

Abstract
Maternal obesity is associated with an increased risk of obesity and metabolic disease in offspring. Increasing evidence suggests that the placenta plays an active role in fetal programming. In this study, we used a mouse model of diet-induced obesity to demonstrate that the abnormal metabolic milieu of maternal obesity sets the stage very early in pregnancy by altering the transcriptome of placenta progenitor cells in the preimplantation (trophectoderm [TE]) and early postimplantation (ectoplacental cone [EPC]) placenta precursors, which is associated with later changes in placenta development and function. Sphingolipid metabolism was markedly altered in the plasma of obese dams very early in pregnancy as was expression of genes related to sphingolipid processing in the early placenta. Upregulation of these pathways inhibits angiogenesis and causes endothelial dysfunction. The expression of many other genes related to angiogenesis and vascular development were disrupted in the TE and EPC. Other key changes in the maternal metabolome in obese dams that are likely to influence placenta and fetal development include a marked decrease in myo and chiro-inositol. These early metabolic and gene expression changes may contribute to phenotypic changes in the placenta, as we found that exposure to a high-fat diet decreased placenta microvessel density at both mid and late gestation. This is the first study to demonstrate that maternal obesity alters the transcriptome at the earliest stages of murine placenta development.
Keywords: angiogenesis, developmental origins of health and disease, gene expression, metabolism, placenta, trophectoderm
Summary Sentence
Obesity in a mouse model leads to alterations in the maternal metabolome and early placenta transcriptome as well as changes in vascularity later in gestation which may provide a mechanism for decreased fetal growth.
Introduction
The rate of obesity has more than doubled in the past 35 years, with one-third of adults in the Unites States now classified as obese [1]. Not surprisingly, this overall increase in the prevalence of obesity has been accompanied by an equivalent rise in the number of obese pregnant women. While obese women are more likely to give birth to large for gestational age babies [2], they may also give birth to infants that are small for gestational age (SGA) [3], and recent studies show that SGA infants born to obese women face higher rates of neonatal morbidity than appropriately grown infants [4]. Moreover, higher maternal weight before pregnancy is associated with the development of obesity, insulin resistance, and dyslipidemia in the offspring [5].
The mechanisms underlying the development of these diseases later in life remain to be fully elucidated. However, it is becoming more evident that the placenta is playing an active role in the programming of offspring of obese mothers. Survival and growth of the fetus are critically dependent on the placenta, which acts as the interface between the maternal and fetal circulation to maintain fetal homeostasis and supply nutrients for optimal fetal growth. Importantly, nutrient transport and hormonal secretion can be altered by the placenta in response to alterations in the maternal environment such as obesity, hypoxia, and inflammation [6].
The window of time during which a fetus is exposed to an obese milieu is wide, from conception through birth. Therefore, attempts to elucidate the impact of maternal obesity at different developmental stages are paramount to understanding this complex process. While the mouse model of diet-induced obesity using a high-fat diet does not directly mimic a Western diet, it is the most common animal model used to mimic morbid obesity in humans and results in very similar features of human obesity, i.e. insulin and leptin resistance and hyperlipidemia [7–10]. Using this mouse model of diet-induced obesity, we previously showed that marked maternal obesity is associated with fetal growth restriction and abnormal placentation and ultimately leads to the development of obesity and impaired glucose tolerance in the offspring, similar to what has been observed in human pregnancies complicated by morbid obesity [11]. Further, in embryo transfer studies, we found that exposure to maternal obesity prior to implantation was sufficient to result in placental dysfunction and fetal growth restriction, suggesting that the early embryonic period represents a critical window of susceptibility [11]. At embryonic day 6.5 (e6.5), implantation is fully established and the ectoplacental cone (EPC) is formed, but its cells are still pluripotent. Therefore, the window between implantation at e4.5 and formation of the EPC at e6.5 provides an ideal time to investigate early alterations of the placenta that are direct consequences of maternal exposures.
The mechanisms underlying abnormal placenta function and development in maternal obesity have been extensively studied [6, 11, 12], yet the precise mechanisms are not fully understood. Maternal metabolic function is altered in the obese state [13], which is likely to have a major impact on the developing conceptus. Complex diseases, such as obesity, require the use of integrated approaches to fully understand the way in which the condition alters normal metabolic and physiologic processes resulting in adverse outcomes [14]. Integration of “-omic” data may identify potential causal pathways underlying abnormal placenta function and later development of obesity and metabolic disorders in the offspring. Therefore, we hypothesized that obesity prior to pregnancy and during very early pregnancy alters the maternal metabolome which in turn adversely impacts placenta development as evidenced by changes in the transcriptome, and leads to downstream effects on placenta development.
Materials and Methods
Animals and Diet
All experimental procedures involving the use of live animals were approved by the Institutional Animal Care and Use Committee review boards of the Children's Hospital of Philadelphia and the University of Pennsylvania. Female C57BL/6J mice (JAX stock #000664) at 4 weeks were housed (five animals per cage) and given free access to feed and water. Animals were randomly selected to be fed one of two diets: either standard control diet (Lab Diet 5015) or a high-fat diet (HFD; Harlan TD.06414). The control diet is 4.7 kcal/gm, with calories provided by 19.8% protein, 54.1% carbohydrates, and 26.1% fat (∼37% saturated, 40% monounsaturated, and 23% polyunsaturated). The HFD is 5.1 kcal/gm, with calories provided by 18.4% protein, 21.3% carbohydrates, and 60.3% fat (37% saturated, 47% monounsaturated, and 16% polyunsaturated). Female animals were permitted to feed ad libitum for at least 12 weeks prior to mating and were continued on control or HFD throughout gestation. Females were mated with control male C57BL/6J mice, and mating was confirmed via presence of a vaginal plug (e0.5).
Transcriptome analysis of trophectoderm (e4.5)
Four days after mating, blastocysts were harvested. The uterine horn of the pregnant mouse was removed from the peritoneal cavity and placed in cold calcium and magnesium-free phosphate buffered saline (PBS). A 30 G needle was attached to a 1 mL syringe filled with PBS, and the needle was inserted into the end of the uterine horn. Blastocysts were flushed from the uterine horn into a 60 mm dish filled with cold PBS. Under a dissecting microscope, microneedles were used to dissect the trophectoderm (TE) mechanically from the abembryonic pole. Trophectoderm from five blastocysts were pooled and preserved in RNAlater Stabilization Solution (Ambion) to produce a single sample. Trophectoderm cDNA from four replicates of either control or HFD-derived blastocysts was utilized for gene expression profiling. Total RNA was isolated from TE (PicoPure RNA Isolation Kit, Arcturus KIT0202). Complementary DNA was generated and linearly amplified (Ovation PICO WTA System, Nugen). A subsequent round of linear amplification was performed (TransPlex WTA Kit, Rubicon) to generate an adequate sample for microarray hybridization. Samples were hybridized to the GeneChip Mouse Exon 1.0ST array (Affymetrix 901168) and scanned (GeneChip Scanner 3000 7G System, Affymetrix). Probe intensity data were normalized and summarized to transcript cluster ID level using RMA as implemented in the Partek Genomics Suite. Absence of embryonic tissue in samples was confirmed by lack of expression of octamer-binding transcription factor 4 (Oct4) using quantitative PCR. Pairwise comparisons were made between groups and false discovery rates (FDR) were calculated along with adjusted P values by the Benjamini and Hochberg step-up method. Ingenuity Pathway Analysis (IPA) software was used to evaluate functional pathways.
Transcriptome analysis of EPC (e6.5)
Postimplantation embryos were isolated at embryonic day 6.5 (e6.5). The uterine horn of the pregnant mouse was removed from the peritoneal cavity and placed in cold calcium and magnesium-free PBS. The decidua was freed from the muscular uterine lining, and the embryo was shelled out from the decidua using forceps under a dissecting microscope. Reichert's membrane was removed [15], and embryos were divided at the embryonic/extraembryonic junction (circumferential groove) [16]. The embryonic portion, or epiblast, was placed in 50 μl extraction buffer (Arcturus PicoPure RNA Isolation Kit), incubated at 42°F for 30 min, and then snap frozen and stored at –140°F. Similarly, the extraembryonic portion which includes the EPC and extra-embryonic ectoderm [17] was separately processed in the same manner. Total RNA was isolated from tissues using the Arcturus PicoPure RNA Isolation Kit (Thermo Fisher), including an on-column DNase digestion (Qiagen). RNA quality was assessed using the Agilent Bioanalyzer RNA 6000 Pico Kit (Agilent) ensuring all RNA Integrity Numbers were >8. Real-time quantitative reverse-transcription polymerase chain reaction (qPCR) was used on the RNA extracted from the corresponding epiblast to determine the sex of the embryo using TaqMan probes for inactive X specific transcripts (Xist) as a positive control (Mm01232884_m1) and eukaryotic translation initiation factor 2, subunit 3, structural gene Y-linked (Eif2s3y) (Mm01210630_m1) as a marker of male sex (Applied Biosystems). Ribosomal and mitochondrial RNA were removed from total RNA with the RiboGone—Mammalian kit (Clontech). RNA-Seq libraries were prepared on individual sexed EPC (n = 5 for female embryos and n = 4 for male embryos from each diet group) using the SMARTer Stranded RNA-Seq kit (Clontech). Twenty-five million 75 base pair (bp) paired-end reads were obtained for each biologic replicate on the NextSeq500 platform (Illumina), aligned to the mm10 genome using STAR [18]. Genes were counted using featureCounts [19] with Gencode M9, and differential expression was done using EdgeR and corrected for multiple testing using the Bonferroni correction [20]. qPCR was used to replicate RNA-Seq findings on a separate cohort of EPCs derived from control and obese mice (n = 10 EPCs in each group, total 20 mice). Select differentially expressed genes involved in early vascular development (core 1 synthase, glycoprotein-N-acetylgalactosamine 3-beta-galactosyltransferase, 1 (C1galt1), endothelial PAS domain protein 1 (Epas), platelet derived growth factor, alpha (Pdgfa), and transforming growth factor, beta receptor II (Tgfbr2)) identified in RNA-Seq studies were examined with qPCR using TaqMan probes (Mm00473987, Mm01236112, Mm01205760, Mm03024091).
