

Abstract
In the past few decades, great conceptual and technological advances have been made in the field of toxicology, but animal model–based research still remains one of the most widely used and readily available tools for furthering our current knowledge. However, animal models are not perfect in predicting all systemic toxicity in humans. Extrapolating animal data to accurately predict human toxicities remains a challenge, and researchers are obligated to question the appropriateness of their chosen animal model. This paper provides an assessment of the utility of the methionine- and choline-deficient (MCD) diet fed animal model in reflecting human nonalcoholic steatohepatitis (NASH) and the potential risks of adverse drug reactions and toxicities that are associated with the disease. As a commonly used NASH model, the MCD model fails to exhibit most metabolic abnormalities in a similar manner to the human disease. The MCD model, on the other hand, closely resembles human NASH histology and reflects signatures of drug transporter alterations in humans. Due to the nature of the MCD model, it should be avoided in studies of NASH pathogenesis, metabolic parameter evaluation, and biomarker identification. But it can be used to accurately predict altered drug disposition due to NASH-associated transporter alterations.
Keywords: NASH, animal models, adverse drug reactions, MCD model
The review by Dr Choudhuri et al. deftly summarizes critical conceptual and technological advances that precede our understanding of toxicology and gives a comprehensive overview of the past and future development of toxicology. Toxicological research in animal models plays a critical role in expanding our current knowledge, even though there is concern as to whether animal data successfully recapitulates human toxicities. In this paper, we will share our perspective on the selection and validation of animal models to address specific research questions in the toxicological research related to nonalcoholic steatohepatitis (NASH).
Experimental animals have been extensively used in scientific research to understand particular biological phenomena and provide insight to human biological processes and human diseases. As a powerful investigative tool, utilization of animals greatly expanded the scope and complexity of biomedical research, such as generating fundamental knowledge of biological or pathological mechanisms, evaluating efficacy and safety of preclinical drugs and medical devices, and assessing the risk of industrial and consumer products as well as environmental toxicants (Rollin, 2014). The long history of using animal models in toxicology has demonstrated their utility, but also revealed that animal models are less than perfect in reflecting human diseases or in predicting the fate of drugs and chemicals and their subsequent effects in humans. A study regarding worldwide laboratory animal use estimated that about 115.3 million animals were used in 2005, which the authors considered a substantial underestimation (Taylor et al., 2008). Meanwhile, a total of $14 billion is spent on animal experimentation worldwide every year and about $2.8 billion of that is spent on toxicological studies (Hartung, 2009). Considering the tremendous investment in animal-based toxicological research, it is crucial to answer the question of how useful the current animal models are and how reliable they are. Olson et al. (2000) reported that only 43% of toxic effects that occurred in humans were correctly predicted in rodent models, which increased to 63% when non-rodent animals were included. Low concordance of toxicity between humans and animals demonstrates that the toxicological community faces a great challenge in animal-to-human extrapolation of experimental results.
General toxicities or adverse drug reactions (ADRs) in clinical drugs are generally categorized into 2 types: intrinsic (type A) and idiosyncratic (type B). Intrinsic ADRs are dose-dependent, and the risk of toxicity increases with increasing drug dose. Increasing dose, however, does not necessarily increase the risk of idiosyncratic ADRs, because they are generally associated with factors that are unique to individuals or certain subpopulations, such as genetic predispositions, age, sex, nutritional status, immune system, and underlying disease (Iasella et al., 2017; Pirmohamed et al., 1998; Ulrich, 2007). Though idiosyncratic ADRs are usually considered to be dose-independent, in some situations they result from altered pharmacokinetics but not pharmacodynamics of particular drugs. In other words, preexisting conditions in patients that lead to alterations of absorption, distribution, metabolism, and excretion (ADME) processes and effect drug metabolism and disposition can also increase the likelihood of ADRs. In this case, predictable type A ADRs may present in particular individuals or subpopulations within the normally safe therapeutic dose range. Current animal models are moderately reliable in predicting type A ADRs, and therefore most type A ADRs of drugs can be identified in preclinical animal studies or human clinical trials. Type B ADRs, however, usually are not revealed until a drug has been widely used, which suggests that current preclinical procedures of drug toxicity and ADR screening in animal models poorly reflect the heterogeneity of the human population. Additionally, identification of risk factors of type B ADRs mostly relies on awareness of potential predisposition conditions in human patients as well as knowledge of the underlying mechanism. This paper aims to present an example of utilizing an animal model in translating observations in human studies and predicting type B ADRs in a particular subpopulation and to provide a perspective of how to use current models to answer appropriate questions.
HIGHLIGHTS AND CHALLENGES IN USING METHIONINE- AND CHOLINE-DEFICIENT MODEL IN NASH RESEARCH
Nonalcoholic steatohepatitis is the progressive stage of the most prevalent chronic liver disease, nonalcoholic fatty liver disease (NAFLD), that affects about 25% of the world population (Younossi et al., 2016). Nonalcoholic steatohepatitis encompasses distinct pathological features such as extensive intracellular lipid deposition, hepatocellular ballooning, and inflammation and can progress to liver cirrhosis and hepatocellular carcinoma (Chalasani et al., 2012). The molecular mechanisms in the development of NASH are not fully understood, though the “multiple hit” hypothesis has been well accepted, which usually encompasses multiple sequential or parallel cytotoxic events, including insulin resistance, hormonal or nutritional perturbation, and gut microbiota alteration, along with genetic or epigenetic factors that collectively promote NASH pathology (Buzzetti et al., 2016). Along with the advancement of understanding of the pathophysiology of NASH, a significant variety of targets have been identified for NASH treatment, which has been thoroughly reviewed recently (Townsend and Newsome, 2017; Wong et al., 2016). Meanwhile, pharmaceutical companies have accelerated the development of specific therapeutics for NASH management, and several pipeline therapies with diverse mechanisms of action are under late-phase clinical studies (Brodosi et al., 2016; Cassidy and Syed, 2016). Demonstrating the safety profiles of these potential NASH drugs will be critical to obtain U.S. Food and Drug Administration (FDA) approval and maintain long-term success of the drugs.
Even though human NASH is highly associated with obesity, one of the widely used animal models of NASH is a nutrient-deficient model, the methionine- and choline-deficient (MCD) diet fed rodent model (Table 1). The MCD diet contains a considerable amount of sucrose (40% of energy and 10% fat) but is deficient in methionine and choline (Dyets, Inc #518810). Choline deprivation in animals significantly impairs hepatic production and secretion of very-low-density lipoprotein (VLDL) (Vance, 2008; Yao and Vance, 1990). In addition, choline plays a major role in mitochondrial membrane integrity, so choline deficiency alters the mitochondrial membrane composition and leads to perturbations in mitochondrial bioenergetics and fatty acid β-oxidation (Corbin and Zeisel, 2012). In one mechanistic study, it has been shown that the MCD diet can significantly increase hepatic fatty acid uptake and reduce VLDL secretion (Rinella et al., 2008). As an essential amino acid, methionine plays a critical role in protein synthesis and is also the intermediate in S-adenosylmethionine (SAM) and glutathione synthesis, which are 2 important endogenous antioxidants (Lu, 2000). Disruption of hepatic methyl balance in either humans or rodents can lead to changes in SAM content that affects transmethylation reactions and promotes liver injury and development of liver diseases (Mato et al., 2008). In brief, in the MCD model, choline deficiency contributes more to the phenotype of steatosis, whereas methionine deficiency initially promotes oxidative stress and changes in cytokines and adipokines, which are believed to drive the progression of inflammation in the animals (Caballero et al., 2010; dela Peña et al., 2007; Larter et al., 2008; Leclercq et al., 2000). Collectively, multiple mechanisms have been proposed in the MCD model, which mostly disagree with human NASH pathogenesis mechanisms; this model does, however, lead to NASH histology, which is highly comparable with humans. Sprague Dawley rats and C57BL/6J mice on 8 weeks of MCD diet exhibit the phenotype of severe NASH (Canet et al., 2014; Machado et al., 2015). Excessive steatosis and cell ballooning were observed in MCD-fed rodents (Canet et al., 2014). Additionally, MCD-fed animals exhibit inflammation as early as 2 weeks after the onset of diet and present pericellular and perisinusoidal fibrosis in 8–10 weeks (Leclercq et al., 2000). Serum alanine aminotransferase is significantly elevated in MCD-fed rodents, indicating substantial liver injury in these animals (Canet et al., 2014; Machado et al., 2015). Moreover, disruptions of cellular defensive pathways, such as endoplasmic reticulum (ER) stress, oxidative stress, and autophagocytic stress, which have been shown as contributing factors to human NASH progression, are observed in MCD models as well (Machado et al., 2015). In summary, the MCD model has the advantage of effectively and reproducibly introducing severe hepatic steatosis and inflammation in animals, which closely resembles human NASH histology, within a relatively shorter time than other dietary models of NASH (Figure 1).
