

Abstract
Cancer is a genetic disorder, meaning that a plethora of different mutations, whether somatic or germ line, underlie the etiology of the ‘Emperor of Maladies’. Point mutations, chromosomal rearrangements and copy number changes, whether they have occurred spontaneously in predisposed individuals or have been induced by intrinsic or extrinsic (environmental) mutagens, lead to the activation of oncogenes and inactivation of tumor suppressor genes, thereby promoting malignancy. This scenario has now been recognized and experimentally confirmed in a wide range of different contexts. Over the past decade, a surge in available sequencing technologies has allowed the sequencing of whole genomes from liquid malignancies and solid tumors belonging to different types and stages of cancer, giving birth to the new field of cancer genomics. One of the most striking discoveries has been that cancer genomes are highly enriched with mutations of specific kinds. It has been suggested that these mutations can be classified into ‘families’ based on their mutational signatures. A mutational signature may be regarded as a type of base substitution (e.g. C:G to T:A) within a particular context of neighboring nucleotide sequence (the bases upstream and/or downstream of the mutation). These mutational signatures, supplemented by mutable motifs (a wider mutational context), promise to help us to understand the nature of the mutational processes that operate during tumor evolution because they represent the footprints of interactions between DNA, mutagens and the enzymes of the repair/replication/modification pathways.
Keywords: mutation spectra, classification, DNA sequence context, somatic hypermutation, cancer genomics, methylation
Introduction
Mutations provide the raw material for natural selection in evolution, but their rate is maintained at a low level to minimize the reduced fitness that would be associated with numerous deleterious mutations. Multicellular organisms can however dramatically elevate mutation rates in subpopulations of cells in certain chromosomal regions, for example in the variable regions of immunoglobulin genes in B cells. The mutator effect is achieved by the recruitment of editing activation-induced cytidine deaminase (AID) to convert cytosines to uracil, together with error-prone DNA polymerases (pols) that augment the mutator effect by inaccurate repair of the uracils [1, 2]. However, aberrant regulation of the elaborate machinery of region-specific hypermutagenesis under pathological conditions can lead to cancer and other diseases [1, 3, 4]. Strong mutator phenotypes are also caused by errors in the global system of DNA replication and maintenance [5–7]. As a result of these aberrations, many cancer genomes are characterized by a large number of changes to their constitutional genetic information. The underlying mechanisms of genetic change and the factors that determine mutational distribution patterns in tumor genomes are multifaceted and have long been regarded as being refractory to direct investigation. However, recent advances in the field have led to the emergence of precision (or personalized) medicine in a cancer context [8, 9].
Tumorigenesis is a multistep process. It begins with the transformation of a single cell that acquires several of the six hallmarks of cancer [10]. Cells gain these characteristics through numerous mutations [11, 12] caused by errors of DNA replication, the action of exogenous mutagens or endogenous DNA damage [13, 14]. It is likely that the mutator phenotype is a feature of many different cancers [15]. The ensuing genetic assault leads to the activation of oncogenes and inactivation of tumor suppressors, thereby promoting malignancy [4, 16]. Impairment of DNA pols, alterations in nucleotide pools or expression of editing deaminases promote tumors because cells become unable to accurately replicate and repair their DNA [4, 17–19]. The sophisticated machinery of replication and genome maintenance can be damaged by mutations, or altered by physiological conditions, such that it can become a potent mutagenic factor in cancer [20, 21]. The frequencies of single base-pair substitutions, chromosomal rearrangements and changes in gene or chromosomal copy number are greatly enhanced by various environmental and intrinsic mutagens, especially in genetically or developmentally predisposed individuals whose cells are unable to properly maintain genome integrity [22]. These effects are tissue-specific: for example, the inherited lack of mismatch repair and/or the exonuclease domain of replicative DNA pols predisposes to colorectal cancer [10, 23]; abnormal DNA double-strand break (DSB) repair leads to an increase in incidence of breast and ovarian cancer [24]; defects in translesion DNA рol η cause skin cancer [25]. Some of the mutations leading to defective DNA metabolism can predispose to pancreatic cancer [26]. However, compromised DNA maintenance is not the only cause of cancer. At the beginning of this century, it was discovered that in addition to faithful repair, human cells are equipped with powerful mutator machines—proteins that act in a highly mutagenic way. Most prominent are the DNA/RNA editing cytosine deaminases of the AID/APOBEC family [1] and inaccurate translesion synthesis DNA pols [27]. The availability of intrinsic mutators provides an opportunity to create variability ‘on demand’ as an integral part of developmental programs and adaptive responses but clearly poses a threat to genome integrity in case of their faulty regulation causing cancer and other diseases [13, 22, 28, 29].
One powerful approach to understand the mechanisms of mutagenesis in cancer is to analyze the DNA sequence context of mutations in tumors [4, 14, 30–32]. The methodology was introduced in the 1990s in the context of deciphering the mechanisms of somatic hypermutation (SHM) in humoral immunity [30, 33, 34] and hypermutagenesis in retroviruses [35]. Mutations have been found in many types of cancer in DNA sequence contexts that are similar to those associated with mutagen-induced mutagenesis in model systems. It was found that AID (mutations in the WRC motif, the mutable C being underlined, W = A/T, R = A/G) may contribute to gastric and hemopoietic cancers [3, 36], especially in sites subject to cytosine methylation [32]; deaminases participating in innate immunity, APOBEC3A and APOBEC3B (the TCW/WGA motif, the mutable C being underlined, W = A/T) may contribute to solid tumors, including breast, lung and others [37–40]. The genomes of several types of cancer may exhibit signatures of environmental mutagens, e.g. tobacco smoke for lung cancer [41], ultraviolet (UV) radiation for skin cancer and ionizing radiation for many other cancers [42, 43]. These examples will be discussed in more detail in the next chapters.
Complete cancer genomes and genomes of other model systems
Emerging genetic factors predisposing to cancer or connected with sporadic cancer include defects of systems maintaining proper quality of nucleotide pools [44], proofreading by replicative DNA pols, mismatch repair [5, 7] and the misregulation of editing deaminases [45]. The worst appears to be a combination of pool imbalance and pol defects, leading to mutational catastrophe and, likely, cancer [46]. Low replication fidelity or extensive genome editing causes hereditary and sporadic cancer and fuels the acquisition of drug resistance. On the other hand, low replication fidelity renders many cancer cells more sensitive to certain antitumor agents, which could be used as therapeutic tools to contain tumor cells [47, 48].
It is imperative to highlight the point that mutational signatures attributable to each particular cancer type were first found and characterized by means of the extensive use of model organisms, bacteria and yeast [49–56]. Despite enormous progress in our understanding of the mechanisms of mutagenesis, the latest data prompt new questions and stimulate the search for new approaches and methods aimed at addressing these questions. Among the most pressing issues are the mechanisms of mutagenesis in tumor cells. The transient hypermutable phenotype that was described in cancer cells and in cultures of microorganisms is worth comprehensive study [4]. Most of the studies devoted to the mutational process were conducted in haploid organisms. It was previously noticed that mutagenesis in diploid organisms possesses some special features [55, 57, 58].
Somatic mutations in normal tissues
Not only cancer genomes but also the genomes of benign cells acquire somatic mutations during the course of apparently normal development and aging. These mutations arise because of various endogenous factors such as the activity of mobile elements, DNA pol slippage, DNA DSBs, inefficient DNA repair, unbalanced chromosomal segregation and various exogenous factors such as cigarette smoke and UV exposure [59, 60]. The genomes of somatic cells carry a substantial burden of somatic mutations and footprints of exogenous and endogenous mutagenic processes. For instance, comparing the mutational burden in skin fibroblasts from forearm and hip from the same donors, it was ascertained that the UV-induced (primarily C:G > T:A and CC:GG > TT:AA) and endogenous mutation rates per year in exposed skin were >2-fold higher than that in protected areas [61]. In similar vein, the impact of smoking was apparent in lung, and an increased burden of C:G > A:T mutations was detectable at tissue-level resolution in smokers indicating pervasive clonal growth (with implications for field cancerization [62, 63]). Endogenous factors also lead to a context-specific increase in mutation burden. For example, somatic variants in peripheral blood occasionally carried signatures of endogenous mutational processes including AID-driven targeted mutagenesis [63–65]. Spontaneous deamination of methylated cytosine residues appears to be an important source of somatic mutations in benign colon and small intestine but not in liver [66]. Germ line BRCA1 and BRCA2 mutations are associated with increased DNA DSB repair defects and a higher burden of genomic alterations (e.g. amplifications and deletions) in benign tissues [67]. It was however difficult to ascertain the developmental lineage in which the majority of somatic mutations were acquired. Using transcription-coupled repair signatures, Yadav et al. [63] were able to associate somatic mutations with transcriptional profiles of the affected cells and infer that the vast majority of somatic mutations detectable in peripheral blood had probably been acquired in the lymphoid progenitor cells and hematopoietic stem cells.
