

Abstract
Calmodulin (CaM) is a calcium sensor protein that directly interacts with the dual-specificity (lipid and protein) kinase PI3Kα through the SH2 domains of the p85 regulatory subunit. In adenocarcinomas, the CaM interaction removes the autoinhibition of the p110 catalytic subunit of PI3Kα, leading to activation of PI3Kα and promoting cell proliferation, survival, and migration. Here we demonstrate that the cSH2 domain of p85α engages its two CaM-binding motifs in the interaction with the N- and C-lobes of CaM as well as the flexible central linker, and our nuclear magnetic resonance experiments provide structural details. We show that in response to binding CaM, cSH2 exposes its tryptophan residue at the N-terminal region to the solvent. Because of the flexible nature of both CaM and cSH2, multiple binding modes of the interactions are possible. Binding of CaM to the cSH2 domain can help release the inhibition imposed on the p110 subunit, similar to the binding of the phosphorylated motif of RTK, or phosphorylated CaM (pCaM), to the SH2 domains. Amino acid sequence analysis shows that CaM-binding motifs are common in SH2 domains of non-RTKs. We speculate that CaM can also activate these kinases through similar mechanisms.
Graphical Abstract
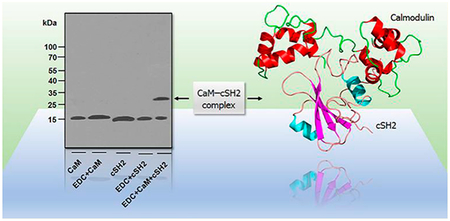
Activation of synthesis of signaling phospholipid phosphatidylinositol 3,4,5-trisphosphate (PIP3) by phosphoinosi-tide-3-kinase (PI3K) through a calcium sensor protein calmodulin (CaM) is emerging as an important regulatory mechanism in adenocarcinomas.1,2 PIP3, a product of phosphorylation of phosphatidylinositol 4,5-bisphosphate (PIP2) by PI3K in response to activation of G-protein-coupled receptors (GPCRs) and receptor tyrosine kinases (RTKs), is critically involved in membrane targeting and activation of protein kinase B (Akt).3 The pleckstrin homology (PH) domain of Akt recognizes increased levels of PIP3 and attaches the kinase to the plasma membrane (PM) where it is fully activated by phosphorylation.4 Akt is phosphorylated primarily by phosphoinositide-dependent kinase 1 (PDK1) and the mammalian target of rapamycin (mTOR) complex II, but also by several other kinases.5 Intriguingly, CaM mediates K-Ras4B-dependent Akt activation stimulated with platelet-derived growth factor (PDGF),6 neuronal survival signaling upstream of Akt,7 and cell proliferation enhancing the activity of PI3K.8
The PIP3-producing catalytic subunits of class IA PI3Ks, including p110α, p110β, and p110δ, are tightly controlled by their association with the regulatory subunits p85α, p85β, p55α, p50α, and p55γ.3 The class IB PI3Ks contain the p110γ catalytic subunit that associates with the regulatory p84/p87 or p101 subunits.9 The catalytic subunits, p110α and p110β, are ubiquitous, while expression of p110δ and p110γ is limited mostly to hematopoietic cells.10 While p110α and p85α are frequently mutated in cancer, somatic mutations of other class I isoforms are less common.11 However, anomalous activation of any isoform can lead to cancer transformation.12 Oncogenic mutations tend to affect the association between the catalytic and regulatory subunits.13,14 The primary role of the regulatory subunits is to prevent undesirable activation of the kinase by inhibiting the basal catalysis. The inhibition is achieved by several key elements of the regulatory subunits, including the N- and C-terminal Src homology (nSH2 and cSH2) domains that are connected by an inter-SH2 (iSH2) coiled-coil domain.15 The iSH2 domain directly interacts with the adaptor-binding domain (ABD) of the catalytic subunit, while nSH2 binds the C2, helical, and kinase domains. The cSH2 domain contacts the C-terminal end of the kinase domain in p110β and p110δ, while it does not interact tightly with p110α.16 Despite the lack of strong association with the p110α catalytic subunit, binding of phosphotyrosine peptides with the pYXXM motif to cSH2 activates the kinase.17,18 Moreover, deletion of cSH2 from p85α activates PI3Kα and Akt, contributes to cellular transformation, and enhances oncogenic effects of nSH2 mutations.19,20 The mechanism of cSH2 regulation of the catalytic activity of p110α involves disruption of the inhibitory contacts with p110.16,19
Ca2+-loaded CaM participates in activation of Akt through PI3K.6,21 However, the mechanism is not fully understood. Formation of a ternary complex among CaM, K-Ras4B, and PI3Kα has been proposed.1,22 Consistent with the model, direct interactions of CaM with K-Ras4B have been observed.23–26 The “ternary complex” hypothesis is appealing because it suggests that the low-affinity association between Ras and p110 of PI3K27 can be stabilized by CaM. CaM also binds the p85α regulatory subunit of PI3Kα primarily through its cSH2 and weakly through nSH2 domains, releasing the autoinhibition of the kinase activity.21 A similar interaction has been observed between CaM and the CaM-binding motif within the SH2 domain of the Src kinase family, leading to removal of the inhibitory intradomain contacts and activation of the kinase.28,29 Activated Src phosphorylates CaM on Tyr99 and potentially alters CaM’s binding to the kinase. Incidentally, recent studies found that CaM phosphorylated mainly on Tyr99 and to some extent on Tyr138 promotes activation of PI3Kα.30,31 Computational studies demonstrated that phosphorylated CaMs (pCaMs) form strong and stable interactions with both nSH2 and cSH2 domains, with pTyr99 in pCaM contributing substantially to the interfaces.32 These observations suggest that CaM may be generally involved in activation of kinases through liberating their catalytic domains from inhibition by SH2 domains. Insight into PI3K activation by CaM has the potential to uncover a universal CaM-dependent regulatory mechanism of kinase function.
In addition to activation of PIP3 production, signaling through GPCRs and RTKs increases intracellular calcium concentrations. Binding of four calcium ions to CaM exposes hydrophobic pockets in its N- and C-lobes and enables interactions with signaling proteins.33 The flexible central linker connecting the globular N- and C-lobes often bends to accommodate the interactions with CaM-binding proteins. This results in a collapsed conformation of CaM that is often observed in its high-affinity protein–protein complexes.34
Here, we investigated the structural determinants of p85 of PI3Kα binding to CaM by nuclear magnetic resonance (NMR). We found that the cSH2 domain contains two CaM-binding motifs positioned in its αB and βD regions (Figure 1), which allow CaM bending and docking to its N- and C-lobes. The intermolecular contacts are accompanied by a conformational change in cSH2 that involves movement of its single tryptophan residue to the protein exterior. We postulate that CaM binding shifts the equilibrium toward the p110α-liberated, active state. CaM-binding motifs are common in the SH2 domains of non-RTKs, suggesting that CaM can play a role in their activation similar to that observed in PI3K.
Figure 1.

