

Abstract
Metatropic dysplasia (MD) is a rare skeletal dysplasia associated with heterozygous mutations in the TRPV4 gene. We describe a 28-month-old boy with knock-knees referred for metabolic investigation suspected of carrying vitamin D-resistant rickets. He has received regular vitamin D prophylaxis at the usual dose. Laboratory investigations revealed normal values for calcium, phosphorus and alkaline phosphatase. He was short (-3.5 SDS), his mental development was normal, and he started to walk at the age of 22 months. The diagnostic clue for the diagnosis of metatropic dysplasia was the presence of the hump back in the upper lumbar and lower thoracic vertebrae, in addition to a long and narrow chest. An X-ray survey of the skeleton revealed platyspondyly, dysplastic metaphyses with dumbbell appearance of the long bones, kyphoscoliosis, and narrow and elongated thorax with short ribs. This is the first patient with MD in the Republic of Macedonia. Knock-knees were the cause of his referral, as a peculiarity of his phenotype. The very presence of the hump back, and the dumbbell appearance of the long bones distinguished the MD from other bone dysplasias with similar characteristics. We believe that the presence of those two features can shorten the path to accurate diagnosis in the crowded field of overlapping skeletal dysplasias. The diagnosis of MD in this patient was further confirmed by the discovery of the mutation c.2396C>T; p.Pro799Leu (P799L) of the TRPV4 gene.
Keywords: A dumbbell appearance, Kyphosis, Metatropic dysplasia, TRPV4 gene mutation
Introduction
Metatropic dysplasia (MD; OMIM 156530) is a bone dysplasia with severe kyphoscoliosis and platyspondyly, severe metaphyseal enlargement, short stature and shortening of long bones [1]. The incidence of this disorder is not known, as less than a hundred cases have been reported so far [2,3].
Transient receptor potential vanilloid 4 channel (TRPV4) is a cation channel, unselectively permeable to calcium and widely expressed and involved in many different physiological processes. Strikingly heterozygous missense mutations of the TRPV4 gene cause several skeletal syndromes: brachyolmia, spondylometaphyseal dysplasia Kozlowski type, metatropic dysplasia, parastremmatic dysplasia, familial digital arthropathy and spondyloepimetaphyseal dysplasia Maroteaux type. On the other hand, heterozygous TRPV4 missense mutations cause peripheral neuropathy, hereditary motor and sensory neuropathy type IIC, congenital distal spinal muscular atrophy, and scapuloperoneal spinal muscular atrophy [4]. A concominant existence of bone dysplasia and neuropathy was also observed: fetal akinesia, understood as a form of neuropathy, was found in four patients with MD [5]. This report describes an unusual presentation of MD in a boy referred for knock-knees presumed to be a rickets sequel.
Patient Report. The proband was born to non consanguineous parents after an uneventful pregnancy, with no infections, smoking, medications or narcotics. The delivery was timely and uneventful. There was no family history of skeletal disorders. This 28-month-old boy with knock-knees was referred for metabolic investigation for a suspected vitamin D-resistant rickets. He got regular vitamin D prophylaxis at an appropriate dose. There was a mid-face hypoplasia with frontal bossing, his mental development was normal, but he started to walk late at the age of 22 months. On physical examination, he had knock-knees, long and narrow thorax, kyphoscoliosis (Figure 1). His height was -3.5 SDS and for his age. Laboratory investigations revealed normal values for calcium, phosphorus and alkaline phosphatase. An X-ray of the skeleton revealed platyspondyly, anteriorly rounded vertebral bodies, dysplastic metaphyses with dumbbell appearance of the long bones, kyphoscoliosis, narrow and elongated thorax with short ribs suggestive for a mild form of MD [Figure 2(A) and 2(B)] There was significant metaphyseal widening of the long bones of the upper [Figure 2(C)] and lower [Figure 2(D)] extremities, halberd-shaped proximal femurs, wide ilia, and hypoplastic acetabular roofs. The epiphyses were flat and quite hypoplastic throughout and the phalanges were short and widened.
Figure 1.
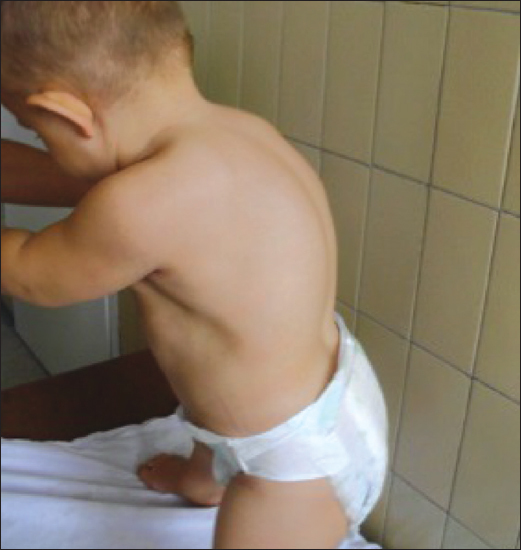
Clinical photograph of the boy: thoracic kyphosis.
