

Abstract
Deep-sea hydrothermal vents are a significant source of dissolved metals to the global oceans, producing midwater plumes enriched in metals that are transported thousands of kilometers from the vent source. Particle precipitation upon emission of hydrothermal fluids controls metal speciation and the magnitude of metal export. Here, we document metal sulfide particles, including pyrite nanoparticles, within the first meter of buoyant plumes from three high-temperature vents at the East Pacific Rise. We observe a zone of particle settling 10–20 cm from the orifice, indicated by stable sulfur isotopes; however, we also demonstrate that nanoparticulate pyrite (FeS2) is not removed from the plume and can account for over half of the filtered Fe (≤0.2 µm) up to one meter from the vent orifice. The persistence of nanoparticulate pyrite demonstrates that it is an important mechanism for near-vent Fe stabilisation and highlights the potential role of nanoparticles in element transport.
There has been much interest recently in the transport mechanisms of metals from hydrothermal vents. Here the authors found that nanoparticulate pyrite is not removed from the plume and can account for over 50% of filtered iron one metre from the vent mouth.
Introduction
The fluids emitted from most deep-sea hydrothermal black smoker vents are enriched in iron, other metals and sulfide up to several orders of magnitude over ambient seawater. Many of these elements are biogeochemically relevant; for example Fe sourced from vents is a globally important component of the oceanic Fe budget1,2, and thus may ultimately affect primary production in the surface ocean3,4. Quantifying the significance of element transport from hydrothermal vents is therefore of broad oceanographic importance.
Determining the potential for element transport away from the vent source is complicated because the vent fluid undergoes a wide spectrum of biogeochemical changes between emission of the fluid and the formation of midwater plumes. In particular, mixing of the reduced, hot and acidic vent fluid with cold, oxygenated seawater leads to the precipitation of many metals in the area surrounding the vents as sulfide or oxide minerals5,6. The formation, aggregation, oxidation and/or stabilisation of particles and nanoparticles in emitted vent fluid directly determines the potential for element transport to the broader oceans. Moreover, although the chemistry of end-member vent fluid and nascent buoyant plumes is highly variable between vents7–9, the chemistry of the non-buoyant plume appears to be consistent, regardless of the initial fluid composition10. This consistency indicates that it is the physical and (bio)geochemical processes in the initial stages of the buoyant plume that exert the primary control on which elements are exported from vents, and that these processes are similar between different black smoker vent fields. In particular, the speciation and solubility of reduced Fe and S are extremely sensitive to oxidation along the steep redox gradients found at the vent orifice, which may result in their loss from the rising fluid. The dynamics of reduced Fe and S are important as these elements dominate fluid chemistry in many systems and therefore affect the speciation of other elements, such as Pb, Co, Cd, Cu and Zn9,11.
Nevertheless, recent work has demonstrated that Fe may be stabilised as nanoparticulate pyrite in the hydrothermal fluid12,13 and by organic ligands within the colder buoyant and non-buoyant plume14–16 and that this Fe is transported hundreds to thousands of kilometers from the local vent area17–20. These findings raise important questions regarding the processes affecting particle formation and transport in the initial mixing zone, including the extent of metal sulfide precipitation and settling. However, systematic studies of particle formation and Fe dynamics within this initial mixing zone are lacking.
In order to elucidate these processes and investigate the potential for Fe transport from the vent sites, we determined the speciation of Fe and S and quantified the abundance of nanoparticulate pyrite within the first meter of buoyant plumes from three hydrothermal vents with high sulphide to iron ratios (ranging from 4 to 10) at the fast-spreading East Pacific Rise (EPR, 9°50′N); P Vent, Bio 9 Vent and Bio Vent. We show that despite a decrease in total Fe and the formation and entrainment of sulfide particles within the plume, nanoparticulate pyrite can comprise up to 60 % of filtered Fe (≤0.2 µm) one meter from the vent orifice, even though it is not detectable at statistically significant concentrations at the point of emission by chemical leaching methods. This, combined with evidence for settling of sulfide particles in the plume from stable sulfur isotope measurements, indicates that physical and geochemical controls on sulfide particle formation and settling have broad implications for the stabilisation of Fe in nanoparticulate pyrite and for metal transport from hydrothermal vents.
Results and Discussion
Fe and S dynamics in the first meter of the buoyant plume
The concentration and speciation of iron and sulphur were measured in samples from three different black smoker vents up to one meter from the vent orifice (Supplementary Figure 1). Within this distance, Fe was predominately (≥80 %) in the ≤0.2 µm fraction (Supplementary Table 1). Despite this, total unfiltered Fe was exponentially removed at P Vent and Bio 9 Vent, particularly in the first 10 centimeters of the buoyant plume, indicating the formation and subsequent rapid settling of larger particles. At Bio Vent, initial Fe concentrations were lower and there was much less Fe removal within the initial stages of the plume (Fig. 1a, Supplementary Table 1).
Fig. 1.