Metabolomic analysis in maternal plasma
Six days after detection of a vaginal plug (e6.5) female mice were briefly anesthetized with isoflurane, and blood was collected via tail clipping (∼0.3 mL/mouse). The blood was collected in EDTA tubes, and samples were centrifuged for 10 min at 1500× g. Plasma from 10 animals on each diet (n = 20) was isolated and stored at –80°C until pregnancy was confirmed by delivery of pups. Samples were sent to Metabolon Inc. (Durham, NC, USA) for analysis and were extracted and prepared using Metabolon's standard solvent extraction method. The extracted samples were split into equal parts for analysis on complementary GC/MS (gas chromatography mass spectrometry) and LC/MS (liquid chromatography mass spectrometry) platforms. Following normalization to volume extracted (plasma), log transformation and imputation of missing values, if any, with the minimum observed value for each compound, Welch two-sample t-test and ANOVA contrasts were used to identify biochemicals that differed significantly between experimental groups. Analysis by two-way ANOVA identified biochemicals exhibiting significant interaction and main effects for experimental parameters of diet. An estimate of the FDR (q-value) was also calculated to take into account for multiple comparisons.
Immunohistochemistry
Pregnant female mice were sacrificed at e12.5 or e18.5 using CO2 and cervical dislocation. The gravid uterus was dissected free and placed in cold PBS on ice. Individual implantation sites were separated, and placentas were dissected from the fetus, divided in half, placed in 10% formalin, and sent to the Children's Hospital of Philadelphia Pathology core for staining with plasmalemma vesicle-associated protein (PLVAP) antibody. PLVAP antibody is a mouse-specific antibody that stains endothelial cells in embryonic and adult tissues and can therefore be used to quantify vascularity via the endothelial cells lining the vascular beds. Placentas were embedded in paraffin, sectioned, and stained with PLVAP antibody (Biorad MCA2539T, RRID AB_1102821, Supplemental Table 1). Staining was performed on a Bond Max automated staining system (Leica Biosystems). The Bond Intense R staining kit (Leica Biosystems DS9263) was used. Standard protocol was followed with the exception of the primary antibody incubation, which was extended to 1 h at room temperature. Avidin Biotin Blocking (Vector Labs SP-2001) and a Peptide Blocking step were added (DAKO X0909). PLVAP antibody was used at a 1:100 dilution, and antigen retrieval was performed with E1 (Leica Biosystems) retrieval solution for 20 min. Biotinylated Anti-Rat Secondary (Vector BA-4001, Supplemental Table 1) was used at a 1:200 dilution. Slides were rinsed, dehydrated through a series of ascending concentrations of ethanol and xylene, then coverslipped. Slides were scanned on a Scanscope CS-O slide scanner at 20× magnification and viewed with Aperio ImageScope software (Leica Biosystems). The Microvessel Analysis V1 algorithm (Leica Biosystems) was used to quantify vascularity. This algorithm detects and quantifies microvessels on slides stained with endothelial markers, such as PLVAP antibody. Microvessel density was calculated by the algorithm as the number of vessels per unit of analysis area in square microns. The analysis area included the entire placenta. Placentas were sexed using genomic DNA isolated from the tails of corresponding fetuses via the QIAamp DNA Mini kit (Qiagen), and qPCR was performed with Taqman probes for Xist (Mm01232884_m1) and sex determining region of Chr Y (Sry) (Mm00441712_s1). Results from the quantification of vascularity were analyzed using two-way ANOVA (PRISM software version 7.0a; GraphPad), and significance was defined as P value ≤ 0.05. At e12.5, one slide each from seven control males, five control females, seven HFD males, and seven HFD females were analyzed. At e18.5, one slide each from five control males, six control females, three HFD males, and five HFD females were analyzed.
Uterine and umbilical artery doppler imaging
All doppler imaging was performed by the Small Animal Imaging Facility at the University of Pennsylvania using the Vevo 2100 Imaging System (FUJIFILM Visual Sonics) as described by Hernandez-Andrade et al. [21]. Mice were anesthetized with 2% isoflurane, and abdominal hair was removed using a chemical hair remover (Nair). Mice were positioned on a heated pad and body temperature and respiratory rate were continuously monitored. Umbilical artery flow was measured at the level of umbilical cord insertion at the abdomen of the fetus. Three measurements of the peak systolic velocity (PSV) and end diastolic velocity (EDV) were taken per cord, and the mean of each was used to calculate the resistive index (RI) using the standard calculation of (PSV-EDV)/PSV. All images were obtained at <60o. At e12.5–13.5, umbilical cord measurements were taken on 12 HFD fetuses and 16 control fetuses (from three litters per diet), and bilateral uterine artery measurements were taken on two control dams and two HFD dams. At e18.5, umbilical cord measurements were taken on six HFD fetuses and four control fetuses (from three litters per diet), and uterine artery measurements were taken on two control dams and three HFD dams. Results were compared using an unpaired t-test (PRISM software version 7.0a; GraphPad).
Results
Maternal, fetal, and placenta characteristics
At the time of mating, mice fed an HFD were significantly heavier, had elevated 2 h glucose tolerance tests (GTT), and increased fasting leptin, as we have previously described [11]. Overall, there were no differences in resorption rates at e6.5 (2.5% control vs 4.5% HFD), litter size at delivery (6.3 control pups vs 7.6 HFD pups), or the male offspring to female offspring ratio between the two groups. Similar to previous studies [11, 22], male and female HFD e18.5 fetuses were significantly growth restricted; however, placentas did not differ in weight (Table 1). We did not assess fertility or fetal malformations in this study; however, previous studies demonstrate that female mice fed a 60% HFD have decreased fertility and higher rates of fetal developmental defects [8, 22, 23].
Table 1.
Weights of control and HFD dams, placentas, and fetuses. Data are presented as mean ± SEM.
| Control Diet | High-Fat Diet | P-value | |
| Dam weight after 12 weeks on diet | 22.41 g ± 0.349 (n = 40) | 29.58 g ± 0.696 (n = 38) | <0.0001 |
| E18.5 fetal weight | 1.163 g ± 0.028 | 1.045 g ± 0.017 | 0.0003 |
| E18.5 female fetus weight | 1.178 g ± 0.033 (n = 22) | 1.040 g ± 0.019 (n = 22) | 0.0007 |
| E18.5 male fetus weight | 1.182 g ± 0.044 (n = 16) | 1.051 g ± 0.028 (n = 21) | 0.0127 |
| E18.5 placenta weight | 0.089 g ± 0.002 | 0.083 g ± 0.002 | 0.067 |
| E18.5 female placenta weight | 0.087 g ± 0.003 (n = 22) | 0.082 g ± 0.004 (n = 22) | 0.2947 |
| E18.5 male placenta weight | 0.095 g ± 0.004 (n = 16) | 0.088 g ± 0.002 (n = 21) | 0.0998 |
Maternal obesity alters transcriptomic pathways related to inflammation and vascular development in the TE
To elucidate the underlying mechanisms linking maternal obesity to altered offspring development and identify pathways that were disrupted at the earliest stage of placenta development, gene expression profiling was performed on TE of e4.5 blastocysts from control and HFD dams. Using an FDR of <0.05, 5285 genes were differentially expressed between HFD and controls (1 gene downregulated and 5284 genes upregulated) (Supplemental Table 2). IPA revealed disruption of over 400 canonical and biologic pathways including endothelin-1 (ET-1) signaling, leptin signaling in obesity, pigment epithelium-derived factor (PEDF) signaling, corticotropin-releasing hormone (CRH) signaling, eNOS signaling, protein kinase A signaling, p38 mitogen-activated protein kinase (p38 MAPK) signaling, fatty acid α-oxidation, and IL-12 signaling and production in macrophages (Supplemental Table 3).
The ET-1 signaling pathway was upregulated in HFD TE. A total of 77 of 181 genes in this pathway were differentially expressed (P = 2.76E-05). Genes such as endothelin 1 (Edn1), fibroblast growth factor receptor 3 (Fgfr3), and mitogen-activated protein kinase 3 (Mapk3) were all significantly upregulated compared to controls. ET-1 is a potent vasoconstrictor, and elevated levels in the placenta are thought to play a role in the development of preeclampsia that is associated with maternal obesity [24].
A total of 42 of 109 genes in the p38 MAPK signaling pathway were differentially expressed leading to predicted activation by an HFD (P = 1.33E-02). This pathway is triggered by stress stimuli such as death ligands, inflammatory cytokines, and oxidative stress and leads to transcription of genes responsible for apoptosis and production of proinflammatory cytokines, such as IL-6 [25].
Importantly, we found that the pathway for leptin signaling in obesity was upregulated in TE of mice fed an HFD (34/84 genes altered, P = 1.08E-02). Genes such as thymoma viral proto-oncogene 1 (Akt1), growth factor receptor bound protein 2 (Grb2), and phosphoinositide-3-kinase regulatory subunit 5 (Pik3r5) were all significantly upregulated.
Maternal obesity alters upstream regulators and regulatory networks
IPA can identify the cascade of upstream transcriptional regulators that may explain changes in gene expression and illuminate the biological activities occurring in the tissue or cell of interest. We identified differential expression of multiple upstream transcriptional regulators including catenin (cadherin associated protein), beta 1 (CTNNB1) (Wnt signaling), p38 MAPK, eph receptor B2 (ERK) 1/2, IL4, toll-like receptor 2 (TLR 2), and CREB binding protein (CREBBP), which were all activated. Important upstream regulators that were inhibited included RE1-silencing transcription factor (REST), protein kinase, AMP-activated, alpha 2 catalytic subunit (PRKAA2), and additional sex combs like 1 (ASXL1). REST represses the expression of the cholinergic gene in non-neuronal cells. Interestingly, the placenta contains large amounts of acetylcholine and choline acetyltransferase, which are thought to regulate blood flow in the placenta [26, 27]. However, its biological functions in the TE are still unclear. PRKAA2 mediates differentiation of trophoblast stem cells [28]. Reduction in activity of PRKAA1/2 inhibits the production of hormones that are necessary for differentiation of trophoblast stem cells, thereby inhibiting further differentiation [29]. ASXL1, a polycomb protein, is obligate for normal embryonic development.
