

Abstract
Objective
To identify the genetic cause of disease in a form of congenital spinal muscular atrophy and arthrogryposis (CSMAA).
Methods
A 2-year-old boy was diagnosed with arthrogryposis multiplex congenita, severe skeletal abnormalities, torticollis, vocal cord paralysis, and diminished lower limb movement. Whole-exome sequencing (WES) was performed on the proband and family members. In silico modeling of protein structure and heterologous protein expression and cytotoxicity assays were performed to validate pathogenicity of the identified variant.
Results
WES revealed a homozygous mutation in the TRPV4 gene (c.281C>T; p.S94L). The identification of a recessive mutation in TRPV4 extends the spectrum of mutations in recessive forms of the TRPV4-associated disease. p.S94L and other previously identified TRPV4 variants in different protein domains were compared in structural modeling and functional studies. In silico structural modeling suggests that the p.S94L mutation is in the disordered N-terminal region proximal to important regulatory binding sites for phosphoinositides and for PACSIN3, which could lead to alterations in trafficking and/or channel sensitivity. Functional studies by Western blot and immunohistochemical analysis show that p.S94L increased TRPV4 activity-based cytotoxicity and resultant decreased TRPV4 expression levels, therefore involves a gain-of-function mechanism.
Conclusions
This study identifies a novel homozygous mutation in TRPV4 as a cause of the recessive form of CSMAA.
Hereditary neuropathies are a clinically and genetically heterogeneous group of diseases with an estimated prevalence of 1:2,500. Clinical manifestations of hereditary neuropathies include slow progressive distal weakness and muscle wasting with or without sensory loss. Hereditary neuropathies are classified into 3 broad categories on the basis of the clinical phenotype. Charcot-Marie-Tooth (CMT) disease or hereditary motor and sensory neuropathy typically exhibits involvement of both motor and sensory systems, hereditary sensory and autonomic neuropathy involves sensory deficits and/or autonomic dysfunction, and distal hereditary motor neuropathy (dHMN) predominantly involves motor deficits. These groups can be further classified into many subtypes depending on electrophysiologic criteria, pathologic defects, mode of inheritance, and molecular genetic defects. These diseases are genetically highly heterogeneous with mutations in at least 80 different genes associated with these subtypes.1,2 Despite this progress, 30%–70% of people affected with neuropathies do not have a genetic diagnosis because of clinical and genetic heterogeneity. Some of the disease symptoms can be controlled with generic drugs. However, the identification of the underlying genetic lesion is necessary for accurate disease prognosis, management, and family planning and may ultimately lead to personalized treatments.
Mutations in the transient receptor potential vanilloid 4 (TRPV4) cation channel gene are a rare cause of dominant inherited axonal neuropathies and skeletal dysplasias.3,4 TRPV4 mutations underlie a wide spectrum of clinical presentation and are associated with dHMN, scapuloperoneal spinal muscular atrophy (SPSMA), congenital spinal muscular atrophy and arthrogryposis (CSMAA), autosomal dominant axonal CMT type 2C, and congenital distal spinal muscular atrophy (dSMA).5 TRPV4-related neuropathies are frequently associated with vocal cord paralysis and occasionally with sensorineural hearing loss. TRPV4 mutations can also result in autosomal dominant skeletal dysplasias. Typically, different mutations in the TRPV4 gene are associated with either neuropathies or skeletal dysplasia; however, some patients exhibit both clinical phenotypes. So far, >20 different mutations in TRPV4 have been identified in patients with neuropathies. However, functional validation of pathogenicity for many of these variants is still lacking, thus preventing a conclusive genetic diagnosis in these patients. To improve the genetic diagnosis of patients affected with neuropathies, we use whole-exome sequencing (WES) combined with functional validation studies to establish the pathogenicity of variants identified by WES. This work has identified a TRPV4 homozygous mutation in a patient presenting with CSMAA. Follow-up functional studies reveal distinct disease mechanisms for different TRPV4 variants and provide novel insights that may inform future therapeutic strategies for these patients.
Methods
Standard protocol approvals, registrations, and patient consents
The proband, both parents, and the unaffected sibling were enrolled, and informed consent was obtained from participants in accord to an institutional review board–approved study at Boston Children's Hospital and Brigham and Women's Hospital.