The burden of somatic mutations in benign cells appears to be substantial [68–70]. According to some estimates [71], almost half of the somatic mutations in cancer genomes have accumulated before neoplastic transformation. This translates into a burden of 10−2–102 mutations per Mb on a genome-wide scale, and 10–100 mutations in protein-coding regions in a single somatic cell in benign human tissues. Although estimates of the rate of stem-cell divisions in adult tissues are controversial and vary widely [72], this is roughly consistent with estimates of the somatic mutation rate (2–10 mutations per diploid genome per cell division) calculated in a number of human cell types including B and T lymphocytes and fibroblasts (reviewed in [73]). Another estimate based on sequencing clonally derived organoids from small intestine, colon and liver of human donors (aged between 3–87 years) suggested that the mutation rate was comparable among progenitor cells in those tissues: ∼40 novel mutations per year, despite the large difference in cancer incidence between these organs [66]. Reliable single-cell data for other tissue types are still relatively sparse. It is not yet known whether stem-cell division rates, and hence, the increase in mutation burden, are constant over the lifetime of the individual. This notwithstanding, these data indicate that as with cancer genomes, the genomes of normal benign cells also carry a substantial burden of somatic mutations during development and aging.
The tissue-level functional consequences of somatic mutations present in a single somatic cell are limited, unless the cell undergoes clonal growth [63, 67, 73]. Clonal growth certainly appears to be widespread in skin and blood. Tissue-level studies that complemented single-cell analyses found that ∼2–30 somatic mutations and 1–8 somatic copy number alterations were detectable at tissue-level resolution (>1% allele frequency) in benign tissues [60, 63, 67]. In some cases, clonally expanded cell populations carried cancer gene mutations (however, this was not obligate, and there were exceptions). For instance, clonal hematopoiesis is often characterized by DNMT3A and TET2 mutations [64, 74]. In benign skin tissues, clones carrying BRAF and TP53 mutations were common [73]. It is possible that such early driver mutations (described in more detail below) have a role in initiating field cancerization and premalignant lesions. Mutational signatures suggested that such mutations tend to propagate under relaxed purifying selection (i.e. nearly neutral and/or positive selection) in nonmalignant tissues [63, 73].
Mutation databases in cancer genetics and potential problems associated with their use
The major cancer genomic databases are listed in Table 1. These databases contain mutations identified in cancer samples using a variety of different methods, e.g. allele-specific polymerase chain reaction (PCR), SNP chip arrays, targeted gene sequencing, whole-exome sequencing and whole-genome sequencing. The heterogeneity of these data is such that it can lead to certain experimental bias, particularly study bias associated with known cancer driver genes and variants. Analysis of mutational signatures, motifs or spectra requires unbiased data sets, such as whole-genome/exome sequences and additional preprocessing. Therefore, databases providing access to data from whole-genome and whole-exome studies, such as the International Cancer Genome Consortium (ICGC), The Cancer Genome Atlas (TCGA), COSMIC WGS, CBioPortal, Pediatric Cancer Data Portal (PCDP) and UCSC Cancer Genomics Browser (listed in Table 1), have been a prerequisite for mutational signature research.
Table 1.
Databases of cancer mutations
| Database | URL | Statistics | Description |
|---|---|---|---|
| COSMIC | http://cancer.sanger.ac.uk/cosmic | 4M coding mutations, 23 489 publications | Curated collection of mutations in cancer includes data from various sources |
| COSMIC-Cell Lines | http://cancer.sanger.ac.uk/cell_lines | Exome sequences of 1015 cancer cell lines | Filtered variants from extensively used cancer cell lines, including NCI-60 set |
| COSMIC-WGS | http://cancer.sanger.ac.uk/wgs | 28 366 samples | Large-scale cancer sequencing projects; genome-wide screens |
| TCGA | https://cancergenome.nih.gov | 32 cancer types | Publicly available catalog of major cancer-causing genomic alterations |
| PCDP | https://www.stjude.org/research/ pediatric-cancer-genome-project.html | 2813 samples from 17 cancer types | Data from PCGP, TARGET, DKFZ, MAGIC, BROAD, etc. |
| ICGC | http://icgc.org | 16 000+ donors, 70 projects and 21 tumor sites | Catalog of major cancer-causing genomic alterations obtained as result of large international projects |
| CCLE | https://portals.broadinstitute.org/ccle/home | 1074 samples | Compilation of genomic data from human cell lines |
| Risctox database | http://risctox.istas.net/en/index.asp? idpagina=607 | 1750 carcinogenic and mutagenic substances | Describes hazardous substances |
| CBioPortal | http://www.cbioportal.org/ | 147 cancer studies | Includes cancer samples from TCGA, ICGC and other sources |
| UCSC Cancer Genomics Browser | http://xena.ucsc.edu/ | 720 data sets including TCGA and CCLE | Collection of cancer genomics data with interactive tools |
In model organisms, mutation accumulation studies involve the genome sequencing of subsequent generations. As described in the previous chapter, these studies reveal both the mutation rate and the mutational spectrum associated with spontaneous mutagenesis. Other mutational studies have focused on controlled experiments exposing cell cultures to certain mutagens or genetically modifying enzymes involved in replication and DNA repair machinery. Human cell lines are cataloged in Biobanks (Table 1), and data are aggregated in databases such as Cancer Cell Line Encyclopedia (CCLE) and COSMIC CLP, whereas for model organisms, the data are scattered around various resources. Many of the known carcinogens are mutagens; when the mutational spectrum of a given mutagen is known, it may be used to identify the likely cause of cancer. Known mutagenic substances and pharmacogenomics data are available in the databases Risctox, COSMIC, CCLE and the UCSC Cancer Genomics Browser (Table 1).
Large-scale cancer genomics projects (Table 1) have generated high volumes of data that, in principle, are invaluable in understanding the biology, initiation and progression of human cancers. One caveat, however, is how to distinguish artifactual DNA damage from the bona fide somatic mutations that occurred in the tumor; Chen et al. [75] have recently reported that mutagenic damage accounts for the majority of the erroneous identification of variants in the low to moderate (1–5%) frequency range in whole (cancer)-genome sequencing studies. If many of the somatic mutations supposedly identified in human cancer genomes are indeed spurious (implying that some of the key data sets used for the analysis of cancer mutational signatures have been compromised from the outset), then some of the conclusions drawn from early studies may have to be revisited once the accuracy and reliability of the mutation data sets have been ascertained.
Mutable motifs: from the DNA context of modifying enzymes and mutagens to mechanisms of mutagenesis
There is no doubt that nucleotide sequence context influences mutation probability [30, 76–85]. Mutable motifs constitute a well-established approach to study mutagenesis because they represent the fingerprints of interactions between DNA and mutagens/repair/replication/modification enzymes, thereby providing clues as to the underlying molecular mechanisms of mutation/recombination [79, 83, 84]. Mutable motifs are usually derived from mutation spectra, sets of data that include the frequency of mutations in a target nucleotide sequence under defined conditions. Mutational spectra are often determined by applying phenotypic selection to an experimental mutagenesis system. Phenotypic selection restricts the mutational spectrum to detectable changes where a mutation has given rise to a phenotypic change. Alternatively, mutations are identified by random sequencing of DNA clones or PCR-amplified DNA molecules. However, this approach only works well when the frequency of mutations is extremely high (roughly in excess of 10−3 per nucleotide). A mutational spectrum is usually displayed with the target nucleotide sequence along a horizontal linear axis, and each mutational variant listed vertically above the nucleotide it replaces (Figure 1, [86]). In other words, a mutational spectrum exhibits the types and frequencies of context-dependent mutations associated with a particular experimental system. It may be either difficult or impossible to integrate mutational spectra originating from different studies. A mutational motif is a generalized representation of mutated nucleotides and their context associated with a mutagenic factor. Motifs often lack quantitative information and serve as qualitative descriptors of most frequently mutated sites, allowing integration of results from different studies. Examples of motifs are the AID motif WRCY/RGYW (the mutable position is underlined, W = A or T, R = purine and Y = pyrimidine) with C to T/G/A mutations [30], and error-prone DNA pol η attributed AID-related mutations (A to G/C/G) at WA/TW motifs [27]. Examples of mutable motifs [27, 30, 32, 40, 58, 77, 83, 87, 88] are shown in Table 2.
Figure 1.

Mutational spectrum of human DNA pol η in the lacZ gene without phenotypic selection [86].
Table 2.