CSH2 domain sequence and structure. (A) Sequence of the cSH2 domain (residues 614–724) taken from the p83α regulatory subunit of PI3Kα. In the sequence, hydrophobic, polar/glycine, positively charged, and negatively charged residues are colored black, green, blue, and red, respectively. Underlined residues denote secondary structure formation. (B) Solution structure of cSH2 of p83α (Protein Data Bank entry 1BFJ). In the structure, the α-helix (blue) and β-sheet (red) secondary structures are shown. The loop regions are denoted by black letters. The nomenclatures for the secondary structure elements were previously reported.18
MATERIALS AND METHODS
Plasmid Construction for Recombinant Proteins.
DNA encoding the wild type Homo sapiens p85α cSH2 domain (111 residues from position 614 to 724) was cloned into pET-21b (Novagen) using the BamHI and HindIII restriction sites through standard polymerase chain reaction (PCR) methods. The hexahistidine tag was added at the C-terminus to ease protein purification. Two mutant plasmids of the H. sapiens p85α cSH2 domain (111 residues from position 614 to 724), cSH2V663K/V667N and cSH2V663K/V667N/L687F/L691V/L696N, were created using a standard PCR mutagenesis protocol. Human full-length CaM DNA (Invitrogen) was cloned into the pET42a expression vector (Novagen). DNA encoding the wild type H. sapiens p85α nSH2 domain (111 residues from position 325 to 435) was also cloned into pET-21b (Novagen) using the BamHI and HindIII restriction sites, and a hexahistidine tag was added at the C-terminus. The sequences of the DNA insert and mutations were confirmed by DNA sequencing before the plasmids were transformed into Escherichia coli BL21(AI) cells.
Expression and Purification of Recombinant Proteins.
The H. sapiens p85α cSH2 domain (111 residues from position 614 to 724) wild type and mutant plasmids were transformed into E. coli One Shot BL21-AI cells (Invitrogen). The transformed cells were grown in LB medium with ampicillin for antibiotic selection at 37 °C and shaken at 220 rpm. cSH2 protein expression was induced at an OD600 of 0.8 with 0.2 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) and 10% (w/v) l-arabinose in the presence of 2% (v/v) ethanol. Cells were harvested after 5 h and frozen at −80 °C.
15N-labeled cSH2 protein ([15N]cSH2) for NMR was produced by using M9 medium (with 1 g/L [15N]ammonium chloride and 4 g/L glucose). For induction, the cell cultures were diluted 1:20 into M9 medium after overnight growth at 37 °C in LB medium. When the OD600 reached 0.8, protein expression was induced at 18 °C with IPTG and l-arabinose and grown while being shaken at 200 rpm for 18 h. Cells were harvested at 5000 rpm for 30 min at 4 °C. The supernatant was discarded, and the cell pellets were frozen at −80 °C.
After thawing, the cell pellet was resuspended, and the cells were lysed by sonication in lysis buffer [50 mM Tris, 150 mM NaCl, 2 mM phenylmethanesulfonyl fluoride, 5 mM β-mercaptoethanol, and 0.1% (w/v) Triton X-100 (pH 7.5)] containing 1 or 2 tablets of EDTA-free complete cocktail (Roche), 50 μg/mL DNaseI, and 200 μg/mL lysozyme. The lysates were centrifuged for 30 min at 20000 rpm and 4 °C. The supernatant was collected and incubated with Ni2+-charged His-Bind resin (Novagen) for 2 h while being gently shaken. The slurry was transferred to a disposable column (Bio-Rad), and the column was washed with wash buffer [50 mM Tris, 150 mM NaCl, 50 mM imidazole, and 5 mM β-mercaptoethanol (pH 7.8)]. Elutions were performed stepwise (100 mM, 200 mM, 300 mM, and 500 M imidazole). An estimated yield of 0.5 mg/L was achieved. All fractions were analyzed on 4 to 20% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) gels. The pure fractions were pooled and concentrated. The typical purity was greater than 95%.
The human full-length CaM plasmid was transformed into BL21-A1 E. coli cells. CaM was expressed and purified as we previously described24 except [15N]CaM for NMR was expressed using M9 medium (with 1 g/L [15N]ammonium chloride). The yield of CaM is ~2 mg/L. The purity of the protein was assessed by SDS–PAGE (purity of >95%). The pure fractions were pooled and concentrated. All the purified proteins were dialyzed into Tris-citrate buffer [50 mM Triscitrate, 50 mM NaCl, 20 mM CaCl2, and 2 mM β-mercaptoethanol (pH 7.5)] for storage.
Two-Step Coupling of Proteins Using EDC and Western Blot Analysis.
For CaM–cSH2 and CaM–nSH2 cross-linking, CaM was exchanged into the activation buffer [0.1 M MES and 0.5 M NaCl (pH 6.0)] at 55 μM (1 mg/mL) and cSH2 (550 μM) or nSH2 (550 μM) into coupling buffer [phosphate-buffered saline (PBS)]. EDC {1-ethyl-3-[3-(dimethylamino)propyl]carbodiimide hydrochloride} (2 mM, 0.4 mg) and 0.6 mg of NHS (5 mM) were added to 1 mL of a CaM solution and incubated for 15 min at room temperature. After the EDC had been quenched with 2-mercaptoethanol (final concentration of 20 mM), 100 μL of cSH2 or nSH2 in coupling buffer was added to the 1 mL of activated CaM. CaM and SH2 were used at an equimolar ratio (final concentration of 50 μM). The proteins were allowed to react for 40 min at room temperature. Using hydroxylamine at a final concentration of 10 mM, the reaction was stopped and the excess quenching reagent was removed with a desalting column.
For Western blot analysis, 2 μL of 6× Laemmli buffer was added to10 μL of the protein samples described above. Samples were boiled at 80 °C for 10 min. Samples (4 μL) were loaded onto a 4 to 20% gradient gel (Bio-Rad) that was subjected to electrophoresis at 140 V for approximately 1 h and then electro-blotted onto a polyvinylidene fluoride (PVDF) membrane (Bio-Rad) at 100 V for 1.5 h. The membrane was blocked with 5% nonfat milk in TBST [20 mM Tris and 150 mM NaCl containing 0.05% Tween 20 (pH 7.4)] for 1 h and then was incubated in the presence of a primary anti-His tag antibody (mouse, Cell Signaling Technology) diluted 1:1000 in 5% milk overnight at 4 °C. After being rinsed, the blot was treated with a secondary antibody (peroxidase-conjugated anti-mouse IgG secondary antibody, GE Healthcare) diluted 1:5000 in 5% milk for 1 h at room temperature. Excess secondary antibody was removed by washing the membranes in TBST three times for 5 min each. Bands were visualized by chemiluminescence using the ECL reagent kit (GE Healthcare) with photographic film.
The cross-linking experiments with CaM–cSH2mutant, including cSH2V663K/V667N and cSH2V663K/V667N/L687F/L691V/L696N mutants, were conducted in parallel and followed the above procedure except that 2 μL samples were loaded onto a 15% SDS–PAGE gel (Bio-Rad). The HRP-conjugated 6*His tag antibody (mouse, Proteintech Co., catalog no. HRP-66005) was diluted 1:10000 in 5% milk and incubated with an electroblotted PVDF membrane at room temperature for 1 h.