Whole-exome sequencing
DNA extraction from blood samples was performed by the Research Connection Biobank Core (Boston Children's Hospital) using the QIAamp DNA Mini kit (Qiagen). WES was performed by the Yale Genome Center. DNA samples from the proband and parents were sent for WES. Samples were prepared as an Illumina sequencing library and enriched for exomic sequences using the Agilent V5 Sureselect kit. The captured libraries were sequenced using Illumina HiSeq 2000 Sequencers at Lab Corp. FASTQs generated from exome sequencing were filtered and aligned, and variants were filtered and annotated, as previously described.6 In short, first, we apply agnostic filtering, and we use pedigree-based inheritance mode filtering for de novo and recessive variants, as well as filter for rare variants only, based on large population databases (gnomAD, gnomad.broadinstitute.org). In the second step, we apply several knowledge-based filters on the genes (e.g., known functional and disease associations, expression level data, and model organism data) and on the variants (e.g., evolutionary conservation and structural constraints). Finally, the variants were prioritized during a crowdsourcing case conference of interdisciplinary audience.6
To validate the TRPV4 variant, PCR products for the proband, unaffected sibling, and parents were analyzed by standard Sanger sequencing (Dana-Farber/Harvard Cancer Center DNA Sequencing Facility).
In silico modeling of TRPV4 mutations
TRPV4 mutations were mapped onto the cryoelectron microscopy (cryoEM) structure of Xenopus tropicalis TRPV4 (PDB ID: 6bdj)7 after aligning the XtTRPV4 and human TRPV4 sequences using Clustal Omega. Figure 2 was generated using PyMOL (Schrodinger).
Figure 2. Structure modeling of TRPV4 mutations.

Structural model based on the cryoEM structure of Xenopus tropicalis TRPV4 (PDB ID: 6bbj). The structure, corresponding to residues 148–788 (human numbering), does not include disordered N- and C-terminal regions. The N-terminal region is schematized as a dotted line for each subunit, with the phosphoinositide-binding domain (PBD; residues 121–125 in human) and proline-rich region (PRR, residues 135–144) indicated. Residue positions for neuropathy-causing mutations (D62N, P97R, R186Q, R232C/S, R237G/L, R269C/H, R315W, R316C/H, and T701I) and disease mutations with mixed phenotypes (G78W, A217S, E278K, S542Y, V620Y, and T740I) are green and yellow, respectively. Compound heterozygous neuromuscular disease mutations N833S and E840K are not illustrated because they are in the disordered cytoplasmic C-terminal region (residues 789–871). The position of p.S94L is indicated in purple.
Cloning of mutant constructs
To generate TRPV4 mutant plasmids, p.S94L, p.R315W, and p.T701I variants were incorporated into TRPV4 cDNA in plasmid pcDNA3.1-TRPV4-FLAG using a Q5 site-directed mutagenesis kit (New England Biolabs). The mutagenesis primer sequences were S94L forward: 5′-TAT GAG TCC TTG GTG GTG CCT-3′, S94L reverse: 5′-TAG GGT GGA CTC CAG CAG-3′, R315W forward: 5′-GGC GGA CAT GTG GCG CCA GGA-3′, R315W reverse: 5′-TTC TTG TGG GGG TTC TCC GTC AGGT-3′, T701I forward: 5′-CTG CTG GTG ATC TAC ATC ATC-3′, and T701I reverse: 5′-GAT GAT GAA GAC CAC GGG-3′. The full coding sequences were confirmed using Sanger sequencing.
Cytotoxicity assay
Human HEK293 cells were cultured in Dulbecco's Modified Eagle Media supplemented with 10% fetal bovine serum. Cells were transfected with the respective TRPV4 or empty vector using Lipofectamine 3,000 (Thermo Fisher Scientific) and cultured in the presence or absence of HC-067047 (5 μM). Cell death analysis was performed 24 hours after transfection using the Cytotoxicity Detection Kit (Roche Diagnostics, Indianapolis, IN) at 25°C according to the manufacturer's instructions as described previously (n = 3).8 Cytotoxicity was calculated by subtracting the absorbance of the background control (normal media) from the absorbance of the experimental samples (supernatant from cells transfected with wild-type [WT] or mutant plasmid). Results are expressed as a percentage relative to the absorbance of the high control (supernatant from cells treated with lysis buffer) as Cytotoxicity (%) = Experimental value-control (normal media)/high control-control (normal media) × 100. Statistical significance was determined using unpaired the Student t test.