Individual genes of interest that were significantly altered in TE of HFD dams include LYR motif containing 4 (Lyrm4) (−27 fold change, q < 0.15), OPA1, mitochondrial dynamin like GTPase (Opa1) (–2.072 fold change, q < 0.15), phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit beta (Pik3cb) (–2.75 fold change, q < 0.15), and fibroblast growth factor receptor 2 (Fgfr2) (–3.428 fold change, q < 0.15). Lyrm4 encodes for an iron–sulfur cluster protein important in oxidative phosphorylation [30]. Opa1 encodes for a mitochondria-shaping protein that controls efficiency of steroidogenesis in trophoblasts [31], and Pik3cb is involved in trophoblast differentiation [32]. The fibroblast growth factor (FGF) signaling pathway has been shown to be critical for trophoblast proliferation and differentiation [33]. Fgfr2 mutant mice display multiple placental defects, including a failure of the labyrinthine layer to develop, and die shortly after implantation [33], therefore downregulation of Fgfr2 in TE of HFD dams is of particular interest.
Maternal obesity alters multiple pathways in EPC
To determine the progression of changes that were identified early in development in HFD TE, we generated strand-specific total RNA-Seq libraries after ribosomal RNA depletion of EPC from mouse embryos at e6.5. 32,193 unique transcripts were identified in the EPC. Using an FDR < 0.05, 350 genes were differentially expressed between control and HFD EPCs (89 genes downregulated and 261 genes upregulated), and using an FDR < 0.1, 595 genes were differentially expressed (188 downregulated and 407 upregulated) (Supplemental Figure 1), (Supplemental Table 4).
We again utilized IPA to highlight key alterations between all (males and females combined) HFD mice and all control mice. Key top canonical pathways disrupted included the signaling pathways of sphingosine-1-phosphate, sphingomyelin metabolism, p21 activated kinases (PAK), ceramide, integrin-linked kinases (ILK), and peroxisome proliferator-activated receptor (PPAR) (Supplemental Table 3).
Sphingosine-1-phosphate (S1P) is a bioactive lipid derived from the metabolism of sphingomyelin [34], and it plays a crucial role in vascular development, immune cell trafficking, and neurogenesis. The sphingosine-1-phosphate signaling pathway was significantly altered in EPC of obese dams (P = 1.9E-03). A total of 10 of 125 genes comprising this pathway were differentially expressed. Many of the genes in this pathway, including Pik3cb, N-acylsphingosine amidohydrolase 1 (Asah1), sphingomyelin phosphodiesterase 1, acid lysosomal (Smpd1), sphingomyelin phosphodiesterase 2, neutral (Smpd2), and growth factor receptor bound protein 2 (Grb2) have been correlated with human obesity and insulin resistance [35–38]. Of note, S1P is elevated in the plasma of obese mice and humans, and plasma S1P concentrations positively correlate with measures of metabolic disease in humans [39].
PAK signaling was activated by maternal obesity (P = 3.63E-04). PAKs are protein kinases involved in a wide array of biologic functions including gene transcription, apoptosis, and cellular morphology [40]. A total of 10 of 101 genes in this pathway were differentially expressed, including Rac/Cdc42 guanine nucleotide exchange factor (GEF) 6 (Arhgef6), fibroblast growth factor receptor 1 (Fgfr1), integrin alpha 2 (Itga2), and platelet derived growth factor receptor, beta polypeptide (Pdgfrb).
ILK signaling was downregulated with 12 of 196 genes differentially expressed including myosin, heavy polypeptide 3, skeletal muscle, embryonic (Myh3) and myelocytomatosis oncogene (Myc) (P = 6.46E-03). ILK is a ubiquitously expressed protein that acts in the critical phases of cell polarization, migration, proliferation, and survival [41]. It is highly expressed in villous cytotrophoblast cells in first trimester human chorionic floating villi, and its activity is increased during syncytialization [42].
The PPAR signaling pathway was inhibited in HFD EPCs with a total of nine genes differentially expressed (P = 9.74E-04). These genes include interleukin 1 receptor-like 2 (Il1rl2), Pdgfrb, tumor necrosis factor receptor superfamily, member 1b (Tnfrsf1b), and nuclear receptor co-repressor 2 (Ncor2). PPARs are known to modulate inflammatory responses and energy homeostasis and are critically important to early placenta development [43, 44].
Additionally, upstream analysis using IPA revealed activation of transforming growth factor, beta 1 (TGF-b1), tumor necrosis factor (TNF), interleukin 6 (IL-6), and SMAD family member 4 (SMAD 4). Examples of individual genes that were significantly altered in EPC of HFD dams include chemokine (C-C motif) receptor-like 2 (Ccrl2), a chemoattractant receptor, which was downregulated 2.46 log2FC in HFD EPCs, and phospholipase A2, group IIF (Pla2g2f), which is involved in lipoprotein modification [45] and was downregulated 2.26 log2FC in HFD EPCs. fibroblast growth factor 4 (Fgf4), which is vital to for the maintenance of undifferentiated trophoblast stem cells, was upregulated 5.08 log2FC, and bone morphogenetic protein 7 (Bmp7), which can induce trophoblast differentiation, was upregulated 1.99 log2FC.
Sex-specific differences in the transcriptome of the EPC
It is well established that the placenta is a sexually dimorphic organ [46]. However, the timing at which this dimorphism first develops is not known nor whether this process occurs prior to the production of sex hormones and is thus dependent only on sex chromosomes. Therefore, we next stratified our RNA-Seq data by sex. Using an FDR of <0.05, there were 14 genes that were differentially expressed between male control and male HFD EPCs, including 3-hydroxybutyrate dehydrogenase, type 1 (Bdh1) which is important in fatty acid catabolism (–1.47 log2FC in HFD males) and coagulation factor II (thrombin) receptor (F2r), which is a thrombin receptor gene (1.42 log2FC in HFD males). By expanding the FDR to < 0.15, we identified 50 genes that were differentially expressed between male control and male HFD EPCs, of which 17 genes were downregulated and 33 were upregulated (Supplemental Table 5). Of major significance, there were no genes that were differentially expressed between female control and female HFD EPCs, suggesting that males are much more susceptible to the effects of maternal obesity.
Canonical pathways altered by an HFD in male EPCs as compared to control male EPCs included pathways previously identified such as axonal guidance, vitamin D receptor/retinoid X receptor (VDR/RXR) activation, PPAR signaling, and PAK signaling, as well as pathways found to be unique to the males such as sertoli cell-sertoli cell junction signaling, ketogenesis, and androgen biosynthesis (Supplemental Table 3). Upstream regulators that were activated included TGF-β1, parathyroid hormone (PTH), and endothelin-1 (EDN1). PPARG and Erb-B2 Receptor Tyrosine Kinase 2 (ERBB2) were repressed by an HFD in EPC of male embryos.
When we compared all female EPCs to all male EPCs, we found 68 genes differentially expressed with an FDR < 0.05. Of these 68 genes, 12 are known to be sex-linked. When we opened the FDR to 0.15 (326 genes, Supplemental Table 6) and performed IPA, we found that several canonical pathways were downregulated in males including glutamate receptor signaling (Supplemental Table 3). Downregulation of this pathway was largely in part to decreased expression of glutamate receptor, metabotropic 1 (Grm1), which had a –6.8 log2FC (FDR 0.05) as compared to females. Inhibited upstream regulators included Sonic hedgehog (SHH) and brain-derived neurotrophic factor (BDNF). Activated upstream regulators included Growth factor receptor bound protein 2 (GRB2) and SRY-box 7 (SOX7).
Multiple pathways overlapped between HFD TE and HFD EPC
There was considerable overlap in canonical pathways altered by an HFD between TE at e4.5 and EPC at e6.5, including VDR/RXR activation, ET-1 signaling, axonal guidance signaling, p38 MAPK signaling, and PPARα/RXRα activation suggesting the importance of these pathways in the development of abnormal placenta function in an obese pregnancy (Figure 1). Additionally, there were changes in expression of genes at e4.5 that persisted at e6.5. For example, several antiangiogenic genes, including angiopoietin 2 (Angpt2), arrestin, beta 1 (Arrb1), collagen, type IV, alpha 1 (Col4a1), collagen, type IV, alpha 2 (Col4a2), serine (or cysteine) peptidase inhibitor, clade E, member 1 (Serpine1), and serine (or cysteine) peptidase inhibitor, clade F, member 1 (Serpinf1) were significantly increased by an HFD (P < 0.05 vs. control) in both TE and EPC.
Figure 1.

Venn diagram of some of the most significant canonical pathways found using IPA that were either unique to TE (e4.5) or EPC (e6.5) or overlapped.
Obesity alters the metabolome in maternal plasma at e6.5
Given that our transcriptome results implicated early changes in lipid metabolism, we next performed metabolic profiling in maternal plasma at e6.5 to investigate whether changes in the maternal metabolic profile correlated with changes in the early placenta transcriptome. The results of global metabolic profiling of maternal plasma demonstrated that an HFD has a profound effect on metabolism, with greater than a third of metabolites measured demonstrating a significant difference between obese and control dams. A total of 522 compounds of known identity were detected in plasma samples. Levels of 192 biochemicals were significantly different between HFD and control samples (P ≤ 0.05); 89 were present in higher levels and 103 were present in lower levels in plasma derived from obese dams compared to controls (Supplemental Table 7).
Principal components analysis revealed distinct separation between the plasma samples derived from the control and obese animals (Figure 2). The large degree of separation between these sample types indicates that global metabolism changed in a significant manner with diet.
Figure 2.

Principal component analysis of plasma samples from control (pink) and HFD (red) pregnant dams at e6.5. N = 10 per diet intervention.
Multiple lipid species in maternal plasma are increased by obesity
Exposure to an HFD resulted in significant changes in the levels of biochemicals involved in fatty acid metabolism. There were significant increases in the levels of several fatty acid species in plasma (Table 2) in the HFD dams. Levels of long-chain fatty acids (LCFA) myristate, myristoleate, palmitate, and stearate were significantly elevated in the plasma of HFD dams. Similar to LCFA, several polyunsaturated fatty acids (PUFA) were also significantly increased by an HFD. Specifically, in HFD maternal plasma, the levels of ω-6 fatty acids (dihomo-linolenate, adrenate, docosapenaenoate, and dihomo-linoleate) were elevated (>2 fold) compared to controls, while the level of ω-3 fatty acids did not differ between the groups.