Examples of mutable motifs
| Test system/mutagen/spectrum | Mutable motif | Comments | Reference |
|---|---|---|---|
| Spontaneous G:C→A:T mutations in human genome | CG | May result from the spontaneous deamination of 5-methylcytosine | [77] |
| Somatic mutations in immunoglobulin genes | RGYW WA | AGYW is more mutable compared with GGYW | [27, 30] |
| TA is more mutable compared with AA | |||
| AID | WRC | In vitro DNA damages | [88] |
| Somatic mutations in many cancers | WRCG | Hybrid motif (WRC+CG) | [32] |
| APOBEC1 | TCW | Expression of rat gene in yeast | [58] |
| APOBEC3G | CCR | Expression of human gene in yeast | [58] |
| APOBEC3A/B | TCW | Expression of human gene in yeast and analysis of mutations in cancer genomes | [27, 30] |
| Hotspots of errors produced by human DNA pol η | WA | In vitro gap filling | [40] |
| Pyrimidine (6-4) pyrimidone photoproducts | YTCA | In vitro DNA damages | [83] |
| Cyclobutane dimers photoproducts | YTT | In vitro DNA damages | [83] |
| Insertions/deletions in human genes | YYYTG | Analysis of human disease genes | [87] |
Note: Hotspot positions are underlined; for some motifs, the exact location of hotspot positions cannot be defined. The standard nomenclature for consensus sequences: R = A/G, Y = T/C, M = A/C, K = G/T, W = A/T, S = G/C, B = T/C/G, D = A/T/G, H = A/T/C, V = A/G/C and N = A/T/G/C.
Several methods are available to analyze mutational spectra represented as a set of aligned sequences (Table 3); these approaches are particularly useful when applied to a set of so-called ‘hotspot’ sites (sites with an elevated frequency of mutations, see [83, 84] for a discussion of hotspot sites). For example, a set of aligned sites can be analyzed to derive a consensus sequence [89] (Table 3) using one of several available approaches as described by Day and McMorris [90, 91]. Methods that rely on arbitrary discrimination between informative and noninformative positions may lead to controversial and/or unreliable results. Simple consensus sequences can be misleading especially when the data set is small; however, they can be reconstructed using any mutational spectrum and any subset of positions.
Table 3.
Putative DNA pol η mutation hotspots in lacZ
| Sequence | Hotspot position | Type of changes | Number of mutations |
|---|---|---|---|
| CAATT | 3 | A→G,T,C | 15,1,1 |
| TTATC | 14 | A→G,C,T | 14,1,1 |
| GTTAT | 15 | T→G,A | 10,5 |
| AAATT | 20 | A→G,T | 11,1 |
| GAAAT | 21 | A→G,T | 16,2 |
| ATAGC | 38 | A→G,T,C | 9,2,1 |
| CATAG | 39 | T→G,A,C | 9,9,2 |
| TCATG | 46 | A→G,T | 13,1 |
| GTAAT | 50 | A→G,T | 16,4 |
| GAATT | 56 | A→G | 17 |
| AAACG | 70 | A→G,T | 18,3 |
| GTAAA | 73 | A→G,T | 14,1 |
| CAACG | 77 | T→C,G | 12 |
| CGACG | 80 | A→G,T | 11,2 |
| WA | Consensus |
||
Note: Hotspot positions are underlined. The mutational spectrum shown in Figure 1 was converted to the complementary orientation.
The binomial test can also be used to study consensus sequences at or near mutation hotspots [92]. In this method, a number NIJ of a nucleotide ‘I’ is calculated in each position ‘J’ in a set of ‘M’ aligned mutation hotspot sequences (Table 3). The probability P(NIJ,M,FI) to find NIJ or more nucleotides ‘I’ in a position ‘J’ is calculated taking a frequency FI of a nucleotide ‘I’ in a target sequence as an expected number of the nucleotide ‘I’ in the position ‘J’. A nucleotide with the lowest probability P(NIJ,M,FI) among all possible nucleotides in a position ‘J’ is accepted as a consensus nucleotide for this position if P(NIJ,M,FI) for this nucleotide is below the significance level, α. It is important to note that α = 0.05 cannot be used to reject or accept a statistical hypothesis owing to the multiplicity of binomial tests; moreover, these tests are strongly interdependent for each position. To estimate the significance level for P(NIJ,M,FI), Malyarchuk et al. [92] developed a resampling procedure, which takes into account the multiplicity of binomial tests.
Multiple regression models can be used for simultaneous analysis of how several neighboring positions influence mutation frequency. The purpose of multiple regression analysis is to learn more about the relationship between several independent (or predictor) variables Xi and a dependent (or criterion) variable Y. Stormo et al. [93] used multiple linear regression analysis to see how nucleotide sequence context affects 2-aminopurine mutagenesis in the lacI gene. The data indicate that the two nucleotides immediately preceding the mutable base strongly affect the frequency of mutation. However, the method assumes a direct linear correlation between the frequency of mutations in detectable positions and factors attributable to the nucleotide sequence context, and that the factors are distributed normally; in general, these assumptions are not valid for experimental mutational spectra. Rogozin and Kolchanov [30] used a heuristic classification approach and a Monte Carlo procedure to build hotspot consensus sequences. This procedure assesses the nonrandomness of nucleotides adjacent to or near a mutation hotspot [30]. Regression trees have also been used to analyze the effect of nucleotide sequence context on mutation frequency [94]. Regression trees are mathematically tenable, do not restrain the number of variables (as do heuristic methods) and are recommended for the study of simulated and real mutation spectra [94]. However, these approaches are based on complex assumptions and need large data sets [94].
Another important computational task is to identify overrepresentation of somatic mutations in known mutable motifs. Usually, the frequency of known mutable motifs for somatic mutations is compared with the frequency of these motifs in the vicinity of the mutated nucleotide. Specifically, for each base substitution, 120 or 150 bases of DNA sequence centered at the mutation are extracted (the DNA neighborhood). This approach has been thoroughly tested and the high accuracy of the analysis demonstrated [38]. The frequency of mutable motifs in the positions of somatic mutations was compared with the frequency of the same motifs in the DNA neighborhood (Figure 2) using Fisher’s exact test (2 × 2 table) and the Monte Carlo test [32, 38].
Figure 2.

Statistical analysis of mutable motifs in sites of somatic mutations and surrounding regions. The excess of mutations in motifs was calculated using the ratio Fm/Fn, where Fm is the fraction of somatic mutations observed in a given mutable motif (the number of mutated motifs divided by the number of mutations), and Fn is the frequency of the motif in the DNA neighborhood of somatic mutations (the number of motif positions divided by the total number of all un-mutated positions in the 120 bp window).
Methods to derive mutational signatures
Cancer genome studies necessitate working with large amounts of data; the obvious problems of analysis of such data were resolved to a large extent by means of the so-called mutational signature technique [16, 31, 95, 96]. As it is usually not possible to define the DNA strand on which a mutation occurred (distinguishing, e.g., C > T mutations from G > A mutations on the opposite strand), there are essentially only six types of substitutions to be analyzed. Similarly, there are 96 context-dependent mutations that consider two nucleotides in the flanking 5′ and 3′ positions of the mutated nucleotide. Analysis of mutational spectra of context-dependent mutations in cancer patients involves pooling all mutations from cancer samples into a discrete distribution according to the mutation types. For multiple patients/samples, their context-dependent mutations can be represented in the form of a nonnegative matrix X, where columns correspond to samples, and rows represent context-dependent mutation types. The problem is to find two nonnegative matrices W and H as a result of decomposition of X ∼ WH, where W corresponds to mutational signatures, and H corresponds to exposure of samples to mutational processes described by the signatures [16]. This so-called nonnegative matrix factorization (NMF) method was introduced in 1999 [97] and was subsequently applied to identify metagenes and pathways in cancer gene expression data [95], most recently being used to derive mutational signatures [16].
There are some variations of this basic technique. For example, Temiz et al. [98] presented a 32 × 12 mutation matrix that captures the nucleotide pattern two nucleotides upstream and downstream of the mutation. In this study, a somatic autosomal mutation matrix (SAMM) representing tumor-specific mutations and mechanistic template mutation matrices (MTMMs) representing oxidative DNA damage, UV-induced DNA damage, (5m)CpG deamination, and APOBEC-mediated cytosine mutation were constructed. MTMMs were mapped to the individual tumor SAMMs to find mutational mechanisms corresponding to each overall mutational pattern. It was found that ∼90% of all tumor genomes had a nearest neighbor from the same tissue of origin. When a distance-dependent six-nearest neighbor classifier was used, ∼10% of the SAMMs had an undetermined tissue of origin, whereas 92% of the remaining SAMMs were assigned to the correct tissue of origin. Thus, although tumors from different tissues may have similar mutation patterns, their SAMMs often display signatures that are characteristic of specific tissues [98].