Western Blot
HEK293 cells were transfected with the respective vector using polyethylenimine (PEI to DNA ratio of 3:1). Cells were incubated for 12 hours at 37°C, then shifted to 33°C, and harvested 48 hours after transfection. Western blots were performed according to standard protocols, using 1:2,000 M2 anti-FLAG (Sigma, F-3165) to detect TRPV4, 1:10,000 anti-GAPDH (Abcam, 8245), and 1:1000 alkaline phosphatase conjugated anti-mouse secondary antibody (Sigma, A3562). Membranes were developed using the 1-Step NBT/BCIP reagent (Thermo Fisher Scientific, 34042). Densitometric analysis was performed with ImageJ (imagej.nih.gov/ij/). Data are presented as the mean ± SD, n = 3, 2-tailed t test, *p < 0.05.
Immunofluorescence analysis
For immunofluorescence, HEK293 cells were plated in 8-chambered slides, and transfections were performed with lipofectamine 3000 (Thermo Fisher Scientific). Twenty-four hours after transfection, cells were fixed in 4% paraformaldehyde, and immunofluorescence was performed as described previously9 using M2 anti-FLAG antibody (Sigma, 1:250) and sodium potassium ATPase antibody (Abcam 76020,1:100). Nuclear staining was performed using DAPI (BioLegend, 422801).
Results
Clinical presentation
The male proband was born to unrelated healthy parents of Puerto Rican descent after 37 weeks of gestation (figure 1A). He has a healthy sister. The proband was delivered by cesarean section because of breech position and arthrogryposis multiplex congenita on ultrasound. At birth, the proband exhibited a right clubfoot, a left congenital vertical talus, and bilateral flexion deformities of the knees and torticollis. X-rays showed dislocated hips with wide proximal femurs. Ultrasound revealed that the patient exhibited dysplastic acetabuli on both sides. An ultrasound of the spine was performed, which showed that the spinal cord was at the L3 level that is slightly lower than the expected level. Bilateral flexible laryngoscopy was performed through the left naris that showed nonobstructive adenoid hypertrophy, normal tongue base, and structurally normal larynx with no evidence of laryngomalacia. Vocal cords were clearly visible and appeared to be immobile bilaterally in the paramedian position. The proband also exhibited inspiratory stridor. Examination of the right and left tympanic membranes showed an evidence of middle ear effusions bilaterally and normal external ear and external auditory canals. Visual reinforced audiometry showed a moderate conductive hearing loss for at least 1 ear at 500 and 2000 Hz. Tympanograms were flat bilaterally consistent with clinical findings.
Figure 1. Identification of TRPV4 homozygous mutation.

(A) Pedigree of the family of the affected patient; the proband is indicated by the arrow. (B) Sanger sequencing pherograms show homozygosity for c.281C>T in TRPV4 in the proband and heterozygosity in both parents. The position of the variant is marked as *. (C) Protein sequence alignment of TRPV4 orthologs showing conservation in the region including p.S94L in higher vertebrates. (D) Schematic diagram of the TRPV4 protein demonstrating localization of p.S94L on the N-terminal intracellular region. Numbers 1–6 correspond to the 6 ankyrin repeats.
At age 2 years, the proband could sit independently and walk on his knees. However, he was unable to stand or walk independently. Except for lower limb movement, the proband has normal speech, fine motor and gross motor skills in his upper limbs, normal hearing, and visual development and exhibits a static condition. EMG examination revealed normal right sural sensory response and normal left ulnar-DV (Digit 5) sensory response. Normal left tibial abductor hallucis and median abductor pollicis brevis motor response for the age was also noted. Concentric needle examination of selected muscles showed fibrillation potentials in the right vastus lateralis muscle with late and fast firing motor unit potentials in left deltoid, right tibialis anterior, and right iliopsoas muscles. The electrophysiologic findings are suggestive of generalized motor axonopathy with coexisting denervation and reinnervation changes.