Plume iron and sulphur geochemistry. a Total unfiltered Fe(II) concentrations in all three plumes and b Total unfiltered AVS concentrations in all three plumes
Total unfiltered Acid Volatile Sulfide (AVS), which comprises free sulfide and less recalcitrant metal sulfides (e.g. FeS, ZnS), demonstrates behaviour consistent with removal within the first 10 centimeters at all three vents (Fig. 1b) and AVS can account for the observed Fe removal. Concentrations of total Chromium Reducible Sulphur (CRS), consisting of more recalcitrant sulfur minerals21 (e.g. FeS2, FeCuS2 and partially S0), are up to an order of magnitude lower than those of AVS and consist of 1–13 % of the total sulphide (Fig. 1b, Supplementary Table 1).
Within the initial first meter of mixing, total unfiltered Fe remains almost exclusively as Fe(II) (Supplementary Table 1). The predominance of Fe(II) is consistent with previous studies which showed that Fe remains reduced at least up to 1.5 m from the vent orifice at the Mid-Atlantic Ridge9,13. Although oxygenated ocean water mixes into the buoyant plume within the first meter with seawater (Table 1), the excess sulphide with respect to Fe present in these fluids keeps Fe reduced due to a catalytic cycle (Eq. 1a, b).
| 1a |
| 1b |
Table 1.
Nanoparticulate pyrite and elemental sulfur concentrations in each sample
| Vent | T | pH | [Mg] (mM) | Distance from orifice (cm) | [Nano py] (µM) | % Fenpy/ Fetotal ≤0.2 µm | [S0]<0.2 (µM) | [S0]>0.2 (µM) | [S0] total (µM) |
|---|---|---|---|---|---|---|---|---|---|
| Bio 9 | 373 | 3.13 | 3.40 | 0 | BDL | — | 30 | 2.3 | 32 |
| 150 | 4.87 | 43.7 | 10 | 43 | 20 | 0.99 | n/a | 0.99 | |
| 79 | 5.31 | 52.2 | 20 | 24 | 20 | BDL | 1.9 | 1.9 | |
| 25 | 5.58 | 51.4 | 50 | 9 | 12 | BDL | 18 | 18 | |
| 10 | 6.28 | 51.1 | 100 | 1 | 6 | 1.3 | 22 | 23 | |
| P Vent | 350 | 3.27 | 5.30 | 0 | BDL | — | 2.4 | 1.7 | 4.2 |
| 240 | 3.93 | 25.9 | 5 | 20 | 4 | 3.2 | 18 | 22 | |
| 210 | 4.01 | 28.3 | 10 | 18 | 6 | 0.41 | 30 | 30 | |
| 120 | 4.87 | 46.0 | 50 | 13 | 6 | 0.62 | 50 | 50 | |
| 35 | 5.73 | 51.5 | 100 | 16 | 30 | 3.8 | 28 | 32 | |
| Bio Vent | 310 | 3.77 | 11.5 | 0 | BDL | — | 4.3 | 10 | 15 |
| 260 | 4.10 | 20.4 | 20 | BDL | — | 1.9 | 17 | 19 | |
| 160 | 4.34 | 32.4 | 30 | BDL | — | BDL | 17 | 17 | |
| 60 | 4.77 | 47.1 | 50 | BDL | — | BDL | 17 | 17 | |
| 10 | 5.79 | 54.0 | 100 | 9 | 60 | BDL | 0.96 | 0.96 |
pH values are shipboard measurements
BDL, below detection limit (nano pyrite 1 µM: S0 0.5 µM)
In addition to the persistence of Fe(II), the presence of filterable and particulate S0 within the plume (Table 1) provides further evidence for such a catalytic cycle. Generally, the particulate fraction of S0 is lowest in the samples taken from the orifice, whereas the <0.2 µm fraction is greatest in these samples. This indicates that S0 is mainly formed from sulfide oxidation in the plume and is not due to entrainment of chimney particles. The S0 in the < 0.2 µm fraction in the end-member samples is expected to be dissolved or nanoparticulate S0, as polysulfides undergo hydrolysis rapidly at high temperature ( ≥150 °C22. The decrease in S0 in the < 0.2 µm fraction is likely due to changes in its solubility over the temperature gradient in the initial plume23.
Formation of pyrite and persistence of pyrite nanoparticles
Significant concentrations of nanoparticulate pyrite (defined operationally as the difference between nitric acid and HCl soluble Fe in the ≤0.2 µm filtered fraction12,13) were measured in all plume samples at Bio 9 and P Vent (Table 1; Supplementary Figure 2). In contrast, pyrite nanoparticles were not statistically detectable in the hot (>350 °C) end-member samples by this leaching method. Pyrite was detected in bulk end-member and plume samples by powder XRD at both Bio 9 and P-Vent, respectively, but was not detected at Bio Vent (Supplementary Table 2).