Mutational signature is an important concept for describing individual mutagenic factors and for quantifying their contribution to mutational spectra in cancer samples. Several computational methods have been proposed for solving the X ∼ WH decomposition problem. The original method of NMF, minimizing the Frobenius norm of decomposition, is available as a Wellcome Trust Sanger Institute Mutational Signature Framework in the form of a MATLAB package [16]. SomaticSignatures is an R Bioconductor package implementing NMF and PCA approaches to signature decomposition from mutational data [99]. The DeconstructSigs R package applies an alternative approach—multiple linear regression models to the reconstruction of signatures [100]. The MutSpec package integrates NMF decomposition into a Galaxy toolbox, enabling genomic data analysis pipelines [101].
A recently developed resource, MutaGene [102], provides a set of tools that allows the exploration of this heterogeneity in terms of the underlying mutagenic processes. The processes are defined based on the concept of mutational signatures obtained by nonsmooth NMF decomposition from available cancer samples. MutaGene can analyze any set of mutations obtained, for instance, from sequencing tumor samples, and identifies the underlying mutagenic processes and the most likely cancer type and subtype for a given sample. Finally, MutaGene applies mutational profiles and signatures as background statistical models for calculating the expected rates of context-dependent mutations for each nucleotide and amino acid in a given gene or corresponding protein, helping to find site-specific cancer-driving events.
An example of a mutational signature is shown in the Figure 3. This signature (Signature 9; http://cancer.sanger.ac.uk/cosmic/signatures) has been found in chronic lymphocytic leukemia and malignant B-cell lymphoma genomes. Signature 9 is characterized by a pattern of mutations that has been attributed to DNA pol η, which has been implicated with the activity of AID during SHM.
Figure 3.

The DNA pol η mutational signature (Signature 9, http://cancer.sanger.ac.uk/cosmic/signatures).
The number of mutational signatures defines the dimensionality of the problem. It is an important parameter because signatures are interpreted as individual mutational processes. An optimal number of signatures are hard to find because a large number of signatures may result in over-fitting, whereas a small number of signatures may result in inaccurate decomposition. A number of approaches have been implemented, for example by Tan and Fevotte [96] in a Bayesian NMF algorithm, and cophenetic correlation inspired by Brunet et al. [95]. To this end, finding a true number of mutagenic processes operating in a set of cancer samples remains an open research problem. Although decomposition into signatures is useful for interpreting the mutagenic processes, there are certain limitations. One of them is the heuristic nature of associations between mutational signatures and molecular mechanisms of mutations. For example, the pol η signature in COSMIC (Figure 3; the Signature 9, http://cancer.sanger.ac.uk/cosmic/signatures) has a higher frequency of T:A > G:C transversions compared with T:A > C:G transitions, although such a pattern has not been observed either in vitro or in vivo [27]. In addition, this pol η mutational signature was not found in follicular lymphoma, although this cancer is associated with the activity of AID [32].
Examples of cancer studies
There are several examples of the successful application of mutable motifs and mutational signatures. As discussed above, several mutations are required for cancer development, and genome sequencing has revealed that many cancers, including breast cancer, have somatic mutational spectra that are dominated by C:G > T:A transitions [40, 103]. Roberts et al. [40] and Burns et al. [103] have shown that APOBEC-mediated mutagenesis is pervasive throughout cancer genomes and correlates with APOBEC mRNA levels. Interestingly, APOBEC3B mRNA is upregulated in most primary breast tumors and breast cancer cell lines. Cancer cells that express high levels of APOBEC3B exhibit twice as many mutations as those that express low levels and are more likely to have mutations in TP53 [103]. Mutation clusters in whole-genome and exome data sets conformed to the stringent criteria indicative of an APOBEC mutation pattern (examples of APOBEC3A and APOBEC3B mutation patterns [56] are shown in the Figure 4). Applying these criteria to somatic mutations from 14 cancer types showed a significant presence of the APOBEC mutation pattern in bladder, cervical, breast, head and neck and lung cancers, reaching 68% of all mutations in some samples [40]. Within breast cancer, the HER2-enriched subtype was clearly enriched for tumors with the APOBEC mutation pattern, suggesting that this type of mutagenesis is functionally linked with cancer development. The APOBEC mutation pattern also extended to cancer-associated genes, implying that ubiquitous APOBEC-mediated mutagenesis is carcinogenesis [40].
Figure 4.
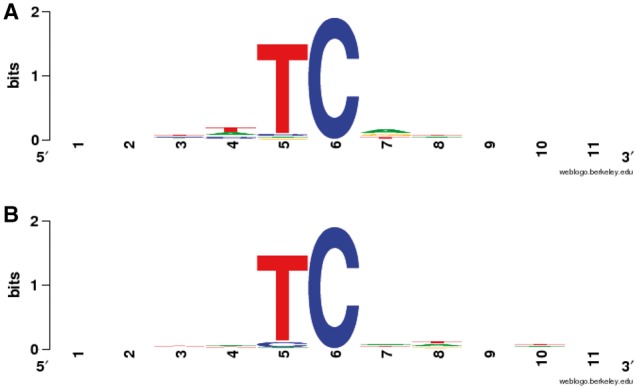
APOBEC3A (A) and APOBEC3B-induced (B) mutation patterns in yeast genomes [56] shown as a logo (weblogo.berkeley.edu). The Position 6 is the mutable position.
Tobacco smoking has been claimed to be associated with an increased risk of at least 17 classes of human cancer. Alexandrov et al. [41] analyzed somatic mutations and DNA methylation in 5243 cancers of those types for which tobacco smoking is associated with an elevated risk [41]. Smoking was found to be associated with an increased mutational burden of multiple distinct mutational signatures that contribute to different extents in different cancers. One of these signatures, mainly but not exclusively found in cancers derived from tissues directly exposed to tobacco smoke, was attributed to misreplication of DNA damage caused by tobacco carcinogens. Other signatures probably reflect the indirect activation of DNA editing by APOBEC cytidine deaminases and of an endogenous clock-like mutational process. These results are consistent with the proposition that smoking increases cancer risk by increasing the somatic mutation load, although direct evidence for this mechanism is still lacking in smoking-related cancer types [41].
Follicular lymphoma is an uncurable cancer characterized by the progressive severity of relapses. The sequence context specificity of mutations in the B cells from a large cohort of follicular lymphoma patients has been analyzed [32]. A substantial excess of mutations was found within a novel hybrid nucleotide motif: the signature of SHM enzyme, AID, which overlaps CG dinucleotides. The prevalence of this hybrid mutational signature in many other types of human cancer was observed, suggesting that AID-mediated, CpG methylation-dependent mutagenesis is a common feature of human tumorigenesis [32]. Analysis of the association between the methylation ratio and somatic mutations in WRCG/CGYW mutable motifs identified a moderate but significant (P < 0.0001) decrease of methylation in the WRCG/CGYW mutation context [32]. Figure 5 shows that the major difference lies within the range of methylation ratios (% of methylated cytosines) of 80 and 100. This finding implies that in follicular lymphoma, the SHM machinery acts at genomic sites containing methylated cytosine. It is consistent with the hypothesis that AID-dependent demethylation occurs preferentially in WRCG/CGYW mutable motifs, so that mutations are one of the outcomes of the multistep demethylation process [32].
Figure 5.

The methylation ratio in WRCG mutable motifs and non-WRCG motifs (YCG/CGR and SNCG/CGNS) [32]. The fraction of motifs in each bin (0–20% methylation ratio, 20–40% methylation ratio, etc.) is shown.
Smith et al. [104] identified a novel signature of accelerated somatic evolution (SASE) marked by a significant excess of clustered somatic mutations localized in a genomic locus, and prioritized those loci that carried the signature in multiple cancer patients. In a pan-cancer analysis of 906 samples from 12 tumor types, SASE was detected in the promoters of several genes, including known cancer genes such as MYC, BCL2, RBM5 and WWOX. Nucleotide substitution patterns consistent with oxidative DNA damage and APOBEC-related local SHM appeared to contribute to this signature in selected gene promoters (e.g. MYC) [104].
Clustering of mutations
Clustering of mutations is characteristic of many DNA-modifying enzymes [105] and may be used as an additional source of information to provide evidential support for the involvement of certain enzymes in generating somatic alterations in cancer. It should be noted that clustering could be because of certain structural or functional features of genomes (e.g. transcription start sites) [57, 106]. Several aspects of a mutational spectrum, including the frequency of nucleotide substitutions, clustering of mutations and hotspots and periodicity of mutational patterns, can be used to understand molecular mechanisms of mutagenesis. Some statistical approaches for analyzing the clustering of mutations are described in [40, 55, 107–109].