Whole-exome sequencing
WES was performed on the proband and parents. The presence of a single affected male individual in this family is consistent with autosomal recessive inheritance, X-linked inheritance, or with a dominant de novo mutation. Therefore, pedigree and population-based filtering was performed to identify potentially damaging homozygous, X-linked, compound heterozygous, and de novo variants. We only considered rare coding and splice variants that have <0.1–1% allele in the general healthy population in the gnomAD database (gnomad.broadinstitute.org) depending on the dominant or recessive inheritance mode that is consistent with the prevalence of the disease. In total, 15 potential genes carried a variant (table e-1, links.lww.com/NXG/A140) that were further prioritized using our knowledge-based and crowdsourcing pipeline as recently reviewed.6 The top candidate was a homozygous missense variant in TRPV4 c.281C>T; (p.S94L). Sanger sequencing confirmed the homozygosity of this variant in the proband (BGM0049-1) and heterozygous inheritance from both parents (BGM0049-2 and BGM0049-3) (figure 1B). The unaffected sister was found to be homozygous for the normal variant (not shown). The other 14 variants/genes could be ruled out by having low population constraints, known unrelated disease associations, unrelated protein functions, and lack of expression in relevant tissues (table e-1), corroborating the potential causal effect of the TRPV4 variant. The missense variant is at a highly conserved position in most of the vertebrate species and is localized to the N-terminus of the TRPV4 protein (figure 1, C and D).
Structural model
A recent cryoEM structure of Xenopus tropicalis TRPV4 was used to model the position of p.S94L and other previously described neuropathy-causing TRPV4 mutations (figure 2).7 Consistent with previous proteolysis protection10 and Nuclear Magnetic Resonance studies,11 the N-terminal region—up to residue 147 (human numbering), thus including p.S94—was disordered and not modeled in the cryoEM structure. However, p.S94L is located N-terminal to 2 important regulatory regions: a phosphoinositide-binding domain; residues 121–125 in humans) that interacts with phosphatidylinositol 4,5-bisphosphate (PIP2)10 and a proline-rich region (PRR; residues 135–144 in humans) that interacts with the SH3 domain of PACSIN3.12 PACSIN3 enhances TRPV4 trafficking to the cellular membrane. PIP2 binding enhances TRPV4 responses to several stimuli, whereas PACSIN3 decreases responses to these same stimuli. This antagonistic interaction between 2 key regulators, PIP2 and PACSIN3, is likely mediated through PACSIN3-mediated structural rearrangements of the PRR.10,13 Therefore, our structural modeling suggests that the p.S94L mutation could potentially affect the regulation and/or sensitivity of TRPV4.
Functional modeling of TRPV4 variants
Functional studies have shown a gain-of-function mechanism for previously known dominant mutations in TRPV4.14 To validate the pathogenicity of the p.S94L variant, functional studies were performed by analyzing the expression levels and localization of the mutant protein. To compare the magnitude of the functional deficits, known neuropathy-causing p.R269C and p.R315W TRPV4 mutants were analyzed in parallel. In addition, a mutation p.T701I has been reported as a cause of neuropathy. p.T701I is localized to the transmembrane domain S6 helix (figure 2), near the vanilloid-binding site in the homologous ion channel TRPV1.15 Currently, there are no functional studies establishing the pathogenicity of this variant in human diseases or TRPV4 protein functions. Therefore, we investigated the effect of p.S94L, p.R269C, p.R314W and p.T701I on the expression level and subcellular localization of TRPV4. To investigate the effect of different variants on protein expression, TRPV4-WT and mutant constructs were transiently expressed in HEK293 cells, and expression was analyzed by Western blot (figure 3). Protein analysis by Western blot showed reduced level of p.S94L protein and reduced levels of p.R269C and p.R315W. In contrast, p.T701I exhibited levels of protein expression similar to those of WT TRPV4 (figure 3, A and B). As previous studies have reported cytotoxic effects of mutant TRPV4 proteins, cytotoxicity analysis was performed in HEK293 cells transfected with control and mutant plasmids. Quantification showed significantly higher levels of cell death in the p.S94L transfected cells in comparison to WT TRPV4-transfected cells. Similarly, p.R269C and p.R315W also resulted in the increased cytotoxicity. Of interest, no changes in cytotoxicity were observed in p.T701I transfected cells in comparison to WT TRPV4-transfected cells. HEK293 cells transfected with control and mutant plasmids were also cultured in the presence of a TRPV4 channel antagonist (HC-067047) that reversed the cytotoxicity effects observed in p. S94L, p.R269C, and p.315W transfected cells (figure 3C). The presence of HC-067047 also resulted in an increased expression of p.S94L, p.R269C, and p.R315W; however, no expression changes were detected for p.701I protein (figure 3B). To examine the subcellular localization of p.S94L and other mutants, immunofluorescence analyses were performed (figure 4). Wild-type TRPV4 primarily localized to the plasma membrane as has been reported for endogenous TRPV4 protein. p.S94L and previously reported p.R269C and p.R315W exhibited localization to the plasma membrane. Interestingly, the p.T701I mutant protein showed a perinuclear localization pattern, and in contrast to WT protein, no protein was detected on the plasma membrane (figure 4).