Table 1.
Metabolic Profile and Pathological Features in Human NASH and MCD Model
| Human NASH | MCD Model | |
|---|---|---|
| Metabolic parameters | ||
| Bodyweight | Highly associated with obesity | 40% weight loss in 8–10 weeks feeding |
| Insulin sensitivity | Systemic insulin resistance | Improved insulin sensitivity, intrahepatic insulin resistance |
| Fast blood glucose | High | Low |
| Triglyceride | High | Low |
| Cholesterol | High | Low |
| Liver injury markers | ||
| ALT | Increase (slightly to moderately) (Schindhelm et al., 2006) | Increase |
| AST | Increase (slightly to moderately) | Increase |
| CK-18 fragments | Increase (Feldstein et al., 2009) | Increase (Anstee et al., 2010) |
| Histology | ||
| Steatosis | Moderate and severe hepatic macrosteatosis characterized by lipid deposition as a single large vacuole within the hepatocyte cytoplasm (Brunt et al., 2011) | Moderate to severe macrovesicular steatosis as human, though microvesicular lipid droplets are common in mice, which is different from typical human histology (Canet et al., 2014) |
| Hepatocellular ballooning | Commonly but not extensively found: cellular enlargement 1.5–2 times the normal hepatocyte diameter, with rarefied cytoplasm (Brunt et al., 2011; Caldwell et al., 2010) | Difficulty in translating concept of cell ballooning in human patients to animal model, though cell enlargement can be found (Lackner, 2011) |
| Inflammation | Mild inflammation and predominantly lobular rather portal based (Brunt et al., 2011) | Fast developed inflammation as early as 2-week feeding, significant hepatocellular inflammation (Leclercq et al., 2007) |
| Fibrosis | “Pericellular” or “chicken-wire” fibrosis (Brunt et al., 2011) | Fibrosis is developed after 6-week feeding, and the chicken-wire fibrosis resembles human phenotype (George et al., 2003) |
| Inflammatory mediators | ||
| TNF-α/TNFR1 | Increase (Abiru et al., 2006) | Increase (Tomita et al., 2006) |
| IL-6 | Increase (Wieckowska et al., 2008) | Increase (Yamaguchi et al., 2010, 2011) |
| Adiponectin | Decrease (Lemoine et al., 2009) | Unchanged or increase (Larter and Yeh, 2008; Leclercq et al., 2007; Schattenberg et al., 2006) |
| Leptin | No significant change (Angulo et al., 2004) | Decrease |
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase, CK-18, cytokeratin-18; TNFR, tumor necrosis factor receptor.
Figure 1.
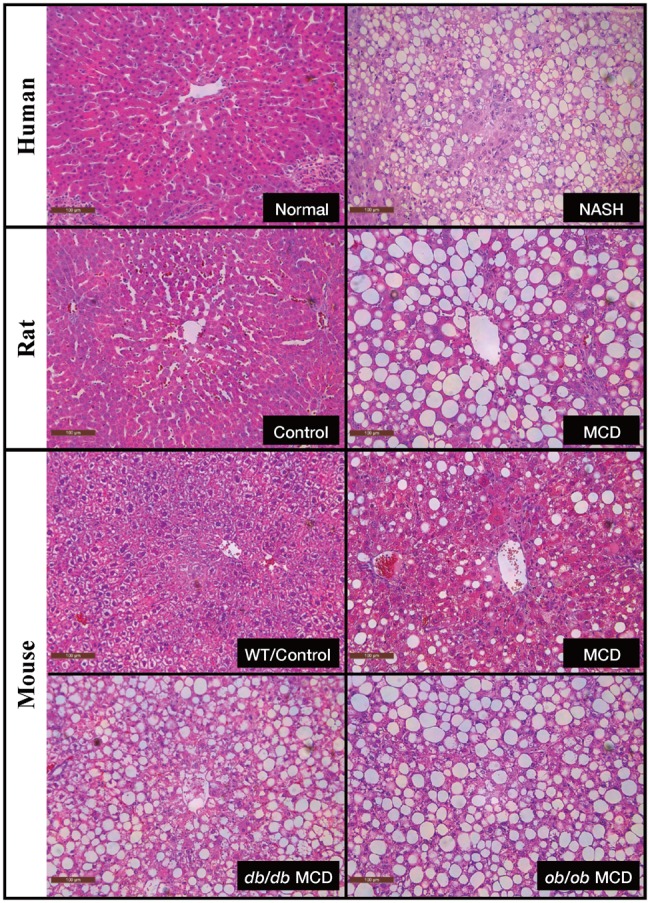
NASH histopathology in human and rodent models. Hematoxylin and eosin (H&E)-stained liver slides from human (normal vs NASH), rat (control vs MCD), and mouse (wild-type control, MCD, db/db MCD, and ob/ob MCD). All of the representative images are shown at × 20 magnification. NASH, nonalcoholic steatohepatitis; MCD, methionine and choline deficient; db/db, Leptin receptor knockout; ob/ob, Leptin knockout.
The utilization of the MCD model in NASH research has been commonly criticized due to its distinct metabolic profiles from human NASH patients. Animals fed on MCD diet have dramatic weight loss, which can be up to 40% in 8–10 weeks of feeding, and along with the loss of white adipose tissue, the liver size decreases in a proportional manner (Canet et al., 2014; Rinella and Green, 2004). Nonalcoholic steatohepatitis is recognized as the hepatic manifestation of metabolic syndrome; systemic insulin resistance is frequent in human NASH patients and has been considered a pivotal factor in driving NAFLD progression (Asrih and Jornayvaz, 2015). In contrast, systemic insulin resistance is absent or even improved in the MCD model (Rinella and Green, 2004), whereas impaired hepatic insulin signaling as well as intrahepatic insulin resistance were observed (Leclercq et al., 2007; Schattenberg et al., 2006). Schattenberg et al. reported JNK-associated intrahepatic insulin resistance as a driving force of steatohepatitis development in MCD mice. The role of peripheral insulin resistance versus hepatic insulin resistance in human NAFLD progression is not fully understood, and therefore the argument of hepatic insulin resistance in the MCD model reflecting human pathogenesis remains questionable. To overcome weight loss and insulin sensitivity in wildtype MCD animals, MCD diets are often fed to genetically modified mice such as the Leptin knockout (ob/ob) mice and the Leptin receptor knockout (db/db) mice to better replicate human NASH (Canet et al., 2014; Clarke et al., 2015; Rinella et al., 2008). The advantage of ob/ob and db/db mouse models is that they attain human characteristics of metabolic syndrome. ob/ob mice fed with 4 weeks of MCD diet show remarkable macrosteatosis, hepatic inflammation, and fibrosis, which is usually more severe than lesions seen in wildtype MCD mice (Clarke et al., 2015; Li et al., 2017a). However, weight loss as well as improved insulin resistance were still observed in ob/ob and db/db mice, meanwhile genetic leptin loss or resistance in humans with obesity are very rare, which is an inevitable limitation of these models. Nevertheless, the wildtype rodents fed with the MCD diet also exhibit lower leptin levels, unchanged or increased adiponectin levels, and decreased triglyceride and cholesterol, all of which contradict the metabolic profile of human disease (Larter and Yeh, 2008; Larter et al., 2008; Rinella and Green, 2004). Furthermore, a recent study showed significant dissimilarities in the metabolomics profiles of MCD rats as compared with human NASH, which further demonstrates that the metabolic context of the MCD model is distinct from human NASH (Han et al., 2017). Due to these limitations, the use of the MCD model to examine the metabolic parameters or study NASH pathogenesis should be discouraged.