In theory, any two mutations that are not distributed randomly can be considered to be clustered [52]. In practice, however, certain thresholds should be used to define cluster borders. Sometimes, when the clusters are prominent, this is rather easy. For example, the sequencing of genomes of certain tumors points to the mechanism where extensive deamination of resected DNA ends by APOBEC enzymes causes formation of strong mutational clusters (termed ‘kataegis’) [110]. Similarly, single-stranded DNA (ssDNA)-specific mutagens cause strand-specific mutation clustering in yeast on DSB repair via homologous recombination [52] or on induction of break-induced replication [111]. An example of a clear cluster is shown schematically on Figure 6A.
Figure 6.

Types of mutational clusters. Horizontal black lines, chromosome. Mutations resulting from damage to the top and bottom DNA strands are shown as lighter (red) and darker (blue) circles, respectively. Clusters are indicated by brackets. (A) Strong, clear cluster resulting from the action of ssDNA-specific mutagen on the resected DNA during DSB repair. (B) Cluster of moderate strength with mixed types of mutations. In this case, clusters of different size can be defined based on the threshold parameters of clustering algorithm (compare two brackets). (C) Six individual clones (e.g. cells, tumors or mutants microorganisms) are shown on top. No apparent clustering is observed except for one clone where two mutations of different types are located close to each other. However, on combining all data sets, prominent and likely strand-specific clustering is detected (bottom). This clustering likely represents the general susceptibility of the corresponding genomic region to the ssDNA-specific mutagen. (D) Example of clustering of intermediate power (compare with scheme on the Panel B). This cluster is found on chromosome X of yeast mutant clone induced by PmCDA1 deaminase [57]. Two clusters can be defined based on the algorithm parameters. Dark (blue) rectangles, heterozygous C >T substitutions, which result from deamination of cytosine in the top DNA strand; lighter (red) rectangles, heterozygous G>A substitutions, which result from deamination of cytosines in the bottom DNA strand. Genomic features, as well as chromosomal coordinates, are shown on top. (E) Example of cluster detected in silico by combining mutational data from independent yeast mutant clones induced by PmCDA1 deaminase. Each individual mutant possesses only a single SNV in this genomic region. However, merging data from several clones reveals a region of susceptibility to the mutagen. Color code and labels are as in the Panel D.
In other cases where mutation numbers are low and/or their densities are either low or high, both the determination of whether clustering is present and the definition of cluster borders require formal mathematical approaches. For example, in a recent study [57], clusters associated with ssDNA vulnerable during transcription initiation have been found. For the most sensitive promoters, every genome contains strong and clear mutational clustering (Figure 6B and D), whereas for the more weakly expressed and better protected genes, clusters can be detected only when combined mutational data sets have been studied. In this case, clusters are defined not in a genetic but rather in a functional way, and depict the profiles of genomic ssDNA vulnerability to specific mutagens (Figure 6C and E).
Large-scale DNA rearrangements, gene expression data and DNA methylation
A variety of large-scale recombination events (duplications, deletions, translocations and inversions) are a characteristic feature of many cancers [112–114]. Some of these events are recurrent and are considered to be signatures of specific cancer subtypes [112, 113]. One well-known example is the BCL2 gene that is involved in translocation with immunoglobulin genes. This translocation is a characteristic feature of follicular lymphoma [115]. Previously, we found a signature of pol η (WA/TW, W = A/T) in follicular lymphoma, which was significant in 5′ untranslated region (UTR) regions (P-value = 0.01) [32]. However, a detailed analysis of pol η mutability suggested that a substantial fraction (24%) of mutated 5′ UTR WA/TW motifs occurred within the BCL2 gene (19 of 28 mutations at A:T bases). After we removed somatic mutations that were identified within the BCL2 5′ UTR region (near the translocation breakpoint), the correlation became insignificant (P-value = 0.11, 60 mutations in WA/TW motifs of 116 mutations at A:T bases) [32]. This is an example of how a single translocation event is able to bias the results of the whole-exome analysis; therefore, such events cannot be ignored.
The expression of genes potentially associated with mutable motifs is also used as an additional feature to delineate proteins involved in mutagenesis as we discussed in the chapter ‘Examples of cancer studies’. However, these data are not always a useful source of information. For example, no correlation between AID mutagenesis and RNA sequencing (RNAseq) expression of AID was found [32]. There have been numerous attempts to use expression data for the analysis of cancer. For example, microarrays have revolutionized breast cancer research by generating various cancer diagnostic and prognostic signatures. Clinically, breast cancer is a highly heterogeneous disease, and gene expression profiling has potentiated the subclassification of tumors into five distinct ‘intrinsic’ subclasses (luminal A, luminal B, ERBB2, basal and normal-like), thereby helping to explain why patients with histologically similar tumors often show different outcomes and responses to therapy [116–119, 120]. Currently, several breast cancer prognostic assays are on the market based on microarray and reverse transcription polymerase chain reaction technologies (Oncotype DX™, MammaPrint®, the H/I ratio test) and their clinical validity and utility extensively studied [121]. MammaPrint®, the first prognostic microarray-based test, received its original FDA approval in 2007 and additional approval for testing in fixed tissues in 2015. Multiple additional expression-based classifiers have been developed [122, 123], and the PAM50 classifier having been translated into a clinical assay (Prosigna™) [124]. Recently, the Personalized Regimen Selection strategy (uses both genetic and clinical variables) was shown to significantly increase response rates for breast cancer patients, especially those with HER2- and ER-negative tumors [125]. In addition to PCR- and microarray-based techniques, the utility of RNAseq-based methods for a variety of breast cancer signatures was demonstrated [121].
Epigenetic modifications, including DNA methylation, play an important role in many gene regulatory processes. Methylation involves two nucleotides, cytosine and adenine, and in humans, it is predominantly found as 5-methylcytosine (5mC) in CpG dinucleotides. CpG constitutes a mutation hotspot in the human genome, both in the germ line and in the soma. This is because of methylation-mediated deamination of 5mC: while cytosine spontaneously deaminates to uracil (which is efficiently recognized as a non-DNA base and removed by uracil-DNA glycosylase), the spontaneous deamination of 5mC yields thymine, thereby creating G•T mismatches whose removal by methyl-CpG-binding domain protein 4 and/or thymine DNA glycosylase followed by base excision repair is inherently less efficient [126]. Recently developed high-throughput techniques such as bisulfite sequencing and DNA methylation arrays have provided data on the methylation status of individual cytosines; these data are deposited in international consortia such as ICGC and TCGA. The patterns of DNA methylation may change as the cell grows and differentiates, and aberrant DNA methylation patterns have been observed in many cancers [127]. In normal tissues, promoter-associated CpG islands remain unmethylated, whereas hundreds of CpG islands in tumors acquire DNA methylation. In the late 1990s, the CpG island methylator phenotype was identified in colorectal cancer [128]. Later, studies have shown that there could be subgroups with similar methylomes even within one cancer type; thus, four different subgroups have been identified in colorectal cancer [129]. At the same time, certain similarities have been detected between the methylomes of different cancer types. In this respect, colorectal, gastric and endometrial cancers have been found to belong to the highly methylated subgroup that is associated with tumors with microsatellite instability and hypermethylation of the MLH1 promoter [130], whereas solid human epithelial tumors and cancer cell lines revealed commonalities and tissue-specific features of the CpG island methylator phenotype [131].
Cancer driver and passenger mutations
A driver is a mutation that directly or indirectly confers a selective advantage on the cell in which it occurs, while a passenger is a mutation that exerts no selective growth advantage on the cell in which it occurs [132]. There is a subtle difference between a driver gene and a driver gene mutation: a driver gene harbors driver gene mutations but may also harbor passenger gene mutations. A driver mutation typically confers on a tumor only a small growth advantage, which may be as low as a 0.4% increase in the difference between cell birth and death rates [133]. More recently, Bozic et al. [134] have shown that the first, and hence most abundant, passenger mutations are influenced by both the mutation rate and by the death–birth ratio of the cancer cells.
It should be appreciated that whereas passenger mutations cannot by definition exert a selective growth advantage, they are not necessarily neutral. Indeed, many are deleterious in terms of their effect on cellular proliferation and cancer progression [135, 136]. It should also be appreciated that while the damaging effect of a nonsynonymous passenger mutation is of the order of 100 times smaller than the effect of a driver mutation, passengers are 100 times more numerous than drivers [136]. The paucity of drivers in a sea of passenger mutations represents a challenge to identify the former [137]. This task is made all the more daunting by the possibility that drivers and passengers are not discrete entities but rather lie along a continuum, which includes latent driver mutations, which ‘behave as passengers but….coupled with other emerging mutations, drive cancer development and drug resistance’ [138]. For most types of cancer, the genomic landscape comprises a small number of ‘mountains’ (genes altered in a high percentage of tumors) and a much larger number of ‘hills’ (genes that are altered much less frequently; see [139]).