Tryptophan Fluorescence Measurements.
Tryptophan fluorescence measurements (TFMs) were performed using an RF-5301PC spectrofluorophotometer (Shimadzu, Kyoto, Japan) at 15 °C. TFMs were performed by exciting the single cSH2 tryptophan residue at 280 nm and scanning the emission fluorescence from 300 to 500 nm. The total volume of the samples was 100 μL, and it was placed in a Greiner LUMITRAC 600 Flat Bottom Black 384-well plate. Binding buffer for TFM contains 50 mM Tris-citrate (pH 7.5), 50 mM NaCl, and 20 mM CaCl2. At first, 100 μL samples containing 10 μM CaM in binding buffer were scanned as a control group. Then, this was followed by addition of cSH2 from 5 to 20 μM CaM with a concentration increment of 5 μM. During the third round, 100 μL samples containing cSH2 protein alone at concentrations of 0, 5, 10, 15, and 20 μM were scanned as another control. Each experiment was performed in triplicate.
NMR 1H–15N HSQC Titrations.
The 1H–15N hetero-nuclear single-quantum coherence (HSQC) NMR experiments were performed on a 900 MHz Avance spectrometer (Bruker Daltonics, Billerica, MA) equipped with a cryogenic probe. All experiments were performed at 15 °C. The buffer conditions were as follows: 10% D2O, 50 mM Tris-citrate (pH 7.5), 50 mM NaCl, 20 mM CaCl2, and 2 mM β-mercaptoethanol. Two-dimensional 1H–15N HSQC spectra of [15N]cSH2 protein were acquired at a protein concentration of 100 μM in the absence or presence of the unlabeled CaM protein at different molar ratios, including 1:0.5, 1:1, 1:1.5, and 1:2 ([15N]-cSH2:CaM). More specifically, to characterize the interactions of CaM with the cSH2 domain, a series of HSQC spectra of [15N]CaM at a protein concentration of 100 μM were collected with gradual addition of cSH2 with a ratio increment of 0.5 until the HSQC peaks no longer changed.
The CaM assignments were as previously published.24 For cSH2, the HSQC peaks were identified according to the NMR assignments of cSH2 published before35 and further confirmed by using a nuclear Overhauser effect spectroscopy (NOESY) experiment. NMR data were processed with NMRPipe.36 By superimposition of the HSQC spectra, the missing HSQC peaks could be identified and assigned to the corresponding cSH2 and CaM residues. The observed amide resonance chemical shift perturbations were calculated using the equation as we have done previously24
| (1) |
where ΔδH and ΔδN are the observed chemical shift changes for 1H and 15N, respectively. The chemical shift changes above average plus one standard deviation were considered statistically significant as commonly done in NMR studies.
The KD was determined using measured changes in 15N line widths or chemical shift perturbations (CSPs) in NMR titration experiments in a nonlinear fitting procedure to the “one-site binding model”37
| (2) |
NMR Three-Dimensional 1H–15N NOESY-HSQC Experiment.
Three-dimensional 150 ms 1H–15N NOESY-HSQC experiments were performed with 100 μM [15N]CaM and 200 μM cSH2 in solutions containing the other components as described above, and spectra were recorded at 15 °C using the same 900 MHz Avance spectrometer equipped with a cryogenic probe. All spectra were processed with NMRPipe,36 and analyzed and assigned with NMRview.38
Molecular Modeling of the CaM–cSH2 Complex.
Docking was performed by the high-ambiguity-driven protein–protein docking HADDOCK web server.39 The initial coordinates of CaM and cSH2 were obtained from Protein Data Bank (PDB) entries 5J03 and 1BFJ for CaM and cSH2, respectively. The CSPs observed for CaM and cSH2 upon complex formation were used to define ambiguous residue interaction restraints (AIRs) at the interface. Active residues were defined as those exhibiting intermolecular NOEs, and passive residues were defined as all other surface accessible residues (relative residue accessible surface area of >55% for either side-chain or backbone atoms). Passive residues were automatically selected for both proteins by the HADDOCK web server. Residues Phe65–Lys94 in CaM were considered semiflexible. The cSH2 semiflexible part is from Glu614 to Arg631. Rigid body energy minimizations generated 1000 structures; 200 structures with the lowest energy were used for semiflexible simulated annealing and explicit water refinement. Clustering of the final 200 structures was performed by the HADDOCK web server. The score of HADDOCK is defined as a weighted sum of intermolecular electrostatic (Elec) and van der Waals (vdW) energies, buried surface area (BSA), desolvation energy (Desolv), and AIR energy. The desolvation energy is calculated using the atomic desolvation parameters.40 The final ensemble of 200 solutions was analyzed and clustered on the basis of the pairwise root-mean-square deviation (RMSD) matrix calculated over the backbone atoms of the interface residues of CaM after fitting on the interface residues of cSH2. This new way of calculating RMSD in HADDOCK results in high values that emphasize the differences between docking solutions. The clustering was performed using a 7.5 Å cutoff.
RESULTS
CaM Binds to the cSH2 Domain of p85.
Earlier, co-immunoprecipitation and affinity chromatography showed that Ca2+-loaded CaM interacts with the p85 regulatory subunit of PI3Kα, enhancing its activity.21 CaM specifically targets both SH2 domains of p85, with the cSH2 domain presenting a higher binding affinity, implying that cSH2 may serve as the main binding site for CaM in the regulation of PI3Kα activation. To further confirm direct binding between recombinant purified CaM and cSH2 and to determine the binding stoichiometry, we incubated CaM with cSH2 at a 1:1 molar ratio in the presence of EDC, a bifunctional cross-linking agent that covalently links amide and carboxyl groups. The cross-linking reaction yielded a 33 kDa 1:1 protein–protein complex that was detected by immunoblotting in the CaM–cSH2 mixture (Figure 2A). This complex was absent in the negative controls of CaM or cSH2 alone. The presence of the complex confirms the direct interaction between CaM and cSH2, consistent with earlier observations. Our cross-linking experiment on CaM with nSH2 failed to yield binary complex formation (Figure 2B), suggesting that the CaM–nSH2 interaction is low-affinity or that the amine and carboxyl groups suitable for EDC cross-linking are lacking in the CaM–nSH2 interface.
Figure 2.

EDC cross-linking shows CaM (50 μM) binding to cSH2 (50 μM). Western blot analysis for (A) CaM–cSH2 and (B) CaM–nSH2 interaction. Lanes 1 and 2 in panels A and B show CaM alone (18 kDa) in the absence and presence of EDC, respectively. Lanes 3 and 4 denote cSH2 (15 kDa) in panel A in nSH2 (13 kDa) in panel B alone in the absence and presence of EDC, respectively. Lane 5 shows CaM incubated with an equimolar amount of cSH2 in panel A and nSH2 in panel B in the presence of EDC. The band representing the 33 kDa CaM–cSH2 complex is marked with an arrow. No CaM–nSH2 complex is observed in panel B.