MCD MODEL REFLECTING ADME ALTERATIONS IN HUMAN NASH
Studies have demonstrated profound hepatic ADME alterations during NASH progression, which influences multiple ADME pathways, including cytochrome P450s (CYPs), UDP-glucuronosyltransferases, sulfotransferases, glutathione-S-transferases, and hepatic drug transporters (Donato et al., 2006; Fisher et al., 2009b; Hardwick et al., 2010, 2011, 2013; Lake et al., 2011; Woolsey et al., 2015) (Table 2). Several studies have reported altered pharmacokinetics in human NAFLD/NASH patients, which further implicates NASH as a risk factor of variable drug response and idiosyncratic ADRs (Canet et al., 2015; Ferslew et al., 2015; Woolsey et al., 2015) (Table 3).
Table 2.
Hepatic ADME Characterization in Human NASH and MCD Models
| Human | MCD Rodent Model | |
|---|---|---|
| Phase I biotransformation | ||
| CYP1A |
|
|
| CYP2B |
|
|
| CYP2C |
|
|
| CYP2D | No change observed (Fisher et al., 2009a,b) |
|
| CYP2E1 |
|
|
| CYP3A4/5 |
|
|
| NQO1 |
|
|
| Phase II biotransformation | ||
| UGTs |
|
|
| SULTs |
|
No report |
| GSTs |
|
|
| Drug transporters | ||
| ABCs |
|
|
| OATPs |
|
|
| OATs | No report |
|
| OCTs | No report |
|
| NTCP |
|
|
Abbreviations: BCRP, breast cancer resistance protein; CAR, constitutive androstane receptor; GST, glutathione S-transferases; MRD, multidrug resistance; NQO1, NAD(P)H dehydrogenase [quinone] 1; NTCP, Na+-taurocholate co-transporting polypeptide; OAT, organic anion transporter; OCT, organic cation transporter; P-gp, P-glycoprotein; SULT, sulfotransferase; UGT-UDP, glucuronosyltransferase.
Table 3.
MCD Model in Predicting Human Pharmacokinetics and ADRs
| Drugs | Human | MCD Model |
|---|---|---|
| Morphine |
|
|
| Acetaminophen |
|
|
| Midazolam |
|
|
| Bupropion |
|
|
Abbreviations: APAP, Acetaminophen; AUC, Area under curve; CYP, Cytochrome p450; M3G, Morphine–glucuronide; MCD, Methionine and choline deficient; NAFLD, Nonalcoholic fatty liver disease; NASH, Nonalcoholic steatohepatitis.
Cytochrome P450s
Cytochrome P450s belong to the largest and the most important drug metabolizing enzyme superfamily, which are involved in the metabolism of the majority of endogenous and exogenous substances. Among the known 57 putatively functional human CYPs, CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4 are involved in metabolism of 78.7% of clinical drugs (Zanger and Schwab, 2013). Alterations in CYP activities can have substantial influence on the pharmacokinetics of substrate drugs, which, depending on drug exposure and the severity of enzyme alterations, may lead to potential compromised therapeutic effects or ADRs. Craig et al. performed systemic characterization of CYPs along NAFLD progression and found significant changes in expression and activity in multiple CYP isoforms, indicating a fundamental transformation of CYP-mediated phase I drug metabolism (Fisher et al., 2009b). Alterations in individual CYP isoforms in response to NAFLD/NASH were also reported by other groups (Aljomah et al., 2015; Aubert et al., 2011; Woolsey et al., 2015) (Table 2). Despite the fact that studies have demonstrated significant CYP changes in NASH, the underlying molecular mechanisms are poorly understood. Considering the complexity of physiological and pathological changes in NAFLD progression, CYPs may respond to multiple factors and cellular pathways, such as proinflammatory cytokines, oxidative stress, and nuclear receptors (Aitken et al., 2006; Hardwick et al., 2010; Lake et al., 2016; Morgan, 2009). For instance, elevated plasma interleukin-6 (IL-6) and tumor necrosis factor alpha (TNF-α) have been confirmed in human NASH studies (Kugelmas et al., 2003). Lipopolysaccharide (LPS) administration, which induces expression of a variety of proinflammatory cytokines, such as IL-6 and TNF-α, can significantly down-regulate hepatic mRNA expression of Cyp1a2, Cyp2c29, Cyp2e1, and Cyp3a11 in mice (Richardson and Morgan, 2005). Collectively, systemic and hepatic inflammation is not only one of the important driving forces of NAFLD progression, but may also play a role as a regulator of CYP activity. The MCD model is similar to human NASH with regard to CYP1A2/Cyp1a2 and CYP2E1/Cyp2e1, but fails to resemble human changes in CYP2C, 2D, and 3A (Leclercq et al., 2000; Li et al., 2017a; Rahman et al., 2007; Weltman et al., 1996; Yamazaki et al., 2007). The disparities in CYP2C, 2D, and 3A could be a reflection of interspecies variabilities in CYP enzymes. Specific CYP isoforms in different species, even those with high sequence identity, may show diverse catalytic activity and specificity. Rat and mouse have comparable expression and activity of CYP1A and CYP2E1 when compared with human, but substantial differences in CYP2C, 2D, and 3A (Bogaards et al., 2000; Martignoni et al., 2006). Studies regarding CYP-mediated drug metabolism in either human NASH or the MCD model are relatively scarce. A study by Woolsey et al. examined CYP3A activity and expression in human NAFLD patients as well as the MCD model and cellular models. This study reported a 2.4-fold increase in plasma midazolam levels in subjects with NASH, though CYP3A4 activity, which was assessed by 4b-hydroxycholesterol, an endogenous CYP3A4/5 marker, had no significant changes (Woolsey et al., 2015). This finding is consistent with a previous ex vivo study of CYP3A4 activity in human NASH (Fisher et al., 2009b). Moreover, they also observed a significant reduction of CYP3A activity and expression in MCD mice and cell culture model (Woolsey et al., 2015). Recently, our group published an in vivo evaluation of Cyp3a activity in the ob/ob MCD model, which used midazolam as a probe drug of Cyp3a. Even though increased midazolam plasma level was observed, the metabolite 1-hydroxy midazolam was also increased, leading to no significant change in the area under curve (AUC) ratio and no change in Cyp3a activity in the ob/ob MCD mice (Li et al., 2017a). Considering that CYP3A isoforms and gene regulation mechanisms greatly differ between rodents and humans, the rodent MCD model may not accurately reflect the human response of CYP3A to NASH, which largely limits the utility of this model in predicting CYP3A-associated drug metabolism. Fisher et al. reported no significant changes in CYP2B6 protein expression and enzymatic activity in human NASH, when enzymatic activity was evaluated by ex vivo incubation of human hepatic microsomes with the CYP2B6 specific substrate bupropion hydroxylase (Fisher et al., 2009b). In a recent study, Cho et al. investigated hepatic Cyp2b1, the rat ortholog of human CYP2B6, and in vivo bupropion disposition in both high-fat diet and MCD diet rats. Cyp2b1 mRNA and protein expression were significantly decreased in MCD rats, whereas the metabolism AUC ratio of bupropion was considerably reduced in MCD rats, suggesting a reduction of in vivo hepatic Cyp2b1-mediated metabolism of bupropion (Cho et al., 2016). Additionally, a study addressed the modulation of xenobiotic disposition and metabolism and increased hepatic exposure to tetrachloethylene and trichloroacetate in MCD mice, which may be associated with a significant reduction of Cyp2b10 expression in MCD mice (Cichocki et al., 2017). Based on the data produced in the MCD model, decreased Cyp2b metabolism in rodent is distinct from unchanged CYP2D6 metabolism in human NASH. In brief, the MCD diet is not an ideal model for modeling CYP alterations or predicting altered drug metabolism and disposition related to CYPs in human NASH.