Table 2.
Fatty acids altered in maternal plasma at e6.5 as a result of diet-induced obesity as detected using liquid chromatography/mass spectroscopy. N = 10 per diet intervention for plasma analyses. Red and green shaded cells indicate P ≤ 0.05 (red indicates that the mean values are significantly higher; green values significantly lower). Light red and light green shaded cells indicate 0.05 < P < 0.10.
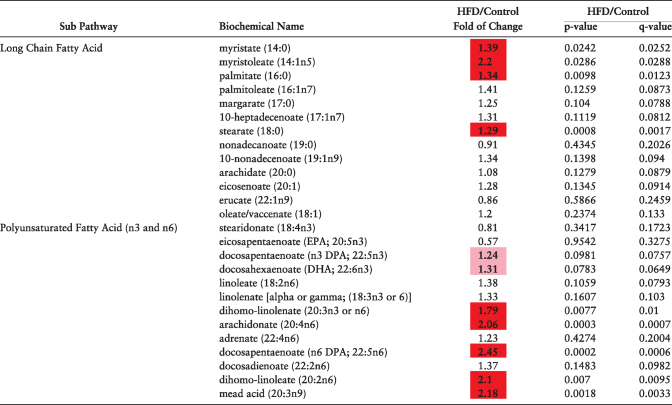
|
When dietary fats are in abundance, animals can metabolize them in multiple ways. They can store them in adipose tissue, shunt them to other complex lipids (phospholipids, sphingolipids), or they can oxidize them for energy production via mitochondrial β-oxidation.
The increases in glycerophospholipids and sphingomyelin species together with decreases in carnitine and its metabolic precursor deoxycarnitine detected in the plasma of obese dams suggest significant alterations in β-oxidation.
Finally, the levels of metabolites involved in sphingolipid metabolism were dramatically altered in the plasma of obese dams. Of the 24 metabolites in this pathway, >50% were significantly higher in HFD dams (Table 3).
Table 3.
Sphingolipid metabolites altered in maternal plasma at e6.5 as a result of diet-induced obesity as detected using liquid chromatography/mass spectroscopy. N = 10 per diet intervention. Red and green shaded cells indicate P ≤ 0.05 (red indicates that the mean values are significantly higher; green values significantly lower). Light red and light green shaded cells indicate 0.05 < P < 0.10.
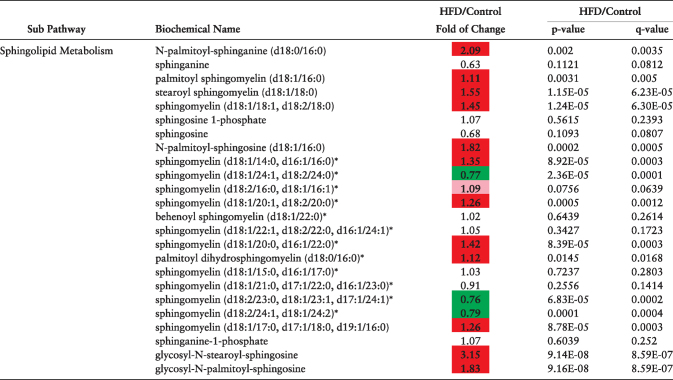
|
Multiple amino acid pathways are influenced by maternal obesity
Plasma of obese dams was enriched in several lysine catabolites (e.g. 2-aminoadipate, 2-oxoadipate, glutarylcarnitine), suggesting that maternal obesity alters lysine availability and/or utilization (Table 4). Acetylated lysine metabolites were also significantly lower in plasma of HFD dams.
Table 4.
Lysine metabolites altered in maternal plasma at e6.5 as a result of diet-induced obesity as detected using liquid chromatography/mass spectroscopy. N = 10 per diet intervention. Red and green shaded cells indicate P ≤ 0.05 (red indicates that the mean values are significantly higher; green values significantly lower). Light red and light green shaded cells indicate 0.05 < P < 0.10.
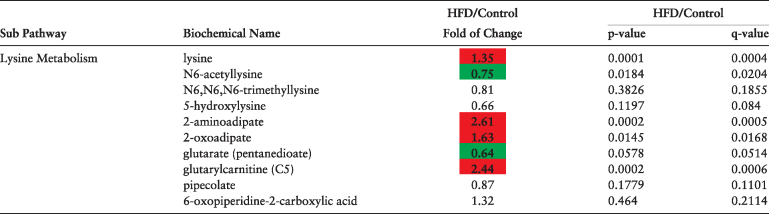
|
Glycine and several glycine metabolites were significantly reduced in plasma of HFD dams. Interestingly, recent studies have shown that low glycine levels are associated with insulin resistance and increased visceral adiposity [47, 48]. Because the utilization of carbohydrates is impaired when insulin resistance is present, the oxidation of amino acids becomes an alternative energy source by entering the citric acid cycle at different points.
Maternal HFD is associated with altered microbial metabolites in plasma
It is well established that the gut microbiota is altered by obesity [49–51]. In our study, HFD dams exhibited alterations in microbial metabolites derived from tyrosine and tryptophan (phenol sulfate, p-cresol sulfate, p-cresol-glucuronide, indolepropionate, and 3-indoxyl sulfate) in their plasma (Figure 3). Further evidence for an altered microbiome was manifested in the bile acid profiles. Bile acids play an important role in lipid metabolism and obesity. While the levels of multiple primary (taurocholate, chenocdeoxycholate) and secondary bile acids (taurodeoxycholate ursodeoxycholate, tauroursodeoxycholate, hyodeoxycholate) were altered by exposure to HFD in plasma, the changes in secondary bile acids were more pronounced. Secondary bile acids are generated when microbiota in the gut deconjugate and dehydroxylate bile acids synthesized by the host. Taken together, these findings strongly suggest that the microbiome is altered by exposure to an HFD, and importantly, the early embryo may be exposed to altered microbial metabolites generated by the maternal microbiome in the setting of maternal obesity.
Figure 3.

Microbial metabolites significantly altered (P < 0.05) using Welch's two-sample t-test in maternal plasma at e6.5 as detected using liquid chromatography/mass spectroscopy. N = 10 per diet intervention.
The results of the above transcriptomic and metabolomic studies are summarized in Table 5.
Table 5.
Summary of major metabolomic and transcriptomic results.
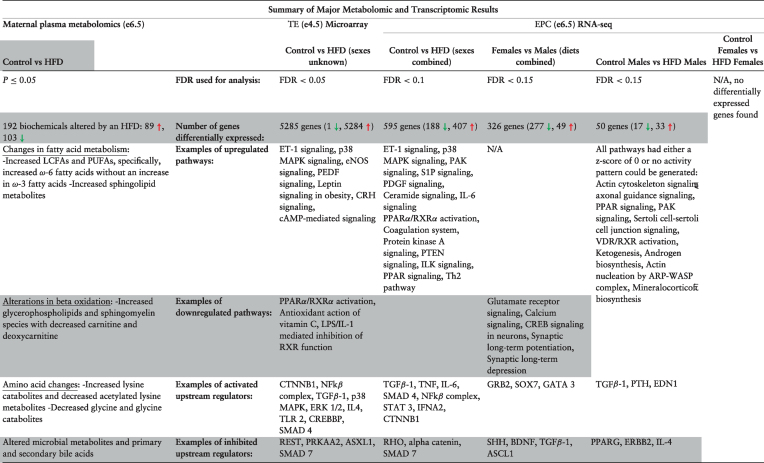
|
Vascular development is impaired in placenta of HFD dams
Given that maternal exposure to an HFD altered several pathways and metabolites vital to vascular development such as sphingosine-1-phosphate, the ET-1 signaling pathway, and sphingolipid metabolism, we sought to explore downstream phenotypic changes in placenta vasculature. Therefore, we next stained mid and late gestation placentas from control and HFD dams with PLVAP antibody to quantify vascularity. At e12.5, placentas of control males had the highest microvessel density, followed by control females and HFD females. HFD males had the lowest microvessel density, which was 31.4% lower than control males (P < 0.05) (Figure 4). The changes in microvessel density persisted throughout the remainder of gestation, and e18.5 male placentas from HFD dams had ∼30% less microvessel density when compared to both control male and control female placentas (P < 0.05). HFD female placentas had ∼20% decreased microvessel density when compared to controls (Figure 5).
Figure 4.
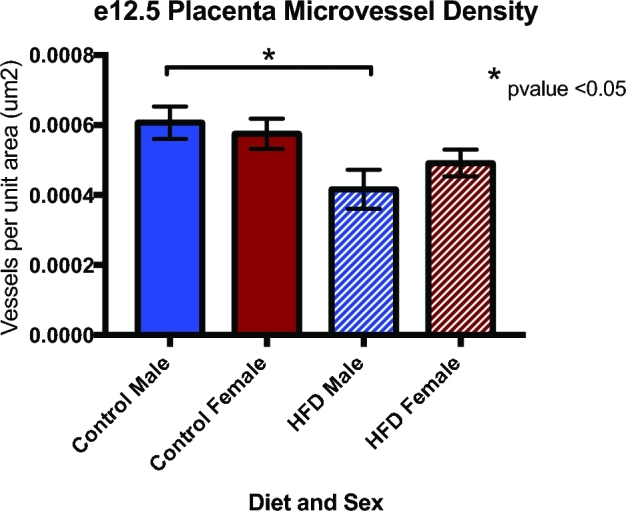
Microvessel density was decreased in male HFD placenta at e12.5 as measured via immunohistochemistry staining with PLVAP antibody (N = 5–7 per group, *P < 0.05).
Figure 5.