Structural Rearrangement of cSH2 upon Binding to CaM.
TFM experiments are commonly used to identify conformational changes by sensing tryptophan residues in protein–protein interactions.41 Upon titration, the transition of a tryptophan residue from the hydrophobic interior to the solvent accessible surface can be detected by the increase in the wavelength or the amplitude of maximum emission of the tryptophan fluorescence. The presence of a single tryptophan residue in cSH2 (Trp624) in the βA region (see the domain structure in Figure 1) and the absence of tryptophan in CaM enable probing of tryptophan fluorescence during the cSH2 conformational change upon binding to CaM. Reference-subtracted tryptophan emission spectra of cSH2 titrated with CaM show an increase in fluorescence intensity (Figure 3). This indicates that the cSH2 domain exhibits a conformational rearrangement due to CaM binding. Addition of cSH2 beyond a 1:2 molar ratio to CaM resulted in protein aggregation and decreased the tryptophan emission (data not shown). Our data suggest that TFM can identify conformational rearrangements within the CaM–cSH2 complex.
Figure 3.

Tryptophan fluorescence suggests conformational rearrangement in cSH2 upon binding CaM. CaM at 10 μM was titrated with cSH2 at molar ratios of 1:0.5 (purple), 1:1 (blue), 1:1.5 (yellow), and 1:2 (red). (A) Intensity of fluorescence emission of the single tryptophan in cSH2 (Trp624) in the wavelength range of 300–500 nm postexcitation at 280 nm. The reference emission traces with no CaM are subtracted. The color of the lines corresponds to the concentration of cSH2. (B) Changes in tryptophan fluorescence intensity of cSH2 at 325 nm during titration with cSH2.
NMR Investigations of Interaction of CaM with cSH2.
We first examined the impact of cSH2 binding on CaM using 15N HSQC NMR spectroscopy (Figure 4A). This technique allows us to determine the intermolecular binding interfaces and dissociation constants through analysis of chemical shifts and 15N line widths. While 1H–15N chemical shifts sense changes in the electromagnetic environment due to binding, 15N line widths report on nuclear relaxation rates that reflect the kinetics of protein–protein complex formation and its tumbling rate. Addition of cSH2 to [15N]CaM at 1:0.5, 1:1, 1:1.5, and 1:2 molar ratios induced CSPs in both N- and C-lobes of CaM (Figure 4B). These CSPs were small but statistically significant with a maximum of 5 ppb in magnitude for Asn137. The effect of cSH2 binding becomes more pronounced on 15N line widths in CaM. Titration of [15N]CaM with cSH2 resulted in a continuous increase in the average 15N line widths from 16.54 ± 5.2 Hz for CaM alone to 19.9 ± 6.5, 21.7 ± 3.9, and 23.5 ± 5.6 Hz at 1:0.5, 1:1, and 1:2 CaM:cSH2 molar ratios, respectively.
Figure 4.

NMR titration of [15N]CaM with cSH2 shows effects of binding on chemical shifts and signal line widths. (A) Superimposition of 15N HSQC spectra of CaM (100 μM) in the absence of unlabeled cSH2 (blue) and in the presence of unlabeled cSH2 at molar ratios of 1:0.5 (red), 1:1 (orange), 1:1.5 (yellow), and 1:2 (green) (CaM:cSH2). Signal disappearances for CaM Ala102 and Val136 at a 1:2 molar ratio (CaM–cSH2) are shown in spectral insets. The red arrows mark the direction of chemical shift changes of CaM Asn137. (B) Normalized HN chemical shift perturbations at molar ratios (CaM–cSH2) from 1:0.5 to 1:2. The red arrows represent the disappearance of resonances in CaM due to binding cSH2, and the horizontal line marks the average value of chemical shift perturbations plus one standard deviation.
Nonlinear regression fitting of the change in line width values to the “one-site binding model”37 yielded a dissociation constant (KD) of 18.7 ± 9.4 μM (Figure 5A). This KD determination was done using measured changes in 15N line widths in NMR titration experiments in a nonlinear regression fitting to the “one-site binding model”.37 Because of their small magnitude, CSPs were difficult to use for KD calculation. However, the CSPs for Lys75, Ala102, and Glu139 produced suitable profiles for the fitting procedure (Figure 5B). The KD resulting from these measurements was 13.9 ± 0.7 μM, similar to the value obtained from the analysis of 15N line widths. Signal broadening beyond detection caused by the intermediate exchange on the NMR time scale was observed for CaM’s N-lobe residues, Gln49, Asp64, and Lys75, at a 1:0.5 molar ratio (Figure 4B, top panel). A further increase in cSH2 concentration until a 1:2 molar ratio was reached resulted in the disappearance of 1H–15N signals for CaM’s N-lob residues, Lys13, Leu32, Ile52, Gly61, Glu67, and Leu69, the residues at the linker region, Thr70 and Met71, and C-lobe residues, Asp93, Ala102, Leu105, His107, Asp122, Glu123, Val136, and Met145 (Figure 4B, bottom panel). Residues in CaM presenting the disappearance of resonances appear to engage in interactions with cSH2 or shift allosterically due to cSH2 binding. Allosteric effects and possible multiple binding modes contribute to chemical shift changes in cSH2 upon binding to CaM, complicating the analysis of CSPs.42 Our results reveal that the N- and C-lobe hydrophobic pockets along with the flexible linker region of CaM participate in cSH2 binding.
Figure 5.

(A) Dissociation constant, KD, measured in NMR shows weak binding affinity for interaction of CaM with cSH2. The graph shows changes in NMR signal intensities measured in the 15N HSQC spectrum of CaM in the presence of different concentrations of cSH2. The KD was determined and optimized by nonlinear regression fitting to a “one-site binding model”.37 (B) KD calculated by NMR CSPs. The KD value, 13.9 ± 0.7 μM, is similar to the value of 18.7 ± 9.4 μM determined from 15N line widths for CaM. The KD value is shown on the graph.
Titration of [15N]cSH2 showed that CaM induced significant line broadening in the HSQC spectrum (Figure 6A), although the CSPs were small as in the [15N]CaM titration (Figure 6B). The 1H–15N signals for cSH2’s residues, Ala658, Cys659, Lys674, Gly680, Phe681, Glu683, Val695, and Gln699, disappeared upon addition of CaM at a 1:0.5 cSH2:CaM molar ratio (Figure 6B, top panel). Further increasing the CaM concentration resulted in a progressive loss of signals from the spectrum. A 2-fold molar excess of CaM led to extensive line broadening in the spectrum of [15N]cSH2, causing the disappearance of signals for Asp620, Glu621, Val626, Gly627, Lys633–Glu635, Arg639, Gly640, Arg642, Gly644, Phe646–Arg649, Ser651, Gln654, Tyr657–Ser660, Val662–Gly665, Val667–Lys674, Tyr679–Glu683, Asn686–Ser689, Leu691–Thr701, Leu703, Ser709–Asn711, Thr713–Ala715, Val718, and Arg723 (Figure 6B, bottom panel). Many residues in cSH2 were involved in signal disappearance, indicating that almost all residues in cSH2 shifted allosterically due to conformational changes upon binding to CaM. Our results suggest that the cSH2 domain is a flexible molecule as is CaM, presenting multiple binding modes of the interaction with CaM through adjustment of its conformation.
