

Abstract
Mucopolysaccharidosis type I (MPS I) is caused by deficiency of α-l-iduronidase (IDUA), a lysosomal enzyme involved in the breakdown and recycling of glycosaminoglycans (GAGs). Although enzyme replacement therapy is available, the efficacy of the treatment for neuropathic manifestations is limited. To facilitate drug discovery and model disease pathophysiology, we generated neural stem cells (NSCs) from MPS I patient-derived induced pluripotent stem cells (iPSCs). The NSCs exhibited characteristic disease phenotypes with deficiency of IDUA, accumulation of GAGs and enlargement of lysosomes, in agreement with the severity of clinical subgroups of MPS I. Transcriptome profiling of NSCs revealed 429 genes that demonstrated a more extensive change in expression in the most severe Hurler syndrome subgroup compared to the intermediate Hurler–Scheie or the least severe Scheie syndrome subgroups. Clustering and pathway analysis revealed high concordance of the severity of neurological defects with marked dysregulation of GAG biosynthesis, GAG degradation, lysosomal function and autophagy. Gene ontology (GO) analysis identified a dramatic upregulation of the autophagy pathway, especially in the Hurler syndrome subgroup. We conclude that GAG accumulation in the patient-derived cells disrupts lysosomal homeostasis, affecting multiple related cellular pathways in response to IDUA deficiency. These dysregulated processes likely lead to enhanced autophagy and progressively severe disease states. Our study provides potentially useful targets for clinical biomarker development, disease diagnosis and prognosis, and drug discovery.
Introduction
Genetic defects impacting lysosomal function constitute a heterogeneous group of ∼50 metabolic disorders of lysosomal storage diseases, with varying clinical consequences that may include ocular, skeletal, visceral and neurological abnormalities (1). Mucopolysaccharidoses type I (MPS I) is caused by mutations in the gene-encoding α-l-iduronidase (IDUA), a lysosomal acid hydrolase (2). IDUA cleaves unsulfated α-l-iduronosidic linkages from complex membrane-associated glycosaminoglycans (GAGs), predominantly consisting of heparan sulfate and dermatan sulfate, which contribute to multiple regulatory functions associated with cell growth, neuronal patterning and development, extracellular matrix (ECM) and as a lubricant/shock-absorber in synovial joints (3). GAGs are long unbranched polysaccharides, containing amino sugars (N-acetylglucosamine or N-acetylgalactosamine) and other sugars (glucuronic acid, iduronic acid or galactose). Over 140 distinct IDUA mutations have been detected in MPS I patients (4–7). Aberrant accumulation of GAGs and secondary glycolipids due to the deficiency of IDUA results in progressive deterioration of central nervous system functions, along with other clinical manifestations including organomegaly, skeletal dysplasia, cardiovascular and ocular pathology (8).
Based on disease severity and progression, MPS I is classified into three clinical subgroups: Hurler, Hurler–Scheie and Scheie syndromes (8). No residual IDUA enzyme activity is detected in Hurler syndrome, the most severe form. Patients with Hurler syndrome exhibit developmental and facial abnormalities, debilitating skeletal abnormalities, corneal clouding, multiple organ dysfunction and progressive neurological defects. These severe pathologies frequently result in premature death. Patients with Hurler–Scheie syndrome display normal intelligence or mild to moderate intellectual disability, facial and skeletal abnormalities, with variable mortality outcome. Scheie syndrome is the mildest form; patients show normal intelligence, no characteristic facial features and usually a normal life expectancy. A wide range of disease foci, including the age of onset and the rate of clinical progression, complicate the classification of patients into specific subtypes. Unpredictable genotype/phenotype combination in MPS I can be attributed, at least in part, to the lack of relevant and reliable biomarkers, confounding prognosis and clinical management. As in several other lysosomal storage diseases, enzyme replacement therapy (ERT) has been approved for treatment of MPS I (9–11). However, limited therapeutic benefit of ERT is evident on neuropathic pathology because of the blood–brain barrier. Small molecule therapy, not currently available, should have an advantage as the drugs can be optimized for penetration of the blood–brain barrier.
Induced pluripotent stem cells (iPSCs) derived from patients offer an emerging paradigm for disease modeling that can be used to generate derivative cells or tissue organoids (12,13), providing new opportunities for drug discovery (14,15). Patient-derived iPSCs are now being utilized for modeling lysosomal storage disorders (16). For example, Hurler syndrome patient-derived iPSCs have been used for IDUA mutation correction and differentiation into hematopoietic cells for the study of potential use in cell therapy (17). Another study employing transplantation of hematopoietic cells into Hurler syndrome patients revealed varying efficacy (18).
Here we report identification of disease phenotype and modeling of three distinct MPS I subgroups using neural stem cells (NSCs) from MPS I patient-derived iPSCs. The patient-derived NSCs displayed different degrees of deficiency of IDUA enzyme activity, GAG accumulation and lysosome enlargement that correlated with the severity of MPS I disease. NSC transcriptome profiling revealed progressively dramatic changes in expression of GAG pathway genes, in concordance with clinical symptoms in the respective MPS I subgroups. Abnormal expression of autophagy-associated genes was clearly evident in Hurler syndrome. Our results suggest potential pathway-specific biomarkers to distinguish MPS I subtypes as well as targets for small molecule drug discovery.
Results
Characterization of iPSCs and NSCs derived from MPS I patients
The reprogrammed iPSC lines were established from fibroblasts of Hurler, Hurler–Scheie and Scheie patients (Fig. 1A). All three patient iPSC lines demonstrated normal morphology (Fig. S1A) and karyotypes (Fig. S1B) and exhibited positive expression of Nanog and Tra1-60, as measured by flow cytometry (Fig. S1C), and of OCT4A, NANOG, SOX2 and SSEA-4, as determined by immunofluorescence staining (Fig. S1D).
Figure 1.

MPS I clone information and functional activity of IDUA and validation of GAG accumulation. (A) The three MPS I patient fibroblasts that were used to generate corresponding iPSC patient lines and clones employed for further studies. (B) IDUA activity in cell lysates from patient-derived NSCs in the presence of serum. For lysates, the nmoles of 4-MU were normalized to a 7-h reaction and equal protein concentrations. Released 4-MU was measured fluorometrically and plotted against a calibration standard. NSCs derived from distinct MPS I subgroups demonstrate varying enzymatic deficiency of IDUA. (C) Immunoblot analysis of MPS I-NSC lysates. The polyclonal IDUA antibody identified a 76 kDa band in control and Scheie lysates but severely reduced protein expression in H/S and H lysates. (D) Quantitative GAG assays to measure overall elevation of GAG in MPS I NSCs. Equal protein concentration was applied in GAG-DMMB assay. Measurements of GAGs plotted against a standard curve prepared using bovine tracheal chondroitin 4-sulfate as a control showed significant increase in GAGs of Hurler cells compared to wild type.
The NSC lines derived from the patient iPSCs showed the characteristic expression of Nestin, PAX6 and SOX2 (Fig. S2A). As predicted, the pluripotency marker OCT4 was undetectable in NSCs (Fig. S2A). The process of iPSC generation and NSC differentiation requires many passages of cell cultures and concurrent handling of multiple cell lines; therefore, short tandem repeat (STR) analysis was performed to validate the final identity of the NSC lines. STR analysis revealed an excellent correlation between the parent fibroblasts and the derivative NSCs (Fig. S2B). Taken together, patient iPSCs and NSCs exhibited high quality with normal growth characteristics and morphology.
Deficiency of IDUA activity in MPS I-NSCs
Mutations in the IDUA gene result in reduction or loss of IDUA enzyme activity in the MPS I patient fibroblasts (19). We therefore measured the IDUA activity in the patient NSCs. Hurler NSCs showed negligible IDUA activity, and correspondingly low IDUA activities were detected in Hurler–Scheie and Scheie NSCs (Fig. 1B). The reduction in IDUA enzyme activity was further confirmed by immunoblot analysis of patient NSCs, with the largest decrease in IDUA protein observed in the Hurler NSCs (Fig. 1C). Our results demonstrate an excellent concordance between the IDUA activity and IDUA protein levels in patient-derived NSCs, which corresponds to the severity of clinical phenotypes in MPS I.
GAG accumulation in NSCs from MPS I patients
IDUA deficiency results in the accumulation of sulfated GAGs, such as heparan and dermatan sulfate, in the lysosomes of MPS I patients (3). We therefore examined GAGs in patient NSCs using a spectrophotometric assay that results in a metachromatic shift in the absorption maximum of 1, 9-dimethylmethylene blue (DMMB) dye upon binding with sulfated GAGs (20). Hurler NSCs demonstrated significantly higher accumulation of GAGs compared to the Hurler–Scheie and Scheie NSCs, which exhibited smaller increases in GAGs (Fig. 1D).