Figure 3. Influence of TRPV4 mutations on protein expression level and cytotoxicity.

(A–B) Western blot (A) and quantification based on 3 experimental repeats (B) from HEK293 cells transiently expressing TRPV4-FLAG, p.S94L-FLAG, p.R269C-FLAG, p.R315W-FLAG, and p.T701I-FLAG or empty vector collected 48 hours after transfection and incubated in the absence or presence of HC-067047 (5 μM). Cell lysates were probed with antibodies against FLAG tag to detect transfected wild-type (WT) and mutant TRPV4 proteins and GAPDH as the loading control. (C) Cytotoxicity analysis in cells transfected with control or mutant TRPV4 plasmids. Differences in the protein levels or cytotoxicity were considered significant with *p < 0.05.
Figure 4. Subcellular localization of mutant TRPV4 proteins.
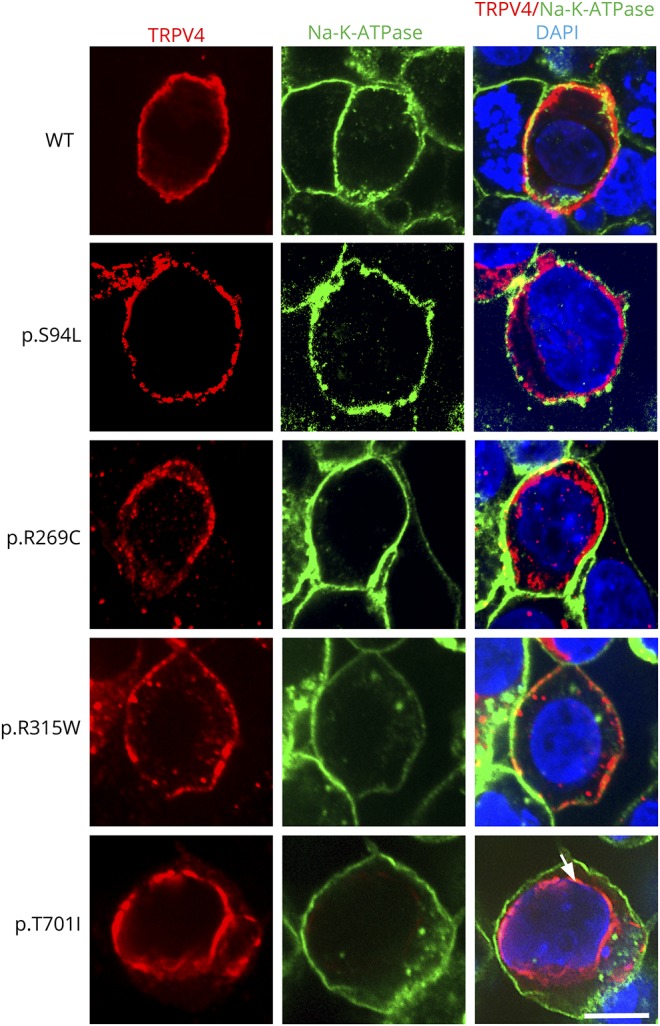
Representative images of HEK293 cells transiently expressing TRPV4-Flag, p.S94L-Flag, p.R269C-Flag, p.R315W-Flag, and p.T701I proteins. Immunofluorescence was performed with anti-Flag tag antibody to detect TRPV4-Flag proteins and Na-K-ATPase to label the plasma membrane. Nuclei were stained with DAPI. Scale bar = 20 μm.