Figure 6.

NMR titration of [15N]cSH2 with CaM shows the effects of binding on chemical shifts and signal line widths. (A) Superimposition of 15N HSQC spectra of cSH2 (100 μM) in the absence of unlabeled CaM (blue) and in the presence of unlabeled CaM at molar ratios of 1:0.5 (red), 1:1 (orange), 1:1.5 (yellow), and 1:2 (green) (cSH2:CaM). Signal disappearances for cSH2 Phe681 at a 1:2 molar ratio (cSH2:CaM) are shown in spectral insets. The red arrows mark the direction of chemical shift changes of cSH2 Thr645. (B) Normalized HN chemical shift perturbations at molar ratios (cSH2:CaM) from 1:0.5 to 1:2. The red arrows represent the disappearance of resonances in cSH2 due to binding CaM, and the horizontal line marks the average value of chemical shift perturbations plus one standard deviation.
Interfacial Contacts between CaM and cSH2.
To identify direct binding sites between cSH2 and CaM, we performed a 15N-edited NOESY-HSQC experiment on [15N]CaM alone and in the presence of cSH2 at a CaM:cSH2 molar ratio of 1:2. This approach was preferred over the filtered NOESY to identify less ambiguous intermolecular 1HN–1HN NOEs because of its higher sensitivity. Changes in the NOE patterns for CaM upon binding to cSH2 were examined, and potential intermolecular NOEs were identified between Met109 in CaM, which does not exhibit significant chemical shift changes during the titration, and cSH2 residues Gln654 and Cys656 (Figure 7). To determine the structure of the CaM–cSH2 complex, we employed the HADDOCK software39 in conjunction with the intermolecular NOEs. Three initial 1HN–1HN NOE restraints, including the linker and both lobes of CaM, were assigned to CaM Glu67:cSH2 Leu694, CaM Met71:cSH2 Thr701, and CaM Lys148:cSH2 Thr675 pairs. Iterative docking and analysis of the NOESY spectra allowed assignment of additional intermolecular NOEs, involving 1HN protons. The linker region of CaM is flexible in solution43,44 and was defined as flexible during the docking. In addition, the N-terminal portion of cSH2 was defined as flexible to allow repositioning of Trp624 in cSH2 upon complex formation, as observed in the tryptophan fluorescence experiments (Figure 3). Letting the N-terminus of cSH2 adjust its structure was also necessary to accommodate the observed NOEs between Ile9 in CaM and cSH2 residues Glu614, Leu616, and Pro617.
Figure 7.

15N-edited NOESY-HSQC spectra exhibit intermolecular contacts between CaM (100 μM) and cSH2 (200 μM). (A) Selected regions show NOEs between amide groups of CaM Met109 from a three-dimensional 15N NOESY experiment acquired on 0.1 mM 15N-labeled CaM in the presence of 0.2 mM unlabeled cSH2. (B) The absence of intermolecular NOEs in the reference 15N-edited NOESY-HSQC spectrum of CaM alone is evident. Uniquely assigned intermolecular NOEs are labeled. Blue boxes denote dramatic signal broadening of the CaM peak during the HSQC titrations.
The model structure of the CaM–cSH2 complex using HADDOCK in conjunction with intermolecular NOEs presents a compact structure with all three CaM domains, the linker and both lobes, involved in the interaction with cSH2 (Figure 8A). The predicted structure by HADDOCK is one of the best possible models representing the CaM–cSH2 complex. Other complex structures with different dimeric interfaces can also be possible due to multiple binding modes of the interactions between CaM and cSH2. For the predicted structure, we observed a salt bridge at the CaM–cSH2 dimeric interface, Arg74–Glu621, and three intermolecular hydrogen bonds, Asn53–Thr701, Glu54–Asn686, and Glu127–Asn673. In addition, the hydrophobic interactions occurred for the intermolecular residue pairs, Met36–Leu687, Ala57–Leu696, Leu69–Leu616, and Met71–Leu687, and the hydrophilic interactions for the Gln41–Tyr685, Thr79–Thr677, Gln143–Thr675, Gln143–Thr677, and Thr146–Tyr679 pairs. It is noteworthy that allosteric effects and multiple binding modes precluded the use of all observed NOEs in the docking. In total, 19 of 43 intermolecular NOEs were mutually exclusive with the rest of the restraints and incompatible with the model of the CaM–cSH2 complex (Table S1). For example, the 1HN of Ile9 in CaM exhibits NOEs with 1Hβ protons of residues Glu614, Leu616, and Pro617 in cSH2, while only the contacts with residues Glu614 and Leu616 could be simultaneously satisfied (Figure 8B).
Figure 8.

(A) Predicted structure for the CaM–cSH2 interaction (middle). Four different types of interactions are highlighted, representing the residue pairs for the salt bridge (top left), hydrogen bond (top right), hydrophobic (bottom left), and hydrophilic (bottom right) interactions. Transparent surface representations for the hydrophobic and hydrophilic interactions denote clustered residue pairs. (B) Interacting residue pairs by NOEs mapped on the model structure of the CaM–cSH2 complex (left) and highlights of these residue pairs (right). The dotted circles denote clustered residue pairs on the CaM domains.
Mutations in the cSH2 Domain Abrogated Interaction.
In addition to PI3K, the only other reported example of binding of CaM to SH2 is activation of Src kinase.28 This interaction plays a critical role in the resistance of pancreatic cancer to Fas death receptor-induced apoptosis. The mechanism of the interaction has been deduced from the results of site-directed mutagenesis and peptide inhibition studies.28,29 The SH2 domain of Src binds CaM through its basic 1–5–10 motif, stimulates phosphorylation of Src on Tyr416, and leads to activation of the kinase. We analyzed the amino acid sequences of the cSH2 domain and found two potential sequences have the characteristics of CaM-binding motifs.45,46 Sequences of the CaM-binding motif in cSH2 domain are Val663–Ile672 (VDGEVKHCVI) and Leu687–Leu696 (LYSSLKELVL) (Figure S1). They both contained the 1–5–10 CaM-binding motif, where the numbers 1, 5, and 10 designate the positions of conserved hydrophobic residues.46 The second motif is located at the CaM–cSH2 interaction interface according to our predicted model, while the first motif is not (Figure 9A). The CaM–cSH2mutant cross-linking experiment shows that cSH2V663K/V667N (cSH2mutant‑1) could form a complex with CaM, but the amount of complex decreased when CaM interacts with cSH2V663K/V667N/L687F/L691V/L696N (cSH2mutant‑2). In both cases, the mutations affected the cross-linking efficiency in comparison with the wild type control (Figure S2). Substitution of one CaM-binding motif had a weaker effect on protein–protein interactions than substitution of both CaM-binding motifs (Figure 9B). These results fit the model in which cSH2 interacts with the C-terminus of CaM through its one CaM-binding motif; at the same time, the N-terminus and linker part of CaM also contribute forces in binding cSH2.
