for corresponding analysis of liver and skeletal muscle tissue. (B–D) Comparison of each organelle marker across phenotypically unique organelle populations from the same sample and across biological replicates. The number of specific antibodies varies across tissue but is consistent across replicates. Error represented as 95% confidence intervals of the median. B = brain, L = liver, M = skeletal muscle, R1 = biological replicate 1, and R2 = biological replicate 2.
Mass cytometry measurements allow for the calculation of the number of antibodies of each type bound to each organelle (eqs 1 and 2, Figure 3B-D). Taking the number of DDD-Tb molecules per organelle as an indicator of relative total membrane volume, the number of antibodies of each type per organelle, normalized by membrane volume, provides additional insight into autophagy progression. Whether monitoring the TOMM22 marker (mitochondria → mitochondria associated with the autophagosome → mitochondria associated with the autolysosome, Figure 3B), LAMP2 marker (lysosome → autolysosome, Figure 3C), or LC3 marker (autophagosome, phagophore → autolysosome, Figure 3D), the relative abundance of each marker tends to decrease with autophagy progression, supporting a general increase of membrane content with a consistent number of markers per organelle. This decrease displays different rates for brain (B), liver (L), and muscle (M), which may indicate custom responses to tissue-specific, autophagy-relevant stimuli such as nutrient availability. For LAMP2+ organelles (lysosomes, Figure 3B), the molar ratio of anti-LAMP2 antibody: DDD-Tb molecule per event is lower for LC3+-LAMP2+ organelles representing autolysosomes (autolyso) and autolyso-mito events relative to lysosomes. This trend is less pronounced in organelles from liver. For TOMM22+ organelles (mitochondria, Figure 3C), the molar ratio of anti-TOMM22 antibody: DDD-Tb molecule per event was lower for dual-positive LC3+-TOMM22+ organelles representing autophagosomes associated with mitochondria (automito), and even lower for triple positive LC3+-LAMP2+-TOMM22+ organelles representing autolysosomes associated with mitochondria (autolyso-mito). This may be the result of an increase of lipid-rich membrane stained with DDD-Tb as multiple organelles associate, but the unchanging abundance of TOMM22 proteins per organelle. For LC3+ organelles (phagophores, autophagosomes, and autolysosomes, Figure 3D), the molar ratio of LC3 tag: DDD-Tb molecule is higher for autophagy organelles prior to the association with lysosomes (autolyso), or mitochondria (automito and autolyso-mito). It is worth noting that the molar ratio of anti-LC3 antibody: DDD-Tb is higher for automito suggesting that a more extensive LC3-decorated membrane is required for processing mitochondria (mitophagy) than for other forms of autophagy. Thus, comparison of the molar ratios of specific antibody markers: DDD-Tb molecules between organelle types is a quantitative measure of autophagy progression in each tissue type (see Figure S-5 for non-normalized antibody count per organelle).
Multiplexed Analysis of Organelle Interactions.
The true power of multiparameter individual organelle analysis lies in the ability to detect heterogeneity in phenotypically identified organelle populations. Because organelles often traverse entire biochemical pathways, organelle heterogeneity may influence individual cell heterogeneity through significant changes in function. For example, heterogeneous changes in intracellular mitochondrial membrane potential have been associated with dysregulated gene expression of MFN1, MFN2, and OPA1 which normally facilitate mitochondrial fusion events. The dysregulation of these genes has been associated with a number of neurodegenerative diseases.23–25 In order to assess interpopulation organelle heterogeneity, organelles were clustered using the t-SNE algorithm,26 such that organelles containing similar normalized levels of each antibody type, were close to one another in high-dimensional space. On the basis of relative cluster position, two-dimensional coordinates are assigned to each organelle allowing for the two-dimensional projection of multidimensional data (Figure 4). Each column represents the two-dimensional projection of a given marker, while each row represents different combinations of organelle specific markers. Panels for triple positive organelles (Figure 4, first row) represent autolysosomes associated with mitochondria, likely engaged in mitophagy. The second row of panels represent autophagosomes associated with mitochondria, while the third row of panels represent autolysosomes sans mitochondria. The large variation in color (heat map) of each panel is indicative of interpopulation heterogeneity. As expected, it is evident that even phenotypically similar organelles are not identical. Subpopulations of autolysosomes (Figure 4, third row) show fairly low heterogeneity as the decile classification for each marker is relatively similar (orange dashed ovals). This is not necessarily the case for the autophagosomes associated with mitochondria (Figure 4, second row). The circled cluster (blue dashed circles) indicates a relatively high abundance of the autophagosome marker LC3, but moderate abundance of TOMM22, a marker for mitochondria. The relative size of this cluster is very heterogeneous as indicated by the wide color variation in DDD-Tb signal. Taken together, we could speculate that this particular cluster is a collection of autophagosomes at distinct points of closure around mitochondria. This same reasoning could be employed in the analysis of autophagosomes anticipating that they are engaged in mitophagy (Figure 4, first row), in that the large variation in the circled subpopulation (black dashed circles) could represent different stages of degradation of mitochondria and release of LC3.