Enlargement of lysosomes in patient NSCs
Lysosomal enlargement due to the accumulation of GAGs, a characteristic phenotype in MPS I patients (21), has previously been observed in patient-derived fibroblasts by a LysoTracker® dye staining assay (22). We used the same assay to examine lysosomes in MPS I patient NSCs. Augmented LysoTracker® staining was observed in the NSCs of all MPS I patients, with Hurler NSCs showing the highest levels, Hurler–Scheie NSCs yielding an intermediate level and Scheie NSCs the lowest levels (Fig. 2A). Imaging analysis demonstrated similar values as depicted in LysoTracker® labeling experiments (Fig. 2B). These results suggest enlarged lysosomes in the patient NSCs by accumulation of GAGs.
Figure 2.
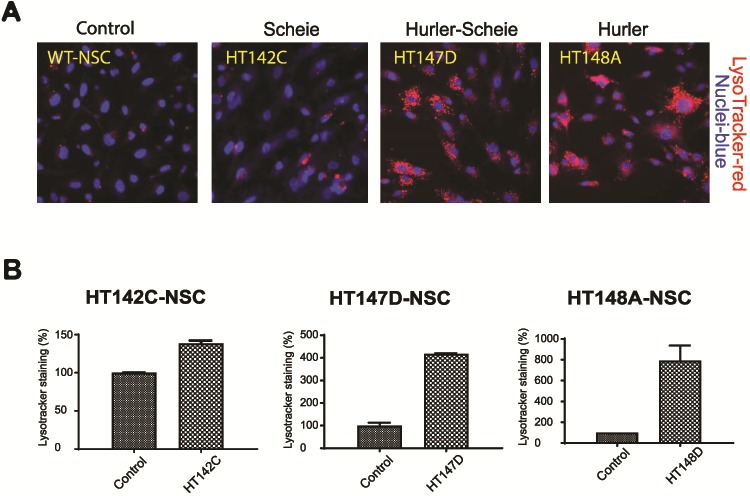
Validation of lysosome accumulation. (A) Variable degree of lysosomal accumulation displayed in patient NSCs in the presence of serum. LysoTracker® staining in MPS I-NSCs in presence of serum exhibit enlarged lysosomes. In comparison to the control line, Hurler cells displayed abundant lysosomal accumulation, whereas H/S was intermediate and Scheie weaker. (B) Quantitative In Cell analysis correlation with enlarged lysosomal phenotypic staining visualized in the NSC cells.
NSC transcriptome analysis
To elucidate molecular mechanisms associated with the pathogenesis of distinct MPS I phenotypes and identify targets for biomarker and drug discovery, we generated global transcriptomes by RNA-sequencing (RNA-seq) analysis of three independent, biological replicates of the control and patient NSCs. Gene level expression values were generated with the analysis pipeline (Fig. S3A) on GRCh38 assembly using Ensembl v82 annotation. The mean sequencing depth (plus or minus the standard deviation) of RNA-seq libraries was 36 ± 2.34 million reads, with 92 ± 1.45% alignment to the human genome and 82 ± 1.17% alignment to the transcriptome (Fig. S3B). We filtered out genes that exhibited low expression in all samples or had high variance within replicates to produce a high confidence data set for further analysis. Principal component analysis (PCA) of the remaining 8995 genes revealed a high degree of precision in biological replicates. Disease severity accounted for the first principal component encompassing 65.3% of the experimental variance (Fig. S3C). Pairwise Pearson correlation validated the low variance among the replicates and recapitulated PCA data (Fig. S3D).
Differential expression (DE) analysis resulted in 3036 genes that showed significant change in expression in the patient NSCs compared to the control, and enhanced DE was concordant with disease severity (Fig. 3A). Interestingly, 429 genes exhibited DE in NSCs of all three patient subgroups, whereas 1274 genes showed DE only in Hurler syndrome. To examine enrichment of specific pathways associated with the severity of MPS I phenotypes, we assembled the gene level z-scores into 52 clusters using affinity propagation and further sequestered these clusters into 17 super clusters (SCs) (Fig. 3B, Fig. S4). We then performed biological process gene ontology (GO) analysis using the genes from each of the 17 SCs (Fig. 3C). While specific metabolic pathways seem to be altered in distinct syndromes, SC10 included the genes that are highly associated with the most severe form of MPS I, the Hurler syndrome. Genes in SC10 are enriched for Golgi transport, endoplasmic reticulum stress, autophagy and vacuole organization. These data are consistent with progressive buildup of specific GAGs in the lysosomes, leading to secondary accumulation of other downstream products and formation of intracellular vacuoles and lesions, which are characteristics of severe MPS I disease.
Figure 3.
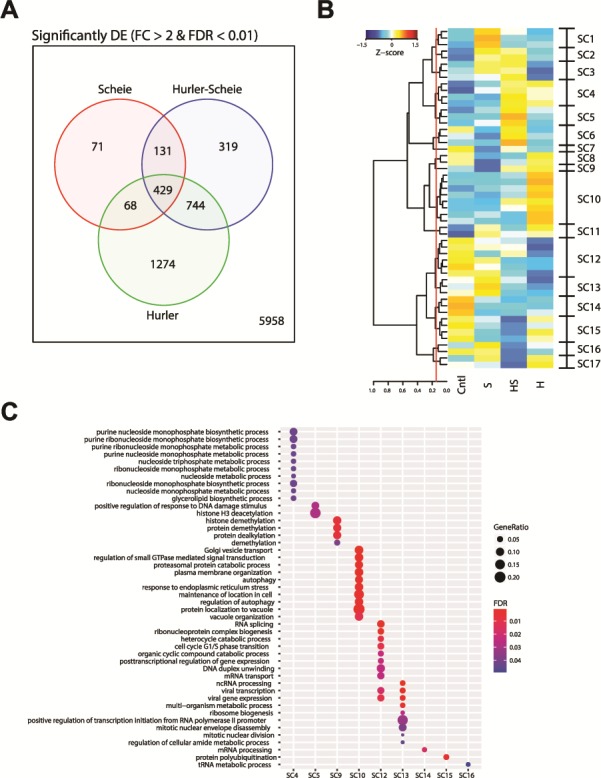
DE, clustering and GO analysis of MPS I. (A) Number of significantly differentially expressed genes in MPS I subtypes when compared to control samples. Genes were considered significant if they had >2-fold change and a false discovery rate less than 1%. (B) Heatmap of the exemplars from the affinity propagation clustering revealing 52 clusters. The dendrogram of the exemplars was cut at a height of 0.145 (red line) to group into 17 SCs for further analysis. (C) The top biological processes from GO analysis results for the genes from the SCs. Only SCs having significantly enriched GO results of less 5% FDR are shown.
Abnormal GAG homeostasis
Given that the biological function of IDUA is hydrolysis of complex GAGs, we first looked at the impact of IDUA deficiency on the expression of genes associated with GAG biosynthesis and degradation pathways (REACTOME database; R-HSA-2022928 and R-HSA-2024096) among DE genes. Only four DE genes belonged to the GAG pathway in the mildest MPS I subgroup (Scheie), with nine genes revealing altered expression in intermediate (Hurler–Scheie) and 21 in the most severe form (Hurler) (Fig. 4A). A heatmap of DE GAG genes highlights their dramatically higher expression in Hurler NSCs (Fig. 4B), with the exception of glucuronyltransferase B3GAT1 (which transfers the glucuronic acid to the tetrasaccharide linker sequence prior to attachment of the GAG disaccharide units) and 3-O-sulfotransferase HS3ST3B1 (responsible for sulfation of heparan sulfate). Expression changes in genes for GAG biosynthesis (Fig. 4C) and GAG degradation (Fig. 4D) were validated by qRT-PCR analysis. Further immunoblot analysis of selected corresponding proteins confirmed the changes in the expression in NSCs (Fig. 4E).
Figure 4.