Recently, it was suggested that only a small number of driver mutations are required for progression of normal tissues into tumor [72]. A high proportion of cancer driver events occur in noncoding regions, and a similarly large fraction affects protein-coding regions. Possible molecular mechanisms of mutation occurrence at the DNA level have been described in previous sections, whereas the effects of cancer missense mutations on proteins have been reliably established only in a few cases. Establishing such effects on protein activity, stability, dynamics and binding would certainly facilitate our understanding of driver events in cancer. Several distinct properties are characteristic of cancer-associated proteins: tumor suppressors in cancer frequently harbor destabilizing mutations that preferably occur within the core of the protein; the enhanced activity of oncogenes is often linked with mutations at functional sites [140]; cancer mutations cluster in three-dimensional space [141, 142] in both oncogenes and tumor suppressors [141]; cancer missense mutations largely affect protein-binding interfaces [143–145]; and the transforming effect of mutations is directly proportional to their frequency in cancer samples [146, 147].
Different in silico approaches have been developed that aim to detect driver genes or sites that acquire significantly more mutations than expected from the background mutational models. An unbiased testing and comparison of these methods is an issue because methods are trained on all available experimental data sets of cancer mutations and their transforming effects, and such data sets are scarce [148]. There are several methods that can distinguish cancer-associated mutations from neutral polymorphisms, but there is no existing method that can accurately distinguish driver mutations from passenger mutations.
In general, the somatic evolution of cancers is expected to be characterized by weak purifying selection in most genes and substantial positive selection in some ‘cancer’ genes that are likely to contain driver mutations [149, 150]. The latter possibility is of particular interest because the positive selection of somatic mutations in cancers flags up that the change in function of the respective genes is relevant for tumorigenesis, leading to the recognition of previously undetected oncogenes and other genes associated with cancer. We shall discuss this in more detail in the next chapter.
There have been numerous attempts to build a census of human cancer genes [149, 151, 152]. Back in 2004, Futreal et al. [151] published a ‘Census of human cancer genes’, which aimed to list all genes that are causally implicated in tumorigenesis. This Census has been kept up to date and currently includes 602 entries (http://cancer.sanger.ac.uk/census/). This implies that >2% of all human genes are implicated via mutation in cancer. Of these, ∼90% have somatic mutations in cancer, 20% have germ line mutations that predispose to cancer and 10% harbor both somatic and germ line mutations. A second resource, the Network of Cancer Genes (http://ncg.kcl.ac.uk/), contains 1053 ‘cancer genes’ whose possible involvement in cancer has been inferred by statistical means. An important direction for this avenue of research has been the development of predictive models for cancer-associated genes that could accelerate their identification, although ubiquitously overexpressed genes could be marked as nonspecific cancer-associated genes when delineating genes that are specific to certain types of cancer [153]. The number of genes recognized as being cancer-associated is likely to increase as new techniques are devised to search for them [154, 155].
One important direction of research lies with attempts to identify the underlying mechanisms of driver mutation generation. For example, analysis of the APOBEC3A/B signature associated with driver mutations suggested that APOBEC signature mutations themselves contribute to carcinogenesis in samples with a strong mutation pattern associated with ABOBEC3A/B [40]. Furthermore, many of the APOBEC3A/B signature mutations that are likely to be driver mutations occurred in genes that are highly mutated in various databases and are also present in the Census of human cancer genes [40]. In lung cancer, despite sustained carcinogen exposure, subclonal mutations showed a relatively lower burden of smoking-related mutations, accompanied by an increase in APOBEC-associated mutations, suggesting that mutagenic processes also evolve over the course of tumor development and that APOBEC-mediated mutagenic processes play a role in subclonal genetic heterogeneity in some tumors [156].
Selectionist and neutralist models of evolution in cancer
There is a widely held presumption that subclone dynamics in human cancers are dominated by strong selection, but this may not be invariably true. Thus, for example, Williams et al. [157] found that subclonal mutant allele frequencies of 323 of 904 cancers of 14 types followed a simple power-law distribution predicted by neutral growth. As the tumor grows, a large number of cell lineages are formed, and intratumoral heterogeneity increases, while the allele frequency of the new heterogeneous mutations rapidly decreases because of expansion. Thus, after malignant transformation, individual subclones with distinct mutational patterns grow at similar rates, coexisting with one another within the tumor for long periods of time, as a consequence of the lack of stringent selection. In malignancies identified as evolving neutrally, all clonal selection appears to have occurred before the onset of cancer growth rather than in later-arising subclones, resulting in numerous passenger mutations that account for the intratumoral heterogeneity.
These data concur with the ‘big bang’ model of cancer growth of Sottoriva et al. [158]. This model of colorectal cancer growth envisages tumors growing predominantly as a single expansion producing numerous intermixed subclones that are not subject to stringent selection. On the assumption of a neutralist model of tumor growth, cancer sequencing data can be used to measure, in each individual, both the in vivo mutation rate and the order and timing of mutations. Uchi et al. [159] noted that known driver mutations were observed frequently among early acquired mutations in colorectal cancer but rarely among the late-acquired mutations. Little evidence was found to support the view that selection had shaped the intratumoral heterogeneity, which was much more likely to have been generated by neutral evolution. The extremely high level of genetic diversity evident in a single hepatocellular carcinoma (>100 million coding region mutations estimated in the entire tumor) has provided further evidence for the occurrence of ‘non-Darwinian cell evolution’ in cancer [160]. A total of 286 regions of this tumor were sequenced, and the lack of any evidence for selection was consistent with a model of big bang-like growth.
Gao et al. [161] investigated copy number evolution in patients with triple-negative breast cancer. They sequenced 1000 single cells from tumors in 12 patients and identified one to three major clonal subpopulations in each tumor that shared a common evolutionary lineage. For each tumor, these authors also identified a minor subpopulation of nonclonal cells. Phylogenetic analysis and mathematical modeling showed that these data were hard to explain by the gradual accumulation of copy number events. These data therefore challenge the paradigm of gradual evolution, showing that the majority of copy number aberrations were acquired at the earliest stages of tumor evolution, in short punctuated bursts, followed by stable clonal expansions to form the tumor mass. Thus, at least in some cases, a saltationist model of evolution may be relevant to cancer [162]. A compromise between a gradualist model and salutatory evolution may well be found in the application of punctuated equilibrium to cancer evolution—periods of stasis punctuated by sudden and dramatic changes [163].
We may conclude that natural selection would not be expected to bias/change mutational patterns to a large extent, and such effects are anticipated to be negligible. Periods of sudden and dramatic changes [163] might be associated with bursts of mutations introduced by error-prone mutational mechanisms, processes reflected in mutational signatures and by mutable motifs; this is likely to be a promising avenue for future research.
Concluding remarks
There are numerous examples of the successful application of mutational signatures and mutable motifs to studies of molecular mechanisms of mutagenesis. The level of success achieved in the context of the AID/APOBEC protein family certainly supports the notion that mutable motifs and mutational signatures are useful tools with which to study molecular mechanisms of mutations in cancer. Such an approach is likely to be helpful in understanding the biology, initiation and progression of human cancers. One potential caveat, however, is how to distinguish artifactual DNA lesions from the bona fide somatic mutations that occurred in the tumor [75]. This important issue requires further investigation.
There is evidence for the presence of circulating tumor DNA (ctDNA) in early cancers [164, 165]. However, the fraction of tumors that shed detectable levels of ctDNA, by tumor stage and type, is not known [165]. The studies to date have small numbers of samples and use a variety of measurement techniques that are often not comparable [164, 165]. In general, the potential for mutational signatures and mutable motifs to provide new cancer biomarkers or drug targets is unclear. For example, AID-related WRC/GYW and WRCG/CGYW mutable motifs for 22 individual follicular lymphoma patient exomes were analyzed (Supplementary Table S3 from Rogozin et al. [32]). A significant excess of mutations in both motifs was found for 13 patients [32]. This finding suggests that the mutational processes associated with AID are active in follicular lymphoma to an extent detectable with sensitive statistical tests in samples with limited numbers of mutations; however, the sensitivity of this test is not high. This notwithstanding, in combination with other mutational signatures, methylation patterns and other biomarkers, this approach may have some value.
There is much room for improvement in our ascertainment of mutable motifs and mutational signatures. Theoretically, various computational approaches can be used to analyze aligned sequences of mutation hotspots. Many techniques have been developed for the analysis of functional signals including information content, weight matrices, perceptron, k-tuple frequencies, discriminant analysis, hidden Markov models, linguistic approaches and neural network models. These methods are well established and have been tested on different types of data, but all of these methods require large data sets.
