

Abstract
Motivation
DNA methylation is important for gene silencing and imprinting in both plants and animals. Recent advances in bisulfite sequencing allow detection of single nucleotide variations (SNVs) achieving high sensitivity, but accurately identifying heterozygous SNVs from partially C-to-T converted sequences remains challenging.
Results
We designed two methods, BayesWC and BinomWC, that substantially improved the precision of heterozygous SNV calls from ∼80% to 99% while retaining comparable recalls. With these SNV calls, we provided functions for allele-specific DNA methylation (ASM) analysis and visualizing the methylation status on reads. Applying ASM analysis to a previous dataset, we found that an average of 1.5% of investigated regions showed allelic methylation, which were significantly enriched in transposon elements and likely to be shared by the same cell-type. A dynamic fragment strategy was utilized for DMR analysis in low-coverage data and was able to find differentially methylated regions (DMRs) related to key genes involved in tumorigenesis using a public cancer dataset. Finally, we integrated 40 applications into the software package CGmapTools to analyze DNA methylomes. This package uses CGmap as the format interface, and designs binary formats to reduce the file size and support fast data retrieval, and can be applied for context-wise, gene-wise, bin-wise, region-wise and sample-wise analyses and visualizations.
Availability and implementation
The CGmapTools software is freely available at https://cgmaptools.github.io/.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
DNA methylation is an epigenetic marker that has been extensively studied in mammals (Smith and Meissner, 2013) and plants (Feng et al., 2010; Law and Jacobsen, 2010; Matzke and Mosher, 2014) and is associated with the activities of genes and transposon elements (TEs) (Jones, 2012). Many technologies have been developed for profiling genome-wide DNA methylation (Plongthongkum et al., 2014). Bisulfite conversion of DNA followed by sequencing (BS-seq) is currently the gold standard for constructing DNA methylomes (Roadmap Epigenomics Consortium et al., 2015; Stricker et al., 2017). Comprehensive DNA methylation analyses provide novel insights into epigenetics regulation (Kawakatsu et al., 2016; Schultz et al., 2015).
Single nucleotide variants (SNVs) and DNA methylation variants contributed to the diversities of the genome and epigenome, respectively. SNVs can be explored with epigenome-wide association study (EWAS) (Do et al., 2017), and allele-specific DNA methylation (ASM) (Xie et al., 2012) calculations. Additionally, demining SNVs from BS-seq data is useful in many applications. However, in BS-seq data, SNVs are confounded with C-to-T converted cytosines, making it more difficult to distinguish SNVs from deaminations, mutations and sequencing errors. Several tools have been developed for SNV calling in BS-seq data (Gao et al., 2015a; Liu et al., 2012), which have achieved good sensitivities. Both Bis-SNP (Liu et al., 2012) and BS-SNPer (Gao et al., 2015a) use Bayesian strategies and predict explicit genotypes through maximum posterior probabilities. It is impossible to always precisely predict an exact genotype from BS-seq data because an observation of read counts may be derived from different genotypes based on C-to-T conversion of unmethylated cytosine. It is still challenging to accurately predict SNVs from BS-seq data.
DNA methylations can be allele-specific and can mediate the expression of imprinted genes. Previous studies have found that ASM is prevalent throughout the genome (Shoemaker et al., 2010; Zhang et al., 2009). Identifying ASM-mediated gene expression requires advanced tools for ASM calling. Fang et al. designed a statistical model to predict ASM without genotype information (Fang et al., 2012). However, this approach cannot distinguish between the differentially methylated regions (DMR) among subpopulations of sample cells and ASMs of parental origin. Gao et al. developed SMAP which supports ASM analyses (Gao et al., 2015b), utilizing SNVs predicted by Bis-SNP.
Concerning tissue-specific regulation of DNA methylation (Ziller et al., 2013), DMR analyses have been widely applied (Akalin et al., 2012b; Almeida et al., 2016; Saito et al., 2014; Sun et al., 2014; Wang et al., 2015; Wen et al., 2016). In some cases, DNA methylomes were constructed based on reduced representation bisulfite sequencing (RRBS) libraries (Meissner et al., 2005), which is a cost-efficient approach to measure a fraction of CpG sites across samples (Li et al., 2014; Meng et al., 2016). It is always difficult to identify bona fide DMRs in low-coverage libraries and discontinuously covered regions especially in RRBS data.