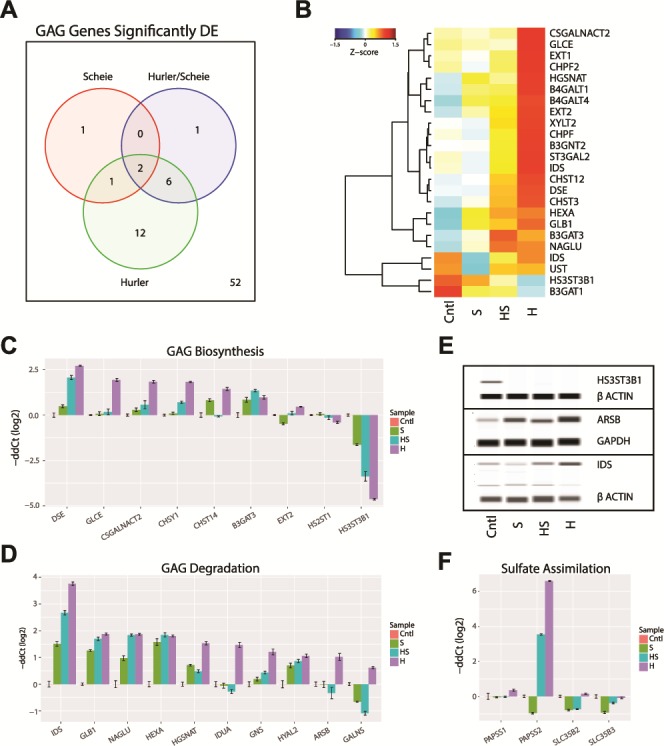
GAG dysregulation in MPS I. (A) GAG biosynthesis and degradation genes from REACTOME pathways (R-HSA-2022928 and R-HSA-2024096) significantly differentially expressed (DE) in the MPS I isotypes. Genes had a >2-fold change and <1% FDR when compared to control samples. (B) The significantly DE GAG gene changes in expression are shown as a heatmap. The Z-score of the gene expression is shown. (C and D) Validation of GAG biosynthesis and degradation genes by qRT-PCR. (E) Immunoblot validation of selected GAG genes. (F) Validation of GAG sulfate assimilation genes by qRT-PCR.
The activated sulfate, 3′-phosphoadenosine 5′-phosphosulfate (PAPS), is generated by the sulfotransferase genes PAPSS1 and PAPSS2 in the cytosol and transported to the Golgi by sulphotransferase genes SLC35B2 and SLC35B3. We performed qRT-PCR to evaluate these genes; only PAPSS2 expression was significantly enhanced and only in the NSCs derived from patients with intermediate and severe forms of MPS I (Fig. 4F).
Dysregulation of the lysosomal pathway
We then examined the impact of GAG accumulation on transcriptional dynamics of lysosomal pathway genes. A majority of genes from the Kyoto Encyclopedia of Genes and Genomes (KEGG) lysosome pathway (hsa04142) revealed higher expression in NSCs correlating with the severity of the disease (Fig. 5A). DE of key lysosomal genes observed in RNA-seq data was validated by qRT-PCR (Fig. 5B). The glycosidase genes (NAGA and GBA) were upregulated in all MPS I subgroups, whereas the protease genes (TPP1, LGMN and CTSC) showed an increased expression level in concordance with disease severity. Augmented expression of the membrane marker genes (LAMP1, SCARB2 and CD68) was detected in the most severe Hurler NSCs. Immunoblot analysis of NSC protein extracts revealed higher expression of lysosomal protease LGMN (Fig. 5C) and of membrane markers SCARB2 and LAMP1 (Fig. 5D) in Hurler–Scheie and Hurler subgroups. We also validated increased LAMP1 and LAMP2 levels in the patient cells using immunofluorescence staining (Fig. 5E). Taken together, our data demonstrate a broad impact on lysosomal genes/proteins with increasing GAG accumulation in more severely affected patient NSCs.
Figure 5.
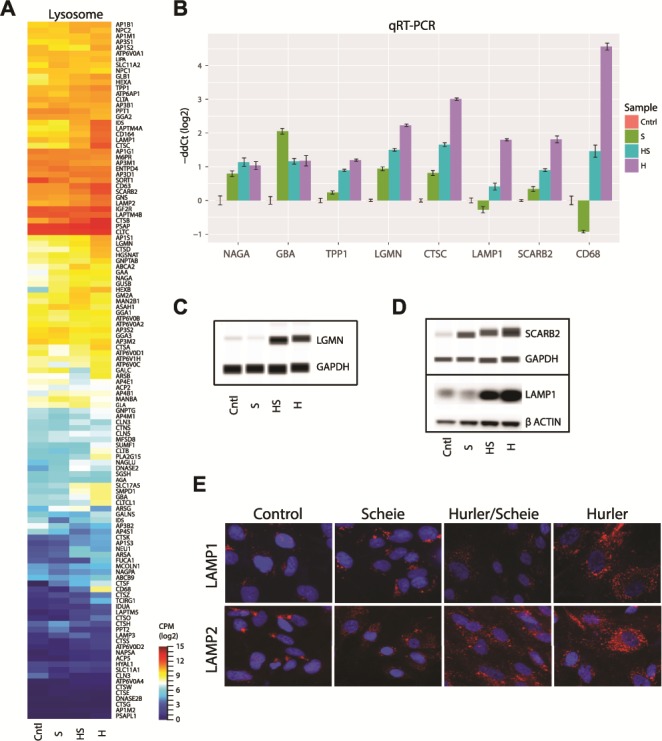
Dysregulation of the lysosome function in MPS I. (A) Expression of genes from KEGG lysosome pathway (hsa04142). Expression level is log2 CPM. (B) Validation of lysosome genes by qRT-PCR. (C and D) Immunoblot validation of selected lysosome genes. (E) Immunohistochemical analysis of lysosome membrane proteins in the NSCs from the MPS I isotypes.
DE of ECM Genes in MPS I Disease
Surface GAGs, in concert with ECM proteins such as collagen, fibronectin and laminin, provide mechanical support to cells and tissues. A query of genes in the ECM Proteoglycan pathway (REACTOME, R-HSA-3000178) identified a broad increase in expression, especially in NSCs of the most severe kind of MPS I disease, the Hurler subgroup (Fig. 6A). DE analysis of several key proteoglycan (PG) genes confirmed their dramatic upregulation with increasing disease severity (Fig. 6B), with the notable exception of Neurocan (NCAN), which exhibited lower expression in Hurler NSCs. We validated changes in CD44 and SDC4 at the protein level by immunoblot analysis and immunofluorescence staining (Fig. 6C and D). Thus, PGs appear to play a significant role in the pathogenesis of MPS I.
Figure 6.
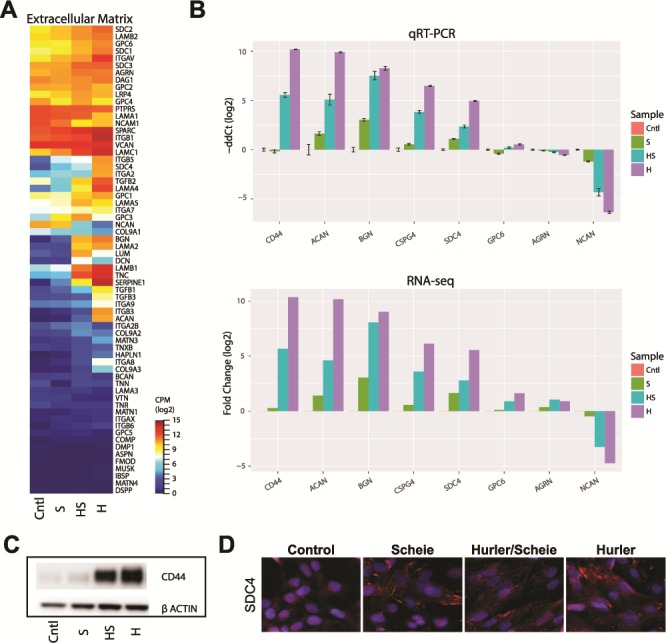
Evaluation of ECM PG disruption. (A) RNA-seq expression of genes from the REACTOME ECM Proteoglycan pathway (R-HSA-3000178). Expression values are log2 CPM. (B) Selected gene validation by qRT-PCR (upper panel). RNA-seq DE results from RNA-seq are shown (lower panel). (C) Immunoblot validation of glycoprotein CD44. (D) Immunohistochemical analysis of syndecan-4 PG protein in the NSCs from the MPS I subgroups.
Autophagy pathway
Malfunctions in autophagy have been reported in several lysosomal storage diseases (23). Therefore, we analyzed DE genes in the autophagy pathway. GO analysis of 1274 genes in Hurler NSCs (see Fig. 3A) identified enrichment of several interesting biological pathways, including autophagy (Fig. 7A). We then looked at the NSC RNA-seq data for DE autophagy genes contained in KEGG (hsa04140) and GO autophagy (GO:0006914) pathways (Fig. 7B). Notably, a vast majority of the autophagy genes exhibited dramatically higher expression in Hurler NSCs compared to other subgroups of MPS I. We validated the expression of several key autophagy genes by qRT-PCR (Fig. 7C) and confirmed the dysregulation of corresponding proteins of the phagophore and autophagosome by immunoblot analysis (Fig. 7D). Our findings corroborate previous reports showing abnormal autophagy in lysosomal storage disease (24,25).