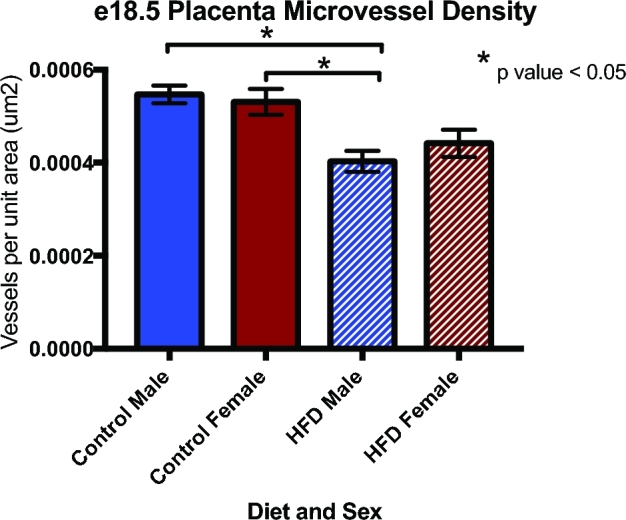
Microvessel density remained decreased in male HFD placentas at e18.5 as measured via immunohistochemistry staining with PLVAP antibody (N = 3–6 per group, *P < 0.05).
Given that we found changes in microvessel density of HFD placentas, we sought to determine whether an HFD altered arterial flow and resistance in the placenta and uterus. Umbilical artery and uterine artery dopplers were performed on HFD and control dams and fetuses at e12.5 and e18.5. The average RI measured in the umbilical artery of fetuses at e12.5 was slightly increased in the HFD group as compared to the controls (0.8731 ± 0.017 vs 0.8552 ± 0.013, P = 0.408). At e18.5, the RI of the umbilical artery in the control group was 0.9031 ± 0.0107 vs 0.917 ± 0.0113 in the HFD group (P = 0.421). The average RI of the uterine arteries in the control group at e12.5 was 0.6097 ± 0.0532 vs 0.6713 ± 0.1254 in the HFD group (P = 0.635), and at e18.5, the control group uterine artery average RI was 0.5634 ± 0.0424 vs 0.5954 ± 0.0059 in the HFD group (P = 0.398). Therefore, the RI was slightly, but not significantly, elevated in the uterine arteries of dams fed an HFD by 10.1% at e12.5 and 5.7% at e18.5. Additionally, the RI of the umbilical arteries of HFD fetuses also trended upward slightly compared to controls by 2.1% at e12.5 and 1.5% at e18.5. However, these results were not statistically significant.
Discussion
In this study, we demonstrated that maternal obesity alters the maternal metabolome very early in pregnancy and is associated with marked changes in expression of genes involved in key pathways such as sphingolipid metabolism, angiogenesis, and fatty acid metabolism in cells that will eventually give rise to the placenta. While multiple studies have demonstrated that obesity in pregnancy alters placenta structure and function [11] [52–54], our data surprisingly show that the abnormal metabolic milieu of maternal obesity sets the stage very early in pregnancy by altering the transcriptome of placenta progenitor cells in the TE and EPC, which in turn are associated with profound implications for placenta development and function. In the mouse, by embryonic day 4 (e4), the totipotent cells of the morula differentiate into two distinct lineages: the outer layer of cells, the TE, develops into the extraembryonic tissue, and the inner cell mass gives rise to the fetal structures [55]. The multipotent layer of TE cells anchors to the uterus and divides to form the extraembryonic ectoderm and EPC which then forms the spongiotrophoblast and trophoblast giant cells [56]. Therefore, the window between embryonic days 4 and 6.5 provides an ideal time to investigate early alterations of the placenta that are direct consequences of maternal exposures.
Not surprisingly, an HFD induced marked elevations in saturated fatty acids, including palmitate, myristoleate, myristate, and stearate, in plasma of obese dams in early pregnancy. Levels of these pro-inflammatory fatty acids are elevated in obese humans and are linked to the development of insulin resistance and other metabolic disorders that accompany obesity [57]. Additionally, we observed a disrupted balance of ω-6:3 ratio in obese dams. ω-6 fatty acids are pro-inflammatory and enhance adipogenesis, whereas ω-3 fatty acids are associated with improved insulin action and decreased adiposity [58–60]. We have previously shown that this pro-inflammatory metabolic milieu in the mother results in marked oxidative stress in the preimplantation embryo [61], and prevention of oxidative stress precludes the development of obesity in the offspring, thus demonstrating the central role of oxidative stress and inflammation in the programming of the offspring [61].
Perhaps most striking was our finding of markedly altered sphingolipid metabolism in plasma of obese dams very early in pregnancy. This directly correlated with changes in gene expression regulating sphingosine-1-phosphate and ceramide signaling pathways in EPC, which persisted in placenta at e12.5 as shown in our previous studies [11]. Upregulation of these pathways inhibit angiogenesis and result in endothelial dysfunction by abolishing vascular endothelial growth factor (VEGF) or insulin-stimulated endothelial nitric oxide synthase activation and subsequent nitric oxide production from endothelial cells [62]. Indeed, many of these key pathways were disrupted in TE and EPC. Further, excess ceramides play a major role in lack of remodeling of the maternal spiral arteries that occurs in preeclampsia, which is associated with maternal obesity [63]. Thus, changes in maternal sphingolipid metabolism could be a mechanism through which obesity results not only in inflammation and insulin resistance in the mother but also activates a transcription program in placenta progenitors resulting in abnormal vascularization in placenta of obese dams.
Multiple phospholipids, including 1-stearoyl-2-arachidonoyl-GPC, were elevated in maternal plasma of obese dams at e6.5. This key phospholipid is an important regulator of p38 MAPK activity under conditions of cell stress and lipotoxicity [64], and the p38 MAP kinase pathway was significantly elevated in EPC at e6.5. The p38 group of MAP kinases serve as a nexus for signal transduction and play a vital role in numerous biological processes including inflammation and placental angiogenesis [65], thus providing another link between changes in the maternal metabolome and changes in gene expression in the EPC.
HFD dams exhibited differences in several lysine metabolites compared to controls. Acetylated lysine molecules are commonly found in histone proteins; the acetylation status of lysine in histones is correlated with active gene expression as acetylation of lysine effectively removes the unmodified amino acid's positive charge and hence reduces the interaction between the histone tail and nucleosome [66]. Additionally, high-throughput proteomic assays have demonstrated that lysine acetylation of proteins involved in immune modulation and metabolism occurs which can alter function [67]. Therefore, changes in acetylation of lysine could have profound effects on development-related gene expression patterns and post-translational effects by altering the function of proteins involved in metabolic signaling.
Another key change in the maternal metabolome of obese dams that is likely to influence placenta and fetal development was a marked decrease in myo and chiro-inositol. Inositols are a family of simple carbohydrates, and these two stereoisomers play a key role in the insulin pathway, acting synergistically as insulin-sensitizing agents through the enhancement of glucose peripheral tissue uptake and glycogen synthesis [68]. Of note, myo-inositol/D-chiro-inositol supplementation in pregnant mice with a metabolic-like syndrome phenotype improves blood pressure, glucose tolerance, and leptin levels and prevents increased maternal weight gain [69]. Myo-inositol also regulates endothelial function by decreasing oxidative stress, enhancing endothelial nitric oxide synthase and nitric oxide activity [70]. Thus, low levels of these important metabolites may contribute to insulin resistance associated with maternal obesity.
Multiple studies have demonstrated that maternal obesity results in a pro-inflammatory state in the placenta [71, 72]. These studies have all been performed in term or near term placenta, yet our transcriptome data showing activation of multiple inflammatory pathways and genes including IL-6 signaling, suggest that this process occurs very early in gestation. The inflammatory state in TE and EPC of HFD dams is likely due to the marked changes in lipid species such as palmitate, omega-6 fatty acids, sphingolipids, and their metabolites, like eicosanoids, that are significantly elevated in the plasma of obese dams during early pregnancy.
There was significant overlap in the gene expression pathways that were altered by an HFD in TE and EPC, implying that these particular pathways may be especially important to early placenta development (Figure 1). These pathways include ET-1 signaling, p38 MAPK signaling, as well as PPARα/RXRα activation. ET-1 is an extremely potent vasoconstrictor that acts directly on vascular smooth muscle cells and has been linked to diseases such as hypertension and atherosclerosis [73]. It is well known that obesity is associated with endothelial dysfunction, and ET-1-mediated vasoconstriction is increased in obese humans [74]. Additionally, elevated circulating plasma ET-1 levels have been positively correlated with preeclampsia [75] and the production of reactive oxygen species that promote apoptosis of trophoblasts [76]. This correlates with the finding that placentas of mice with diet-induced obesity display both oxidative stress and decreased number of trophoblasts [77]. These changes may be another mechanism by which an HFD leads to decreased microvessel density later in gestation.
The PPARα/RXRα activation pathway was predicted to be inhibited by an HFD in both TE and EPC, and the more generalized PPAR signaling pathway was also predicted to be inhibited, though this was only statistically significant in EPC. This was an unexpected finding given that the physiologic actions of fatty acids are mediated by PPARs. However, it is possible that an overabundance of free fatty acids leads to a subsequent downregulation of these pathways. PPARs are highly expressed in the placenta and thought to be involved in trophoblast differentiation and function [78], and deficiencies of both PPARγ and PPARδ result in lethal placenta defects [44]. The placentas of PPARγ null mice display vascular anomalies and poor differentiation of the trophoblast [78]. Additionally, in human studies, gestational diabetes mellitus has been associated with a downregulation in term placental PPARα, PPARγ, and RXRα [79]
In addition to ET-1 and PPAR signaling, there were many other genes and pathways altered by an HFD in early placenta that are related to angiogenesis and vascular development, including the PEDF signaling pathway and the pathway for leptin signaling in obesity. The PEDF signaling pathway was activated by an HFD in TE. PEDF is a widely expressed glycoprotein that acts as a potent antiangiogenic factor [80]. Huppertz et al. recently identified PEDF secretion as a mechanism by which human trophoblasts restrain angiogenesis and vascularization later in pregnancy [81]. PEDF binds to VEGFR2 and blocks the stimulatory effect of VEGF. These changes in key genes regulating vascular development in the TE and EPC were correlated with changes we found in microvessel density in mid and late gestation placenta, suggesting that the mechanism underlying abnormal placenta vascularization occurs prior to placentation in the very early embryo.