Published DNA methylomes are useful resources for extracting interesting features and further exploration (Guo et al., 2014, 2016). As DNA methylomes at the single-nucleotide resolution are usually very large, challenges in storing, sharing and processing large DNA methylomes datasets call for better methodological and computational tools (Laird, 2010). Several web servers have been developed to display collected DNA methylome data, such as MethBase (Song et al., 2013), NGSmethDB (Hackenberg et al., 2011) and MethBank (Zou et al., 2015). However, most of these databases only report CpG methylation levels and have limited access to coverage information and non-CG methylations (Feng et al., 2010; Guo et al., 2016). Moreover, retrieval of DNA methylome data from the internet is always a bottleneck due to bandwidth limitations. Most tools for aligning BS-seq data (Guo et al., 2013; Krueger and Andrews, 2011; Xi and Li, 2009) can produce SAM format (Li et al., 2009) files but use variant formats to store methylation information, which is a barrier for sharing the data. Several downstream pipelines for BS-seq data analysis have been developed (Benoukraf et al., 2013; Chen et al., 2014; Gao et al., 2015b; Liao et al., 2015; Luu et al., 2016; Park et al., 2014; Song et al., 2013; Sun et al., 2013; Warden et al., 2013), but most of these can carry out only a limited set of analyses.
Here, we propose novel methods for identifying SNVs from BS-seq data by introducing wild-card genotypes specifically defined for BS-seq data. Our methods have significantly improved the precision of heterozygous SNV calls. We also developed ASM analysis pipelines based on heterozygous SNV calls, and provided functionality for visualizing allele-specific methylation across reads. We applied SNV and ASM analyses to a previous dataset, showing that an average of 1.5% regions are ASM, and characterized the distributions and cell-type specificities of ASM regions. We also developed a dynamic-fragment DMR finding method that is especially suitable for RRBS and low coverage WGBS libraries. Applying the method to a published cancer dataset, we were able to find DMRs that are related to key genes involved in tumorigenesis. Moreover, we integrated 40 functions into the CGmapTools package, which provides advances for downstream analysis of BS-seq data.
2 Materials and methods
2.1 SNV calling strategies with wildcards
The ATCGmap format provides read counts on the Watson strands and the Crick strands at each base-pair, which we denoted as the ATCGmap table (Fig. 1A). Using the ATCGmap table, CGmapTools can compute a genotype. As C may be converted to U in BS-seq data, the presence of a T in a read may indicate either a T or C in the unconverted genome. For example, if the ATCGmap table only has a T on the Watson strand, the site could arise from genotypes such as TT, CC or TC genotypes (Fig. 1B). Therefore, we used wildcards to denote this ambiguity in predicted genotypes (Y to refer to either T or C, R to refer to either A or G) (Table 1). Although the wildcards are associated with ambiguous genotypes, sometimes we can still estimate whether it is an SNV, and even whether it is a heterozygous SNV. When both strands have high coverages, we can resolve this ambiguity and compute an exact genotype. Here, we provide two strategies for SNV calling, (i) a Bayesian model with a wildcard (BayesWC) strategy, and (ii) a Binomial model with wildcard (BinomWC) strategy.
Fig. 1.
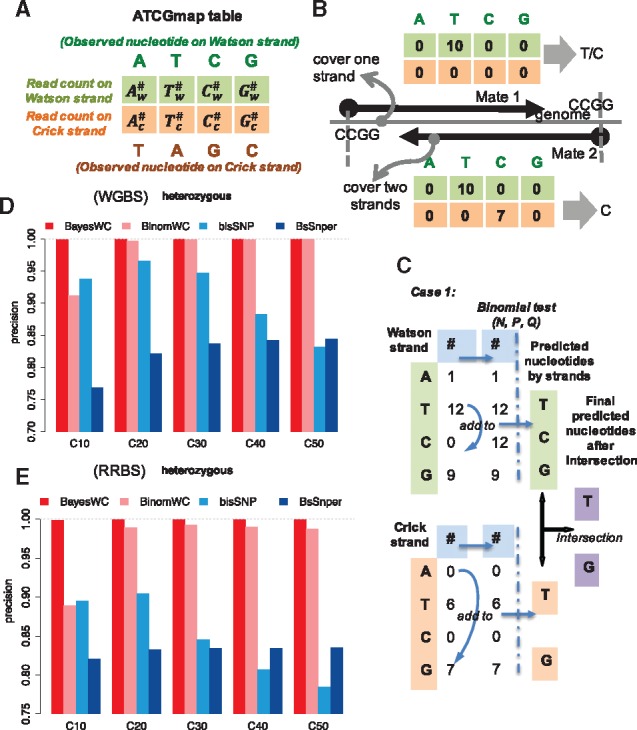
SNV calling from BS-seq data by introducing wild-card genotypes. (A) Definition of an ATCGmap table for one position. is the read count of the Watson strand supporting the position as A on the Watson strand. is the read count of the Crick strand support the position as A on the Watson strand (T on the Crick strand). (B) Examples for genotype prediction from an ATCGmap table. Taking RRBS as an example, the upper case only covers one strand, and the read counts could be either from genotype T or from genotype C, considering the effects of bisulfite conversion, and therefore, introducing a wildcard in the genotype is necessary. The lower case has high coverage on both strands, and information from the reverse strand helps the inference of an explicit genotype. (C) The schema for the BinomWC strategy when both strands have sufficient coverages. Ambiguous read counts are added to corresponding positions in the table, and a binomial test is used to select a set of nucleotides from each strand; then the intersection of the two sets is used as the final predicted genotype. (D) The precision analysis for heterozygous SNV calling in simulated WGBS datasets for four strategies. The average coverage levels are 10×, 20×, 30×, 40× and 50×. (E) The precision analysis for heterozygous SNV calling in simulated RRBS datasets for four strategies
Table 1.