Evidence for bulk pyrite formation comes from the stable isotope fractionation between total unfiltered AVS and CRS at P Vent and Bio 9, which are consistent with partial isotopic equilibrium between pyrite and H2S through an FeS precursor24 (Eq. 2; Fig. 2).
| 2 |
Fig. 2.
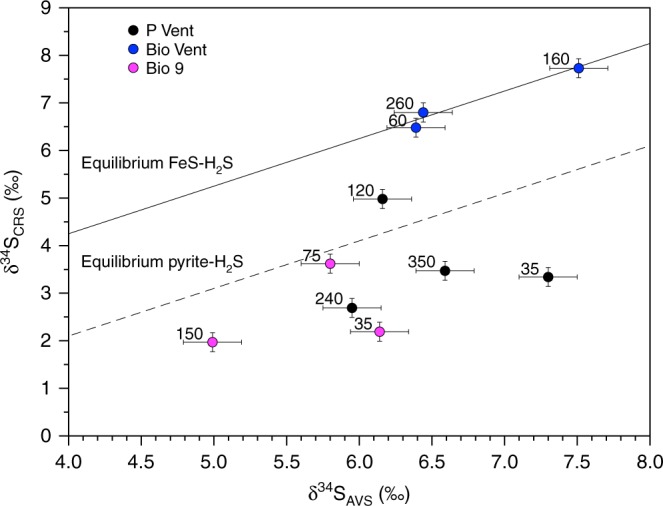
Sulphur isotopic composition of AVS and CRS in the buoyant plume. Paired δ34SAVS and δ34SCRS values from plume samples showing the expected values based upon isotopic equlibration between FeS–H2S and pyrite–H2S (the experimental equilibrium values were taken from Syverson et al.24 and were determined for T = 350 °C. We note that it is not certain what the isotope fractionation is at lower temperatures). Note that CRS values are not available for all AVS samples from Fig. 2 due to small sample size. Labels next to the data points represent the in situ temperature (°C) at which the samples were taken
The FeS−H2S pathway is expected to be the predominant mechanism for pyrite formation in buoyant vent plumes based on pH, reactant availability and reaction kinetics25. The rate of the reaction presented in Eq. 2 plateaus above 125 °C as the reaction is diffusion controlled26 and decreases with decreasing H2S concentration and at temperatures lower than 125 °C. Thus, the most significant pyrite formation should occur near the vent orifice, and the nanoparticulate pyrite detected higher in the plume has likely persisted throughout the cooling and rise of the higher temperature fluid. At high temperatures, nucleation rates are high, favouring the formation of nanoparticles27, and the rapidly cooling temperatures and decreasing reactant concentrations during mixing limit further growth, and thereby settling, of the nanoparticles. Interestingly, in contrast to the chimney pyrite from Bio 9 analysed by Rouxel et al.28, the S isotope composition for the total unfiltered fluid samples measured here from Bio 9 and P Vent fall mostly below the pyrite-H2S equilibration line. This may be due to formation of pyrite (e.g., CRS) at high temperatures, followed by removal of light H2S from the solution, either due to metal sulphide precipitation or degassing of H2S, both of which would leave the remaining AVS pool isotopically heavy.
In contrast to Bio 9 and P Vent, statistically significant concentrations of nanoparticulate pyrite were only observed in one sample at Bio Vent (Table 1), one meter from the orifice in the buoyant plume. Pyrite was also not detected in bulk samples via XRD (Supplementary Table 2). The apparent isotopic fractionation between AVS and CRS falls directly on the FeS−H2S equilibration line29, indicating that metal monosulphides, rather than FeS2 are the major particles forming at Bio Vent due to the lower Fe concentrations (Fig. 1a; Supplementary Table 1) and cooler temperature of the emitted fluid (i.e. 310 °C vs. ≥350 °C). Importantly, at all vent sites, the distribution of nanoparticulate pyrite in the plume (Table 1) does not indicate that it is removed within the first meter. At P vent, for example, concentrations of nanoparticulate pyrite are nearly constant as temperature decreases.
Sulphide precipitation and implications for element export
The value of total unfiltered δ34SAVS over the plume for P Vent and Bio 9 decreases and then increases again with increasing distance from the vent orifice (Fig. 3). These isotopic differences are larger than the typical variability for end-member samples30,31. This pattern is not consistent with fractionation caused by reaction or particle loss from the plume due to mass balance considerations; however, it does correspond to the portion of particulate Fe, as well as the visual particle density in the samples (Supplementary Figure 3, Supplementary Table 1). Although the isotope fractionation between sulfide and FeS is small (≤0.250/00), there is a fractionation of 2.70/00 between dissolved H2S and HS−, by which HS− is lighter32. FeS and other metal sulfides within the plume are expected to be formed by reaction of metals with HS−, rather than H2S as the pH increases. The pK1 of H2S in seawater at 150 °C is 6.0833, so that the pH of the fluids after 10–20 cm begins to approach the pK1. Furthermore, in situ pH is typically higher than the shipboard pH; the in situ pH in the end-member fluid at P Vent was previously measured to be about 5.534, compared with our shipboard measurement of 3.27 (Table 1).