The canonical pathway for leptin signaling in obesity was also activated by an HFD in TE. Leptin is an abundant circulating adipokine, and while it is widely known for its role in energy homeostasis and metabolism, it also participates in a myriad of other processes including immune function and inflammation [82]. Activation of leptin receptors on endothelial cells leads to endothelial cell dysfunction via increased oxidative stress responses as well as increases in TNF-α and IL-6 [82], both of which were also predicted to be activated by an HFD in EPC. In the more mature placenta, leptin is produced by syncytiotrophoblast and controls growth of the placenta. High levels of leptin in placentas from obese pregnancies reduce cytotrophoblast proliferation and placental growth factor (PlGF) production and are thought to contribute to the development of preeclampsia [75].
In addition to changes in placenta development and function, obesity may impact fetal development by altering microbial metabolites and molecules to which the fetus is exposed. In our study, multiple microbial metabolites were altered in plasma of HFD dams. Several studies have shown that the composition of gut microbiota is altered by obesity [49–51]; however, the functional implications of an altered microbiome on fetal growth and development are not fully understood. Gomez de Aguero et al. showed that maternal microbiota-derived compounds are transferred from the mother to the offspring and shape the immune system of the offspring [83]. Therefore, gaining better understanding of the compounds present in maternal plasma and, thus potentially transferred to the offspring, is paramount. The obese maternal plasma profiles showed altered levels of microbial-derived metabolites produced from amino acids (tyrosine and tryptophan), which have been suggested to interact with host signaling pathways and affect host immunity [84]. Indole, a major tryptophan-derived microbial metabolite we found to be present in higher levels in plasma of HFD dams, has been shown to interact with inflammation-related processes in the human host [85]. Therefore, in addition to altered placenta function, our studies suggest that obesity alters the composition of biochemicals to which the embryo is exposed. This will not only affect fetal development, but also impact long-term offspring outcomes.
Male offspring are known to be more susceptible to the effects of intrauterine exposures such as maternal obesity and diabetes [86, 87], and it is possible that the placenta plays a key role in this finding. It is recognized that there are sex differences in gene expression in normal placenta, which have been attributed to sex hormones, and it is also known that maternal HFD can affect gene expression in a sex-specific manner [88]. However, given our previous findings that a pregestational exposure to obesity also results in sex-specific differences in long-term outcomes between males and females [11], it is possible that sex-specific developmental programming is occurring even prior to the expression of sex hormones.
When we compared all male EPCs to all female EPCs, we found alterations in several interesting pathways, many of which are related to placenta and vascular development. Glutamate receptor signaling was downregulated in males. Though glutamate signaling was previously thought to be limited to the central nervous system, it is now known that functional glutamate receptors are found in many different organs, including skin and placenta [89]. Grm1 is upregulated in some forms of cancer, and it has been found to enhance angiogenic signaling [90]. SHH and BDNF, both upstream regulators, were inhibited in males as compared to females, and these two regulators also play important roles in placenta formation and angiogenesis. Shh helps to control placenta labyrinth development, and Shh null mice have pronounced defects in yolk sac vasculature [91]. Additionally, studies have shown Shh to be indirectly angiogenic by upregulating Vegf, angiopoietin 1 (Angpt1), and Angpt2 [92]. Of note, Vegfc was downregulated in males, which is consistent with deactivation of the SHH pathway.
BDNF is neurotrophin thought to play key roles in placenta angiogenesis, cytotrophoblast proliferation, and protection of endothelial progenitor cells via increased expression of superoxide dismutase, and alterations have been implicated in a wide variety of diseases [93]. Lower levels of BDNF have been found in both placenta and cord blood of women with preeclampsia [94], and BDNF was recently shown to be sexually dimorphic in term human placentas [95]. Baseline differences between male and female EPC may be one explanation for the increased susceptibility of males to the effects of maternal obesity.
As mentioned above, we found that exposure to an HFD decreased microvessel density at both mid and late gestation. Notably, this decrease was most pronounced in the placentas of male fetuses. Additionally, we found increased uterine artery resistance in HFD dams and a very slight trend toward increased umbilical artery resistance in fetuses of dams fed an HFD. However, these results were not statistically significant. Given that the histological changes we noted in microvessel density were more significant in males, and we were not able to determine the sex of the fetuses undergoing doppler imaging, we speculate that this may be why the increase in RI in the uterine and umbilical arteries was not statistically significant.
In conclusion, our data demonstrate that diet-induced obesity in the female mouse profoundly affects the maternal plasma metabolome and alters gene expression pathways in the TE and EPC that are vital to placenta development and necessary to support the developing embryo. This is the first study to demonstrate that maternal obesity alters the transcriptome at the earliest stages of murine placenta development. IPA highlighted significant clustering of genes involved in sphingolipid metabolism, vascular development, and inflammation, and we speculate that these early changes are directly associated with the decreased placenta microvessel density we observed later in gestation. These alterations may represent some of the many complex ways in which maternal obesity exerts its effects on the offspring via the placenta.
Supplementary data
Supplemental Table 1. Antibodies used for immunohistochemistry staining.
Supplemental Table 2. Gene expression profiling of trophectoderm from e4.5 blastocysts from control and HFD dams. Using an FDR of ≤0.05, 5285 genes were differentially expressed between HFD and controls (1 gene downregulated and 5284 genes upregulated).
Supplemental Table 3. Canonical pathways with P ≤ 0.05 for all transcriptomic analyses.
Supplemental Figure 1. Volcano plot of all genes sequenced, comparing all control EPC samples to all HFD EPC samples at e6.5. Noted in red are genes with an FDR ≤0.15.
Supplemental Table 4. RNA-Seq of e6.5 EPC, comparing all control EPCs to all HFD EPCs. Using an FDR of ≤0.1, 595 genes were differentially expressed (188 downregulated and 407 upregulated).
Supplemental Table 5. RNA-seq of e6.5 EPCs, comparing male control EPCs to male HFD EPCs. Using an FDR of ≤0.15, there were 50 genes that were differentially expressed (17 downregulated and 33 upregulated).
Supplemental Table 6. RNA-seq of e6.5 EPCs, comparing all female EPCs to allmale EPCs. Using an FDR of ≤0.15, there were 326 genes differentially expressed (277 downregulated and 49 upregulated).
Supplemental Table 7. Global metabolic profiling of e6.5 maternal plasma. Levels of 192 biochemicals were significantly different between HFD and control samples (P ≤ 0.05); 89 were present in higher levels, and 103 were present in lower levels.
Acknowledgments
The authors would like to acknowledge John Tobias, Teri Ord, Metabolon Inc, the Children's Hospital of Philadelphia Pathology core, University of Pennsylvania Small Animal Imaging Facility, and the University of Pennsylvania Epigenetics Program for their help with this research.
Notes
Conference Presentation: Presented in part at the 2016 American Society for Reproductive Medicine, Salt Lake City, Utah, the 2017 Pediatric Academic Societies Meeting, San Francisco, California, and the 2017 Society for Reproductive Investigation Meeting, Orlando, Florida.
Edited by Dr. Sarah Kimmins, PhD, McGill University
Footnotes
Grant Support: Funding for this research was provided by K12 HD000849-28 (KEO RSDP), T32 HD040135 (KEO), T32HD060556 (TS), R01DK062965 (RAS).
Conflict of Interest. The authors have declared that no conflict of interest exists.