Wildcard symbol table for ambiguous genotypes
| Ambiguous GN symbol | Possible genotypes | Hete- or Homo-zygous | Sure to be SNV if reference is |
|---|---|---|---|
| Y | TT/TC/CC | Not sure | A, G |
| R | AA/AG/GG | Not sure | T, C |
| A, Y | AT/AC | Heterozygous | A, T, C, G |
| C, Y | CT/CC | Not sure | A, T, G |
| G, Y | GT/GC | Heterozygous | A, T, C, G |
| T, Y | TT/TC | Not sure | A, C, G |
| A, R | AA/AG | Not sure | T, C, G |
| C, R | CA/CG | Heterozygous | A, T, C, G |
| G, R | GA/GG | Not sure | A, T, C |
| T, R | TA/TG | Heterozygous | A, T, C, G |
Note: The wildcard characters are defined as: Y = T/C and R = A/G.
2.2 BayesWC strategy
In BayesWC, we assume the genome is diploid. Thus, the posterior probability of a genome type is
| (1) |
where are the observed read counts in the ATCGmap table
| (2) |
and the genotype is either homozygous, or heterozygous, , that is
| (3) |
where
| (4) |
and
| (5) |
The prior is defined as
| (6) |
and
| (7) |
The posterior can be noted as the product of the posteriors of each observed genotype, that is
| (8) |
where
| (9) |
and are the nucleotides of the two alleles.
Let us suppose the rate for miscalling one nucleotide as another is , then the rate for correctly calling a nucleotide is . Thus we can draw a table for the likelihood (Supplementary Table S1).
We introduce wildcard genotypes as
| (10) |
where Y and R are the wildcard symbols for genotype.
For the wildcard genotype, we calculate the posteriors as seen in the following examples:
| (11) |
| (12) |
where the coefficients were selected considering and . Finally, a genotype with the highest posterior from the exact genotype set and wildcard genotype set is selected as the predicted genotype.
2.3 BinomWC strategy
Alternatively, we propose a Binomial-wildcard strategy, a modified version of the previous SNV-calling method for BS-seq data (Orozco et al., 2015). The basic idea is to predict the genotype from the ATCGmap table using a binomial distribution. (i) When both strands have sufficient reads (≥10×), the nucleotides are called on each strand first and are then intersected between the two strands (Fig. 1C). (ii) When only one strand has sufficient reads, the nucleotides will be predicted from the high-coverage strand, and an ambiguous genotype may be introduced (Supplementary Fig. S1A). (iii) When neither strand has sufficient reads, the counts on the two strands are merged as a six-element vector for genotype-calling (Supplementary Fig. S1B).
2.4 Evaluation of SNV calling methods
A 50 Mbp reference genome was used, from which we generated a diploid genome with rates of homozygous and heterozygous SNVs of 0.1%. We generated 100-bp reads and aligned both whole-genome bisulfite-sequencing (WGBS) and RRBS libraries following the methods of Guo et al. (2014). The precisions and recalls were evaluated under different coverage levels for BayesWC (command: cgmaptools snv −m bayes - -dynamicP), BinomWC (command: cgmaptools snv −m binom), Bis-SNP (v0.69, default parameters) and BS-SNPer (parameters: - -minhetfreq 0.1 - -minhomfreq 0.85 - -minquali 15 - -mincover 0.4 - -maxcover 1000 - -minread2 2 - -errorate 0.02 - -mapvalue 20).
2.5 Allele-specific methylation regions
To find ASM regions, we first identified heterozygous SNVs using BayesWC. SNVs with ambiguous genotypes were discarded. Then, ASM regions were computed using the ‘asr’ mode of CGmapTools. All regions linked by heterozygous SNV sites through mapped reads were investigated. ASM regions should satisfy the following criteria: (i) at least two CpG sites are covered; (ii) the mCG level on the hypo-methylated allele is ≤0.2 and ≥0.8 on the hyper-methylated allele and (iii) corrected P-values from multiple t-tests should be ≤0.05. In the enrichment study of different genomic elements, at least one base of overlap is considered to be overlapping. A hypergeometric test was performed for enrichment between ASM regions and specific genomic regions. All heterozygous SNV-linked regions were used as background. When investigating the tissue specificity of ASM, the hypergeometric test was performed to evaluate the overlap of ASM regions between two samples, where the ASM regions in two samples were considered to be overlapped if two SNVs were within a distance of 500 bp.