Discussion
The distal hereditary motor neuropathies (dHMNs) are a genetically heterogeneous group of diseases characterized by distal lower motor neuron weakness. This is in contrast to CMT disease and the hereditary sensory neuropathies where sensory involvement forms a major component of the disease. Many forms of dHMN can exhibit a minor sensory component, and there is an overlap between axonal forms of CMT (CMT2) and dHMN. Autosomal dominant mutations in TRPV4 are associated with diverse clinical presentations including dHMN, SPSMA, CSMAA, and autosomal dominant axonal CMT type 2C (CMT2C) disease. TRPV4 mutations also result in several forms of autosomal dominant skeletal dysplasias. The proband exhibited arthrogryposis associated with distal muscle weakness with normal sensory response and therefore clinically diagnosed as CSMAA, a subtype of dHMN. Congenital arthrogryposis could result from a defect in skeletal or neuromuscular system. As both skeletal and neurologic deficiencies were present in the proband during early childhood, it is hard to predict which of these contributed to primary disease pathology. TRPV4 mutations are typically associated with either neuropathies or skeletal dysplasia; however, the presence of both skeletal and neurologic phenotypes has been seen in several previously reported cases.16 This suggests that genotype-phenotype association in TRPV4-related diseases is not stringent and may lead to both skeletal and neurologic abnormalities.
Several functional studies have demonstrated a gain-of-function mechanism for mutant TRPV4-mediated neuropathies.14 Mutant TRPV4 proteins frequently exhibit normal localization; however, they demonstrate increased calcium channel activity at both basal and activated levels. This results in increased intracellular calcium concentration leading to cytotoxicity.
Following a combination of WES and functional analysis, we identified a homozygous missense variant, p.S94L, in TRPV4 as a cause of a severe form of dHMN. This serine residue is highly conserved in mammals, and only 6 heterozygous and no homozygous individuals are reported in a control population of 246210 people (Genome Aggregation Database, gnomAD), implicating that this as a crucial amino acid. Here, we demonstrate that the TRPV4 p.S94L variant resulted in increased cytotoxicity, as previously reported for other neuropathy-causing pathogenic TRPV4 variants. A limitation of this work is that these studies were performed in HEK293 cells rather than neuronal-like cells. However, HEK293 cells have commonly been used to determine aberrant channel function for dominant TRPV4 mutations, allowing comparisons of our results with previous work.3 Furthermore, p.S94L mutation resulted in reduced levels of the mutant protein that were rescued by the TRPV4 channel antagonist as previously observed for other mutations in neuronal cells.14 The rescue of protein expression levels by the antagonist suggests that it is an increase in the channel activity of the p.S94L variant that causes its reduced expression levels. Previous work has also identified reduced stability of many dominant variants in the intracellular ankyrin repeat domain of TRPV4.17 A recent study also identified biallelic heterozygous mutations located in the C-terminus of the TRPV4 channel in 2 siblings with neuropathy associated with severe intellectual disability.18 These mutant proteins demonstrated normal membrane localization; however, they showed reduced channel function. The severe phenotype in the proband is consistent with the observed cytotoxicity of the p.S94L variant in the HEK293 cells.