References
- 1. Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of Obesity in the United States, 2009–2010. NCHS Data Brief 2012;(82):1–8. [PubMed] [Google Scholar]
- 2. Marchi J, Berg M, Dencker A, Olander EK, Begley C. Risks associated with obesity in pregnancy, for the mother and baby: a systematic review of reviews. Obes Rev 2015; 16(8):621–638. [DOI] [PubMed] [Google Scholar]
- 3. Deshmukh VL, Jadhav M, Yelikar K. Impact of HIGH BMI on pregnancy: maternal and foetal outcome. J Obstet Gynecol India 2016; 66(S1):192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yao R, Park BY, Caughey AB. The effects of maternal obesity on perinatal outcomes among those born small for gestational age. J Matern Fetal Neonatal Med 2017; 30(12):1417–1422. [DOI] [PubMed] [Google Scholar]
- 5. O’Reilly JR, Reynolds RM. The risk of maternal obesity to the long-term health of the offspring. Clin Endocrinol 2013; 78(1):9–16. [DOI] [PubMed] [Google Scholar]
- 6. Dimasuay KG, Boeuf P, Powell TL, Jansson T. Placental responses to changes in the maternal environment determine fetal growth. Front Physiol 2016; 7:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang CY, Liao JK. A mouse model of diet-induced obesity and insulin resistance. Methods Mol Biol 2012; 821:421–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Skaznik-Wikiel ME, Swindle DC, Allshouse AA, Polotsky AJ, McManaman JL. High-fat diet causes subfertility and compromised ovarian function independent of obesity in mice1. Biol Reprod 2016; 94(5):108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bibi S, Kang Y, Du M, Zhu MJ. Maternal high-fat diet consumption enhances offspring susceptibility to DSS-induced colitis in mice. Obesity 2017; 25(5):901–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Boudoures AL, Chi M, Thompson A, Zhang W, Moley KH. The effects of voluntary exercise on oocyte quality in a diet-induced obese murine model. Reproduction 2016; 151(3):261–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sasson I, Vitins AP, Mainigi MA, Moley KH, Simmons RA. Pre-gestational vs gestational exposure to maternal obesity differentially programs the offspring in mice. Diabetologia 2015; 58(3):615–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Howell KR, Powell TL. Effects of maternal obesity on placental function and fetal development. Reproduction 2017; 153(3):R97–R108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hellmuth C, Lindsay KL, Uhl O, Buss C, Wadhwa PD, Koletzko B, Entringer S. Association of maternal prepregnancy BMI with metabolomic profile across gestation. Int J Obes 2017; 41(1):159–169. [DOI] [PubMed] [Google Scholar]
- 14. Benton SJ, Ly C, Vukovic S, Bainbridge SA. Andree Gruslin award lecture: Metabolomics as an important modality to better understand preeclampsia. Placenta 2017; 60(suppl 1):S32–S40. [DOI] [PubMed] [Google Scholar]
- 15. Shea K, Geijsen N. Dissection of 6.5 dpc mouse embryos. J Vis Exp 2007; (2):160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Downs KM, Davies T. Staging of gastrulating mouse embryos by morphological landmarks in the dissecting microscope. Development 1993; 118(4):1255–1266. [DOI] [PubMed] [Google Scholar]
- 17. Tam PP, Rossant J. Mouse embryonic chimeras: tools for studying mammalian development. Development 2003; 130(25):6155–6163. [DOI] [PubMed] [Google Scholar]
- 18. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013; 29(1):15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014; 30(7):923–930. [DOI] [PubMed] [Google Scholar]
- 20. Datta S, Xia X-Q, Bhattacharjee S, Jia Z. Advances in statistical medicine. Comput Math Methods Med 2014; 2014:316153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hernandez-Andrade E, Ahn H, Szalai G, Korzeniewski SJ, Wang B, King M, Chaiworapongsa T, Than NG, Romero1 R. Evaluation of utero-placental and fetal hemodynamic parameters throughout gestation in pregnant mice using high-frequency ultrasound. Ultrasound Med Biol 2014; 40(2):351–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jungheim ES, Schoeller EL, Marquard KL, Louden ED, Schaffer JE, Moley KH. Diet-induced obesity model: abnormal oocytes and persistent growth abnormalities in the offspring. Endocrinology 2010; 151(8):4039–4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wu LL, Dunning KR, Yang X, Russell DL, Lane M, Norman RJ, Robker RL. High-fat diet causes lipotoxicity responses in cumulus–oocyte complexes and decreased fertilization rates. Endocrinology 2010; 151(11):5438–5445. [DOI] [PubMed] [Google Scholar]
- 24. Spradley FT, Palei AC, Granger JP. Immune mechanisms linking obesity and preeclampsia. Biomolecules 2015; 5(4):3142–3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Han HJ, Lee YJ. Insulin stimulates Ca2+ uptake via PKC, cAMP, and p38 MAPK in mouse embryonic stem cells. Life Sci 2005; 76(25):2903–2919. [DOI] [PubMed] [Google Scholar]
- 26. Wessler I, Kilbinger H, Bittinger F, Unger R, Kirkpatrick CJ. The non-neuronal cholinergic system in humans: expression, function and pathophysiology. Life Sci 2003; 72(18–19):2055–2061. [DOI] [PubMed] [Google Scholar]
- 27. Sastry BV. Human placental cholinergic system. Biochem Pharmacol 1997; 53(11):1577–1586. [DOI] [PubMed] [Google Scholar]
- 28. Carey EA, Albers RE, Doliboa SR, Hughes M, Wyatt CN, Natale DR, Brown TL. AMPK knockdown in placental trophoblast cells results in altered morphology and function. Stem Cells Dev 2014; 23(23):2921–2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhong W, Xie Y, Abdallah M, Awonuga AO, Slater JA, Sipahi L, Puscheck EE, Rappolee DA. Cellular stress causes reversible, PRKAA1/2-, and proteasome-dependent ID2 protein loss in trophoblast stem cells. Reproduction 2010; 140(6):921–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lim SC, Friemel M, Marum JE, Tucker EJ, Bruno DL, Riley LG, Christodoulou J, Kirk EP, Boneh A, DeGennaro CM, Springer M, Mootha VK et al. Mutations in LYRM4, encoding iron–sulfur cluster biogenesis factor ISD11, cause deficiency of multiple respiratory chain complexes. Hum Mol Genet 2013; 22(22):4460–4473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wasilewski M, Semenzato M, Rafelski SM, Robbins J, Bakardjiev AI, Scorrano L. Optic atrophy 1-dependent mitochondrial remodeling controls steroidogenesis in trophoblasts. Curr Bio 2012; 22(13):1228–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kent LN, Konno T, Soares MJ. Phosphatidylinositol 3 kinase modulation of trophoblast cell differentiation. BMC Dev Biol 2010; 10(1):97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kunath T, Yamanaka Y, Detmar J, MacPhee D, Caniggia I, Rossant J, Jurisicova A. Developmental differences in the expression of FGF receptors between human and mouse embryos. Placenta 2014; 35(12):1079–1088. [DOI] [PubMed] [Google Scholar]
- 34. Mendelson K, Evans T, Hla T, Sphingosine 1-phosphate signalling. Development 2014; 141(1):5–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang K, Li WD, Zhang CK, Wang Z, Glessner JT, Grant SF, Zhao H, Hakonarson H, Price RA. A genome-wide association study on obesity and obesity-related traits. PLoS One 2011; 6(4):e18939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Le Stunff C, Dechartres A, Mariot V, Lotton C, Trainor C, Miraglia Del Giudice E, Meyre D, Bieche I, Laurendeau I, Froguel P, Zelenika D, Fallin D et al. Association analysis indicates that a variant GATA-binding site in the PIK3CB promoter is a Cis-acting expression quantitative trait locus for this gene and attenuates insulin resistance in obese children. Diabetes 2008; 57(2):494–502. [DOI] [PubMed] [Google Scholar]
- 37. Kolak M, Gertow J, Westerbacka J, Summers SA, Liska J, Franco-Cereceda A, Orešič M, Yki-Järvinen H, Eriksson P, Fisher RM. Expression of ceramide-metabolising enzymes in subcutaneous and intra-abdominal human adipose tissue. Lipids Health Dis 2012; 11(1):115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Desbuquois B, Carre N, Burnol AF. Regulation of insulin and type 1 insulin-like growth factor signaling and action by the Grb10/14 and SH2B1/B2 adaptor proteins. FEBS J 2013; 280(3):794–816. [DOI] [PubMed] [Google Scholar]
- 39. Kowalski GM, Carey AL, Selathurai A, Kingwell BA, Bruce CR. Plasma sphingosine-1-phosphate is elevated in obesity. PLoS One 2013; 8(9):e72449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jaffer ZM, Chernoff J. p21-activated kinases: three more join the Pak. Int J Biochem Cell Biol 2002; 34(7):713–717. [DOI] [PubMed] [Google Scholar]
- 41. Widmaier M, Rognoni E, Radovanac K, Azimifar SB, Fässler R. Integrin-linked kinase at a glance. J Cell Sci 2012; 125(8):1839–1843. [DOI] [PubMed] [Google Scholar]
- 42. Butler TM, Elustondo PA, Hannigan GE, MacPhee DJ. Integrin-linked kinase can facilitate syncytialization and hormonal differentiation of the human trophoblast-derived BeWo cell line. Reprod Biol Endocrinol 2009; 7(1):51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stienstra R, Duval C, Müller M, Kersten S. PPARs, obesity, and inflammation. PPAR Res 2007; 2007:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Barak Y, Sadovsky Y, Shalom-Barak T. PPAR signaling in placental development and function. PPAR Res 2008; 2008:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sato H, Kato R, Isogai Y, Saka G-I, Ohtsuki M, Taketomi Y, Yamamoto K, Tsutsumi K, Yamada J, Masuda S, Ishikawa Y, Ishii T et al. Analyses of group III secreted phospholipase A2 transgenic mice reveal potential participation of this enzyme in plasma lipoprotein modification, macrophage foam cell formation, and atherosclerosis. J Biol Chem 2008; 283(48):33483–33497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Myatt L, Maloyan A. Obesity and placental function. Semin Reprod Med 2016; 34(01):042–049. [DOI] [PubMed] [Google Scholar]
- 47. Lustgarten MS, Price LL, Phillips EM, Fielding RA. Serum glycine is associated with regional body fat and insulin resistance in functionally-limited older adults. PLoS One 2013; 8(12):e84034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cheng Y, Meng Q, Wang C, Li H, Huang Z, Chen S, Xiao F, Guo F. Leucine deprivation decreases fat mass by stimulation of lipolysis in white adipose tissue and upregulation of uncoupling protein 1 (UCP1) in brown adipose tissue. Diabetes 2010; 59(1):17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ley RE, Bäckhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci USA 2005; 102(31):11070–11075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006; 444(7122):1027–1131. [DOI] [PubMed] [Google Scholar]
- 51. Murphy EF, Cotter PD, Healy S, Marques TM, O'Sullivan O, Fouhy F, Clarke SF, O’Toole PW, Quigley EM, Stanton C, Ross PR, O’Doherty RM et al. Composition and energy harvesting capacity of the gut microbiota: relationship to diet, obesity and time in mouse models. Gut 2010; 59(12):1635–1642. [DOI] [PubMed] [Google Scholar]