Figure 7.
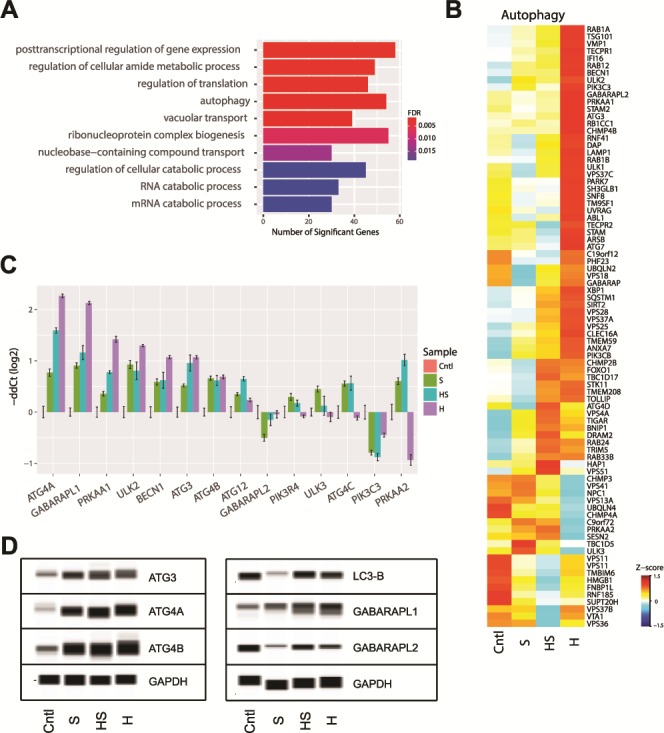
Evaluation of autophagy in MPS I. (A) Top terms enriched from GO analysis of the genes significantly differentially expressed in MPS I-Hurler NSCs. (B) Evaluation of gene expression change in KEGG (hsa04140) and GO autophagy (GO:0006914) pathways. Z-score of RNA-seq expression is shown. (C) Validation of selected autophagy genes by qRT-PCR. (D) Immunoblot validation of autophagy genes.
Proposed gene expression panel for biomarkers of MPS I
A robust panel of biomarkers for MPS 1 validation would be extremely valuable for the in vivo animal model study and clinical trials for drug development discovery. We implemented a filtering scheme with the goal of assembling a short list of genes that would provide an expression signature for the more severe forms of MPS I. We required potential biomarkers to show: >4-fold change in Hurler compared to the control, expression <10 CPM (counts per million) in control and >30 CPM in Hurler. This filtering scheme produced a list of 260 genes (Fig. S5). Ten percent of these genes were featured previously in Figures 4–7, 8A. Several of these genes were further validated by qRT-PCR (Fig. 8B).
Figure 8.
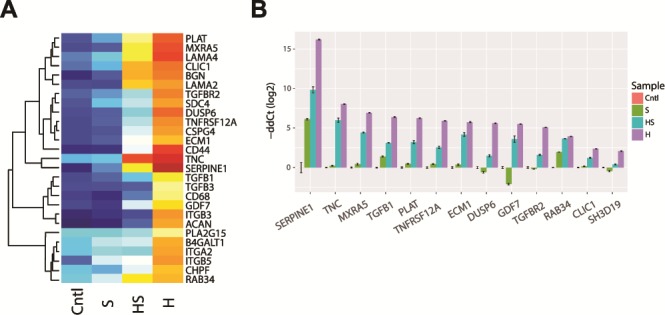
A proposed biomarker panel for genetic diagnosis and developing treatments. (A) Signature genes, identified by stringent expression filtering (Cntl < 10 CPM, Hurler > 30 CPM and DE > 16-fold) and based on pathways investigated in Figures 4–7. Genes indicated in bold were validated by qRT-PCR. (B) Validation of selected top candidate signature genes by qRT-PCR.
Possible model of disease severity
Transcriptome analysis of NSCs from three distinct MPS I subgroups suggests progressive changes in specific pathways and a possible mechanism of progression to more severe disease phenotypes (Fig. 9). We propose that IDUA deficiency leads to progressive accumulation of GAGs in the lysosomes, which in turn alters GAG biosynthesis in the Golgi and disrupts protein sulfation and several major metabolic pathways. Subsequently, these changes impact key steps in fusion of the autophagosomes with lysosomes and protein degradation. Impaired turnover and recycling of GAGs leads to dysfunction of ECM glycoproteins and regulators, collagens, PGs and accumulation of autophagosomes, eventually causing the clinical phenotypes of neurodegenerative disease observed in MPS I patients.
Figure 9.
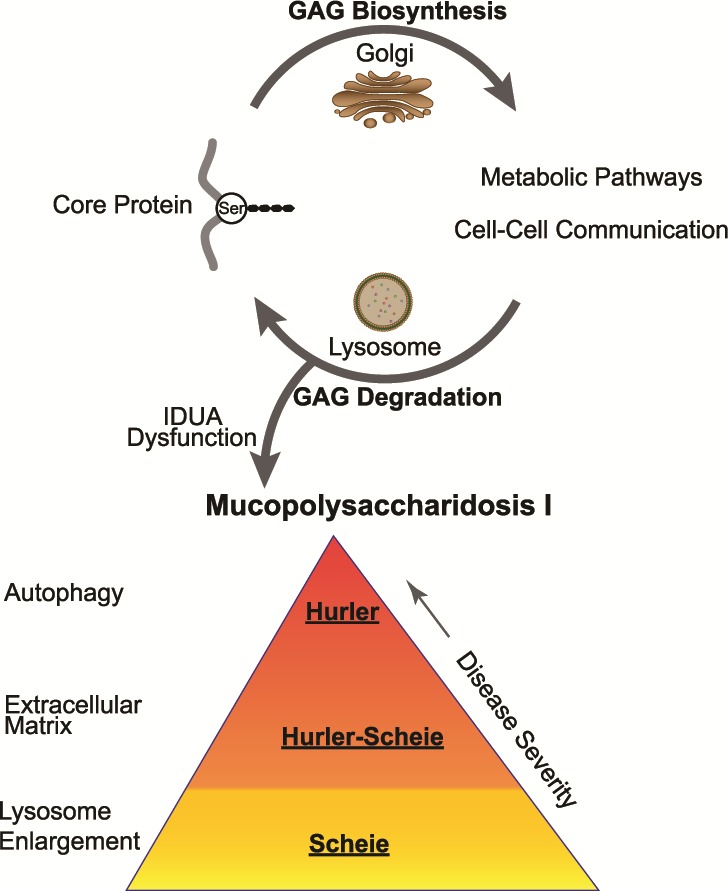
A model of MPS I pathogenesis illustrating the disruption of the normal GAG biosynthesis and degradation cycle by IDUA deficiency. Decrease in loss of IDUA enzyme activity leads to MPS I. The Scheie subgroup manifests a mild lysosome accumulation. Progressively more dysfunction is observed in the ECM of the intermediate disease state, i.e. Scheie–Hurler subgroup. Finally, major dysfunction of autophagy manifests in the most severe disease state, the Hurler subgroup.
Discusssion
Clinical manifestations of MPS I include a progressive deterioration of central nervous system. Accurate discrimination of distinct MPS I clinical subgroups is complicated by significant commonalities including IDUA deficiency, GAG accumulation and enlargement of lysosomes. Yet, unambiguous and early diagnosis of the severe MPS I phenotypes will greatly facilitate optimal strategies for clinical management, including treatment design and evaluation of efficacy. Patient cells derived from the iPSCs of different MPS I subgroups can serve as useful cell-based models for characterization of disease phenotypes and study of MPS I pathophysiology. In our present study, we developed NSCs from patient fibroblast-derived iPSCs to model distinct subgroups of MPS I. This data set permitted systematic dissection of the molecular phenotypes and pathways associated with each specific MPS I subgroup. Our studies revealed that reduced IDUA enzyme activity, accumulation of GAGs and enlargement of lysosomes in the patient-derived NSCs are concordant with the clinical severity of mild, medium and severe symptoms in the Scheie, Hurler–Scheie and Hurler subgroups, respectively (Fig. 9). Thus, the patient-derived NSCs can serve as disease models for investigating the pathophysiology of MPS 1.