Multidrug Resistance–Associated Proteins: MRP2 and MRP3
The ATP-binding cassette (ABC) family of transporters consists of a variety of proteins that use the energy of ATP hydrolysis and actively transport xenobiotics and endobiotics across cellular membranes. The ABCC subfamily of ABC transporters are also known as multidrug resistance–associated proteins (MRPs), which are all efflux transporters. In the liver, these efflux transporters reside on the sinusoidal and canalicular membranes of the hepatocyte and are responsible for substrate efflux into blood and bile (Klaassen and Aleksunes, 2010). Multidrug resistance–associated protein 2 is an efflux transporter on the hepatic canalicular membrane that shuttles xenobiotic conjugates into the bile, whereas MRP3 is a sinusoidal efflux transporter responsible for transport into blood circulation and shares a wide range of substrate specificity with MRP2. Multidrug resistance–associated proteins 2 and 3 levels are significantly increased in human NASH, whereas evidence showed that during NASH progression, some of the MRP2 protein was mislocalized away from the canalicular membrane and led to compromised MRP2 function in human patients (Hardwick et al., 2011). Similar Mrp2 and Mrp3 alterations were also found in MCD models. Increased Mrp2 and Mrp3 protein levels were universally found in all MCD-fed rodent models, whereas mislocalization of Mrp2 was also confirmed in MCD rats (Canet et al., 2014; Dzierlenga et al., 2015, 2016). These disease-induced alterations in MRP2/MRP3 efflux system have been associated with significant pharmacokinetic changes in clinical drugs. Barshop et al. (2011) defined an altered acetaminophen (APAP) glucuronidation and altered APAP-Gluc disposition in children with NAFLD, although the actual underlying mechanism was not described. Furthermore, Canet et al. (2015) demonstrated that a significant increase in serum and urinary levels of APAP-Gluc only presented in NASH patients, the advanced stage of NAFLD, and the study further established the mechanistic connection between the loss of MRP2 function and induced MRP3 function and altered APAP-Gluc disposition. These findings were consistent with a previous animal study, which reported significant induction of plasma and urinary APAP-Gluc and reduction of biliary APAP-Gluc in MCD rats administered APAP (Lickteig et al., 2007a,b). Morphine-3-glucuronide (M3G) and morphine-6-glucuronide (M6G) are 2 major metabolites of morphine in humans, which mostly rely on MRP2 and MRP3 for hepatobiliary disposition. A human study showed that morphine glucuronide systemic exposure and bile acid serum concentrations were substantially higher in NASH patients. A follow-up study further established a quantitative relationship between NASH severity score and the exposure of M3G, which was attributed to reduced biliary excretion and increased basolateral efflux of M3G (Ferslew et al., 2015; Pierre et al., 2017). Meanwhile, a parallel morphine disposition study in MCD rats also showed increased systemic exposure to M3G, but decreased biliary excretion and hepatic accumulation of M3G (Dzierlenga et al., 2015). Other than clinical drugs that were characterized in both humans and rodents, several studies reported changed disposition of MRP2/MRP3 substrate drugs, such as ezetimibe, methotrexate, and pemetrexed, in MCD-fed rodents (Dzierlenga et al., 2016; Hardwick et al., 2012, 2014). Collectively, current research has well documented MRP2 and MRP3 alterations in NASH and potential clinical outcomes in increased systemic toxicities and decreased hepatic drug efficacy. Furthermore, the MCD model precisely reflects molecular alterations of MRP2 and MRP3 in human NASH and successfully repeats changes in MRP2/MRP3-mediated drug dispositions in human NASH. In terms of promoting precision medicine in clinical practice, NASH should be critically considered in MRP2/MRP3 substrate drug prescriptions. Considering the wide range of these transporters, the MCD model can be a useful model in the preclinical evaluation of individual substrates to predict ADRs and mechanistic studies of variable drug responses in NASH.
Organic Anion–Transporting Polypeptides: OATP1B1, OATP1B3, and OATP2B1
Organic anion–transporting polypeptides (OATPs) belong to the superfamily of solute carriers and mediate the uptake of a broad range of compounds into cells (Kim, 2003). Organic anion–transporting polypeptide substrates include endogenous substances such as bile salts, hormones, and steroid conjugates as well as clinical drugs like the HMG-CoA-reductase inhibitors (statins), cardiac glycosides, anticancer agents like methotrexate, and antibiotics like rifampicin (König et al., 2006). Among 11 human OATP transporters, OATP1B1, OATP1B3, and OATP2B1 are predominantly expressed on sinusoidal membrane of hepatocytes and play particular roles in hepatic drug pharmacokinetics (Kalliokoski and Niemi, 2009; Smith et al., 2005). Additionally, OATP1B1, OATP1B3, and OATP2B1 exhibit great overlap in substrate specificity, which provides a compensatory system that maintains general uptake capacity if one transporter’s function is compromised or lost (Kalliokoski and Niemi, 2009). In human NASH, OATP1B1 expression is increased 3-fold, whereas OATP1B3 expression is decreased 10-fold, though OATP2B1 is not significantly changed (Clarke et al., 2014b). In MCD rats, studies reported decreased Oatp1a1, 1a4, and 1b2 protein expression levels (Canet et al., 2014; Clarke et al., 2014a; Fisher et al., 2009a). An increase in plasma concentration of bromosulfophthalein, a common compound used in liver function test, indicated impaired function of hepatic uptake transporters in the MCD rats (Fisher et al., 2009a). Furthermore, a study reported an increase in exposure to simvastatin hydroxyl and potential myopathy toxicity in MCD rats, which may be associated with an overall downregulation of OATP transporters (Clarke et al., 2014a). In the MCD mice, Oatp1a4 expression is increased, whereas Oatp1a1, 1b2, and 2b1 are significantly decreased, which moderately reflects the OATP alteration pattern in human NASH to some extent (Canet et al., 2014; Clarke et al., 2014b). Moreover, Clarke et al. demonstrated a gene-by-environment effect on pravastatin disposition in MCD-fed Oatp1b2 knockout mice, which provided mechanistic insight into the occurrence of statin-induced ADRs in the general population and particularly in the subpopulation of NASH patients (Clarke et al., 2014b). So far, studies regarding NASH impact on OATP substrate drugs in actual human patients are still absent, and therefore future studies into the pharmacokinetics of OATP substrates in human NASH are required to substantialize this effect as well as to evaluate the current animal models.