3 Results
3.1 Improved precision of heterozygous SNV calls
To evaluate the performances of our SNV calling methods, we compared BayesWC and BinomWC with Bis-SNP (Sun et al., 2013) and BS-SNPer (Gao et al., 2015a) on simulated WGBS and RRBS datasets. For WGBS, the results showed that BayesWC and BinomWC outperformed the other two methods in terms of the precision for heterozygous SNV calls (Fig. 1D). In particular, BayesWC achieves approximately 99% precisions for both heterozygous and homozygous SNV calls (Fig. 1D and Supplementary Fig. S2). For homozygous SNV calls, BayesWC has comparable precisions with Bis-SNP, but outperforms Bis-SNP on recalls when the average coverage is higher than 30× (Supplementary Fig. S2). Even for low coverage data, BayesWC achieves high precision; the trade-off is that recalls are low compared with other methods. For RRBS, our results showed that BayesWC also has high precisions for heterozygous SNV calls with different coverage ranges (Fig. 1E), and achieves similar recalls as Bis-SNP, but lower recalls when coverage is low. Compared with BS-SNPer, BayesWC has much higher precisions and comparable recalls (Supplementary Fig. S3).
3.2 Pervasive allele-specific DNA methylations are enriched in transposons
Based on the precisely predicted heterozygous SNVs, CGmapTools provides a function to identify ASM regions. Based on a previously published cohort of DNA methylomes (Guo et al., 2016), we computed SNVs (Supplementary Table S2) and predicted ASM regions using CGmapTools. A considerable portion of heterozygous SNV linked regions showed asymmetric methylation levels on two alleles in both humans and mice (Supplementary Fig. S4). Accordingly, we defined ASM regions with strict thresholds. In human, 1.50% of the regions, on average, are defined as ASM regions, and the number is 1.45% in mice (Fig. 2A). Furthermore, we found that ASM regions in both humans and mice are enriched on L1 repeats; Alu elements are also enriched for ASM regions in humans; LTR elements are enriched in ASM regions in mice (Fig. 2B).
Fig. 2.
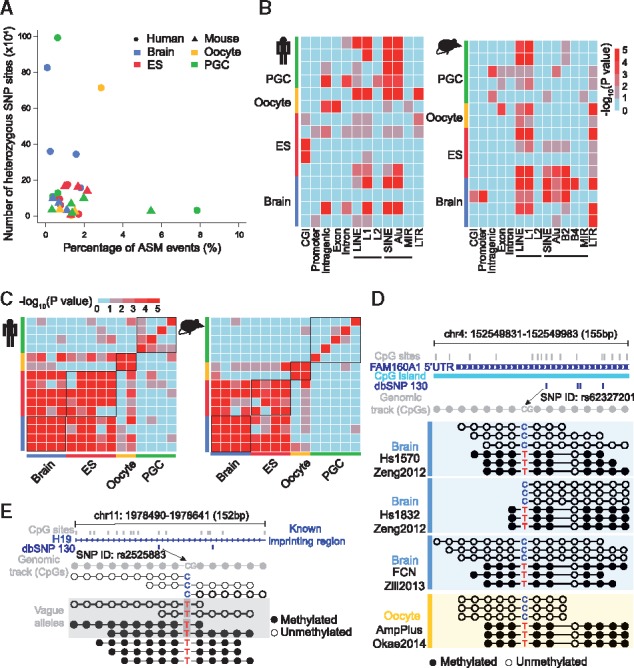
Allele-specific DNA methylation in humans and mice. (A) Scatter plot showing the percentage of ASM events and the number of heterozygous SNVs defined in both human and mouse samples. Round dots represent human samples and triangles represent mouse samples. (B) Enrichment analysis of ASM events in genomic elements showing different genomic bias within species. Colours indicate significance levels of enrichment by the hypergeometric test. (C) Heatmap showing the consistency of ASM among cell-types in both human (left panel) and mouse (right panel). Colours indicate significance level of consistency from low to high using the hypergeometric test. (D) Representative locus of ASM linked by a known SNP site in dbSNP130 located at 5’ UTR of FAM160A and in a CpG island. Reads linked by two heterozygous alleles were representatively selected in the Tanghulu plot for three brain samples and one oocyte sample. (E) Representative ASM locus linked by a heterozygous SNV site with C to T transition disrupting the CpG context of one allele, which is located in a known imprinting gene, H19. Reads linked by T, identified with a grey rectangular background, were ambiguous reads that could not be assigned to allele C or T due to bisulfite conversion. Open circles, unmethylated CpG sites; filled circles, methylated CpG sites
By evaluating the pairwise overlap of ASM regions for all samples, we found that the ASM regions were significantly overlapped within the same cell type, such as oocytes, embryonic stem cells (ESC) and neurons. Moreover, the ASM regions of ESCs and neurons were found to be similar (Fig. 2C).
CGmapTools also provides a function for visualizing the DNA methylation states on individual reads. A Tanghulu plot was drawn for an ASM region in the 5′ UTR of gene FAM160A1, which contains a CpG-TpG SNV located within a CpG island (Fig. 2D). Interestingly, different samples of brain neurons and oocytes all showed that the CpG allele is hypo-methylated and the TpG allele is hyper-methylated. As discussed above, a T in a read that maps onto the Watson strand could either originate from the conversion of an un-methylated C or a T, and therefore the read T is marked as ambiguous when shown in the Tanghulu plot (Fig. 2E).