Transcriptome profiling of the patient NSCs revealed 3021 differentially expressed genes in the patient cells; of these, 1227 showed higher expression with disease severity, whereas 806 were down-regulated in all disease groups compared to the controls. Of key significance is the altered expression of genes in the GAG pathway. Normal functioning of the GAG pathway is influenced by enzymes that regulate the biosynthesis of heparan and dermatan sulfate (HS/DS) GAGs in three key steps: chain initiation, polymerization and a series of modifications including N-deacetylation and N-sulfation, epimerization and variable O-sulfation. Many diseases arise as a result of inborn errors of GAG biosynthesis (GAG_SIEMS) and defects in linker biosynthesis, sugar transport, GAG polymerases, GAG sulfation and epimerization (26). MPS I has significant GAG accumulation resulting from the deficiency in IDUA enzyme function that may cause malfunction in the GAG pathway.
We observed upregulation of a number of biosynthesis genes such as D-Glucuronyl C5-Epimerase (GLCE), Chondroitin Sulfate N-Acetylgalactosaminyltransferase 2 (CSGALNACT2), Chondroitin Sulfate Synthase 1 (CHSY1), Carbohydrate Sulfotransferase 14 (CHST14) and Dermatan Sulfate Epimerase (DSE), thereby resulting in altered heparan sulfate PGs and chondroitin and dermatan sulfate (CS/DS) PG expression in MPS I both at the PG core and the GAG chains.
Down-regulated genes in the GAG biosynthesis pathway include those encoding Heparan Sulfate 2-O-Sulfotransferase 1 (HS2ST1) and Heparan Sulfate-Glucosamine 3-Sulfotransferase 3B1 (HS3ST3B1) proteins that catalyze the addition of sulfate groups in heparan sulfate. We inferred that the down regulation of these genes was related to expression levels of two synthetases, 3′-Phosphoadenosine 5′-Phosphosulfate Synthase 1 (PAPPS1) and 3′-Phosphoadenosine 5′-Phosphosulfate Synthase 2 (PAPPS2), which catalyze the transfer of activated sulfur from the universal donor 3′-phosphoadenosine 5′-phosphosulfate (PAPS) to acceptor molecules. PAPPS2 was upregulated in Hurler–Scheie and Hurler NSCs whereas PAPPS1 was not affected. The transporter genes encoding Adenosine 3′′-phospho 5′′-phosphosulfate transporter 2 and 3, SLC5B2 and SLC5B3, regulate the transfer of the sulfonate group from PAPS to the substrate in the cytosol or Golgi; however, no significant change was observed in the expression of these genes.
Sulfate maintenance genes, e.g. synthetase PAPPS2, sulfotransferase CHST14 and several sulfatases such as iduronate 2-sulfatase (IDS), glucosamine (N-acetyl)-6-sulfatase (GNS) and arylsulfatase B (ARSB), were upregulated in Hurler cells. The impact of elevated ARSB in Hurler cells could result in aberrant sulfation of GAGs leading to buildup in the lysosome. Sulfation is essential for proper fetal development. Disturbances in sulfate metabolism are linked to neurological, skeletal and reproductive defects (27). Key enzymes involved in CS/DS linkage region, chain elongation and modification were highly affected in comparison to the HS chain processing enzymes. We suggest that cellular systems are attempting to compensate for the lack of proper GAGs (due the accumulation of GAGs in lysosomes) by synthesizing more enzymes to overcome the shortage of functional GAGs.
Expression of genes encoding lysosomal proteins was significantly altered in the MPS I patient cells. We observed the upregulation of genes for lysosomal membrane proteins that are involved in lysosomal stability, scavenger proteins and autophagy. Lysosomal-associated membrane proteins (LAMP1 and LAMP2), which are responsible for maintaining lysosomal integrity and pH to protect the membrane surface from autodigestion, were upregulated in Hurler NSCs. Lysosomal integral membrane protein-2 (LIMP II) (28) (encoded by SCARB2), an essential protein for normal biogenesis and maintenance of lysosomes, was elevated. Similarly, CD68, a transmembrane glycoprotein belonging to the lysosomal/endosomal-associated LAMP family and a member of the scavenger receptor family, also displayed increased expression levels. The increased expression of these genes may assist in the removal of cellular debris, promote phagocytosis and mediate recruitment and activation of macrophages. Enhanced expression of GM130, a Golgi matrix protein, was observed by immunostaining. Aberrant expression of GM130 could lead to neuron dysfunction and death as seen in MPS type IIIB (29).
Increased expression of some of these genes observed in the data could result in blockage of lysosomal function, eventually causing a higher expression level in many autophagy genes. Defective autophagy is observed in many lysosomal storage diseases, probably due to defective lysosome–autophagosome fusion resulting from an abnormal lysosomal membrane composition and disrupted lysosomal function. Significantly reduced expression was evident for Translation initiation factor IF-1 (INFA1) and Phosphatidylinositol 3-Kinase Catalytic Subunit Type 3 (PIK3C3) both of which are involved in the initiation of autophagosomes.
We also found elevated expression of many genes encoding autophagy proteins, including the core ATG4A gene, PRKAA1, which is an upstream regulator, E2 like enzyme Atg3 that facilitates LC3/GABARAP lipidation and the Atg8 subfamily γ-aminobutyric acid (GABA)-receptor-associated proteins (GABARAPs). GABA Type A receptor-associated protein like 1 (GABARAPL1) is the most predominant ATG8 homologue in the central nervous system; it was highly expressed in Hurler cells. LC3B is involved in elongation of the phagophore membrane, whereas GABRAPL1 is essential for a later stage in autophagosome maturation and plays a distinct role in autophagy by binding to mutated proteins that form aggregates in neurodegeneration.
Dysregulation of autophagy can further exacerbate lysosomal substrate accumulation and neurodegeneration. Normally, GAG precursors are recycled; however, in MPS I, recycling of GAGs is inhibited and the useful GAGs in the patient cells are significantly reduced due to the deficiency in IDUA enzyme activity. Therefore, the expression changes of these genes may indicate that the cellular system tries to compensate for the consequences of IDUA deficiency by synthesizing more GAG precursors de novo. This further increases the lysosomal GAG accumulation and enlarges lysosomes. As substrates accumulate, the lysosomes swell and expand into the cytoplasm. As a consequence, the enlarged cells lead to organomegally.
GAGs are essential components of ECM, bound covalently to form PGs that are important for the bone organic matrix. The MPS I patients exhibit skeletal pathology due to the buildup of these undegraded GAGs; defects in both skeletal and soft connective tissue are also a symptom of MPS I patients. Our data suggest that observed skeletal defects in MPS I patients coincide with tissue distribution and expression level of PGs. We identified higher expression of the major PGs present in the ECM of bone, biglycan (BGN), which has affinity for transforming growth factor-β (TGFβ), and Aggrecan (ACAN), which is an integral part of the ECM in cartilaginous tissue. Another PG, Syndecan 4 (SDC4), was significantly upregulated. Notably, SDC4’s interaction with fibronectin is modulated by tenascin-C (TNC), which together with chondroitin sulfate PG 4 (CSPG4) showed higher expression.
Some CD44 isoforms exist as heparan sulfate PGs that involve cell adhesion, cell migration and cell–cell interaction. CD44/aggrecan are believed to be a major component of the cartilage matrix and associated with growth factor regulation in inflammatory arthritis (30). CD44 was substantially elevated in Hurler cells with less change in Hurler–Scheie and close to normal levels in Scheie. The only CS/DS PG that was significantly reduced in all three patient cells was NCAN, which is a major component of the brain ECM and is also abundantly expressed in developing rat retina. Down regulation of NCAN might play a role in cortical folding leading to impaired cognitive function and early visual processing (31). No dramatic gene expression changes were observed in Glypicans/GPI-anchored PGs (GPC6), which are heparan sulfate PGs bound to the plasma membrane. We observed significant gene overexpression in PGs that have chondroitin sulfate or both CS and heparan sulfate chains when compared to heparan bearing PGs. However, Agrin, an HS PG (AGRN) was somewhat down-regulated in all patient NSCs. String network analysis further shows interaction between AGRN, and laminin α 4 (LAMA4), which is also upregulated significantly. We note that AGRN and LAMA4 are required for maintenance of neuromuscular junctions (32).