CONCLUSIONS
Overall, although an ideal animal model or in vitro model of NAFLD/NASH has yet to be discovered, especially for studies of pathogenic mechanisms and metabolic signature profiling, the current MCD model provides an auxiliary tool to study this major human disease, particularly with regard to potential drug ADRs and toxicities associated with drug transporter alterations during disease progression. Mechanistically, NASH-induced membrane transporter alterations are likely more associated with morphological changes on a cellular basis rather than metabolic pathogenic abnormalities in NASH; therefore, the MCD model, which most closely resembles human NASH histopathology, shows advantage in reflecting changes in drug transporters. In this case, alterations in rodent transporter function in response to the MCD diet closely resemble the alterations in human NASH and provide the pathological context that can reflect potential risk factors in altering pharmacokinetics or pharmacodynamics in the NASH subpopulation.
Investment in drug discovery and development in NASH are growing rapidly. It is foreseeable that medication therapy will become a crucial part of NASH management. As we come to better understand the potential for altered ADME processes in NASH and other inflammatory disease states, animal models that accurately reflect these changes will be invaluable in identifying potential ADRs. Therefore, animal model selection and development remain a major priority to researchers in this field.
Extrapolation of animal-to-human results is always challenging, because there is no perfect animal model that predicts the pharmacokinetics and pharmacodynamics of all chemicals in humans. Although there are many reasons to reject the MCD model for various applications, the similarity of alterations in ADME processes, particularly those of drug disposition mediated by MRP2/MRP3 and OATPs, make it a useful tool in predicting NASH-associated ADRs in this sensitive population. This publication was supported by National Institutes of Health [Grants HD062489 and GM123643], and the National Institute of Environmental Health Science [Grant ES007091 and ES006694].
REFERENCES
- Abiru S., Migita K., Maeda Y., Daikoku M., Ito M., Ohata K., Nagaoka S., Matsumoto T., Takii Y., Kusumoto K., et al. (2006). Serum cytokine and soluble cytokine receptor levels in patients with non-alcoholic steatohepatitis. Liver Int. 26, 39–45. [DOI] [PubMed] [Google Scholar]
- Aguilar-Olivos N. E., Carrillo-Córdova D., Oria-Hernández J., Sánchez-Valle V., Ponciano-Rodríguez G., Ramírez-Jaramillo M., Chablé-Montero F., Chávez-Tapia N. C., Uribe M., Méndez-Sánchez N. (2015). The nuclear receptor FXR, but not LXR, up-regulates bile acid transporter expression in non-alcoholic fatty liver disease. Ann. Hepatol. 14, 487–493. [PubMed] [Google Scholar]
- Aitken A. E., Richardson T. A., Morgan E. T. (2006). Regulation of drug-metabolizing enzymes and transporters in inflammation. Annu. Rev. Pharmacol. Toxicol. 46, 123–149. 10.1146/annurev.pharmtox.46.120604.141059 [DOI] [PubMed] [Google Scholar]
- Aljomah G., Baker S. S., Liu W., Kozielski R., Oluwole J., Lupu B., Baker R. D., Zhu L. (2015). Induction of CYP2E1 in non-alcoholic fatty liver diseases. Exp. Mol. Pathol. 99, 677–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angulo P., Alba L. M., Petrovic L. M., Adams L. A., Lindor K. D., Jensen M. D. (2004). Leptin, insulin resistance, and liver fibrosis in human nonalcoholic fatty liver disease. J. Hepatol. 41, 943–949. [DOI] [PubMed] [Google Scholar]
- Anstee Q. M., Concas D., Kudo H., Levene A., Pollard J., Charlton P., Thomas H. C., Thursz M. R., Goldin R. D. (2010). Impact of pan-caspase inhibition in animal models of established steatosis and non-alcoholic steatohepatitis. J. Hepatol. 53, 542–550. [DOI] [PubMed] [Google Scholar]
- Asrih M., Jornayvaz F. R. (2015). Metabolic syndrome and nonalcoholic fatty liver disease: Is insulin resistance the link? Mol. Cell. Endocrinol. 418, 55–65. 10.1016/j.mce.2015.02.018 [DOI] [PubMed] [Google Scholar]
- Aubert J., Begriche K., Knockaert L., Robin M. A., Fromenty B. (2011). Increased expression of cytochrome P450 2E1 in nonalcoholic fatty liver disease: Mechanisms and pathophysiological role. Clin. Res. Hepatol. Gastroenterol. 35, 630–637. 10.1016/j.clinre.2011.04.015 [DOI] [PubMed] [Google Scholar]
- Barshop N. J., Capparelli E. V., Sirlin C. B., Schwimmer J. B., Lavine J. E. (2011). Acetaminophen pharmacokinetics in children with nonalcoholic fatty liver disease. J. Pediatr. Gastroenterol. Nutr. 52, 198–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogaards J. J. P., Bertrand M., Jackson P., Oudshoorn M. J., Weaver R. J., Van Bladeren P. J., Walther B. (2000). Determining the best animal model for human cytochrome P450 activities: A comparison of mouse, rat, rabbit, dog, micropig, monkey and man. Xenobiotica 30, 1131–1152. [DOI] [PubMed] [Google Scholar]
- Brodosi L., Marchignoli F., Petroni M. L., Marchesini G. (2016). NASH: A glance at the landscape of pharmacological treatment. Ann. Hepatol. 15, 673–681. [DOI] [PubMed] [Google Scholar]
- Brunt E. M., Kleiner D. E., Wilson L. A., Belt P., Neuschwander-Tetri B. A. (2011). Nonalcoholic fatty liver disease (NAFLD) activity score and the histopathologic diagnosis in NAFLD: Distinct clinicopathologic meanings. Hepatology 53, 810–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzzetti E., Pinzani M., Tsochatzis E. A. (2016). The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 65, 1038–1048. 10.1016/j.metabol.2015.12.012 [DOI] [PubMed] [Google Scholar]
- Caballero F., Fernández A., Matías N., Martínez L., Fucho R., Elena M., Caballeria J., Morales A., Fernández-Checa J. C., García-Ruiz C. (2010). Specific contribution of methionine and choline in nutritional nonalcoholic steatohepatitis: Impact on mitochondrial S-adenosyl-L-methionine and glutathione. J. Biol. Chem. 285, 18528–18536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell S., Ikura Y., Dias D., Isomoto K., Yabu A., Moskaluk C., Pramoonjago P., Simmons W., Scruggs H., Rosenbaum N., et al. (2010). Hepatocellular ballooning in NASH. J. Hepatol. 53, 719–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canet M. J., Hardwick R. N., Lake A. D., Dzierlenga A. L., Clarke J. D., Cherrington N. J. (2014). Modeling human nonalcoholic steatohepatitis-associated changes in drug transporter expression using experimental rodent models. Drug Metab. Dispos. 42, 586–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canet M. J., Merrell M. D., Hardwick R. N., Bataille A. M., Campion S. N., Ferreira D. W., Xanthakos S. A., Manautou J. E., Hesham A-Kader H., Erickson R. P., et al. (2015). Altered regulation of hepatic efflux transporters disrupts acetaminophen disposition in pediatric nonalcoholic steatohepatitis. Drug Metab. Dispos. 43, 829–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassidy S., Syed B. A. (2016). Nonalcoholic steatohepatitis (NASH) drugs market. Nat. Rev. Drug Discov. 15, 745–746. 10.1038/nrd.2016.188 [DOI] [PubMed] [Google Scholar]