A previous study reported that ASM is often enriched at heterozygous SNVs that are found at CpG dinucleotides (Shoemaker et al., 2010). We analyzed the location preference of ASM regions genome-widely in a set of high-coverage DNA methylomes. We found that the percentages of ASM regions vary across cell types, with a minimum rate in primordial germ cells (PGCs) in both human and mouse (Supplementary Fig. S5).
3.3 Identify DMRs with dynamic fragmentation strategy
Both differentially methylated site (DMS) analyses and DMR analyses are supported. As WGBS is still resources-intensive, RRBS is a more popular method that reduces the cost per sample and is widely applied in clinical studies. Because the regions covered by RRBS libraries are fragmented, we propose a dynamic fragmentation strategy for identifying DMRs between a pair of samples. First, only CpG sites covered by sufficient reads (≥5×) in both samples are selected for DMR analysis. Background fragments are dynamically defined using the criteria that they contain a minimum number of CpGs, a maximal length of bases, and a maximal distance between two adjacent CpGs (Fig. 3A). An unpaired t-test is carried out to compare the methylation levels of shared CpG sites within each background fragment. Finally, DMRs are selected from background fragments by the thresholds of P-values and delta methylation levels.
Fig. 3.
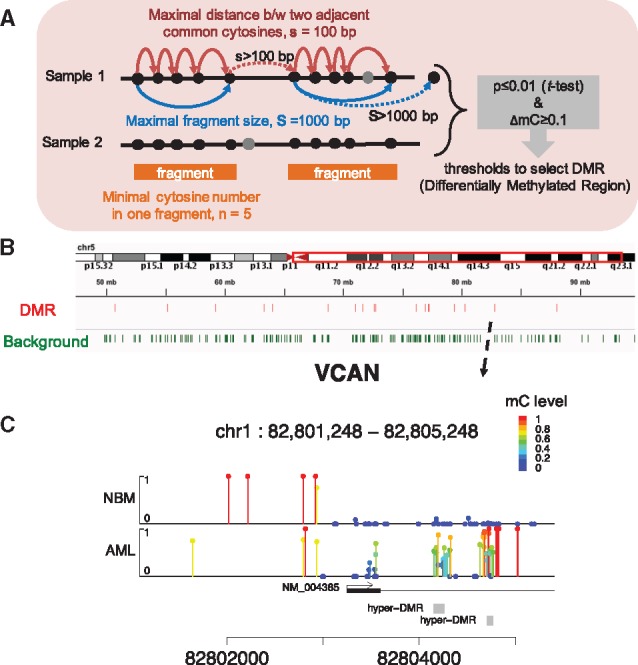
Differentially methylated region analysis in CGmapTools. (A) Schematic presentation for defining dynamic fragments. First, only sites covered by two samples are selected; Second, the genome is scanned by defining fragments with the minimal cytosine (usually CpG) counts n, the maximal fragment size S, and the maximal distance between two adjacent cytosines. Grey circles indicate cytosine sites that were only covered by one sample. Then, a t-test is applied to compare between methylation levels of cytosines in each fragment. Solid arrows indicate extending of a fragment, and dotted arrows indicate terminating the extension of a fragment. (B) Graphical presentation of the DMR and dynamic fragments (background) in a region on chr5. Data were from an eRRBS dataset. (C) Lollipop plot for the DMRs in the promoter region of gene VCAN. The arrow indicates the position in (B). The site-specific methylation levels are represented both by the height of bars. From the figure, two dynamic fragments (grey boxes) are reported as DMRs, which are hyper-methylated in AML
We applied this DMR analysis to samples from acute myelogeneous leukemia (AML) and normal CD34+ bone marrow (NBM) control samples obtained from a published RRBS dataset (Akalin et al., 2012a). A total of 53061 dynamic regions were selected, of which 2961 regions were DMRs (P ≤ 0.001 and ΔmC ≥ 0.5) (Fig. 3B), and 2161 DMRs were hyper-methylated in AML samples.
To visualize the DNA methylation levels in a local region, we designed a Lollipop plot to distinguish unmethylated sites from low-coverage sites. The gene VCAN is reported to be associated with tumorigenesis and cancer relapse (Du et al., 2013). Using the gene VCAN as an example, the Lollipop plot showed two dynamically determined regions located downstream of the transcription starting site, which were hyper-methylated in AMLs (Fig. 3C). Another example was shown for a DMR in the promoter of the oncogene TRIM59 (Supplementary Fig. S6).