Fig. 3.
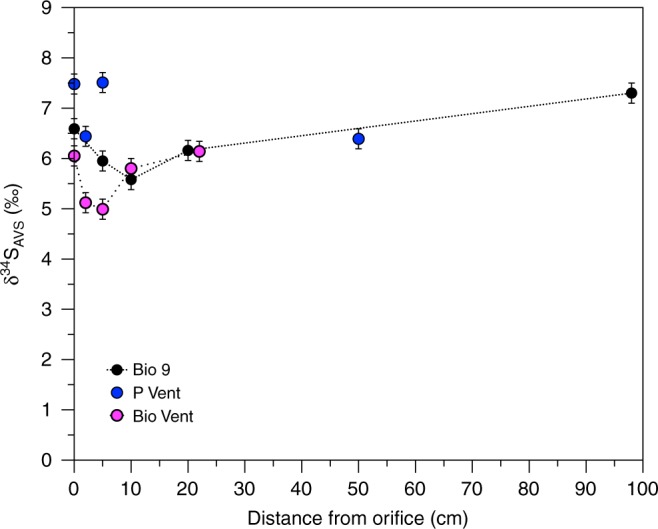
Sulphide isotopic composition in the buoyant plume. δ34SAVS (unfiltered) with increasing distance from the vent orifice in the plume. Error bars represent one standard deviation
Metal sulfide particles should therefore be isotopically lighter than coexisting H2S. If the distribution of these particles were homogenous throughout the plume, there would be no apparent difference in δ34SAVS, but both visual (Supplementary Figure 3) and Fe distribution data (Supplementary Table 1) demonstrate that particle distribution is not homogenous. Therefore, the more depleted δ34SAVS values must reflect a concentration of particles (and thereby isotopically light sulfide) within the plume within the first 10–20 cm at intermediate temperatures (100–250 °C). Although higher particle concentrations are due in part to the increased amount of particle formation at cooler temperatures and higher pH35, the δ34SAVS patterns can only be explained if bulk particles are concentrated in this area by settling from higher in the plume. Particle settling is induced as the particles grow with distance from the orifice. Simultaneously, the upward flow decreases, changing the buoyancy (Fig. 4). This observation indicates a critical point within the nascent plume that is decisive for the quantity and type of particles and elements that will be transported further up in the water column to the neutrally buoyant plume, where they can then be transported laterally.
Fig. 4.
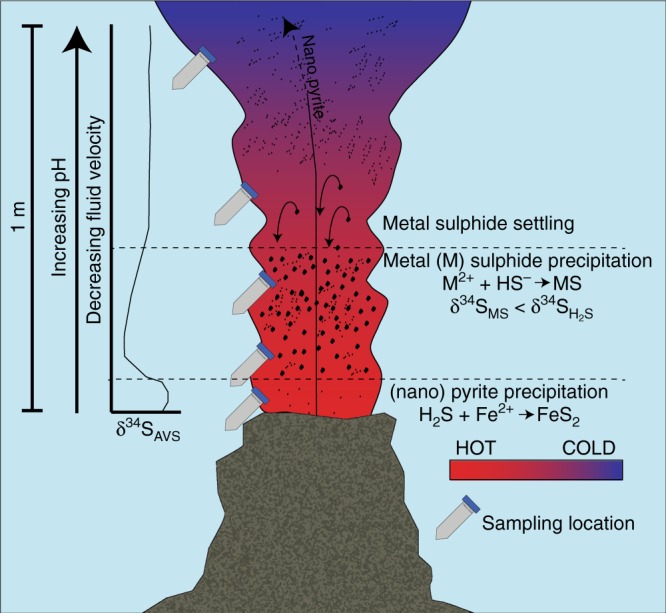
Particle settling and isotope effects in the buoyant plume. Conceptual model of particle settling within the initial buoyant plume and the resulting isotopic effects
The location and extent of this zone will be affected by the physical characteristics of a given vent, including the temperature of the emitted fluids and the size and flow rate of the plume. For example, the isotopic composition of AVS in the Bio Vent plume does not follow the same trend observed at Bio 9 and P Vent. This is consistent with the visually lower flow and particle concentration in this plume, as well as the observed lower removal of Fe and higher ratio of sulfide to Fe, compared to Bio 9 and P Vent (Supplementary Table 1). These characteristics suggest moreover that major Fe and particle removal took place at an earlier point along the fluid flow pathway, for example within the chimney.
For all vents, nanoparticulate pyrite is a significant portion (up to 60%) of ≤0.2 µm Fe at 1 m above the vent orifice and above this particle concentration zone. The lack of evidence for removal of nanoparticulate pyrite in any of the three plumes, despite settling of bulk particles, indicates that pyrite nanoparticles stabilise Fe with respect to particle growth, within the ≤0.2 µm filtered fraction, limiting the extent of Fe that settles from the plume. Due to their small size12 and resistence to oxidation36, pyrite nanoparticles may thus mediate Fe export from the vents to the global oceans.