The diagnosis of MD in this patient was further confirmed by direct DNA sequencing of the TRPV4 gene and the discovery of the c.2396C>T, p.Pro799Leu (P799L) mutation in a heterozygous state (Figure 3). As the parents did not carry the mutation, the mutation in the proband has arisen as a de novo event.
Figure 2 .
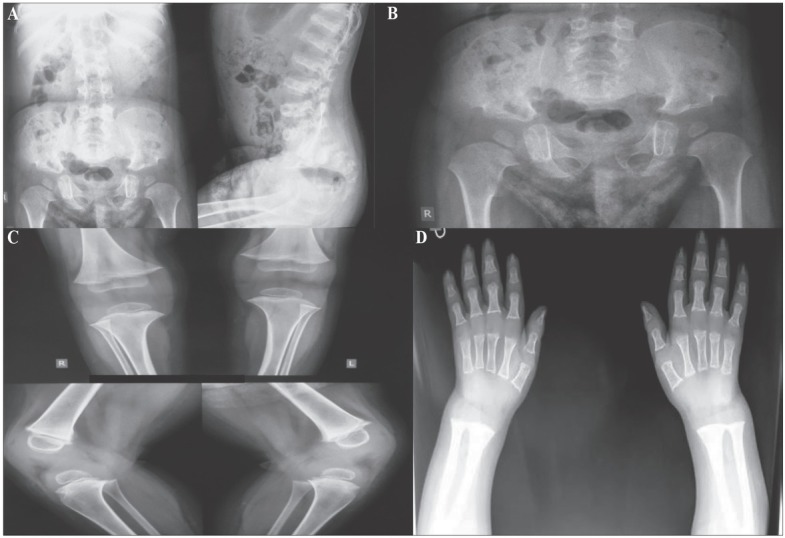
A) AP spine showing platyspondyly. Lateral spine showing kyphosis, narrow chest. The ribs are narrow posteriorly, but widened and irregular anteriorly and markedly sclerotic. There is a gibbous deformity. B) Short sacro-iliac notches. Hypoplasia of the bodies of iliac bones. The shape of the iliac bone is unique, a notch between the shortened iliac body and prominent crescent shaped iliac wing resembles a halberd. Shortened tubular bones with flared metaphyses. C) The chondroosseous junction is smooth at the knee, but there are cartilaginous remnants in the distal femora. Carpal ossification is significantly retarded. D) The middle and distal phalanges are short. Degenerative joint disease is found in the proximal interphalangeal joints. Hypoplastic/dysplastic carpal bones. Short metacarpals and phalanges with flared ends.
Figure 3.
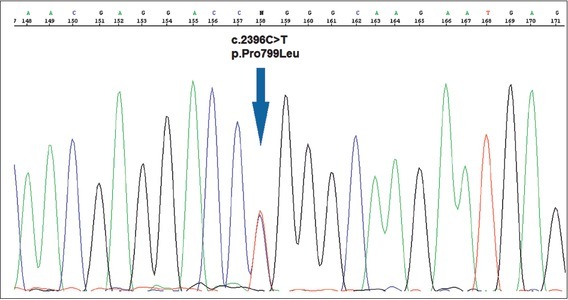
The c.2396C>T; p.Pro799Leu (P799L) mutation in the TPRV4 gene.
Discussion
The clinical diagnosis of a bone dysplasia can be rather complex. There are not only many overlapping skeletal dysplasias, but there are complex and overlapping phenotypes in the frame of one disease. There are three types of MD: 1) a nonlethal autosomal recessive form; 2) a nonlethal dominant form, and 3) a lethal form with possible autosomal recessive inheritance. However, the distinction between these three types has proven to be difficult.