The p.S94L mutation is located in the N-terminal intracellular region upstream of the ankyrin repeat domain. Previous studies have shown that dominant mutations in this region result in neuropathy in affected individuals.19 In comparison to previously known TRPV4 variants such as p.R269C and p.R315W, higher levels of p.S94L TRPV4 protein were detected in transfected HEK293 cells (figure 3, A and B). Similarly, we observed a lower level of cytotoxicity in p.S94L transfected cells in comparison with p.R269C and p.R315W transfected cells (figure 3C). Furthermore, the facts that the proband's parents who are heterozygous for the TRPV4 p.S94L variant did not exhibit any phenotype and that 6 heterozygous individuals are reported in the gnomAD database suggest reduced penetrance of this variant in the heterozygous state. Nonetheless, many heterozygous TRPV4 variants exhibit reduced penetrance in adult carriers, and for this reason, the parents of the proband were counseled to routine follow-up in a neurology clinic. Of interest, most human disease-causing variants studied in this work exhibited a reduced protein expression level, whereas the p.T701I variant altered the cellular localization of TRPV4 protein from the plasma membrane to the perinuclear area. This mislocalization may result in altered calcium channel function in affected patients. Together, these results demonstrate that different mutations in TRPV4 affect the channel function by different mechanisms. Understanding the functional alterations in mutant TRPV4 channels is essential not only for determining the pathogenicity of novel variants but also for the design of specific therapies as both agonists and antagonists of TRPV4 channels are available.
Data availability
The research material supporting this publication can be accessed by contacting the corresponding author at “vgupta@research.bwh.harvard.edu.”
Acknowledgments
The authors thank the Brigham Genomics Medicine for clinical and genomic data analyses. Brigham Genomics Medicine acknowledgments and full collaborator list are provided in Appendix 2.
Glossary
- CMT
Charcot-Marie-Tooth
- cryoEM
cryoelectron microscopy
- CSMAA
congenital spinal muscular atrophy and arthrogryposis
- dHMN
distal hereditary motor neuropathy
- PIP2
phosphatidylinositol 4,5-bisphosphate
- PRR
proline-rich region
- SPSMA
scapuloperoneal spinal muscular atrophy
- TRPV
transient receptor potential vanilloid
- WES
whole-exome sequencing
- WT
wild-type
Appendix 1. Author contributions

Appendix 2. Brigham Genomics Medicine co-investigators
Study funding
This work was supported by K01 AR062601 (VAG) and the Eleanor, Miles Shore Fellowship for Scholars in Medicine and Brigham and Women's Hospital Career Development Award (VAG), a Brigham Biomedical Research Institute Director's Transformative Award (Brigham Genomic Medicine), and the American Heart Association 16GRNT27250119 (RG).
Disclosure
J. Velilla, M.M. Marchetti, A. Toth-Petroczy, C. Grosgogeat, A.H. Bennett, N. Carmichael, and E. Estrella report no disclosures. B.T. Darras has served on the scientific advisory boards of Hoffman-La Roche, Cytokinetics, Inc, BMS, Inc, Sarepta Therapeutics, Biogen, AveXis, and PTC Therapeutics; has received travel funding and/or speaker honoraria from Biogen; has received publishing royalties from UpToDate; and has received research support from PTC Therapeutics, Valerion Therapeutics (MTM), PTC Pharmaceuticals, Sarepta Therapeutics, Biogen, Summit, AveXis, Roche, Fibrogen, Santhera, Cytokinetics, NIH/NINDS, SMA Foundation, Muscular Dystrophy Association, and the Slaney Family Fund for SMA. N.Y. Frank holds patents assigned to Brigham and Women's Hospital, Boston, MA, and licensed to Ticeba GmbH (Heidelberg, Germany) and Rheacell GmbH & Co. KG (Heidelberg, Germany); serves as an advisor to Veritas Genetics, Inc. (Danvers, MA); and her spouse Dr. Markus Frank serves as a scientific advisor to Ticeba GmbH and Rheacell GmbH & Co. KG. J. Krier has received research support from the NIH. Rachelle Gaudet's spouse is employed by and holds stock/stock options in Bristol-Myers Squibb, and she has received research support from the NIH. V.A. Gupta was supported by K01 AR062601 (VAG) and the Eleanor and Miles Shore Fellowship for Scholars in Medicine and Brigham and Women's Hospital Career Development Award (VAG). Disclosures available: Neurology.org/NG.