3.4 Additional features of CGmapTools
3.4.1 CGmap is used as the standard format
CGmapTools is a downstream analysis package following the alignment of BS-seq reads. Because the CGmap and ATCGmap formats report comprehensive information associated with methylomes and are suitable for further processing, we use them as standard formats in CGmapTools. Similar to the output formats of BS-Seeker2 (Guo et al., 2013), CGmapTools also provides functions to generate the two formats directly from BAMs. The ATCGmap format provides readable information for all four nucleotides on both strands. The CGmap format only provides read counts for CpG dinucleotides, which can then be used for most DNA methylation analyses (Fig. 4). Inspired by the BAM format (Li et al., 2009), we designed binary formats, ATCGbz and CGbz, to store sorted DNA methylome as compressed binary file formats. Requiring much less space than BAM files, they also save spaces compared to compressed ATCGmap/CGmap formats (Supplementary Fig. S7). Another advantage of the binary formats is that they support fast retrieval from the disk. To be extendable, programs in CGmapTools are implemented as command-line tools, so that users can easily integrate them to their own customized pipelines.
Fig. 4.
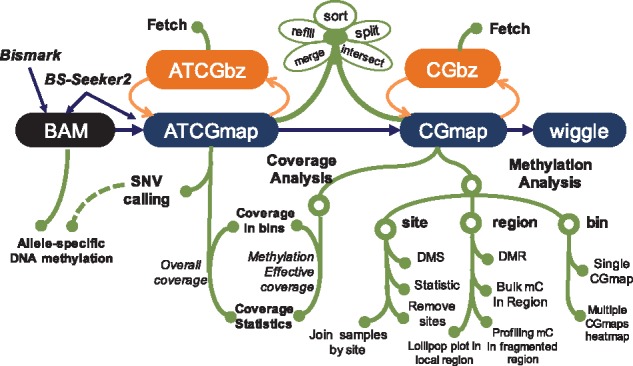
Flowchart for CGmapTools. CGmapTools accepts BAM file from BS-Seeker2 or Bismark, produces ATCGmap and CGmap files, and provides a set of functions derived from the two formats, such as SNV calling, coverage analysis, and methylation analysis. CGmapTools also defines the binary formats ATCGbz and CGbz, supporting rapid retrieval of data from large DNA methylome datasets
3.4.2 Versatile analyses and visualization functions
Regarding file processing, CGmapTools provides functions for converting the file formats, and manipulating files, such as file sorting, merging, intersecting, splitting and patching up missing information, and so on. CGmapTools provides versatile functions for visualizing DNA methylomes. For a single sample, CGmapTools generates pie chart plots for methylation contributions in different sequence contexts (Fig. 5A), and also generates bar plots for bulk methylations (Fig. 5B) and methylation level distribution (Fig. 5D) and methylation profiles across the genome (Fig. 5D). As Guo et al. previously proposed, CW (W is A or T) is a distinct methylation context from CC in mammals (Guo et al., 2016), CGmapTools reports DNA methylations in CG, CHG and CHH contexts for plant studies, as well as CG, CW and CC contexts for mammalian studies.
Fig. 5.
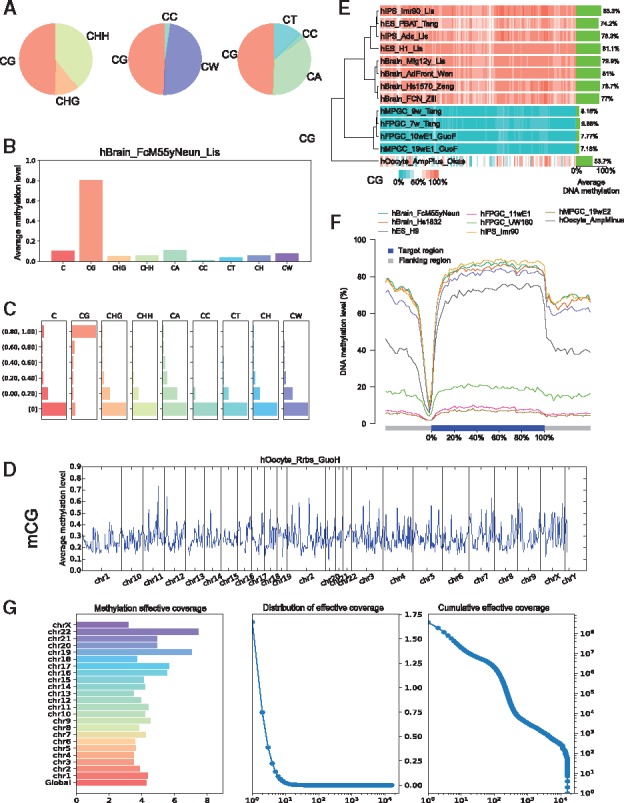
Graphs generated by CGmapTools. (A) Pie chart plots for DNA methylation contributions by different contexts in the sample hBrain_FcM55yNeun_Lis. (B) Bar plots for bulk DNA methylations in different contexts. (C) Distribution plot for DNA methylations in different contexts. (D) Distribution plots for mCG are shown in bins across the whole genome for single sample hOocyte_Rrbs_GuoH. (E) Heatmap plot for DNA methylation in bins across multiple samples. Average methylation levels of CpG are shown on the right, and a hierarchical clustering tree is built based on Spearman’s correlation coefficients. (F) Distribution plot of CpG methylation levels in fragmented regions across gene bodies. (G) The chromosome-wide MEC (left), density plot of MEC, and cumulative distribution of MEC (right) in AML sample
For multiple samples, CGmapTools generates heatmaps showing the DNA methylation levels in windows across chromosomes (Fig. 5E). To view the mCG levels across gene bodies, CGmapTools also provides functions to profile methylation level distributions across genes or across a panel of specified regions (Fig. 5F).