- Chalasani N., Younossi Z., Lavine J. E., Diehl A. M., Brunt E. M., Cusi K., Charlton M., Sanyal A. J. (2012). The diagnosis and management of non-alcoholic fatty liver disease: Practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology 55, 2005–2023. [DOI] [PubMed] [Google Scholar]
- Cho S.-J., Kim S.-B., Cho H.-J., Chong S., Chung S.-J., Kang I.-M., Lee J. I., Yoon I.-S., Kim D.-D. (2016). Effects of nonalcoholic fatty liver disease on hepatic CYP2B1 and in vivo bupropion disposition in rats fed a high-fat or methionine/choline-deficient diet. J. Agric. Food Chem. 64, 5598–5606. [DOI] [PubMed] [Google Scholar]
- Chtioui H., Semela D., Ledermann M., Zimmermann A., Dufour J.-F. (2007). Expression and activity of the cytochrome P450 2E1 in patients with nonalcoholic steatosis and steatohepatitis. Liver Int. 27, 764–771. [DOI] [PubMed] [Google Scholar]
- Cichocki J. A., Furuya S., Konganti K., Luo Y.-S., McDonald T. J., Iwata Y., Chiu W. A., Threadgill D. W., Pogribny I. P., Rusyn I. (2017). Impact of nonalcoholic fatty liver disease on toxicokinetics of tetrachloroethylene in mice. J. Pharmacol. Exp. Ther. 361, 17.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke J. D., Dzierlenga A. L., Nelson N. R., Li H., Werts S., Goedken M. J., Cherrington N. J. (2015). Mechanism of altered metformin distribution in nonalcoholic steatohepatitis. Diabetes 64, 3305.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke J. D., Hardwick R. N., Lake A. D., Canet M. J., Cherrington N. J. (2014a). Experimental nonalcoholic steatohepatitis increases exposure to simvastatin hydroxy acid by decreasing hepatic organic anion transporting polypeptide expression. J. Pharmacol. Exp. Ther. 348, 452–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke J. D., Hardwick R. N., Lake A. D., Lickteig A. J., Goedken M. J., Klaassen C. D., Cherrington N. J. (2014b). Synergistic interaction between genetics and disease on pravastatin disposition. J. Hepatol. 61, 139–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbin K. D., Zeisel S. H. (2012). Choline metabolism provides novel insights into nonalcoholic fatty liver disease and its progression. Curr. Opin. Gastroenterol. 28, 159–165. 10.1097/MOG.0b013e32834e7b4b [DOI] [PMC free article] [PubMed] [Google Scholar]
- dela Peña A., Leclercq I. A., Williams J., Farrell G. C. (2007). NADPH oxidase is not an essential mediator of oxidative stress or liver injury in murine MCD diet-induced steatohepatitis. J. Hepatol. 46, 304–313. [DOI] [PubMed] [Google Scholar]
- Donato M. T., Lahoz A., Jiménez N., Pérez G., Serralta A., Mir J., Castell J. V., Gómez-Lechón M. J. (2006). Potential impact of steatosis on cytochrome P450 enzymes of human hepatocytes isolated from fatty liver grafts. Drug Metab. Dispos. 34, 1556–1562. [DOI] [PubMed] [Google Scholar]
- Dzierlenga A. L., Clarke J. D., Hargraves T. L., Ainslie G. R., Vanderah T. W., Paine M. F., Cherrington N. J. (2015). Mechanistic basis of altered morphine disposition in nonalcoholic steatohepatitis. J. Pharmacol. Exp. Ther. 352, 462–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzierlenga A. L., Clarke J. D., Klein D. M., Anumol T., Snyder S. A., Li H., Cherrington N. J. (2016). Biliary elimination of pemetrexed is dependent on Mrp2 in rats: Potential mechanism of variable response in nonalcoholic steatohepatitis. J. Pharmacol. Exp. Ther. 358, 246–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldstein A. E., Wieckowska A., Lopez A. R., Liu Y.-C., Zein N. N., McCullough A. J. (2009). Cytokeratin-18 fragment levels as noninvasive biomarkers for nonalcoholic steatohepatitis: A multicenter validation study. Hepatology 50, 1072–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferslew B. C., Johnston C. K., Tsakalozou E., Bridges A. S., Paine M. F., Jia W., Stewart P. W., Barritt A. S., Brouwer K. L. R. (2015). Altered morphine glucuronide and bile acid disposition in patients with nonalcoholic steatohepatitis. Clin. Pharmacol. Ther. 97, 419–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher C. D., Lickteig A. J., Augustine L. M., Oude Elferink R. P. J. J., Besselsen D. G., Erickson R. P., Cherrington N. J. (2009a). Experimental non-alcoholic fatty liver disease results in decreased hepatic uptake transporter expression and function in rats. Eur. J. Pharmacol. 613, 119–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher C. D., Lickteig A. J., Augustine L. M., Ranger-Moore J., Jackson J. P., Ferguson S., Cherrington N. J. (2009b). Hepatic cytochrome P450 enzyme alterations in humans with progressive stages of nonalcoholic fatty liver disease. Drug Metab. Dispos. 37, 2087–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George J., Pera N., Phung N., Leclercq I., Yun Hou J., Farrell G. (2003). Lipid peroxidation, stellate cell activation and hepatic fibrogenesis in a rat model of chronic steatohepatitis. J. Hepatol. 39, 756–764. 10.1016/S0168-8278(03)00376-3 [DOI] [PubMed] [Google Scholar]
- Han J., Dzierlenga A. L., Lu Z., Billheimer D. D., Torabzadeh E., Lake A. D., Li H., Novak P., Shipkova P., Aranibar N., et al. (2017). Metabolomic profiling distinction of human nonalcoholic fatty liver disease progression from a common rat model. Obesity 25, 1069–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick R. N., Clarke J. D., Lake A. D., Canet M. J., Anumol T., Street S. M., Merrell M. D., Goedken M. J., Snyder S. A., Cherrington N. J. (2014). Increased susceptibility to methotrexate-induced toxicity in nonalcoholic steatohepatitis. Toxicol. Sci. 142, 45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick R. N., Ferreira D. W., More V. R., Lake A. D., Lu Z., Manautou J. E., Slitt A. L., Cherrington N. J. (2013). Altered UDP-glucuronosyltransferase and sulfotransferase expression and function during progressive stages of human nonalcoholic fatty liver disease. Drug Metab. Dispos. 41, 554–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick R. N., Fisher C. D., Canet M. J., Lake A. D., Cherrington N. J. (2010). Diversity in antioxidant response enzymes in progressive stages of human nonalcoholic fatty liver disease. Drug Metab. Dispos. 38, 2293–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick R. N., Fisher C. D., Canet M. J., Scheffer G. L., Cherrington N. J. (2011). Variations in ATP-binding cassette transporter regulation during the progression of human nonalcoholic fatty liver disease. Drug Metab. Dispos. 39, 2395–2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick R. N., Fisher C. D., Street S. M., Canet M. J., Cherrington N. J. (2012). Molecular mechanism of altered ezetimibe disposition in nonalcoholic steatohepatitis. Drug Metab. Dispos. 40, 450–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartung T. (2009). Toxicology for the twenty-first century. Nature 460, 208–212. 10.1038/460208a [DOI] [PubMed] [Google Scholar]