Previous work has shown the widespread presence of FeS2 nanoparticles in the end-member fluid of a variety of different hydrothermal settings with different geological (back arc basin, slow and fast spreading centers) and chemical characteristics (Fe to sulphide ratios ranging from 0.1 to 10)12,13. Here, using a combination of chemical leaching, mineralogical and stable isotope methods, we demonstrated that these nanoparticles serve to stabilise Fe during the extensive mixing and particle precipitation that occurs immediately upon fluid emission from the vent orifice. This study shows that particle formation, settling and buoyancy between the hot vent fluids and the neutrally buoyant plume directly affect metal transport from the initial mixing zone. The formation of nanoparticulate pyrite at high temperatures and limits on its growth due to rapidly cooling temperatures during mixing act to stabilise the hydrothermally derived Fe as nanoparticulate pyrite and prevent further particle growth and oxidation. The data presented here demonstrate for the first time the persistance of nanoparticulate pyrite throughout a zone of major particle growth and settling, and suggest the possibility for continued nanoparticle transport, including transport from the vent sites.
Methodology
Sampling
Samples were taken using the Human Operated Vehicle Alvin II operated by the RV Atlantis at three different vent sites (P Vent, Bio 9 and Bio Vent) along the East Pacific Rise 9°N during March and April 2017. The in situ temperature of each sample was taken with the high-temperature probe attached to the titanium Major Sampler. One sample was taken within the vent orifice, then subsequent samples were taken from first meter of the rising plume in order to capture a broad temperature gradient. Great care was taken by the Alvin pilots to hold the nozzle of the sampler in the centre of the plume during sampling.
Sample processing
The samples were processed immediately upon recovery shipboard, typically 2–4 h after they were taken. The shipboard pH and temperature were measured immediately and samples were fixed for either shipboard or shorebased analyses.
Fe concentration and speciation
For Fe samples, triplicate aliquots of 10 mL (0.2 µm filtered and unfiltered) were added to centrifuge tubes containing 0.5 mL HCl (trace metal grade). Iron measurements were performed after sequential leaching with HCl and HNO312. Each acid leach was allowed to react for at least 8 hours. Spectrophotometric measurements were made onboard the ship using the ferrozine method of Stookey37 at a wavelength of 562 nm. Samples were buffered in 2.5 M ammonium acetate prior to addition of the ferrozine reagent. After the initial analysis of the HCl leach, the samples were treated with hydroxylamine hydrochloride as a reducing agent to measure total iron. Fe(III) was determined by the difference between these numbers. After the HNO3 leach, hydroxylamine was added simultaneously with the ferrozine reagent to measure total Fe. Pyrite concentrations were determined by subtracting total Fe from the sum of Fe(II) and Fe(III) determined after the HCl leach. The detection limit of this method was 100 nM.
Sulphide speciation and concentration
Samples for sulfide analysis (AVS/CRS) were preserved by adding 2 mL NaOH (0.5 M) and 2 mL zinc acetate (0.1 M) to 2 mL of sample, which was then frozen and stored at −20 °C. Measurements were made on shore using the distillation method of Fossing and Jørgensen21. Samples preserved in NaOH and zinc acetate were defrosted and the entire sample was transferred to a bubbler system along with 12 mL Ar-purged MQ water. In the bubbler, samples were purged with Ar for an additional 10 min. 0.5 mL of 3 M HCl (trace metal grade) was then added via syringe to the system and evolved H2S was captured in 20 mL 1 M trace metal clean NaOH. The base de-protonates the H2S gas, converting it to HS− and allowing direct quantification via UV-Vis spectroscopy (HS− peak at 230 nm). Reaction time prior to analysis was 1.5 hours. This initial measurement represents the Acid Volatile Sulfide fraction. Test tubes containing a fresh 20 mL aliquot of 1 M trace metal clean NaOH were then placed in-line with the bubbler system and 0.5 mL of 1 M Cr(II) solution in 1 M HCl (prepared using a Jones reduction column) was injected into the sample via syringe. Samples were again bubbled for 1.5 h prior to analysis of evolved H2S as HS− trapped in NaOH. This second analysis represents the concentration of Chromium-Reducible Sulfide.
Elemental sulphur
Sample aliquots of 30–50 mL were filtered through a 0.2 µm Millipore GTTP filter into a zinc acetate solution to fix free sulfide and prevent oxidation during the extraction. The filtrate was then extracted shipboard in 5 mL of toluene for 1.5 h38. The toluene layer was separated and stored at −20 °C for later analysis. The filters were also stored at −20 °C for later extraction and analysis onshore. Elemental sulfur was quantified by HPLC using a C-18 column and 98% methanol 2% water as the eluent with UV detection at 230 nm. The retention time for S8 was approximately six minutes. The detection limit for this method is 0.5 µM.