Figure 4.

Two-dimensional cluster analysis of heterogenous autophagy organelle subpopulations. t-SNE plots from brain tissue show phenotypically identified populations of organelles. Distributions of specific markers on individual organelles were binned into deciles and color-coded according to their assigned decile. Wide variation in color across specific markers in t-SNE plots indicate heterogeneous subpopulations which may have significantly different functions. See Figures S-6 and S-7 for corresponding analysis of liver and skeletal muscle tissue, respectively.
CONCLUSIONS
Further analysis of these heterogeneous organelle populations will be possible using an expanded antibody panel to include other markers of macroautophagy and mitophagy, such as the ubiquitous nucleoporin p62, PTEN-induced putative kinase 1, and Parkin targets will enhance our understanding of both macro-and mitophagy. This type of high-dimensional individual organelle analysis could illuminate functional, morphological, or phenotypic variation that drive disease development. This analysis used 6 of nearly 50 available mass channels, leaving room for many additional markers. We expect this multiparameter technique to expand our understanding of subcellular organelle variation in the progression of disease across many tissue and organelle types.
Supplementary Material
ACKNOWLEDGMENTS
We thank Nick Livezey for assistance metallating the didodecyl-DTPA reagent and interpretation of the 1H NMR of the nonmetalated ligand. H.M.G.B. and E.A.A. thank the National Institutes of Health (Grants RO1-AG020866 and T32-GM008347).
Footnotes
Notes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.8b02790.
Table S-1, Metal-labeled antibody panel characterization; Figure S-1, intra-assay precision assessed via technical replicates; Figure S-2, organelle event gating strategy; Figure S-3, low signal threshold calculations; Table S-2, calculated low signal threshold values; Figure S-4, characterization of the abundance of phenotypically unique organelle populations; Figure S-5, comparison of the number of specific marker antibodies bound to each organelle type; Figure S-6, multi-dimensional t-SNE analysis of organelle from liver tissue; and Figure S-7, multi-dimensional t-SNE analysis of organelles from skeletal muscle tissue (PDF)
REFERENCES
- (1).Valm AM; Cohen S; Legant WR; Melunis J; Hershberg U; Wait E; Cohen AR; Davidson MW; Betzig E; Lippincott-Schwartz J Nature 2017, 546 (7656), 162–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Rubinsztein DC; Marino G; Kroemer G Cell 2011, 146 (5), 682–95. [DOI] [PubMed] [Google Scholar]
- (3).Cuervo AM; Bergamini E; Brunk UT; Dröge W; Ffrench M; Terman A Autophagy 2005, 1 (3), 131–140. [DOI] [PubMed] [Google Scholar]
- (4).Seglen PO; Eskelinen E-L; Reggiori F; Baba M; Kovacs AL; et al. Autophagy 2011, 7 (9), 935–956. [DOI] [PubMed] [Google Scholar]
- (5).Satori CP; Arriaga EA Anal. Chem 2013, 85 (23), 11391–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Muratore KA; Grundhofer HM; Arriaga EA Anal. Chem 2016, 88 (23), 11691–11698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Rajotte D; Stearns CD; Kabcenell AK Cytometry 2003, 55A (2), 94–101. [DOI] [PubMed] [Google Scholar]