Further analysis revealed significant dysregulation of a number of genes involved in TGFβ signaling pathway in Hurler cells. TGFβ is a multifunctional protein that plays an important role in skeletal development in vivo including cell proliferation, cellular differentiation and other functions (33). A majority of the TGFβ ECM pathway genes exhibited dramatically enhanced expression in Hurler NSCs compared to control cells that displayed negligible expression as seen by expression profiling and qPCR. Genes included were members of the TGFβ family, specifically TGFβ1, growth differentiation factor 7 (GDF7)—a secreted ligand of the TGFβ superfamily of proteins, TGFβR2 and the inhibitory SMAD7. However, GDF7 and TGFBR2 were down-regulated in Scheie NSCs. TGFβ1 activation in turn stimulates secretion of a protease inhibitor, plasminogen activator inhibitor-1 (Serpine 1/PAI-1), which acts as an inhibitor of tissue-type plasminogen activator (PLAT). Serpine 1 was upregulated significantly in all NSCs, whereas PLAT expression was highest in Hurler, moderate in Hurler–Scheie, lowest in Scheie and negligible in control NSCs. Loss of IDUA probably leads to dysregulated TGFβ signaling at different levels, which may contribute to the IDUA deficiency phenotypes.
Hurler syndrome is the most severe clinical form of MPS I, exhibiting the most differentially expressed genes involved in the GAG synthesis and degradation, ECM, the autophagy pathway and inflammation. Disturbances in the functional GAG chain biosynthesis and degradation can induce a cascade of molecular abnormalities following GAG accumulation in lysosomes that results in ECM dysregulation, and ultimately autophagy dysregulation, apoptosis (Fig. 9). These results support our hypothesis that the accumulation of lysosomal GAG (HS and DS) causes the elevation of gene expression in the GAG biosynthesis and autophagy pathway genes. Possibly, the dysregulation of these pathways exacerbate the MPS I lysosomal storage pathology.
In conclusion, we have demonstrated that patient iPSCl-derived NSCs exhibit the characteristic MPS I phenotypes of reduced IDUA enzyme activity, GAG accumulation and enlarged lysosomes. These cells can be used for further study of disease pathophysiology and as a cell-based disease model system for drug discovery. We note that patient iPSCs and their derivative NSCs can exhibit variations in characteristics, especially because of associated genetic mutation and epigenome changes. Therefore, further investigations involving additional NSC lines from patients with distinct IDUA mutations are needed to substantiate our findings.
Our results can be used to customize a signature gene expression panel for diagnosis of MPS I, permitting faster and more accurate distinctions between Hurler, Hurler–Scheie and Scheie subgroups rather than the current practice of diagnosing based on symptoms alone. The results of observed gene expression changes can be further studied to develop a set of genetic in vivo diagnostic biomarkers and for endpoints for clinical studies, as well as for the identification of potential drug targets for therapy.
Materials and Methods
Materials
LysoTracker® Red DND-99 dye (cat #L7528) and Hoechst 33342 nuclear dye (cat # H3570) were purchased from Thermo Fisher Scientific Life Sciences (Waltham, MA), 96-well black clear bottom plates (cat #655090) from Greiner Bio-One (Monroe, NC), Matrigel® (cat #354277) from Corning (San Jose, CA), Rho-kinase (ROCK) Inhibitor Y27632 (cat # Y0503) from Sigma-Aldrich (St. Louis, MO) and Blyscan™ Sulfated GAG assay, (cat # NC9922400) from Thermo Fisher. All taqMan probes for the genes of interest and inventoried were ordered from Applied Biosystems with FAM and GAPDH-Vic labeled, respectively.
iPSC and NSC cultures
Control human iPSC-derived NSCs were obtained from xCell Science (Novato, CA). All NSCs were cultured in StemPro® NSC SFM media (Thermo Fisher cat #A10509-01) consisting of KnockOut DMEM/F-12 basal medium, 0.2% StemPro NSC SFM supplement,0.02 μg/ml FGF Basic Recombinant Human and 0.02 μg/ml EGF Recombinant Human.
For generating MPS I syndrome-specific iPSCs, we obtained the following skin-derived fibroblasts from Coriell Cell Repository (Camden, NJ) (Fig. 1A): GM00415, Hurler Syndrome, 4-year-old male, mutation not listed; GM00963, Hurler–Scheie Syndrome, 5-year-old Caucasian, no mutation listed; GM01256, Scheie Syndrome, 12-year-old male carrying compound heterozygous mutations including a G > A transition in intron 5, in position −7 from exon 6 (IVS5AS-7G > A) and a W > X at nucleotide 1293 in exon 9 [Trp402Ter (W402X)]. Fibroblast lines were reprogrammed to iPSCs using the non-integrating CytoTune-iPSC Sendai Reprogramming Kit (Thermo Fisher), as described (34).
Karyotyping of iPSCs was performed by WiCell Research Institute (Cytogenetics Laboratory, Madison, WI). For immunostaining with stem cell markers, iPSC colonies were treated with 0.5 mM EDTA and plated in Greiner 96 well Matrigel® matrix-coated plates. The cells were fixed with 4% paraformaldehyde and Hoechst stain for 20 min at room temperature and then permeabilized with 0.3% Triton-X for 15 min, blocked in cell staining buffer (BioLegend cat #420201) for 2–3 h, and incubated overnight at 4°C with the primary antibody. The following primary antibodies were used for pluripotency markers: OCT-4A (cat #2840, 1:400), NANOG (cat #4903, 1:400), SOX2 (cat #3579, 1:400), all three from Cell signaling (Danvers, MA); SSEA-4 (cat #sc-21704-1:100) was purchased from Santa Cruz Biotechnology (Dallas, TX). Cells were washed with phosphate-buffered saline (PBS) and incubated with secondary Goat anti-Mouse IgG H&L, (DyLight® 594) pre-adsorbed (Abcam cat #ab96881) Goat anti-rabbit IgG H–L (DyLight® 594) preabsorbed (Abcam cat #ab96901) antibodies. After incubation, cells were rinsed twice with PBS and immediately imaged using IN Cell Analyzer 2000 (GE Healthcare, Waukesha, WA) with 20× objective lens and filter sets for DAPI and Texas Red. All secondary antibodies were tested for cross-reactivity and non-specific immunoreactivity.
The iPSCs were expanded and differentiated into NSCs using the PSC Neural Induction medium (Thermo Fisher, cat #A1647801). Briefly, high-quality human iPSCs were rinsed with PBS and dislodged with 0.5 mM EDTA for 4–5 min. Cells were triturated, counted, and seeded in freshly Matrigel®-coated 6-well plates at 1 × 106 cells/ml in Essential 8 medium supplemented with 10 μM ROCK Inhibitor to prevent cell death. The following day, cells were induced in PSC Neural Induction medium and cultures were continued for 5–7 days. NSCs were harvested and expanded for five passages in Neural Induction medium containing neurobasal medium and 1× neural induction supplement (Thermo Fisher, cat #A1647801) and later switched to StemPro® NSC SFM media. NSCs were stained for neuronal and pluripotency markers, as described. MPS I disease patient cell lines (both fibroblast and NSC cell types) were sent out to Wicell for profiling of recommended STR markers to show relatedness between cell lines, identify the cells as human and examine chromosomal aberrations.
LysoTracker® staining and image analysis
NSCs were seeded at 8000 cells per well, in 100 μl medium in 96-well plates for the LysoTracker® imaging experiments. After 3 days in medium containing 10% FBS, cells were incubated in 100 μl per well 50 nM LysoTracker® dye at 37°C for 1 h. The assay plates were imaged with live cell staining or fixed in 100 μl per well 4% paraformaldehyde solution containing 1 μg/ml Hoechst 33342 in PBS and incubated at room temperature for 30 min. After washing twice with PBS, the image acquisition was carried out in an IN Cell Analyzer 2000 (GE Healthcare). The DAPI (Ex = 350 ± 50, Em = 455 ± 50 nm) and Texas Red (TR) (Em = 545 ± 20, Ex = 593 ± 20 nm) filter sets were used to visualize Hoechst nuclear staining and LysoTracker® staining, respectively. Nine fields of images per well were recorded with a 40× objective.
Image analysis was conducted using IN Cell Analyzer 2000 software (GE Healthcare, version 3.7). The Multi-Target Analysis protocol was used for quantitation of Hoechst stained nuclei and LysoTracker® stained lysosomes. Nuclei were segmented using the Top Hat segmentation method with a minimum area set at 150 microns and a sensitivity set at 50. Lysosomes were identified as ‘Organelles’ within the analysis software and were segmented using the Multiscale top-hat algorithm. Settings for lysosome detection were: identify granules ranging in size of 2–10 μm (3–13 pixels), and a sensitivity setting of 60. Total organelle intensity was calculated by multiplying the mean intensity per granule by the total area of the organelles. A single contrast setting for image analysis was applied using the imaging analysis software included in the IN Cell Analyzer 2000. Results in the figures are expressed as the mean of triplicates ± the standard deviation (SD) unless they are otherwise specified.