- 52. Ramsay JE, Ferrell WR, Crawford L, Wallace AM, Greer IA, Sattar N. Maternal obesity is associated with dysregulation of metabolic, vascular, and inflammatory pathways. J Clin Endocrinol Metab 2002; 87(9):4231–4237. [DOI] [PubMed] [Google Scholar]
- 53. Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M, Shimomura I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest 2004; 114(12):1752–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rajasingam D, Seed PT, Briley AL, Shennan AH, Poston L. A prospective study of pregnancy outcome and biomarkers of oxidative stress in nulliparous obese women. Am J Obstet Gynecol 2009; 200(4):395.e1–395.e9. [DOI] [PubMed] [Google Scholar]
- 55. Wang H, Dey SK. Roadmap to embryo implantation: clues from mouse models. Nat Rev Genet 2006; 7(3):185–199. [DOI] [PubMed] [Google Scholar]
- 56. Cross JC. Genes regulating embryonic and fetal survival. Theriogenology 2001; 55(1):193–207. [DOI] [PubMed] [Google Scholar]
- 57. Bush NC, Basu R, Rizza RA, Nair KS, Khosla S, Jensen MD. Insulin-mediated FFA suppression is associated with triglyceridemia and insulin sensitivity independent of adiposity. J Clin Endocrinol Metab 2012; 97(11):4130–4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pisani DF, Ghandour RA, Beranger GE, Le Faouder P, Chambard JC, Giroud M, Vegiopoulos A, Djedaini M, Bertrand-Michel J, Tauc M, Herzig S, Langin D et al. The ω6-fatty acid, arachidonic acid, regulates the conversion of white to brite adipocyte through a prostaglandin/calcium mediated pathway. Mol Metab 2014; 3(9):834–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Simopoulos AP. The importance of the omega-6/omega-3 fatty acid ratio in cardiovascular disease and other chronic diseases. Exp Biol Med (Maywood) 2008; 233(6):674–688. [DOI] [PubMed] [Google Scholar]
- 60. Blasbalg TL, Hibbeln JR, Ramsden CE, Majchrzak SF, Rawlings RR. Changes in consumption of omega-3 and omega-6 fatty acids in the United States during the 20th century. Am J Clin Nutr 2011; 93(5):950–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sen S, Simmons RA. Maternal antioxidant supplementation prevents adiposity in the offspring of Western diet-fed rats. Diabetes 2010; 59(12):3058–3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yazama H, Kitatani K, Fujiwara K, Kato M, Hashimoto-Nishimura M, Kawamoto K, Hasegawa K, Kitano H, Bielawska A, Bielawski J, Okazaki T. Dietary glucosylceramides suppress tumor growth in a mouse xenograft model of head and neck squamous cell carcinoma by the inhibition of angiogenesis through an increase in ceramide. Int J Clin Oncol 2015; 20(3):438–446. [DOI] [PubMed] [Google Scholar]
- 63. Melland-Smith M, Ermini L, Chauvin S, Craig-Barnes H, Tagliaferro A, Todros T, Post M, Caniggia I. Disruption of sphingolipid metabolism augments ceramide-induced autophagy in preeclampsia. Autophagy 2015; 11(4):653–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Koeberle A, Pergola C, Shindou H, Koeberle SC, Shimizu T, Laufer SA, Werz O. Role of p38 mitogen-activated protein kinase in linking stearoyl-CoA desaturase-1 activity with endoplasmic reticulum homeostasis. FASEB J 2015; 29(6):2439–2449. [DOI] [PubMed] [Google Scholar]
- 65. Zarubin T, Han J. Activation and signaling of the p38 MAP kinase pathway. Cell Res 2005; 15(1):11–18. [DOI] [PubMed] [Google Scholar]
- 66. Spange S, Wagner T, Heinzel T, Krämer OH. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int J Biochem Cell Biol 2009; 41(1):185–198. [DOI] [PubMed] [Google Scholar]
- 67. Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009; 325(5942):834–840. [DOI] [PubMed] [Google Scholar]
- 68. Croze ML, Soulage CO. Potential role and therapeutic interests of myo-inositol in metabolic diseases. Biochimie 2013; 95(10):1811–1827. [DOI] [PubMed] [Google Scholar]
- 69. Ferrari F, Facchinetti F, Ontiveros AE, Roberts RP, Saade MM, Blackwell SC, Sibai BM, Refuerzo JS, Longo M. The effect of combined inositol supplementation on maternal metabolic profile in pregnancies complicated by metabolic syndrome and obesity. Am J Obstet Gynecol 2016; 215(4):503.e1–503.e8. [DOI] [PubMed] [Google Scholar]
- 70. Nascimento NRF, Lessa LM, Kerntopf MR, Sousa CM, Alves RS, Queiroz MG, Price J, Heimark DB, Larner J, Du X, Brownlee M, Gow A et al. Inositols prevent and reverse endothelial dysfunction in diabetic rat and rabbit vasculature metabolically and by scavenging superoxide. Proc Natl Acad Sci USA 2006; 103(1):218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Challier JC, Basu S, Bintein T, Minium J, Hotmire K, Catalano PM, Hauguel-de Mouzon S. Obesity in pregnancy stimulates macrophage accumulation and inflammation in the placenta. Placenta 2008; 29(3):274–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Saben J, Lindsey F, Zhong Y, Thakali K, Badger TM, Andres A, Gomez-Acevedo H, Shankar K. Maternal obesity is associated with a lipotoxic placental environment. Placenta 2014; 35(3):171–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Rocha NG, Templeton DL, Greiner JJ, Stauffer BL, DeSouza CA. Metabolic syndrome and endothelin-1 mediated vasoconstrictor tone in overweight/obese adults. Metabolism 2014; 63(7):951–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Tesauro M, Schinzari F, Rovella V, Di Daniele N, Lauro D, Mores N, Veneziani A, Cardillo C. Ghrelin restores the endothelin 1/nitric oxide balance in patients with obesity-related metabolic syndrome. Hypertension 2009; 54(5):995–1000. [DOI] [PubMed] [Google Scholar]
- 75. Spradley FT, Palei AC, Granger JP. Increased risk for the development of preeclampsia in obese pregnancies: weighing in on the mechanisms. Am J Physiol Regul Integr Comp Physiol 2015; 309(11):R1326–R1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Fiore G, Capasso A. Effects of vitamin E and C on placental oxidative stress: an in vitro evidence for the potential therapeutic or prophylactic treatment of preeclampsia. Med Chem 2008; 4(6):526–530. [DOI] [PubMed] [Google Scholar]
- 77. Liang C, DeCourcy K, Prater MR. High–saturated-fat diet induces gestational diabetes and placental vasculopathy in C57BL/6 mice. Metabolism 2010; 59(7):943–950. [DOI] [PubMed] [Google Scholar]
- 78. Fournier T, Tsatsaris V, Handschuh K, Evain-Brion D. PPARs and the placenta. Placenta 2007; 28(2-3):65–76. [DOI] [PubMed] [Google Scholar]
- 79. Holdsworth-Carson SJ, Lim R, Mitton A, Whitehead C, Rice GE, Permezel M, Lappas M. Peroxisome proliferator-activated receptors are altered in pathologies of the human placenta: gestational diabetes mellitus, intrauterine growth restriction and preeclampsia. Placenta 2010; 31(3):222–229. [DOI] [PubMed] [Google Scholar]
- 80. Ho TC, Chen SL, Yang YC, Liao CL, Cheng HC, Tsao YP. PEDF induces p53-mediated apoptosis through PPAR gamma signaling in human umbilical vein endothelial cells. Cardiovasc Res 2007; 76(2):213–223. [DOI] [PubMed] [Google Scholar]
- 81. Loegl J, Nussbaumer E, Hiden U, Majali-Martinez A, Ghaffari-Tabrizi-Wizy N, Cvitic S, Lang I, Desoye G, Huppertz B. Pigment epithelium-derived factor (PEDF): a novel trophoblast-derived factor limiting feto-placental angiogenesis in late pregnancy. Angiogenesis 2016; 19(3):373–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Adya R, Tan BK, Randeva HS, Differential effects of leptin and adiponectin in endothelial angiogenesis. J Diab Res 2015; 2015:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Gomez de Aguero M, Ganal-Vonarburg SC, Fuhrer T, Rupp S, Uchimura Y, Li H, Steinert A, Heikenwalder M, Hapfelmeier S, Sauer U, McCoy KD, Macpherson AJ. The maternal microbiota drives early postnatal innate immune development. Science 2016; 351(6279):1296–1302. [DOI] [PubMed] [Google Scholar]
- 84. Boulange CL, Neves AL, Chilloux J, Nicholson JK, Dumas ME. Impact of the gut microbiota on inflammation, obesity, and metabolic disease. Genome Med 2016; 8(1):42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Venkatesh M, Mukherjee S, Wang H, Li H, Sun K, Benechet AP, Qiu Z, Maher L, Redinbo MR, Phillips RS, Fleet JC, Kortagere S et al. Symbiotic bacterial metabolites regulate gastrointestinal barrier function via the xenobiotic sensor PXR and Toll-like receptor 4. Immunity 2014; 41(2):296–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Flanagan DE, Moore VM, Godsland IF, Cockington RA, Robinson JS, Phillips DI. Fetal growth and the physiological control of glucose tolerance in adults: a minimal model analysis. Am J Physiol Endocrinol Metab 2000; 278(4):E700–E706. [DOI] [PubMed] [Google Scholar]
- 87. De Blasio MJ, Dodic M, Jefferies AJ, Moritz KM, Wintour EM, Owens JA. Maternal exposure to dexamethasone or cortisol in early pregnancy differentially alters insulin secretion and glucose homeostasis in adult male sheep offspring. Am J Physiol Endocrinol Metab 2007; 293(1):E75–E82. [DOI] [PubMed] [Google Scholar]
- 88. Gabory A, Ferry L, Fajardy I, Jouneau L, Gothié JD, Vigé A, Fleur C, Mayeur S, Gallou-Kabani C, Gross MS, Attig L, Vambergue A et al. Maternal diets trigger sex-specific divergent trajectories of gene expression and epigenetic systems in mouse placenta. PLoS One 2012; 7(11):e47986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Prickett TD, Samuels Y. Molecular pathways: dysregulated glutamatergic signaling pathways in cancer. Clin Cancer Res 2012; 18(16):4240–4246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Wen Y, Li J, Koo J, Shin SS, Lin Y, Jeong BS, Mehnert JM, Chen S, Cohen-Sola KA, Goydos JS. Activation of the glutamate receptor GRM1 enhances angiogenic signaling to drive melanoma progression. Cancer Res 2014; 74(9):2499–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Pan YB, Gong Y, Ruan HF, Pan LY, Wu XK, Tang C, Wang CJ, Zhu HB, Zhang ZM, Tang LF, Zou CC, Wang HB et al. Sonic hedgehog through Gli2 and Gli3 is required for the proper development of placental labyrinth. Cell Death Dis 2015; 6(2):e1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Pola R, Ling LE, Silver M, Corbley MJ, Kearney M, Blake Pepinsky R, Shapiro R, Taylor FR, Baker DP, Asahara T, Isner JM. The morphogen Sonic hedgehog is an indirect angiogenic agent upregulating two families of angiogenic growth factors. Nat Med 2001; 7(6):706–711. [DOI] [PubMed] [Google Scholar]
- 93. Dhobale M. Neurotrophins: Role in adverse pregnancy outcome. Int J Dev Neurosci 2014; 37:8–14. [DOI] [PubMed] [Google Scholar]
- 94. D'Souza V, Patil V, Pisal H, Randhir K, Joshi A, Mehendale S, Wagh G, Gupte S, Joshi S. Levels of brain derived neurotrophic factors across gestation in women with preeclampsia. Int J Dev Neurosci 2014; 37:36–40. [DOI] [PubMed] [Google Scholar]
- 95. Prince CS, Maloyan A, Myatt L. Maternal obesity alters brain derived neurotrophic factor (BDNF) signaling in the placenta in a sexually dimorphic manner. Placenta 2017; 49:55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.