- (8).Daniele JR; Heydari K; Arriaga EA; Dillin A Anal. Chem 2016, 88 (12), 6309–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Bendall SC; Simonds EF; Qiu P; Amir AD; Krutzik PO; Finck R; Bruggner RV; Melamed R; Trejo A; Ornatsky OI; Balderas RS; Plevritis SK; Sachs K; Pe’er D; Tanner SD; Nolan GP Science 2011, 332 (6030), 687–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Bandura D; Baranov V; Ornatsky O; Antonov A; Kinach R; Lou X; Pavlov S; Dick J; Tanner S; Vorobiev S Anal. Chem 2009, 81 (16), 6813–6822. [DOI] [PubMed] [Google Scholar]
- (11).Lou X; Herrera I; Zhang G; Kinach R; Ornatsky O; Baranov V; Nitz M; Winnik MA Angew. Chem., Int. Ed 2007, 46 (32), 6111–6114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Leipold MD; Ornatsky O; Baranov V; Whitfield C; Nitz M Anal. Biochem 2011, 419 (1), 1–8. [DOI] [PubMed] [Google Scholar]
- (13).Chinopoulos C; Zhang SF; Thomas B; Ten V; Starkov AA Methods Mol. Biol 2011, 793, 311–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Frezza C; Cipolat S; Scorrano L Nat. Protoc 2007, 2 (2), 287–95. [DOI] [PubMed] [Google Scholar]
- (15).Anderson AB; Xiong G; Arriaga EA J. Am. Chem. Soc 2004, 126 (30), 9168–9169. [DOI] [PubMed] [Google Scholar]
- (16).O’Gorman MRG; Thomas J Cytometry 1999, 38 (2), 78–80. [PubMed] [Google Scholar]
- (17).Keeney M; Gratama JW; Chin-Yee IH; Sutherland DR Cytometry 1998, 34 (6), 280–283. [PubMed] [Google Scholar]
- (18).Behbehani GK; Bendall SC; Clutter MR; Fantl WJ; Nolan GP Cytometry, Part A 2012, 81A (7), 552–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Kielar F; Tei L; Terreno E; Botta MJ Am. Chem. Soc 2010, 132 (23), 7836–7837. [DOI] [PubMed] [Google Scholar]
- (20).Satori CP; Henderson MM; Krautkramer EA; Kostal V; Distefano MD; Arriaga EA Chem. Rev 2013, 113 (4), 2733–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Ju WK; Kim KY; Noh YH; Hoshijima M; Lukas TJ; Ellisman MH; Weinreb RN; Perkins GA Glia 2015, 63 (5), 736–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Koike M; Shibata M; Waguri S; Yoshimura K; Tanida I; Kominami E; Gotow T; Peters C; von Figura K; Mizushima N; Saftig P; Uchiyama Y Am. J. Pathol 2005, 167 (6), 1713–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Delettre C; Lenaers G; Griffoin J-M; Gigarel N; Lorenzo C; Belenguer P; Pelloquin L; Grosgeorge J; Turc-Carel C; Perret E; Astarie-Dequeker C; Lasquellec L; Arnaud B; Ducommun B; Kaplan J; Hamel CP Nat. Genet 2000, 26, 207. [DOI] [PubMed] [Google Scholar]
- (24).Chen H; Chomyn A; Chan DC J. Biol. Chem 2005, 280 (28), 26185–92. [DOI] [PubMed] [Google Scholar]
- (25).Zuchner S; Mersiyanova IV; Muglia M; Bissar-Tadmouri N; Rochelle J; Dadali EL; Zappia M; Nelis E; Patitucci A; Senderek J; Parman Y; Evgrafov O; Jonghe PD; Takahashi Y; Tsuji S; Pericak-Vance MA; Quattrone A; Battologlu E; Polyakov AV; Timmerman V; Schroder JM; Vance JM Nat. Genet 2004, 36 (5), 449–451. [DOI] [PubMed] [Google Scholar]
- (26).van der Matten LH; Geoffrey. Journal of Machine Learning Research 2008, 9, 2579–2605. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.