It should be noted that the analysis of mutations is rather a classification problem than discriminant analysis (commonly used in bioinformatics, e.g. analysis of splicing signals) with well-defined training (learning) and control (test) sets. This necessarily imposes certain restrictions on the interpretation of results, and conclusions should still be regarded as hypotheses/observations rather than proven facts; in many cases, such conclusions will require further experimental validation. However, all attempts to assign mutational signatures to known human carcinogenic exposures or endogenous mechanisms of mutagenesis [79] should still be appreciated for what they are: the first tentative attempts to found a vital new branch of enquiry in cancer genomics. Such studies will certainly add significantly to our knowledge of mutagenesis in human cancers.
Key Points
Cancer genomes are highly enriched with mutations of different kinds.
The DNA sequence context and distribution of mutations represent the signatures of mutational processes that can be deconvoluted into individual components.
These mutational signatures, supplemented by mutable motifs (a wider descriptor of mutation context), represent the footprints of interactions between DNA, mutagens and the enzymes of the repair/replication/modification pathways.
It has become clear that it is possible to acquire an understanding of the underlying mutational mechanisms in cancer by indirectly analyzing DNA sequences of whole genomes of tumor cells.
Funding
The Intramural Research Program of the National Library of Medicine at the National Institutes of Health (to I.B.R., A.G. and A.R.P.); the National Institutes of Health Intramural Research Program of the National Eye Institute (to E.P.); Boettcher Foundation, American Cancer Society, P30 CA072720 (to S.D.); Nebraska Department of Health and Human Services LB506, grant 2017-48 (to Y.I.P.); and Qiagen Inc through a License Agreement with Cardiff University (to D.N.C.).
Biographies
Igor B. Rogozin is Staff Scientist at the National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, USA. He has been Adjunct Lecturer at the Foundation for Advanced Education in Science, USA, the Johns Hopkins University, USA and the Novosibirsk State University, Russia and Senior Researcher at the Institute of Cytology and Genetics, Novosibirsk, Russia.
Youri I. Pavlov is Laboratory Head and Professor of Genetics at the Eppley Institute for Cancer Research, University of Nebraska Medical Center, USA.
Alexander Goncearenco is Research Fellow at the National Center for Biotechnology Information, National Institutes of Health, USA.
Subhajyoti De is Assistant Professor of Pathology Medical Informatics (Systems Biology) at Rutgers University Cancer Institute, USA.
Artem G. Lada is Postdoctoral Fellow at the Department Microbiology and Molecular Genetics, University of California, Davis, USA.
Eugenia Poliakov is Staff Scientist at the Laboratory of Retinal Cell and Molecular Biology, National Eye Institute, National Institutes of Health, USA.
Anna R. Panchenko is Lead Scientist, Head of Computational Biophysics Group at the National Center for Biotechnology Information, National Institutes of Health, USA.
David N. Cooper is Professor of Human Molecular Genetics at the Institute of Medical Genetics, Cardiff University, UK.
References
- 1. Neuberger MS, Harris RS, Di Noia J, et al.Immunity through DNA deamination. Trends Biochem Sci 2003;28:305–12. [DOI] [PubMed] [Google Scholar]
- 2. Zanotti KJ, Gearhart PJ.. Antibody diversification caused by disrupted mismatch repair and promiscuous DNA polymerases. DNA Repair 2016;38:110–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Matsumoto Y, Marusawa H, Kinoshita K, et al.Helicobacter pylori infection triggers aberrant expression of activation-induced cytidine deaminase in gastric epithelium. Nat Med 2007;13:470–6. [DOI] [PubMed] [Google Scholar]
- 4. Roberts SA, Gordenin DA.. Hypermutation in human cancer genomes: footprints and mechanisms. Nat Rev Cancer 2014;14:786–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Prolla TA. DNA mismatch repair and cancer. Curr Opin Cell Biol 1998;10:311–16. [DOI] [PubMed] [Google Scholar]
- 6. Beckman RA, Loeb LA.. Genetic instability in cancer: theory and experiment. Semin Cancer Biol 2005;15:423–35. [DOI] [PubMed] [Google Scholar]
- 7. Rayner E, van Gool IC, Palles C, et al.A panoply of errors: polymerase proofreading domain mutations in cancer. Nat Rev Cancer 2016;16:71–81. [DOI] [PubMed] [Google Scholar]
- 8. de Bono JS, Ashworth A.. Translating cancer research into targeted therapeutics. Nature 2010;467:543–9. [DOI] [PubMed] [Google Scholar]
- 9. Deng X, Nakamura Y.. Cancer precision medicine: from cancer screening to drug selection and personalized immunotherapy. Trends Pharmacol Sci 2017;38:15–24. [DOI] [PubMed] [Google Scholar]
- 10. Hanahan D, Weinberg RA.. The hallmarks of cancer. Cell 2000;100:57–70. [DOI] [PubMed] [Google Scholar]
- 11. Waddell N, Pajic M, Patch AM, et al.Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015;518:495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Watson IR, Takahashi K, Futreal PA, et al.Emerging patterns of somatic mutations in cancer. Nat Rev Genet 2013;14:703–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Salk JJ, Fox EJ, Loeb LA.. Mutational heterogeneity in human cancers: origin and consequences. Annu Rev Pathol 2010;5:51–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Alexandrov LB, Nik-Zainal S, Wedge DC, et al.Signatures of mutational processes in human cancer. Nature 2013;500:415–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Loeb LA. Human cancers express a mutator phenotype: hypothesis, origin, and consequences. Cancer Res 2016;76:2057–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Alexandrov LB, Nik-Zainal S, Wedge DC, et al.Deciphering signatures of mutational processes operative in human cancer. Cell Rep 2013;3:246–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kunkel TA. Considering the cancer consequences of altered DNA polymerase function. Cancer Cell 2003;3:105–10. [DOI] [PubMed] [Google Scholar]
- 18. Lange SS, Takata K, Wood RD.. DNA polymerases and cancer. Nat Rev Cancer 2011;11:96–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Preston BD, Albertson TM, Herr AJ.. DNA replication fidelity and cancer. Semin Cancer Biol 2010;20:281–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shlien A, Campbell BB, de Borja R, et al.Combined hereditary and somatic mutations of replication error repair genes result in rapid onset of ultra-hypermutated cancers. Nat Genet 2015;47:257–62. [DOI] [PubMed] [Google Scholar]
- 21. Waisertreiger IS, Liston VG, Menezes MR, et al.Modulation of mutagenesis in eukaryotes by DNA replication fork dynamics and quality of nucleotide pools. Environ Mol Mutagen 2012;53:699–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Roberts SA, Gordenin DA.. Clustered and genome-wide transient mutagenesis in human cancers: hypermutation without permanent mutators or loss of fitness. Bioessays 2014;23:745–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fishel R, Kolodner RD.. Identification of mismatch repair genes and their role in the development of cancer. Curr Opin Genet Dev 1995;5:382–95. [DOI] [PubMed] [Google Scholar]
- 24. Scully R. Role of BRCA gene dysfunction in breast and ovarian cancer predisposition. Breast Cancer Res 2000;2:324–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Masutani C, Kusumoto R, Yamada A, et al.The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase eta. Nature 1999;399:700–4. [DOI] [PubMed] [Google Scholar]
- 26. Wood LD, Hruban RH.. Genomic landscapes of pancreatic neoplasia. J Pathol Transl Med 2015;49:13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rogozin IB, Pavlov YI, Bebenek K, et al.Somatic mutation hotspots correlate with DNA polymerase eta error spectrum. Nat Immunol 2001;2:530–6. [DOI] [PubMed] [Google Scholar]
- 28. Jackson AL, Loeb LA.. The mutation rate and cancer. Genetics 1998;148:1483–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Loeb LA, Springgate CF, Battula N.. Errors in DNA replication as a basis of malignant changes. Cancer Res 1974;34:2311–21. [PubMed] [Google Scholar]
- 30. Rogozin IB, Kolchanov NA.. Somatic hypermutagenesis in immunoglobulin genes. II. Influence of neighbouring base sequences on mutagenesis. Biochim Biophys Acta 1992;1171:11–18. [DOI] [PubMed] [Google Scholar]
- 31. Alexandrov LB, Stratton MR.. Mutational signatures: the patterns of somatic mutations hidden in cancer genomes. Curr Opin Genet Dev 2014;24:52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rogozin IB, Lada AG, Goncearenco A, et al.Activation induced deaminase mutational signature overlaps with CpG methylation sites in follicular lymphoma and other cancers. Sci Rep 2016;6:38133.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bachl J, Steinberg C, Wabl M.. Critical test of hot spot motifs for immunoglobulin hypermutation. Eur J Immunol 1997;27:3398–403. [DOI] [PubMed] [Google Scholar]