Figure 9.

(A) CaM-binding motifs in cSH2 (left) and highlights of the residues involved in the motifs (right). The first motif (colored green surface) involves the sequence Val663–Ile672 (VDGEVKHCVI), and the second motif (blue surface) contains the sequence Leu687–Leu696 (LYSSLKELVL). In the right panel, an asterisk represents the residues to be selected for the mutant experiments. (B) Western blot analysis for the interaction of CaM (50 μM) with cSH2mutants (50 μM). EDC cross-linking shows that both cSH2V663K/V667N (cSH2mutant-1) and cSH2V663K/V667N/L687F/L691V/L696N (cSH2mutant-2) mutants weaken the CaM–cSH2 interaction. For the results of cSH2mutant-2 on the left-hand side, lanes 1 and 2 show 50 μM CaM alone (18 kDa) in the absence and presence of EDC, respectively. Lanes 3 and 4 denote 50 μM cSH2mutant-2 alone (15 kDa) in the absence and presence of EDC, respectively. Lane 5 shows 50 μM CaM incubated with an equimolar amount of cSH2mutant-2 in the presence of EDC. The band representing the 33 kDa CaM–cSH2mutant-2 complex is marked with an arrow. The same for cSH2mutant-1 is shown on the right-hand side.
DISCUSSION
Although CaM regulates non-RTKs,21,28,45 detailed insight into its mechanism of action is in its infancy. On the basis of the available data relating to PI3K activation, it seems plausible that CaM might act to release inhibitory interactions between the SH2 domains and the p110 subunit3 through either CaM phosphorylation on Tyr9932 or by unmodified CaM.21 In addition to PI3K, the only other reported example of binding of CaM to SH2 is found in activation of Src kinase.28 This interaction plays a critical role in the resistance of pancreatic cancer to Fas death receptor-induced apoptosis. The CaM–SH2 domain binding mechanism has been deduced by site-directed mutagenesis and peptide inhibition studies.28,29 The SH2 domain of Src binds CaM through its basic 1–5–10 motif, stimulates phosphorylation of Src on Tyr416, and leads to activation of the kinase. To test if known CaM-binding motifs45,46 are common in SH2 domains of non-RTKs, we analyzed the amino acid sequences of 20 different kinases.47 We found that SH2 domains in all but four kinases (nSH2 of SYK, ITK/TSK, cytoplasmic tyrosine-protein kinase BMX, and nSH2 of p85α) contained 1–5–8–14, 1–8–14, or 1–5–10 CaM-binding motifs, where the numbers 1, 5, 8, 10, and 14 designate the positions of conserved hydrophobic residues.46 This suggests that CaM may broadly regulate non-RTKs through their SH2 domains.
The sequences of SH2 domains in mammalian class IA PI3K adaptor subunits reveal that two CaM-binding motifs are present in the cSH2 domain of p55γ, while the cSH2 domain of p85β contains only one CaM-binding motif. Each of the nSH2 domains of p55γ and p85β possesses two CaM-binding motifs. While nSH2 of p85α lacks CaM-binding motifs, two 1–5–10 CaM-binding motifs are present in cSH2 of p85α. p85α, p55α, and p50α are encoded by the same gene, PIK3R1.48 They are produced from mRNAs transcribed using alternative promoters; thus, the cSH2 domains of p55α and p50α have CaM-binding motifs identical to that of p85α, and neither has nSH2 CaM-binding motifs. In cSH2 of p85α, the first motif includes residues Val663–Ala676 in βD, and the other motif ranges from Leu687 to Gln699 in αB. We demonstrated that the second motif in αB of cSH2 participates in the interaction with CaM (Figure 8). This interaction involves bending of CaM to accommodate contacts between the hydrophobic pockets of both lobes and the hydrophobic αB helix of cSH2. Additional contacts are made between CaM and the second CaM-binding motif in strand βD of cSH2. This is similar to the interactions observed between CaM and the single CaM-binding motif in the SH2 domain of Src.29 CaM may bind proteins at multiple surfaces. Inspection of the PDB illustrates that for small target peptides, as well as proteins, CaM prefers α-helices. Both CaM and cSH2 are flexible molecules, suggesting that their interactions may present multiple binding modes of the interactions, and exploration of conformational ensemble states on a broad free energy landscape.
The activation of PI3Kα via SH2 domains is through the release of their autoinhibitory interactions with the p110 catalytic subunit, thereby together with K-Ras4B (and likely KRas4A49,50) activating the PI3Kα. Our recent work showed that pCaM binds stably to both nSH2 and cSH2 via conformational selection in a manner similar to that of the RTK pYXXM motif,51 suggesting that pCaM may be one of the key factors in PI3Kα activation.32 CaM without phosphorylation can bind cSH2 (and weakly nSH221), releasing the autoinhibition by competing with the p110 interaction and shifting the landscape to the open state.
Because of the presence of CaM-binding motifs in SH2 domains of p85α, p55α, p50α, p85β, and p55γ, we speculate that CaM can also interact with these isoforms. In fact, the in-cell experiments reporting on binding of CaM to PI3K did not distinguish between different isoforms of this kinase.21 In the crystal structure of the lipid kinase p110β/p85β, cSH2 makes an inhibitory contact with the C-terminal region of the p110β kinase domain.16,18 By affecting the C-terminal helix of p110β, cSH2 might also interfere with the binding of the kinase to the PM. Notably, the equivalent C-terminal helix in the primordial class III PI3K Vps34 interacts with the membrane phospholipids, and its deletion decreases the activity of the kinase.52 The region of cSH2 important for binding p110 is occupied by the N-terminal lobe of CaM. Thus, we propose that CaM may sequester cSH2 from the kinase domain of PI3Kβ and release the inhibition of the catalytic activity. The nSH2 domains in p85β and p55γ contain CaM-binding motifs and may similarly contribute to interaction with CaM.
CONCLUSIONS
To conclude, we identified CaM’s engaging its N- and C-lobes in interaction with two CaM-binding motifs of cSH2 in p85α. In response to CaM binding, cSH2 disengages from p110 and undergoes significant conformational changes, which expose the tryptophan residue. The nSH2 domain of p85α lacks the CaM-binding motifs, suggesting that CaM engages in binding nSH2 with a different interface. The β and δ isoforms of PI3K may also interact with CaM, because the nSH2 domains of p85β and p55γ also contain the CaM-binding motifs and may also participate in CaM-induced release of autoinhibition in the β and δ isoforms of PI3K, suggesting that CaM may activate these kinases through similar mechanisms.
Supplementary Material
Funding
The authors gratefully acknowledge the support from the National Natural Science Foundation of China (81322046, 81473137, and 21778037) and the Shanghai Rising-Star Program (13QA1402300) to J.Z. This project has been funded in whole or in part with Federal funds from the Frederick National Laboratory for Cancer Research, National Institutes of Health, under Contract HHSN261200800001E to R.N., H.J., and M.Z. and by National Cancer Institute Grant R01 CA188427 to V.G. The authors gratefully acknowledge financial support from the China Scholarship Council to G.W. for one year’s study abroad at the University of Illinois at Chicago.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.7b01130.