- Iasella C. J., Johnson H. J., Dunn M. A. (2017). Adverse drug reactions: Type A (intrinsic) or type B (idiosyncratic). Clin. Liver Dis. 21, 73–87. 10.1016/j.cld.2016.08.005 [DOI] [PubMed] [Google Scholar]
- Kalliokoski A., Niemi M. (2009). Impact of OATP transporters on pharmacokinetics. Br. J. Pharmacol. 158, 693–705. 10.1111/j.1476-5381.2009.00430.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim R. B. (2003). Organic anion‐transporting polypeptide (OATP) transporter family and drug disposition. Eur. J. Clin. Invest. 33, 1–5. [DOI] [PubMed] [Google Scholar]
- Klaassen C. D., Aleksunes L. M. (2010). Xenobiotic, bile acid, and cholesterol transporters: Function and regulation. Pharmacol. Rev. 62, 1–96. 10.1124/pr.109.002014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- König J., Seithel A., Gradhand U., Fromm M. F. (2006). Pharmacogenomics of human OATP transporters. Naunyn Schmiedebergs Arch. Pharmacol. 372, 432–443. [DOI] [PubMed] [Google Scholar]
- Kugelmas M., Hill D. B., Vivian B., Marsano L., McClain C. J. (2003). Cytokines and NASH: A pilot study of the effects of lifestyle modification and vitamin E. Hepatology 38, 413–419. [DOI] [PubMed] [Google Scholar]
- Lackner C. (2011). Hepatocellular ballooning in nonalcoholic steatohepatitis: The pathologist’s perspective. Expert Rev. Gastroenterol. Hepatol. 5, 223–231. [DOI] [PubMed] [Google Scholar]
- Lake A. D., Chaput A. L., Novak P., Cherrington N. J., Smith C. L. (2016). Transcription factor binding site enrichment analysis predicts drivers of altered gene expression in nonalcoholic steatohepatitis. Biochem. Pharmacol. 122, 62–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake A. D., Novak P., Fisher C. D., Jackson J. P., Hardwick R. N., Billheimer D. D., Klimecki W. T., Cherrington N. J. (2011). Analysis of global and absorption, distribution, metabolism, and elimination gene expression in the progressive stages of human nonalcoholic fatty liver disease. Drug Metab. Dispos. 39, 1954–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larter C. Z., Yeh M. M. (2008). Animal models of NASH: Getting both pathology and metabolic context right. J. Gastroenterol. Hepatol. 23, 1635–1648. 10.1111/j.1440-1746.2008.05543.x [DOI] [PubMed] [Google Scholar]
- Larter C. Z., Yeh M. M., Williams J., Bell-Anderson K. S., Farrell G. C. (2008). MCD-induced steatohepatitis is associated with hepatic adiponectin resistance and adipogenic transformation of hepatocytes. J. Hepatol. 49, 407–416. [DOI] [PubMed] [Google Scholar]
- Leclercq I. A., Farrell G. C., Field J., Bell D. R., Gonzalez F. J., Robertson G. R. (2000). CYP2E1 and CYP4A as microsomal catalysts of lipid peroxides in murine nonalcoholic steatohepatitis. J. Clin. Invest. 105, 1067–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leclercq I. A., Lebrun V. A., Stärkel P., Horsmans Y. J. (2007). Intrahepatic insulin resistance in a murine model of steatohepatitis: Effect of PPARγ agonist pioglitazone. Lab. Investig. 87, 56–65. [DOI] [PubMed] [Google Scholar]
- Lemoine M., Ratziu V., Kim M., Maachi M., Wendum D., Paye F., Bastard J. P., Poupon R., Housset C., Capeau J., et al. (2009). Serum adipokine levels predictive of liver injury in non-alcoholic fatty liver disease. Liver Int. 29, 1431–1438. 10.1111/j.1478-3231.2009.02022.x [DOI] [PubMed] [Google Scholar]
- Li H., Clarke J. D., Dzierlenga A. L., Bear J., Goedken M. J., Cherrington N. J. (2017a). In vivo cytochrome P450 activity alterations in diabetic nonalcoholic steatohepatitis mice. J. Biochem. Mol. Toxicol. 31, e21840.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Mark C. J., Clarke J. D., Billheimer D., Xanthakos S. A., Lavine J. E., Erickson R. P., Cherrington N. J. (2017b). Pediatric cytochrome P450 activity alterations in nonalcoholic steatohepatitis. Drug Metab. Dispos. 45, 1317.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lickteig A. J., Fisher C. D., Augustine L. M., Aleksunes L. M., Besselsen D. G., Slitt A. L., Manautou J. E., Cherrington N. J. (2007a). Efflux transporter expression and acetaminophen metabolite excretion are altered in rodent models of nonalcoholic fatty liver disease. Drug Metab. Dispos. 35, 1970–1978. [DOI] [PubMed] [Google Scholar]
- Lickteig A. J., Fisher C. D., Augustine L. M., Cherrington N. J. (2007b). Genes of the antioxidant response undergo upregulation in a rodent model of nonalcoholic steatohepatitis. J. Biochem. Mol. Toxicol. 21, 216–220. [DOI] [PubMed] [Google Scholar]
- Lu S. C. (2000). S-Adenosylmethionine. Int. J. Biochem. Cell Biol. 32, 391–395. 10.1016/S1357-2725(99)00139-9 [DOI] [PubMed] [Google Scholar]
- Machado M. V., Michelotti G. A., Xie G., de Almeida T. P., Boursier J., Bohnic B., Guy C. D., Diehl A. M., Sookoian S. C. (2015). Mouse models of diet-induced nonalcoholic steatohepatitis reproduce the heterogeneity of the human disease. PLoS One 10, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martignoni M., Groothuis G. M., de Kanter R. (2006). Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition and induction. Expert Opin. Drug Metab. Toxicol. 2, 875–894. 10.1517/17425255.2.6.875 [DOI] [PubMed] [Google Scholar]
- Mato J. M., Martínez-Chantar M. L., Lu S. C. (2008). Methionine metabolism and liver disease. Annu. Rev. Nutr. 28, 273–293. [DOI] [PubMed] [Google Scholar]
- Morgan E. (2009). Impact of infectious and inflammatory disease on cytochrome P450? Mediated drug metabolism and pharmacokinetics. Clin. Pharmacol. Ther. 85, 434–438. 10.1038/clpt.2008.302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okushin K., Tsutsumi T., Enooku K., Fujinaga H., Kado A., Shibahara J., Fukayama M., Moriya K., Yotsuyanagi H., Koike K. (2016). The intrahepatic expression levels of bile acid transporters are inversely correlated with the histological progression of nonalcoholic fatty liver disease. J. Gastroenterol. 51, 808–818. [DOI] [PubMed] [Google Scholar]
- Olson H., Betton G., Robinson D., Thomas K., Monro A., Kolaja G., Lilly P., Sanders J., Sipes G., Bracken W., et al. (2000). Concordance of the toxicity of pharmaceuticals in humans and in animals. Regul. Toxicol. Pharmacol. 32, 56–67. [DOI] [PubMed] [Google Scholar]