3.4.3 Evaluate the coverages in BS-seq library
Read coverage is an important factor when estimating DNA methylation levels and fetching SNV calls. SNV calls depend on all nucleotides (A, T, C and G), whereas DNA methylation levels only depend on T and C read counts aligned to cytosines. Thus, we defined overall coverage (OAC) as the average read coverage on all nucleotides on both strands, which are calculated from the ATCGmap file. We also defined methylation-effective coverage (MEC), as the average read coverage only for cytosines, which can be calculated from the CGmap file. Generally, the MEC is slightly higher than half of the OAC (Supplementary Table S2). CGmapTools also provides multiple tools for visualizing the distributions of coverages (Fig. 5G).
4 Discussion
In this study, we use the novel methods BayesWC and BinomWC, which significantly improved the precisions of heterozygous SNV calls compared to previous tools, and retained comparable recalls. To deeply explore the DNA methyomes at the allelic level, we provide pipelines utilizing accurate heterozygous calls and de novo exploration of the SNV-related ASM regions. To deeply explore the DNA methyomes at the allelic level, we provide pipelines utilizing accurate heterozygous calls and de novo exploration of the SNV-related ASM regions. Our study showed that ASM regions are significantly enriched for transposon elements.
Beyond the functionalities described above, we designed CGmapTools as an integrated DNA methylome analyses package, with the advantages of, (i) implementation of a dynamic fragmentation strategy for exploring DMR in low-coverage data; (ii) use of standard ATCGmap/CGmap formats for ease of sharing methylomes; (iii) implementation as command-line tools that are convenient for parallel processing and to be extended; (iv) support for instant retrieval based on binary file formats and (v) user-friendly functions for visualizing methylomes at multiple levels, such as designing a Tanghulu plot for visualizing the methylation status on original reads, and designing a Lollipop plot to reveal both low-coverage cytosines and un-methylated cytosines in a local region. Finally, CGmapTools provides advanced and resourceful solutions to the computational challenges in analysing BS-seq data.
Supplementary Material
Acknowledgements
We thank Petko Fiziev for kindly providing code for converting BAM format to CGmap format. We thank Prof. Hong Wu for suggestions on visualization.
Funding
This work has been supported by National Key Research and Development Program of China [grant number 2016YFD0100801] and the National Natural Science Foundation of China [grant number 31701415 and 91435204].
Conflict of Interest: none declared.
References
- Akalin A. et al. (2012a) Base-pair resolution DNA methylation sequencing reveals profoundly divergent epigenetic landscapes in acute myeloid leukemia. PLoS Genet., 8, e1002781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akalin A. et al. (2012b) methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol., 13, R87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida D. et al. (2017) Efficient detection of differentially methylated regions using DiMmeR. Bioinformatics (Oxford, England), 33, 549–551. [DOI] [PubMed] [Google Scholar]
- Benoukraf T. et al. (2013) GBSA: a comprehensive software for analysing whole genome bisulfite sequencing data. Nucleic Acids Res., 41, e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G.G. et al. (2014) BisQC: an operational pipeline for multiplexed bisulfite sequencing. BMC Genomics, 15, 290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roadmap Epigenomics Consortium et al. (2015) Integrative analysis of 111 reference human epigenomes. Nature, 518, 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do C. et al. (2017) Genetic–epigenetic interactions in cis: a major focus in the post-GWAS era. Genome Biol., 18, 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du W.W. et al. (2013) Roles of versican in cancer biology–tumorigenesis, progression and metastasis. Histol. Histopathol., 28, 701–713. [DOI] [PubMed] [Google Scholar]
- Fang F. et al. (2012) Genomic landscape of human allele-specific DNA methylation. Proc. Natl. Acad. Sci. U S A., 109, 7332–7337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S. et al. (2010) Conservation and divergence of methylation patterning in plants and animals. Proc. Natl. Acad. Sci. U S A., 107, 8689–8694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S. et al. (2015a) BS-SNPer: SNP calling in bisulfite-seq data. Bioinformatics, 31,4006–4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S. et al. (2015b) SMAP: a streamlined methylation analysis pipeline for bisulfite sequencing. GigaScience, 4, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W. et al. (2013) BS-Seeker2: a versatile aligning pipeline for bisulfite sequencing data. BMC Genomics, 14, 774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W. et al. (2014) Characterizing the strand-specific distribution of non-CpG methylation in human pluripotent cells. Nucleic Acids Res., 42, 3009–3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W. et al. (2016) Mammalian non-CG methylations are conserved and cell-type specific and may have been involved in the evolution of transposon elements. Sci. Rep., 6, 32207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackenberg M. et al. (2011) NGSmethDB: a database for next-generation sequencing single-cytosine-resolution DNA methylation data. Nucleic Acids Res., 39, D75–D79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P.A. (2012) Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet., 13, 484–492. [DOI] [PubMed] [Google Scholar]