Figures S1 and S2 and Table S1 (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Nussinov R, Muratcioglu S, Tsai CJ, Jang H, Gursoy A, and Keskin O (2015) The Key Role of Calmodulin in KRAS-Driven Adenocarcinomas,. Mol. Cancer Res 13, 1265–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Nussinov R, Wang G, Tsai CJ, Jang H, Lu S, Banerjee A, Zhang J, and Gaponenko V (2017) Calmodulin and PI3K Signaling in KRAS Cancers. Trends Cancer 3, 214–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Burke JE, and Williams RL (2015) Synergy in activating class I PI3Ks. Trends Biochem. Sci 40, 88–100. [DOI] [PubMed] [Google Scholar]
- (4).Calleja V, Laguerre M, Parker PJ, and Larijani B (2009) Role of a novel PH-kinase domain interface in PKB/Akt regulation: structural mechanism for allosteric inhibition. PLoS Biol 7, e1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Manning BD, and Toker A (2017) AKT/PKB Signaling: Navigating the Network. Cell 169, 381–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Liao J, Planchon SM, Wolfman JC, and Wolfman A (2006) Growth factor-dependent AKT activation and cell migration requires the function of c-K(B)-Ras versus other cellular ras isoforms. J. Biol. Chem 281, 29730–29738. [DOI] [PubMed] [Google Scholar]
- (7).Cheng A, Wang S, Yang D, Xiao R, and Mattson MP (2003) Calmodulin mediates brain-derived neurotrophic factor cell survival signaling upstream of Akt kinase in embryonic neocortical neurons. J. Biol. Chem 278, 7591–7599. [DOI] [PubMed] [Google Scholar]
- (8).Zhao L, Zhao Q, Lu R, Fu Z, Zhu Z, Jia J, Wang S, Shi L, Jian X, and Yao Z (2008) Effects of tyroserleutide on gene expression of calmodulin and PI3K in hepatocellular carcinoma. J. Cell. Biochem 103, 471–478. [DOI] [PubMed] [Google Scholar]
- (9).Amzel LM, Huang CH, Mandelker D, Lengauer C, Gabelli SB, and Vogelstein B (2008) Structural comparisons of class I phosphoinositide 3-kinases. Nat. Rev. Cancer 8, 665–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Kok K, Geering B, and Vanhaesebroeck B (2009) Regulation of phosphoinositide 3-kinase expression in health and disease. Trends Biochem. Sci 34, 115–127. [DOI] [PubMed] [Google Scholar]
- (11).Kang S, Denley A, Vanhaesebroeck B, and Vogt PK (2006) Oncogenic transformation induced by the p110beta, -gamma, and -delta isoforms of class I phosphoinositide 3-kinase. Proc. Natl. Acad. Sci. U. S. A 103, 1289–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Edling CE, Selvaggi F, Buus R, Maffucci T, Di Sebastiano P, Friess H, Innocenti P, Kocher HM, and Falasca M (2010) Key role of phosphoinositide 3-kinase class IB in pancreatic cancer. Clin. Cancer Res 16, 4928–4937. [DOI] [PubMed] [Google Scholar]
- (13).Miled N, Yan Y, Hon WC, Perisic O, Zvelebil M, Inbar Y, Schneidman-Duhovny D, Wolfson HJ, Backer JM, and Williams RL (2007) Mechanism of two classes of cancer mutations in the phosphoinositide 3-kinase catalytic subunit. Science 317, 239–242. [DOI] [PubMed] [Google Scholar]
- (14).Burke JE, Perisic O, Masson GR, Vadas O, and Williams RL (2012) Oncogenic mutations mimic and enhance dynamic events in the natural activation of phosphoinositide 3-kinase p110alpha (PIK3CA). Proc. Natl. Acad. Sci. U. S. A 109, 15259–15264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Backer JM (2010) The regulation of class IA PI 3-kinases by inter-subunit interactions,. Curr. Top. Microbiol. Immunol 346, 87–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Zhang X, Vadas O, Perisic O, Anderson KE, Clark J, Hawkins PT, Stephens LR, and Williams RL (2011) Structure of lipid kinase p110beta/p85beta elucidates an unusual SH2-domain-mediated inhibitory mechanism. Mol. Cell 41, 567–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Holt KH, Olson L, Moye-Rowley WS, and Pessin JE (1994) Phosphatidylinositol 3-kinase activation is mediated by high-affinity interactions between distinct domains within the p110 and p85 subunits. Mol. Cell. Biol 14, 42–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Breeze AL, Kara BV, Barratt DG, Anderson M, Smith JC, Luke RW, Best JR, and Cartlidge SA (1996) Structure of a specific peptide complex of the carboxy-terminal SH2 domain from the p85 alpha subunit of phosphatidylinositol 3-kinase. EMBO J 15, 3579–3589. [PMC free article] [PubMed] [Google Scholar]
- (19).Hofmann BT, and Jucker M (2012) Activation of PI3K/Akt signaling by n-terminal SH2 domain mutants of the p85alpha regulatory subunit of PI3K is enhanced by deletion of its c-terminal SH2 domain. Cell. Signalling 24, 1950–1954. [DOI] [PubMed] [Google Scholar]
- (20).Jucker M, Sudel K, Horn S, Sickel M, Wegner W, Fiedler W, and Feldman RA (2002) Expression of a mutated form of the p85alpha regulatory subunit of phosphatidylinositol 3-kinase in a Hodgkin’s lymphoma-derived cell line (CO). Leukemia 16, 894–901. [DOI] [PubMed] [Google Scholar]
- (21).Joyal JL, Burks DJ, Pons S, Matter WF, Vlahos CJ, White MF, and Sacks DB (1997) Calmodulin activates phosphatidylinositol 3-kinase. J. Biol. Chem 272, 28183–28186. [DOI] [PubMed] [Google Scholar]
- (22).Nussinov R, Muratcioglu S, Tsai CJ, Jang H, Gursoy A, and Keskin O (2016) K-Ras4B/calmodulin/PI3Kalpha: A promising new adenocarcinoma-specific drug target?,. Expert Opin. Ther. Targets 20, 831–842. [DOI] [PubMed] [Google Scholar]
- (23).Villalonga P, Lopez-Alcala C, Bosch M, Chiloeches A, Rocamora N, Gil J, Marais R, Marshall CJ, Bachs O, and Agell N (2001) Calmodulin binds to K-Ras, but not to H- or N-Ras, and modulates its downstream signaling. Mol. Cell. Biol 21, 7345–7354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Abraham SJ, Nolet RP, Calvert RJ, Anderson LM, and Gaponenko V (2009) The hypervariable region of K-Ras4B is responsible for its specific interactions with calmodulin,. Biochemistry 48, 7575–7583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Wang MT, Holderfield M, Galeas J, Delrosario R, To MD, Balmain A, and McCormick F (2015) K-Ras Promote Tumorigenicity through Suppression of Non-canonical Wnt Signaling. Cell 163, 1237–1251. [DOI] [PubMed] [Google Scholar]