Stable sulfur isotope measurements
Unfiltered 5–50 mL samples fixed in zinc acetate were distilled via the two-step AVS/CRS procedure outlined above to separate AVS and CRS. Evolved sulfide was trapped as ZnS and was converted to Ag2S upon addition of AgNO3 (1 M). The precipitate was aged one week, washed with 18.2 MΩ water (MilliQ) and NH4OH (1 M), then dried overnight at 60 °C. Ag2S was converted to SF6 by reaction with excess F2 at 300 °C for at least 10 hours in Ni alloy reaction chambers. The SF6 was then purified cryogenically and by preparative gas chromatography. Following purification, stable sulphur isotopic measurements were conducted on a Finnigan MAT 253 dual inlet mass spectrometer39.
Isotopic composition is presented in permil using standard δ notation relative to VCDT (Eq. 3)
| 3 |
in which 34R = 34S/32S.
XRD
Samples for XRD analysis were prepared by the centrifugation of 300–700 mL of unfiltered fluid13. Samples were then capped with UHP nitrogen and frozen. For analysis, pelleted samples were resuspended in Milleq ® water and evaporated under nitrogen onto zero diffraction wafers by MTI. Samples were run on a Panalytical X’Pert3 Powder XRD using a Cu Kα source at 40 kV and 45 mA, and scanned from 5–70° 2θ. Three scans were performed with a 0, −1, and + 1 degree wobble in order to avoid preferential orientiation; final scans were an average of all three scans. HighScore Plus software was used for peak identification and fitting, with International Center for Diffraction Data 2014 used for sample ID. Mineralogy as determined from XRD was checked for compatibility with elemental results obtained from SEM/EDS. SEM was performed using a Tescan VP-SEM in high vacuum mode at an accelerating potential of 20 kV.
Mg concentrations
Samples for Mg concentrations were filtered shipboard (0.2 µm) and acidified in 0.75 M nitric acid. Onshore, samples were diluted between 1:100 and 1:2000, using sample temperature as an estimate of Mg. Samples were analyzed on a Thermo Electron Corporation Finnigan Element XR Inductively Coupled Plasma Mass Spectrometer (ICP-MS) in low resolution mode, using a rhodium internal standard. Dilution preparation and sample analysis were conducted under trace metal clean conditions.
Supplementary information
Acknowledgements
This work was supported by a grant from the Marine Geology and Geophysics Program of the U.S. National Science Foundation (OCE-1558712) to G.W.L. and both a Kreitman Postdoctoral Fellowship (BGU) and a Marie Curie Fellowship to A.J.F. (MSCA SedSulphOx 746872). We thank the DSV Alvin group and the Captain and crew of the R/V Atlantis for their excellent work. André Pellerin is thanked for assistance with Fig. 4.
Author contributions
A.J.F. conceived the study, participated in sampling, conducted analyses, interpreted the data and wrote the initial draft of the paper, E.R.E. participated in sampling and conducted analyses, A.G. conceived the study, conducted analyses and participated in the interpretation of the results, G.W.L. funded the project, planned and lead the cruise and participated in the interpretation of the results, A.K. participated in the interpretation of the results, M.Y. participated in sampling and conducted analyses. All authors participated in the preparation of the final version of manuscript and have approved it.
Data Availability
All data generated or analysed during this study are included in this published article (and its supplementary information files).
Code Availability
No custom code was used in the production or analysis of these data.
Competing interests
The authors declare no competing interests.
Footnotes
Journal peer review information: Nature Communications thanks Drew Syverson and the other anonymous reviewer for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information accompanies this paper at 10.1038/s41467-019-09580-5.