- Kawakatsu T. et al. (2016) Epigenomic diversity in a global collection of Arabidopsis thaliana accessions. Cell, 166, 492–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger F., Andrews S.R. (2011) Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics, 27, 1571–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird P.W. (2010) Principles and challenges of genomewide DNA methylation analysis. Nat. Rev. Genet., 11, 191–203. [DOI] [PubMed] [Google Scholar]
- Law J.A., Jacobsen S.E. (2010) Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet., 11, 204–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. et al. (2009) The sequence alignment/map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y. et al. (2014) An epigenetic signature in peripheral blood associated with the haplotype on 17q21.31, a risk factor for neurodegenerative tauopathy. PLoS Genet., 10, e1004211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao W.-W. et al. (2015) MethGo: a comprehensive tool for analyzing whole-genome bisulfite sequencing data. BMC Genomics, 16, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y. et al. (2012) Bis-SNP: combined DNA methylation and SNP calling for Bisulfite-seq data. Genome Biol., 13, R61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luu P.-L. et al. (2016) P3BSseq: parallel processing pipeline software for automatic analysis of bisulfite sequencing data. Bioinformatics, 33, 428–431. [DOI] [PubMed] [Google Scholar]
- Matzke M.A., Mosher R.A. (2014) RNA-directed DNA methylation: an epigenetic pathway of increasing complexity. Nat. Rev. Genet., 15, 394–408. [DOI] [PubMed] [Google Scholar]
- Meissner A. et al. (2005) Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res., 33, 5868–5877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Q. et al. (2016) Systems nutrigenomics reveals brain gene networks linking metabolic and brain disorders. EBioMedicine, 7, 157–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orozco L.D. et al. (2015) Epigenome-wide association of liver methylation patterns and complex metabolic traits in mice. Cell Metab., 21, 905–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park Y. et al. (2014) MethylSig: a whole genome DNA methylation analysis pipeline. Bioinformatics, 30, 2414–2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plongthongkum N. et al. (2014) Advances in the profiling of DNA modifications: cytosine methylation and beyond. Nat. Rev. Genet., 15, 647–661. [DOI] [PubMed] [Google Scholar]
- Saito Y. et al. (2014) Bisulfighter: accurate detection of methylated cytosines and differentially methylated regions. Nucleic Acids Res., 42, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz M.D. et al. (2015) Human body epigenome maps reveal noncanonical DNA methylation variation. Nature, 523, 212–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker R. et al. (2010) Allele-specific methylation is prevalent and is contributed by CpG-SNPs in the human genome. Genome Res., 20, 883–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith Z.D., Meissner A. (2013) DNA methylation: roles in mammalian development. Nat. Rev. Genet., 14, 204–220. [DOI] [PubMed] [Google Scholar]
- Song Q. et al. (2013) A reference methylome database and analysis pipeline to facilitate integrative and comparative epigenomics. PLoS One, 8, e81148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stricker S.H. et al. (2017) From profiles to function in epigenomics. Nat. Rev. Genet., 18, 51–66. [DOI] [PubMed] [Google Scholar]
- Sun D. et al. (2014) MOABS: model based analysis of bisulfite sequencing data. Genome Biol., 15, R38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S. et al. (2013) MethyQA: a pipeline for bisulfite-treated methylation sequencing quality assessment. BMC Bioinformatics, 14, 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z. et al. (2015) swDMR: a sliding window approach to identify differentially methylated regions based on whole genome bisulfite sequencing. PLoS One, 10, e0132866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warden C.D. et al. (2013) COHCAP: an integrative genomic pipeline for single-nucleotide resolution DNA methylation analysis. Nucleic Acids Res., 41, e117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Y. et al. (2016) Detection of differentially methylated regions in whole genome bisulfite sequencing data using local Getis-Ord statistics. Bioinformatics (Oxford, England), 32, 3396–3404. [DOI] [PubMed] [Google Scholar]
- Xi Y., Li W. (2009) BSMAP: whole genome bisulfite sequence MAPping program. BMC Bioinformatics, 10, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W. et al. (2012) Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome. Cell, 148, 816–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. et al. (2009) Non-imprinted allele-specific DNA methylation on human autosomes. Genome Biol., 10, R138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziller M.J. et al. (2013) Charting a dynamic DNA methylation landscape of the human genome. Nature, 500, 477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou D. et al. (2015) MethBank: a database integrating next-generation sequencing single-base-resolution DNA methylation programming data. Nucleic Acids Res., 43, D54–D58. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.