- Pierre V., Johnston C., Ferslew B., Brouwer K., Gonzalez D. (2017). Population pharmacokinetics of morphine in patients with nonalcoholic steatohepatitis (NASH) and healthy adults. CPT Pharmacometrics Syst. Pharmacol. 6, 331–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirmohamed M., Breckenridge A. M., Kitteringham N. R., Park B. K. (1998). Adverse drug reactions. BMJ 316, 1295–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman S. M., Schroeder-Gloeckler J. M., Janssen R. C., Jiang H., Qadri I., Maclean K. N., Friedman J. E. (2007). CCAAT/enhancing binding protein beta deletion in mice attenuates inflammation, endoplasmic reticulum stress, and lipid accumulation in diet-induced nonalcoholic steatohepatitis. Hepatology 45, 1108–1117. [DOI] [PubMed] [Google Scholar]
- Richardson T. A., Morgan E. T. (2005). Hepatic cytochrome P450 gene regulation during endotoxin-induced inflammation in nuclear receptor knockout mice. J. Pharmacol. Exp. Ther. 314, 703–709. 10.1124/jpet.105.085456 [DOI] [PubMed] [Google Scholar]
- Rinella M. E., Elias M. S., Smolak R. R., Fu T., Borensztajn J., Green R. M. (2008). Mechanisms of hepatic steatosis in mice fed a lipogenic methionine choline-deficient diet. J. Lipid Res. 49, 1068–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinella M. E., Green R. M. (2004). The methionine-choline deficient dietary model of steatohepatitis does not exhibit insulin resistance. J. Hepatol. 40, 47–51. 10.1016/j.jhep.2003.09.020 [DOI] [PubMed] [Google Scholar]
- Rollin B. E. (2014). Animal Research. In, Encyclopedia of Global Bioethics. Springer International Publishing, Cham, pp. 1–9. [Google Scholar]
- Schattenberg J. M., Singh R., Wang Y., Lefkowitch J. H., Rigoli R. M., Scherer P. E., Czaja M. J. (2006). JNK1 but not JNK2 promotes the development of steatohepatitis in mice. Hepatology 43, 163–172. [DOI] [PubMed] [Google Scholar]
- Schindhelm R. K., Diamant M., Dekker J. M., Tushuizen M. E., Teerlink T., Heine R. J. (2006). Alanine aminotransferase as a marker of non-alcoholic fatty liver disease in relation to type 2 diabetes mellitus and cardiovascular disease. Diabetes Metab. Res. Rev. 22, 437–443. [DOI] [PubMed] [Google Scholar]
- Smith N. F., Figg W. D., Sparreboom A. (2005). Role of the liver-specific transporters OATP1B1 and OATP1B3 in governing drug elimination. Expert Opin. Drug Metab. Toxicol. 1, 429–445. 10.1517/17425255.1.3.429 [DOI] [PubMed] [Google Scholar]
- Sugimoto H., Okada K., Shoda J., Warabi E., Ishige K., Ueda T., Taguchi K., Yanagawa T., Nakahara A., Hyodo I., et al. (2010). Deletion of nuclear factor-E2-related factor-2 leads to rapid onset and progression of nutritional steatohepatitis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 298, G283.. [DOI] [PubMed] [Google Scholar]
- Tanaka N., Matsubara T., Krausz K. W., Patterson A. D., Gonzalez F. J. (2012). Disruption of phospholipid and bile acid homeostasis in mice with nonalcoholic steatohepatitis. Hepatology 56, 118–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor K., Gordon N., Langley G., Higgins W. (2008). Estimates for worldwide laboratory animal use in 2005. Altern. Lab. Anim. 36, 327–342. [DOI] [PubMed] [Google Scholar]
- Tomita K., Tamiya G., Ando S., Ohsumi K., Chiyo T., Mizutani A., Kitamura N., Toda K., Kaneko T., Horie Y., et al. (2006). Tumour necrosis factor alpha signalling through activation of Kupffer cells plays an essential role in liver fibrosis of non-alcoholic steatohepatitis in mice. Gut 55, 415–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend S. A., Newsome P. N. (2017). Review article: New treatments in non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 46, 494–507. 10.1111/apt.14210 [DOI] [PubMed] [Google Scholar]
- Ulrich R. G. (2007). Idiosyncratic toxicity: A convergence of risk factors. Annu. Rev. Med. 58, 17–34. 10.1146/annurev.med.58.072905.160823 [DOI] [PubMed] [Google Scholar]
- Vance D. E. (2008). Role of phosphatidylcholine biosynthesis in the regulation of lipoprotein homeostasis. Curr. Opin. Lipidol. 19, 229–234. 10.1097/MOL.0b013e3282fee935 [DOI] [PubMed] [Google Scholar]
- Varela N. M., Quiñones L. A., Orellana M., Poniachik J., Csendes A., Smok G., Rodrigo R., Cáceres D. D., Videla L. A. (2008). Study of cytochrome P450 2E1 and its allele variants in liver injury of nondiabetic, nonalcoholic steatohepatitis obese women. Biol. Res. 41, 81–92. [PubMed] [Google Scholar]
- Weltman M., Farrell G., Liddle C. (1996). Increased hepatocyte CYP2E1 expression in a rat nutritional model of hepatic steatosis with inflammation. Gastroenterology 111, 1645–1653. 10.1016/S0016-5085(96)70028-8 [DOI] [PubMed] [Google Scholar]
- Wieckowska A., Papouchado B. G., Li Z., Lopez R., Zein N. N., Feldstein A. E. (2008). Increased hepatic and circulating interleukin-6 levels in human nonalcoholic steatohepatitis. Am. J. Gastroenterol. 103, 1372–1379. [DOI] [PubMed] [Google Scholar]
- Wong V. W., Chitturi S., Wong G. L., Yu J., Chan H. L., Farrell G. C. (2016). Pathogenesis and novel treatment options for non-alcoholic steatohepatitis. Lancet Gastroenterol. Hepatol. 1, 56–67. [DOI] [PubMed] [Google Scholar]
- Woolsey S. J., Mansell S. E., Kim R. B., Tirona R. G., Beaton M. D. (2015). CYP3A activity and expression in nonalcoholic fatty liver disease. Drug Metab. Dispos. 43, 1484–1490. [DOI] [PubMed] [Google Scholar]
- Yamaguchi K., Itoh Y., Yokomizo C., Nishimura T., Niimi T., Fujii H., Okanoue T., Yoshikawa T. (2010). Blockade of interleukin-6 signaling enhances hepatic steatosis but improves liver injury in methionine choline-deficient diet-fed mice. Lab. Investig. 90, 1169–1178. [DOI] [PubMed] [Google Scholar]
- Yamaguchi K., Itoh Y., Yokomizo C., Nishimura T., Niimi T., Umemura A., Fujii H., Okanoue T., Yoshikawa T. (2011). Blockade of IL-6 signaling exacerbates liver injury and suppresses antiapoptotic gene expression in methionine choline-deficient diet-fed db/db mice. Lab. Investig. 91, 609–618. [DOI] [PubMed] [Google Scholar]
- Yamazaki Y., Kakizaki S., Horiguchi N., Sohara N., Sato K., Takagi H., Mori M., Negishi M. (2007). The role of the nuclear receptor constitutive androstane receptor in the pathogenesis of non-alcoholic steatohepatitis. Gut 56, 565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Z., Vance D. E. (1990). Reduction in VLDL, but not HDL, in plasma of rats deficient in choline. Biochem. Cell Biol. 68, 552–558. 10.1139/o90-079 [DOI] [PubMed] [Google Scholar]
- Younossi Z. M., Koenig A. B., Abdelatif D., Fazel Y., Henry L., Wymer M. (2016). Global epidemiology of nonalcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 64, 73–84. [DOI] [PubMed] [Google Scholar]
- Zanger U. M., Schwab M. (2013). Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 138, 103–141. 10.1016/j.pharmthera.2012.12.007 [DOI] [PubMed] [Google Scholar]
- Zhang Y.-K. J., Yeager R. L., Tanaka Y., Klaassen C. D. (2010). Enhanced expression of Nrf2 in mice attenuates the fatty liver produced by a methionine- and choline-deficient diet. Toxicol. Appl. Pharmacol. 245, 326–334. [DOI] [PMC free article] [PubMed] [Google Scholar]