References
- 1.Tagliabue A, et al. Hydrothermal contribution to the oceanic dissolved iron inventory. Nat. Geosci. 2010;3:252–256. doi: 10.1038/ngeo818. [DOI] [Google Scholar]
- 2.Tagliabue A. More to hydrothermal iron input than meets the eye. Proc. Natl Acad. Sci. USA. 2014;111:16641–16642. doi: 10.1073/pnas.1419829111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tagliabue A, Resing JA. Impact of hydrothermalism on the ocean iron cycle. Philos. Trans. A. 2016;374:2081. doi: 10.1098/rsta.2015.0291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burd BJ, Thomson RE. The importance of hydrothermal venting to water-column secondary production in the northeast Pacific. Deep. Res. Part II Top. Stud. Oceanogr. 2015;121:85–94. doi: 10.1016/j.dsr2.2015.04.014. [DOI] [Google Scholar]
- 5.Feely RA, et al. Composition and sedimentation of hydrothermal plume particles from north Cleft segment, Juan de Fuca Ridge. J. Geophys. Res. 1994;99:4985–5006. doi: 10.1029/93JB02509. [DOI] [Google Scholar]
- 6.Ludford EM, Palmer MR, German CR, Klinkhammer GP. The geochemistry of Atlantic hydrothermal particles. Geophys. Res. Lett. 1996;23:3503–3506. doi: 10.1029/96GL02078. [DOI] [Google Scholar]
- 7.Douville E, et al. The rainbow vent fluids (36 degrees 14’ N, MAR): the influence of ultramafic rocks and phase separation on trace metal content in Mid-Atlantic Ridge hydrothermal fluids. Chem. Geol. 2002;184:37–48. doi: 10.1016/S0009-2541(01)00351-5. [DOI] [Google Scholar]
- 8.Demina LL, Holm NG, Galkin SV, Lein AY. Some features of the trace metal biogeochemistry in the deep-sea hydrothermal vent fields (Menez Gwen, Rainbow, Broken Spur at the MAR and 9°50’N at the EPR): A synthesis. J. Mar. Syst. 2013;126:94–105. doi: 10.1016/j.jmarsys.2012.09.005. [DOI] [Google Scholar]
- 9.Findlay AJ, Gartman A, Shaw TJ, Luther GW. Trace metal concentration and partitioning in the first 1.5m of hydrothermal vent plumes along the Mid-Atlantic Ridge: TAG, Snakepit, and Rainbow. Chem. Geol. 2015;412:117–131. doi: 10.1016/j.chemgeo.2015.07.021. [DOI] [Google Scholar]
- 10.Edmonds HN, German CR. Particle geochemistry in the rainbow hydrothermal plume, mid-atlantic ridge. Geochim. Cosmochim. Acta. 2004;68:759–772. doi: 10.1016/S0016-7037(03)00498-8. [DOI] [Google Scholar]
- 11.Mottl MJ, McConachy TF. Chemical processes in buoyant hydrothermal plumes on the East Pacific Rise near 21°N. Geochim. Cosmochim. Acta. 1990;54:1911–1927. doi: 10.1016/0016-7037(90)90261-I. [DOI] [Google Scholar]
- 12.Yücel M, Gartman A, Chan CS, Luther GW. Hydrothermal vents as a kinetically stable source of iron-sulphide-bearing nanoparticles to the ocean. Nat. Geosci. 2011;4:367–371. doi: 10.1038/ngeo1148. [DOI] [Google Scholar]
- 13.Gartman A, Findlay AJ, Luther GW. Nanoparticulate pyrite and other nanoparticles are a widespread component of hydrothermal vent black smoker emissions. Chem. Geol. 2014;366:32–41. doi: 10.1016/j.chemgeo.2013.12.013. [DOI] [Google Scholar]
- 14.Bennett SA, et al. Deep-Sea Research I Dissolved and particulate organic carbon in hydrothermal plumes from the East Pacific Rise, 9 1 50 0 N. Deep. Res. Part I. 2011;58:922–931. doi: 10.1016/j.dsr.2011.06.010. [DOI] [Google Scholar]
- 15.Sander SG, Koschinsky A. Metal flux from hydrothermal vents increased by organic complexation. Nat. Geosci. 2011;4:145–150. doi: 10.1038/ngeo1088. [DOI] [Google Scholar]
- 16.Kleint C, Hawkes JA, Sander SG, Koschinsky A. Voltammetric investigation of hydrothermal iron speciation. Front. Mar. Sci. 2016;3:1–11. doi: 10.3389/fmars.2016.00075. [DOI] [Google Scholar]
- 17.Saito MA, et al. Slow-spreading submarine ridges in the South Atlantic as a significant oceanic iron source. Nat. Geosci. 2013;6:775–779. doi: 10.1038/ngeo1893. [DOI] [Google Scholar]
- 18.Fitzsimmons JN, Boyle EA, Jenkins WJ. Distal transport of dissolved hydrothermal iron in the deep South Pacific Ocean. Proc. Natl Acad. Sci. USA. 2014;111:16654–16661. doi: 10.1073/pnas.1418778111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Resing JA, et al. Basin-scale transport of hydrothermal dissolved metals across the South Pacific Ocean. Nature. 2015;523:200–203. doi: 10.1038/nature14577. [DOI] [PubMed] [Google Scholar]
- 20.Fitzsimmons JN, et al. Iron persistence in a distal hydrothermal plume supported by dissolved-particulate exchange. Nat. Geosci. 2017;10:195–201. doi: 10.1038/ngeo2900. [DOI] [Google Scholar]