- (26).Jang H, Banerjee A, Chavan T, Gaponenko V, and Nussinov R (2017) Flexible-body motions of calmodulin and the farnesylated hypervariable region yield a high-affinity interaction enabling K-Ras4B membrane extraction. J. Biol. Chem 292, 12544–12559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Pacold ME, Suire S, Perisic O, Lara-Gonzalez S, Davis CT, Walker EH, Hawkins PT, Stephens L, Eccleston JF, and Williams RL (2000) Crystal structure and functional analysis of Ras binding to its effector phosphoinositide 3-kinase gamma. Cell 103, 931–943. [DOI] [PubMed] [Google Scholar]
- (28).Yuan K, Jing G, Chen J, Liu H, Zhang K, Li Y, Wu H, McDonald JM, and Chen Y (2011) Calmodulin mediates Fas-induced FADD-independent survival signaling in pancreatic cancer cells via activation of Src-extracellular signal-regulated kinase (ERK). J. Biol. Chem 286, 24776–24784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Tzou YM, Bailey SK, Yuan K, Shin R, Zhang W, Chen Y, Singh RK, Shevde LA, and Rama Krishna N (2016) Identification of initial leads directed at the calmodulin-binding region on the Src-SH2 domain that exhibit anti-proliferation activity against pancreatic cancer. Bioorg. Med. Chem. Lett 26, 1237–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Benaim G, and Villalobo A (2002) Phosphorylation of calmodulin. Functional implications. Eur. J. Biochem 269, 3619–3631. [DOI] [PubMed] [Google Scholar]
- (31).Stateva SR, Salas V, Benguria A, Cossio I, Anguita E, Martin-Nieto J, Benaim G, and Villalobo A (2015) The activating role of phospho-(Tyr)-calmodulin on the epidermal growth factor receptor,. Biochem. J 472, 195–204. [DOI] [PubMed] [Google Scholar]
- (32).Zhang M, Jang H, Gaponenko V, and Nussinov R (2017) Phosphorylated Calmodulin Promotes PI3K Activation by Binding to the SH2 Domains. Biophys. J 113, 1956–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Kovalevskaya NV, van de Waterbeemd M, Bokhovchuk FM, Bate N, Bindels RJ, Hoenderop JG, and Vuister GW (2013) Structural analysis of calmodulin binding to ion channels demonstrates the role of its plasticity in regulation. Pfluegers Arch 465, 1507–1519. [DOI] [PubMed] [Google Scholar]
- (34).Tidow H, and Nissen P (2013) Structural diversity of calmodulin binding to its target sites. FEBS J 280, 5551–5565. [DOI] [PubMed] [Google Scholar]
- (35).Siegal G, Davis B, Kristensen SM, Sankar A, Linacre J, Stein RC, Panayotou G, Waterfield MD, and Driscoll PC (1998) Solution Structure of the C-terminal SH2 Domain of the p85a Regulatory Subunit of Phosphoinositide 3-Kinase. J. Mol. Biol 276, 461–478. [DOI] [PubMed] [Google Scholar]
- (36).Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, and Bax A (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293. [DOI] [PubMed] [Google Scholar]
- (37).Wemmer DE, and Williams PG (1994) Use of nuclear magnetic resonance in probing ligand-macromolecule interactions. Methods Enzymol 239, 739–767. [DOI] [PubMed] [Google Scholar]
- (38).Kirby NI, DeRose EF, London RE, and Mueller GA (2004) NvAssign: protein NMR spectral assignment with NMRView. Bioinformatics 20, 1201–1203. [DOI] [PubMed] [Google Scholar]
- (39).Dominguez C, Boelens R, and Bonvin AM (2003) HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc 125, 1731–1737. [DOI] [PubMed] [Google Scholar]
- (40).Fernandez-Recio J, Totrov M, and Abagyan R (2004) Identification of protein-protein interaction sites from docking energy landscapes. J. Mol. Biol 335, 843–865. [DOI] [PubMed] [Google Scholar]
- (41).Roy S, and Bhattacharyya B (1995) Fluorescence spectroscopic studies of proteins. Subcell. Biochem 24, 101–114. [DOI] [PubMed] [Google Scholar]
- (42).Abu-Abed M, Das R, Wang L, and Melacini G (2007) Definition of an electrostatic relay switch critical for the cAMP-dependent activation of protein kinase A as revealed by the D170A mutant of RIalpha. Proteins: Struct., Funct., Genet 69, 112–124. [DOI] [PubMed] [Google Scholar]
- (43).Vogel HJ, and Zhang M (1995) Protein engineering and NMR studies of calmodulin. Mol. Cell. Biochem 149–150, 3–15. [DOI] [PubMed] [Google Scholar]
- (44).Wriggers W, Mehler E, Pitici F, Weinstein H, and Schulten K (1998) Structure and dynamics of calmodulin in solution. Biophys. J 74, 1622–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Yap KL, Kim J, Truong K, Sherman M, Yuan T, and Ikura M (2000) Calmodulin target database. J. Struct. Funct. Genomics 1, 8–14. [DOI] [PubMed] [Google Scholar]
- (46).Rhoads AR, and Friedberg F (1997) Sequence motifs for calmodulin recognition. FASEB J 11, 331–340. [DOI] [PubMed] [Google Scholar]
- (47).Park MJ, Sheng R, Silkov A, Jung DJ, Wang ZG, Xin Y, Kim H, Thiagarajan-Rosenkranz P, Song S, Yoon Y, Nam W, Kim I, Kim E, Lee DG, Chen Y, Singaram I, Wang L, Jang MH, Hwang CS, Honig B, Ryu S, Lorieau J, Kim YM, and Cho W (2016) SH2 Domains Serve as Lipid-Binding Modules for pTyr-Signaling Proteins. Mol. Cell 62, 7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Fruman DA (2010) Regulatory subunits of class IA PI3K. Curr. Top. Microbiol. Immunol 346, 225–244. [DOI] [PubMed] [Google Scholar]
- (49).Nussinov R, Tsai CJ, Chakrabarti M, and Jang H (2016) A New View of Ras Isoforms in Cancers,. Cancer Res 76, 18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Chakrabarti M, Jang H, and Nussinov R (2016) Comparison of the Conformations of KRAS Isoforms, K-Ras4A and K-Ras4B, Points to Similarities and Significant Differences. J. Phys. Chem. B 120, 667–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Chan TO, Rodeck U, Chan AM, Kimmelman AC, Rittenhouse SE, Panayotou G, and Tsichlis PN (2002) Small GTPases and tyrosine kinases coregulate a molecular switch in the phosphoinositide 3-kinase regulatory subunit. Cancer Cell 1, 181–191. [DOI] [PubMed] [Google Scholar]
- (52).Miller S, Tavshanjian B, Oleksy A, Perisic O, Houseman BT, Shokat KM, and Williams RL (2010) Shaping development of autophagy inhibitors with the structure of the lipid kinase Vps34. Science 327, 1638–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.