References
- 1.Rossor AM, Polke JM, Houlden H, Reilly MM. Clinical implications of genetic advances in Charcot-Marie-Tooth disease. Nat Rev Neurol 2013;9:562–571. [DOI] [PubMed] [Google Scholar]
- 2.Pareyson D, Saveri P, Pisciotta C. New developments in Charcot-Marie-Tooth neuropathy and related diseases. Curr Opin Neurol 2017;30:471–480. [DOI] [PubMed] [Google Scholar]
- 3.Landouré G, Zdebik AA, Martinez TL, et al. Mutations in TRPV4 cause Charcot-Marie-Tooth disease type 2C. Nat Genet 2010;42:170–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rock MJ, Prenen J, Funari VA, et al. Gain-of-function mutations in TRPV4 cause autosomal dominant brachyolmia. Nat Genet 2008;40:999–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fawcett KA, Murphy SM, Polke JM, et al. Comprehensive analysis of the TRPV4 gene in a large series of inherited neuropathies and controls. J Neurol Neurosurg Psychiatry 2012;83:1204–1209. [DOI] [PubMed] [Google Scholar]
- 6.Haghighi A, Cassa CA, Krier JB, et al. An integrated clinical program and crowdsourcing strategy for genomic sequencing and mendelian disease gene discovery. NPJ Genom Med 2018;3:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deng Z, Paknejad N, Maksaev G, et al. Cryo-EM and X-ray structures of TRPV4 reveal insight into ion permeation and gating mechanisms. Nat Struct Mol Biol 2018;25:252–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sullivan JM, Zimanyi CM, Aisenberg W, et al. Novel mutations highlight the key role of the ankyrin repeat domain in TRPV4-mediated neuropathy. Neurol Genet 2015;1:e29. doi: 10.1212/NXG.0000000000000029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bennett AH, O'Donohue MF, Gundry SR, et al. RNA helicase, DDX27 regulates skeletal muscle growth and regeneration by modulation of translational processes. PLoS Genet 2018;14:e1007226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garcia-Elias A, Mrkonjic S, Pardo-Pastor C, et al. Phosphatidylinositol-4,5-biphosphate-dependent rearrangement of TRPV4 cytosolic tails enables channel activation by physiological stimuli. Proc Natl Acad Sci U S A 2013;110:9553–9558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goretzki B, Glogowski NA, Diehl E, et al. Structural Basis of TRPV4 N Terminus Interaction with Syndapin/PACSIN1-3 and PIP2. Structure 2018;26:1583–1593.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cuajungco MP, Grimm C, Oshima K, et al. PACSINs bind to the TRPV4 cation channel. PACSIN 3 modulates the subcellular localization of TRPV4. J Biol Chem 2006;281:18753–18762. [DOI] [PubMed] [Google Scholar]
- 13.D'hoedt D, Owsianik G, Prenen J, et al. Stimulus-specific modulation of the cation channel TRPV4 by PACSIN 3. J Biol Chem 2008;283:6272–6280. [DOI] [PubMed] [Google Scholar]
- 14.Fecto F, Shi Y, Huda R, Martina M, Siddique T, Deng HX. Mutant TRPV4-mediated toxicity is linked to increased constitutive function in axonal neuropathies. J Biol Chem 2011;286:17281–17291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao Y, Cao E, Julius D, Cheng Y. TRPV1 structures in nanodiscs reveal mechanisms of ligand and lipid action. Nature 2016;534:347–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cho TJ, Matsumoto K, Fano V, et al. TRPV4-pathy manifesting both skeletal dysplasia and peripheral neuropathy: a report of three patients. Am J Med Genet A 2012;158A:795–802. [DOI] [PubMed] [Google Scholar]
- 17.Inada H, Procko E, Sotomayor M, Gaudet R. Structural and biochemical consequences of disease-causing mutations in the ankyrin repeat domain of the human TRPV4 channel. Biochemistry 2012;51:6195–6206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thibodeau ML, Peters CH, Townsend KN, et al. Compound heterozygous TRPV4 mutations in two siblings with a complex phenotype including severe intellectual disability and neuropathy. Am J Med Genet A 2017;173:3087–3092. [DOI] [PubMed] [Google Scholar]
- 19.Fiorillo C, Moro F, Brisca G, et al. TRPV4 mutations in children with congenital distal spinal muscular atrophy. Neurogenetics 2012;13:195–203. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The research material supporting this publication can be accessed by contacting the corresponding author at “vgupta@research.bwh.harvard.edu.”