IDUA enzyme activity
The activity of IUDA was determined by a fluorometric assay using 4-methylumbelliferyl α-L-iduronide (4-MU-iduronide) (Glycosynth, UK, Cat #44076) as the substrate as previously described (19). Human NSCs were grown in the presence of serum for 3 days. The cells were lysed using 1% triton buffer (150 mM NaCl, 50 mM Tris–HCl, pH 8.0, 5 mM EDTA) and total protein was quantified using the BCA assay kit. The IDUA activity from protein lysates was measured following addition of 100 μM 4-MU-iduronide substrate diluted in 50 mM sodium acetate (NaOAc), 150 mM NaCl, and 0.2% Triton X-100 pH 3.5 in a 60 μl reaction volume. The substrate-sample complex was incubated for 7 hours at 37°С and reaction was quenched with 60 μl 2X stop buffer (pH 10). IDUA catalyzed the cleavage of the non-fluorescent substrate (4-MU-iduronide) into a fluorescent product (4-MU). The resulting 4-MU fluorescence was measured using a spectraMax microplate reader with excitation at 365 nm and emission at 450 nm. A calibration curve was performed for each experiment using 50 mM 4-MU stock solution prepared in water. Data was compared to a standard control curve made with a recombinant IDUA standard.
Immunoblotting
NSCs were harvested after 3 days in serum; cellular extracts were prepared by addition of RIPA lysis buffer. Protein concentration was determined by BCA protein assay kit (Thermo Fisher, cat #23225), and 20 μg of total protein was loaded onto an SDS-NuPAGE gel. Protein was transferred using Invitrogen iBlot2. PVDF membranes were incubated with human pAb-IDUA primary antibody followed by incubation in anti-rabbit HRP-conjugated IgG secondary antibody at 1:5000 dilution. Luminata Forte Western HRP substrate and image were performed using the BioSpectrum® Imaging System. For higher throughput protein separation and detection, we used the Wes instrument from Proteinsimple (San Jose, CA) as directed by the manufacturer.
Quantification of GAGs
Equivalent amounts of protein lysate (5 μg), measured by using BCA protein assay kit were digested with 100 μg/ml Proteinase K in 50 mM Tris–HCl, 10 mM NaCl, 3 mM MgCl2, pH 7.9 overnight at 65°C. Proteinase K was inactivated at 90°C for 30 minutes. DNA and RNA were eliminated by DNase (3 U/sample) and RNase overnight at 37°C. Samples were centrifuged at 13,000xg for 10 min. Double-stranded (dsDNA) was quantified using the Quant-iT Picogreen dsDNA reagent from Thermo Fisher (cat #P7589).
The recovered soluble GAG was quantified by following the Blyscan™ sulfated GAG-DMMB assay protocol (Accurate Chemical and Scientific #CLRB3000). GAG-dye complex absorption was measured at A656 nm in a spectramax microplate reader. GAG values were obtained by normalizing against the dsDNA content and plotted against a sulfated GAG standard containing chondroitin 4-sulfate and expressed in μg GAG.
Strand-specific RNA-seq and data analysis
RNeasy Midi kit (cat #75144) from Qiagen (Gaithersburg, MD) was used for total RNA isolation from human NSCs that were cultured in 6-well plates. Bioanalyzer 6100 (Agilent, Santa Clara, CA) was used to assess RNA quality.
Directional RNA-seq were performed with 100 ng of total RNA using TruSeq Stranded mRNA Library Prep Kit (Illumina, San Diego, CA), and 125 base paired-end sequence reads were generated on the HiSeq 2500 platform (Illumina), as previously described (35,36). Quality control, sequence alignment, transcript and gene-level quantification of primary RNA-seq data were accomplished using an established bioinformatics pipeline (36,37). All secondary analyses were performed in the R statistical environment as reported recently (35,36). Genes were kept for further analysis only if there were >10 counts in at least two-thirds of the samples and group replicates to fall within an interdecile range of the mean for their respective group. The data were subjected to TMM normalization by edgeR v3.10.5, and PCA and Pearson correlation were performed with normalized log2 CPM values. PCA analyses were visualized with the pca3d v0.8 package. DE analysis was performed using limma v3.24.15, and genes having greater than a 2-fold change, as compared to control, and a false discovery rate (FDR) < 0.01 were considered to be significantly differentially expressed. Gene expression clustering was performed on Z-scores using Affinity Propagation (AP) with the apcluster v1.4.3 package (38). AP clusters were grouped into SCs with a cut height of 0.145 with a resulting exemplar dendrogram tree. GO enrichment analysis was performed using clusterProfiler v3.2.14 (39). To reduce redundancy associated with GO analysis, we performed semantic similarity comparisons of the enriched GO terms using GOSemSim v2.0.4 (40). Heatmaps of GO term gene lists were generated using the Ward’s hierarchical clustering method on the log2 CPM values.
Quantitative RT-PCR
Quantitative RT-PCR validation of the RNA-seq data was performed using the ViiA7 Real-Time PCR System (Applied Biosystems, Foster City, CA). GAPDH was included as the reference housekeeping gene because of its stable expression under all experimental conditions. TaqMan probes for the genes of interest were ordered from Applied Biosystems with FAM and GAPDH-Vic labeled, respectively. The samples for RNA-seq were also used for qRT-PCR with duplicate technical replicates and triplicate biological replicates for all genes analyzed. All reagents were purchased from Applied Biosystems, the experiment and -∆∆Ct analysis parameters were as stipulated by the vendor.
Supplementary Material
Acknowledgements
Authors thank Dr. DeeAnn Visk for assistance in editing the manuscript. This research was supported by Intramural Research Programs of the National Center for Advancing Translational Sciences and National Eye Institute. Transcriptome analysis utilized the high-performance computational capabilities of the Biowulf Linux cluster at the National Institutes of Health. This research was also supported by the Improved Therapies for MPS I Pilot Grant program of Orphan Disease Center in the University of Pennsylvania.
Conflict of Interest statement. None declared.
Accession Numbers
The RNA-seq data reported in this paper are deposited in NCBI’s Gene Expression Omnibus with the accession: GSE111906. The data are also available at https://neicommons.nei.nih.gov/#/.