- 21.Fossing H, Jorgensen BB. Chromium Reduction Method of bacterial sulfate reduction in sediments: Measurement reduction of a single-step chromium method Evaluation. Biogeochemistry. 1989;8:205–222. doi: 10.1007/BF00002889. [DOI] [Google Scholar]
- 22.Giggenbach WF. Equilibria involving polysulfide ions in aqueous sulfide solutions up to 240°. Inorg. Chem. 1974;13:1724–1730. doi: 10.1021/ic50137a038. [DOI] [Google Scholar]
- 23.Kamyshny A. Solubility of cyclooctasulfur in pure water and sea water at different temperatures. Geochim. Cosmochim. Acta. 2009;73:6022–6028. doi: 10.1016/j.gca.2009.07.003. [DOI] [Google Scholar]
- 24.Syverson DD, Ono S, Shanks WC, Seyfried WE. Multiple sulfur isotope fractionation and mass transfer processes during pyrite precipitation and recrystallization: An experimental study at 300 and 350??C. Geochim. Cosmochim. Acta. 2015;165:418–434. doi: 10.1016/j.gca.2015.06.022. [DOI] [Google Scholar]
- 25.Gartman A, Luther GW. Comparison of pyrite (FeS2) synthesis mechanisms to reproduce natural FeS2 nanoparticles found at hydrothermal vents. Geochim. Cosmochim. Acta. 2013;120:447–458. doi: 10.1016/j.gca.2013.06.016. [DOI] [Google Scholar]
- 26.Rickard D, Luther GW. Kinetics of pyrite formation by the H2S oxidation of iron (II) monosulfide in aqueous solutions between 25 and 125°C: The mechanism. Geochim. Cosmochim. Acta. 1997;61:135–147. doi: 10.1016/S0016-7037(96)00322-5. [DOI] [Google Scholar]
- 27.Hochella MF, Jr., et al. Nanominerals, mineral nanoparticles, and Earth Systems. Science. 2008;319,:1631–1635. doi: 10.1126/science.1141134. [DOI] [PubMed] [Google Scholar]
- 28.Rouxel O, Shanks WC, Bach W, Edwards KJ. Integrated Fe- and S-isotope study of seafloor hydrothermal vents at East Pacific Rise 9-10°N. Chem. Geol. 2008;252:214–227. doi: 10.1016/j.chemgeo.2008.03.009. [DOI] [Google Scholar]
- 29.Ohmoto, H. & Rye, R. O. In Geochemistry of Hydrothermal Ore Deposits (ed. Barnes, H. L.). 509–567 (Wiley, New York 1979).
- 30.Ono S, Shanks WC, Rouxel OJ, Rumble D. S-33 constraints on the seawater sulfate contribution in modern seafloor hydrothermal vent sulfides. Geochim. Cosmochim. Acta. 2007;71:1170–1182. doi: 10.1016/j.gca.2006.11.017. [DOI] [Google Scholar]
- 31.McDermott JM, et al. Identification of sulfur sources and isotopic equilibria in submarine hot-springs using multiple sulfur isotopes. Geochim. Cosmochim. Acta. 2015;160:169–187. doi: 10.1016/j.gca.2015.02.016. [DOI] [Google Scholar]
- 32.Fry B, Gest H, Hayes JM. Sulfur isotope effects associated with protonation of Hs- and volatilization of H2S. Chem. Geol. Isot. Geosci. Sect. 1986;58:253–258. doi: 10.1016/0168-9622(86)90014-X. [DOI] [PubMed] [Google Scholar]
- 33.Hershey JP, Plese T, Millero FJ. The pK1* for the dissociation of H2S in various ionic media. Geochim. Cosmochim. Acta. 1988;52:2047–2051. doi: 10.1016/0016-7037(88)90183-4. [DOI] [Google Scholar]
- 34.Ding, K. & Seyfried, W. E. In situ measurement of pH and dissolved H-2 in mid-ocean ridge hydrothermal fluids at elevated temperatures and pressures. Chem Rev107, 601–622 (2007). [DOI] [PubMed]
- 35.Reed MH. Sulfide mineral precipitation from hydrothermal fluids. Rev. Mineral. Geochem. 2006;61:609–631. doi: 10.2138/rmg.2006.61.11. [DOI] [Google Scholar]
- 36.Gartman A, Iii GWL. ScienceDirect Oxidation of synthesized sub-micron pyrite (FeS 2) in seawater. Geochim. Cosmochim. Acta. 2014;144:96–108. doi: 10.1016/j.gca.2014.08.022. [DOI] [Google Scholar]
- 37.Stookey LL. Ferrozine---a new spectrophotometric reagent for iron. Anal. Chem. 1970;42:779–781. doi: 10.1021/ac60289a016. [DOI] [Google Scholar]
- 38.Findlay AJ, et al. Distribution and size fractionation of elemental sulfur in aqueous environments: The Chesapeake Bay and Mid-Atlantic Ridge. Geochim. Cosmochim. Acta. 2014;142:334–348. doi: 10.1016/j.gca.2014.07.032. [DOI] [Google Scholar]
- 39.Ono S, Wing B, Johnston D, Farquhar J, Rumble D. Mass-dependent fractionation of quadruple stable sulfur isotope system as a new tracer of sulfur biogeochemical cycles. Geochim. Cosmochim. Acta. 2006;70:2238–2252. doi: 10.1016/j.gca.2006.01.022. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analysed during this study are included in this published article (and its supplementary information files).
No custom code was used in the production or analysis of these data.