Typically, the radiological manifestations of dumbbell aspect of the long bones, severe platyspondyly and severe scoliosis, are pointing towards the nonlethal autosomal recessive form of MD [1,6]. Nevertheless, some researchers have found a striking evolution of the skeletal anomalies with amelioration of the size of the long bones and significant improvement of the platyspondyly resulting in almost normal vertebral bodies at 15 years of age, was also reported [1]. On the contrary, Dai et al [7] found that the evolution of body proportion with age was not essential, while only the prominent joints were consistently found in MD. Andreucci et al [2] suggested that the TRPV4 skeletal dysplasias have a phenotypic overlap, even within the same family. To add to the complexity of the phenotype, a likely somatic mosaicism in an L618P patient with mild-er clinical presentation. Therefore, no definite genotype-phenotype correlation could be observed. The degree of activation of the TRPV4 gene was thought to predict the severity of the phenotype. Our patient had the knock-knees as a prominent feature. This feature was not an effect of rickets, as the laboratory investigation and X-rays have proven. This was not found in the other skeletal peculiarities reported as precocious calcification of hyoid and cricoid cartilage, severe hypoplasia of the anterior portion of the first cervical vertebrae, irregular and squared-off calcaneal bones, and microcalcifications in vertebral bodies and epiphyses [1]. In this report the leading sign pointing to the diagnosis of MD in our patient was the humpback (the gibbous) in the upper lumbar and lower thoracic vertebrae.
Fortunately, our patient did not have any of the described complications that can jeopardize the patient’s life. Laryngotracheal dysfunction can compromise aspiration, while the thoracic kyphoscoliosis can progress rapidly and can be out of surgical manageability. The short stature can be in the range of dwarfism 107-135cm. Cervical instability, hearing loss, degenerative joint deformity can add to the misery of the patients [6]. Our patient’s gibbous was not surgically corrected, so far, braces have been used.
Conclusions: This is the first patient with MD in the Republic of Macedonia. Knock-knees were the cause of his referral, as a peculiarity of his phenotype. The very presence of the humpback and the dumbbell appearance of the long bones distinguished the MD from other bone dysplasias with similar characteristics. We believe that the presence of those two features can shorten the path to accurate diagnosis in the crowded field of overlapping skeletal dys-plasias. The diagnosis of MD in this patient was further confirmed by the discovery of the mutation c.2396C>T [p.Pro799Leu (P799L)] of the TRPV4 gene.
Footnotes
Declaration of Interest. The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- 1.Genevieve D, Le Merrer M, Feingold J, Munnich A, Maroteaux P, Cormier-Daire V. Revisiting metatropic dysplasia: Presentation of a series of 19 novel patients and review of the literature. Am J Med Genet A. 2008;146A(8):992–996. doi: 10.1002/ajmg.a.32191. [DOI] [PubMed] [Google Scholar]
- 2.Andreucci E, Aftimos S, Alcausin M, Haan E, Hunter W, Kannu P. TRPV4 related skeletal dysplasias: a phenotypic spectrum highlighted byclinical, radiographic, and molecular studies in 21 new families. Orphanet J Rare Dis. 2011;6:37. doi: 10.1186/1750-1172-6-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bargiacchi S, Della Monica M, Biagiotti R, Andreucci E, Ciabattoni S, Poggi P. Metatropic dysplasia in third trimester of pregnancy and a novel causative variant in the TRPV4 gene. Eur J Med Genet. 2017;60(7):365–368. doi: 10.1016/j.ejmg.2017.04.007. [DOI] [PubMed] [Google Scholar]
- 4.Cho TJ, Matsumoto K, Fano V, Dai J, Kim OH, Chae JH. TRPV4-pathy manifesting both skeletal dysplasia and peripheral neuropathy: a report of three patients. Am J Med Genet A. 2012;158A(4):795–802. doi: 10.1002/ajmg.a.35268. [DOI] [PubMed] [Google Scholar]
- 5.Unger S, Lausch E, Stanzial F, Gillessen-Kaesbach G, Stefanova I, Di Stefano CM. Fetal akinesia in metatropic dysplasia: The combined phenotype of chon-drodysplasia and neuropathy? Am J Med Genet A. 2011;155A(11):2860–2864. doi: 10.1002/ajmg.a.34268. [DOI] [PubMed] [Google Scholar]
- 6.Kannu P, Aftimos S, Mayne V, Donnan L, Savarirayan R. Metatropic dysplasia: clinical and radiographic findings in 11 patients demonstrating long-term natural history. Am J Med Genet A. 2007;143A(21):2512–2522. doi: 10.1002/ajmg.a.31941. [DOI] [PubMed] [Google Scholar]
- 7.Dai J, Cho TJ, Unger S, Lausch E, Nishimura G, Kim OH. TRPV4-pathy, a novel channelopathy affecting diverse systems. J Hum Genet. 2010;55(7):400–402. doi: 10.1038/jhg.2010.37. [DOI] [PubMed] [Google Scholar]