References
- 1. Parenti G., Andria G. and Ballabio A. (2015) Lysosomal storage diseases: from pathophysiology to therapy. Annu. Rev. Med., 66, 471–486. [DOI] [PubMed] [Google Scholar]
- 2. Scott H.S., Bunge S., Gal A., Clarke L.A., Morris C.P. and Hopwood J.J. (1995) Molecular genetics of mucopolysaccharidosis type I: diagnostic, clinical, and biological implications. Hum. Mutat., 6, 288–302. [DOI] [PubMed] [Google Scholar]
- 3. Campos D. and Monaga M. (2012) Mucopolysaccharidosis type I: current knowledge on its pathophysiological mechanisms. Metab. Brain Dis., 27, 121–129. [DOI] [PubMed] [Google Scholar]
- 4. Scott H.S., Litjens T., Nelson P.V., Thompson P.R., Brooks D.A., Hopwood J.J. and Morris C.P. (1993) Identification of mutations in the alpha-L-iduronidase gene (IDUA) that cause Hurler and Scheie syndromes. Am. J. Hum. Genet., 53, 973–986. [PMC free article] [PubMed] [Google Scholar]
- 5. Bunge S., Kleijer W.J., Steglich C., Beck M., Schwinger E. and Gal A. (1995) Mucopolysaccharidosis type I: identification of 13 novel mutations of the alpha-L-iduronidase gene. Hum. Mutat., 6, 91–94. [DOI] [PubMed] [Google Scholar]
- 6. Li P., Wood T. and Thompson J.N. (2002) Diversity of mutations and distribution of single nucleotide polymorphic alleles in the human alpha-L-iduronidase (IDUA) gene. Genet. Med., 4, 420–426. [DOI] [PubMed] [Google Scholar]
- 7. Froissart R., Da Silva I.M. and Maire I. (2007) Mucopolysaccharidosis type II: an update on mutation spectrum. Acta Paediatr., 96, 71–77. [DOI] [PubMed] [Google Scholar]
- 8. Wraith J.E. and Jones S. (2014) Mucopolysaccharidosis type I. Pediatric Endocrinol. Rev., 12(suppl), 102–106. [PubMed] [Google Scholar]
- 9. Xu M., Motabar O., Ferrer M., Marugan J.J., Zheng W. and Ottinger E.A. (2016) Disease models for the development of therapies for lysosomal storage diseases. Ann. N. Y. Acad. Sci., 1371, 15–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Silva E.M., Strufaldi M.W., Andriolo R.B. and Silva L.A. (2016) Enzyme replacement therapy with idursulfase for mucopolysaccharidosis type II (Hunter syndrome). Cochrane Database Syst. Rev., 2, CD008185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Parini R., Deodato F., Di Rocco M., Lanino E., Locatelli F., Messina C., Rovelli A. and Scarpa M. (2017) Open issues in Mucopolysaccharidosis type I-Hurler. Orphanet J. Rare Dis., 12, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sasai Y. (2013) Next-generation regenerative medicine: organogenesis from stem cells in 3D culture. Cell Stem Cell, 12, 520–530. [DOI] [PubMed] [Google Scholar]
- 13. Sterneckert J.L., Reinhardt P. and Scholer H.R. (2014) Investigating human disease using stem cell models. Nat. Rev. Genet., 15, 625–639. [DOI] [PubMed] [Google Scholar]
- 14. Haston K.M. and Finkbeiner S. (2016) Clinical trials in a dish: the potential of pluripotent stem cells to develop therapies for neurodegenerative diseases. Ann. Rev. Pharmacol. Toxicol., 56, 489–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kaewkhaw R., Swaroop M., Homma K., Nakamura J., Brooks M., Kaya K.D., Chaitankar V., Michael S., Tawa G., Zou J. et al. (2016) Treatment paradigms for retinal and macular diseases using 3-D retina cultures derived from human reporter pluripotent stem cell lines. Invest. Ophthalmol. Vis. Sci., 57, ORSFl1-ORSFl11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Borger D.K., McMahon B., Roshan Lal T., Serra-Vinardell J., Aflaki E. and Sidransky E. (2017) Induced pluripotent stem cell models of lysosomal storage disorders. Dis. Mod. Mech., 10, 691–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tolar J., Park I.H., Xia L., Lees C.J., Peacock B., Webber B., McElmurry R.T., Eide C.R., Orchard P.J., Kyba M. et al. (2011) Hematopoietic differentiation of induced pluripotent stem cells from patients with mucopolysaccharidosis type I (Hurler syndrome). Blood, 117, 839–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Aldenhoven M., Wynn R.F., Orchard P.J., O'Meara A., Veys P., Fischer A., Valayannopoulos V., Neven B., Rovelli A., Prasad V.K. et al. (2015) Long-term outcome of Hurler syndrome patients after hematopoietic cell transplantation: an international multicenter study. Blood, 125, 2164–2172. [DOI] [PubMed] [Google Scholar]
- 19. Oussoren E., Keulemans J., Diggelen O.P., Oemardien L.F., Timmermans R.G., Ploeg A.T. and Ruijter G.J. (2013) Residual alpha-L-iduronidase activity in fibroblasts of mild to severe Mucopolysaccharidosis type I patients. Mol. Genet. Metab., 109, 377–381. [DOI] [PubMed] [Google Scholar]
- 20. Barbosa I., Garcia S., Barbier-Chassefiere V., Caruelle J.P., Martelly I. and Papy-Garcia D. (2003) Improved and simple micro assay for sulfated glycosaminoglycans quantification in biological extracts and its use in skin and muscle tissue studies. Glycobiology, 13, 647–653. [DOI] [PubMed] [Google Scholar]
- 21. Keeling K.M., Brooks D.A., Hopwood J.J., Li P., Thompson J.N. and Bedwell D.M. (2001) Gentamicin-mediated suppression of Hurler syndrome stop mutations restores a low level of alpha-L-iduronidase activity and reduces lysosomal glycosaminoglycan accumulation. Hum. Mol. Genet., 10, 291–299. [DOI] [PubMed] [Google Scholar]
- 22. Xu M., Liu K., Swaroop M., Sun W., Dehdashti S.J., McKew J.C. and Zheng W. (2014) A phenotypic compound screening assay for lysosomal storage diseases. J. Biomol. Screen., 19, 168–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Settembre C., Fraldi A., Jahreiss L., Spampanato C., Venturi C., Medina D., Pablo R., Tacchetti C., Rubinsztein D.C. and Ballabio A. (2008) A block of autophagy in lysosomal storage disorders. Hum. Mol. Genet., 17, 119–129. [DOI] [PubMed] [Google Scholar]
- 24. Tessitore A., Pirozzi M. and Auricchio A. (2009) Abnormal autophagy, ubiquitination, inflammation and apoptosis are dependent upon lysosomal storage and are useful biomarkers of mucopolysaccharidosis VI. Pathogenetics, 2, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lieberman A.P., Puertollano R., Raben N., Slaugenhaupt S., Walkley S.U. and Ballabio A. (2012) Autophagy in lysosomal storage disorders. Autophagy, 8, 719–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sasarman F., Maftei C., Campeau P.M., Brunel-Guitton C., Mitchell G.A., and Allard P. (2016) Biosynthesis of glycosaminoglycans: associated disorders and biochemical tests. J. Inherit. Metab. Dis., 39, 173–188. [DOI] [PubMed] [Google Scholar]
- 27. Langford R., Hurrion E. and Dawson P.A. (2017) Genetics and pathophysiology of mammalian sulfate biology. J. Genet. Genomics, 44, 7–20. [DOI] [PubMed] [Google Scholar]
- 28. Gonzalez A., Valeiras M., Sidransky E. and Tayebi N. (2014) Lysosomal integral membrane protein-2: a new player in lysosome-related pathology. Mol. Genet. Metab., 111, 84–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vitry S., Bruyere J., Hocquemiller M., Bigou S., Ausseil J., Colle M.A., Prevost M.C. and Heard J.M. (2010) Storage vesicles in neurons are related to Golgi complex alterations in mucopolysaccharidosis IIIB. Am. J. Path., 177, 2984–2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fujimoto T., Kawashima H., Tanaka T., Hirose M., Toyama-Sorimachi N., Matsuzawa Y. and Miyasaka M. (2001) CD44 binds a chondroitin sulfate proteoglycan, aggrecan. Int. Immunol., 13, 359–366. [DOI] [PubMed] [Google Scholar]
- 31. Raum H., Dietsche B., Nagels A., Witt S.H., Rietschel M., Kircher T. and Krug A. (2015) A genome-wide supported psychiatric risk variant in NCAN influences brain function and cognitive performance in healthy subjects. Hum. Brain Mapp., 36, 378–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Samuel M.A., Valdez G., Tapia J.C., Lichtman J.W. and Sanes J.R. (2012) Agrin and synaptic laminin are required to maintain adult neuromuscular junctions. PloS One, 7, e46663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Santibanez J.F., Quintanilla M., and Bernabeu C. (2011) TGF-beta/TGF-beta receptor system and its role in physiological and pathological conditions. Clin. Sci., 121, 233–251. [DOI] [PubMed] [Google Scholar]
- 34. Beers J., Linask K.L., Chen J.A., Siniscalchi L.I., Lin Y., Zheng W., Rao M. and Chen G. (2015) A cost-effective and efficient reprogramming platform for large-scale production of integration-free human induced pluripotent stem cells in chemically defined culture. Sci. Rep., 5, 11319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kim J.W., Yang H.J., Brooks M.J., Zelinger L., Karakulah G., Gotoh N., Boleda A., Gieser L., Giuste F., Whitaker D.T. et al. (2016) NRL-regulated transcriptome dynamics of developing rod photoreceptors. Cell Rep., 17, 2460–2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hoshino A., Ratnapriya R., Brooks M.J., Chaitankar V., Wilken M.S., Zhang C., Starostik M.R., Gieser L., La Torre A., Nishio M. et al. (2017) Molecular Anatomy of the Developing Human Retina. Dev. Cell., 43, 763–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chaitankar V., Karakulah G., Ratnapriya R., Giuste F.O., Brooks M.J. and Swaroop A. (2016) Next generation sequencing technology and genomewide data analysis: Perspectives for retinal research. Prog. Retin. Eye Res., 55, 1–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bodenhofer U., Kothmeier A. and Hochreiter S. (2011) APCluster: an R package for affinity propagation clustering. Bioinformatics, 27, 2463–2464. [DOI] [PubMed] [Google Scholar]
- 39. Yu G., Wang L.G., Han Y. and He Q.Y. (2012) clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS, 16, 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yu G., Li F., Qin Y., Bo X., Wu Y. and Wang S. (2010) GOSemSim: an R package for measuring semantic similarity among GO terms and gene products. Bioinformatics, 26, 976–978. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.