- 34. Rogozin IB, Sredneva NE, Kolchanov NA.. Somatic hypermutagenesis in immunoglobulin genes. III. Somatic mutations in the chicken light chain locus. Biochim Biophys Acta 1996;1306:171–8. [DOI] [PubMed] [Google Scholar]
- 35. KewalRamani VN, Coffin JM.. Virology. Weapons of mutational destruction. Science 2003;301:923–5. [DOI] [PubMed] [Google Scholar]
- 36. Lu Z, Tsai AG, Akasaka T, et al.BCL6 breaks occur at different AID sequence motifs in Ig-BCL6 and non-Ig-BCL6 rearrangements. Blood 2013;121:4551–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Burns MB, Lackey L, Carpenter MA, et al.APOBEC3B is an enzymatic source of mutation in breast cancer. Nature 2013;494:366–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chan K, Roberts SA, Klimczak LJ, et al.An APOBEC3A hypermutation signature is distinguishable from the signature of background mutagenesis by APOBEC3B in human cancers. Nat Genet 2015;47:1067–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nik-Zainal S, Wedge DC, Alexandrov LB, et al.Association of a germline copy number polymorphism of APOBEC3A and APOBEC3B with burden of putative APOBEC-dependent mutations in breast cancer. Nat Genet 2014;46:487–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Roberts SA, Lawrence MS, Klimczak LJ, et al.An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat Genet 2013;45:970–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Alexandrov LB, Ju YS, Haase K, et al.Mutational signatures associated with tobacco smoking in human cancer. Science 2016;354:618–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Brash DE. UV signature mutations. Photochem Photobiol 2015;91:15–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Behjati S, Gundem G, Wedge DC, et al.Mutational signatures of ionizing radiation in second malignancies. Nat Commun 2016;7:12605.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tsuzuki T, Egashira A, Igarashi H, et al.Spontaneous tumorigenesis in mice defective in the MTH1 gene encoding 8-oxo-dGTPase. Proc Natl Acad Sci USA 2001;98:11456–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rebhandl S, Huemer M, Greil R, et al.AID/APOBEC deaminases and cancer. Oncoscience 2015;2:320–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mertz TM, Sharma S, Chabes A, et al.Colon cancer-associated mutator DNA polymerase delta variant causes expansion of dNTP pools increasing its own infidelity. Proc Natl Acad Sci USA 2015;112:E2467–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Middlebrooks CD, Banday AR, Matsuda K, et al.Association of germline variants in the APOBEC3 region with cancer risk and enrichment with APOBEC-signature mutations in tumors. Nat Genet 2016;48:1330–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gargiulo P, Della Pepa C, Berardi S, et al.Tumor genotype and immune microenvironment in POLE-ultramutated and MSI-hypermutated endometrial cancers: new candidates for checkpoint blockade immunotherapy?. Cancer Treat Rev 2016;48:61–8. [DOI] [PubMed] [Google Scholar]
- 49. Shcherbakova PV, Pavlov YI, Chilkova O, et al.Unique error signature of the four-subunit yeast DNA polymerase epsilon. J Biol Chem 2003;278:43770–80. [DOI] [PubMed] [Google Scholar]
- 50. Beale RC, Petersen-Mahrt SK, Watt IN, et al.Comparison of the differential context-dependence of DNA deamination by APOBEC enzymes: correlation with mutation spectra in vivo. J Mol Biol 2004;337:585–96. [DOI] [PubMed] [Google Scholar]
- 51. Petersen-Mahrt SK, Harris RS, Neuberger MS.. AID mutates E. coli suggesting a DNA deamination mechanism for antibody diversification. Nature 2002;418:99–103. [DOI] [PubMed] [Google Scholar]
- 52. Roberts SA, Sterling J, Thompson C, et al.Clustered mutations in yeast and in human cancers can arise from damaged long single-strand DNA regions. Mol Cell 2012;46:424–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Daee DL, Mertz TM, Shcherbakova PV.. A cancer-associated DNA polymerase delta variant modeled in yeast causes a catastrophic increase in genomic instability. Proc Natl Acad Sci USA 2010;107:157–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lada AG, Dhar A, Boissy RJ, et al.AID/APOBEC cytosine deaminase induces genome-wide kataegis. Biol Direct 2012;7:47; discussion 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lada AG, Stepchenkova EI, Waisertreiger IS, et al.Genome-wide mutation avalanches induced in diploid yeast cells by a base analog or an APOBEC deaminase. PLoS Genet 2013;9:e1003736.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Taylor BJ, Nik-Zainal S, Wu YL, et al.DNA deaminases induce break-associated mutation showers with implication of APOBEC3B and 3A in breast cancer kataegis. Elife 2013;2:e00534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lada AG, Kliver SF, Dhar A, et al.Disruption of transcriptional coactivator Sub1 leads to genome-wide re-distribution of clustered mutations induced by APOBEC in active yeast genes. PLoS Genet 2015;11:e1005217.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lada AG, Krick CF, Kozmin SG, et al.Mutator effects and mutation signatures of editing deaminases produced in bacteria and yeast. Biochemistry 2011;76:131–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. De S. Somatic mosaicism in healthy human tissues. Trends Genet 2011;27:217–23. [DOI] [PubMed] [Google Scholar]
- 60. Martincorena I, Campbell PJ.. Somatic mutation in cancer and normal cells. Science 2015;349:1483–9. [DOI] [PubMed] [Google Scholar]
- 61. Saini N, Roberts SA, Klimczak LJ, et al.The impact of environmental and endogenous damage on somatic mutation load in human skin fibroblasts. PLoS Genet 2016;12:e1006385.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kadara H, Wistuba II.. Field cancerization in non-small cell lung cancer: implications in disease pathogenesis. Proc Am Thorac Soc 2012;9:38–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yadav VK, DeGregori J, De S.. The landscape of somatic mutations in protein coding genes in apparently benign human tissues carries signatures of relaxed purifying selection. Nucleic Acids Res 2016;44:2075–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Genovese G, Kahler AK, Handsaker RE, et al.Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 2014;371:2477–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Holstege H, Pfeiffer W, Sie D, et al.Somatic mutations found in the healthy blood compartment of a 115-yr-old woman demonstrate oligoclonal hematopoiesis. Genome Res 2014;24:733–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Blokzijl F, de Ligt J, Jager M, et al.Tissue-specific mutation accumulation in human adult stem cells during life. Nature 2016;538:260–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Aghili L, Foo J, DeGregori J, et al.Patterns of somatically acquired amplifications and deletions in apparently normal tissues of ovarian cancer patients. Cell Rep 2014;7:1310–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Alexandrov LB, Jones PH, Wedge DC, et al.Clock-like mutational processes in human somatic cells. Nat Genet 2015;47:1402–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hoang ML, Kinde I, Tomasetti C, et al.Genome-wide quantification of rare somatic mutations in normal human tissues using massively parallel sequencing. Proc Natl Acad Sci USA 2016;113:9846–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Milholland B, Auton A, Suh Y, et al.Age-related somatic mutations in the cancer genome. Oncotarget 2015;6:24627–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Tomasetti C, Vogelstein B, Parmigiani G.. Half or more of the somatic mutations in cancers of self-renewing tissues originate prior to tumor initiation. Proc Natl Acad Sci USA 2013;110:1999–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Tomasetti C, Marchionni L, Nowak MA, et al.Only three driver gene mutations are required for the development of lung and colorectal cancers. Proc Natl Acad Sci USA 2015;112:118–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Martincorena I, Roshan A, Gerstung M, et al.Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 2015;348:880–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Jaiswal S, Fontanillas P, Flannick J, et al.Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 2014;371:2488–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Chen L, Liu P, Evans TC Jr, et al.DNA damage is a pervasive cause of sequencing errors, directly confounding variant identification. Science 2017;355:752–6. [DOI] [PubMed] [Google Scholar]
- 76. Benzer S. From the gene to behavior. JAMA 1971;218:1015–22. [PubMed] [Google Scholar]
- 77. Cooper DN, Youssoufian H.. The CpG dinucleotide and human genetic disease. Hum Genet 1988;78:151–5. [DOI] [PubMed] [Google Scholar]
- 78. Coulondre C, Miller JH, Farabaugh PJ, et al.Molecular basis of base substitution hotspots in Escherichia coli. Nature 1978;274:775–80. [DOI] [PubMed] [Google Scholar]
- 79. Hollstein M, Alexandrov LB, Wild CP, et al.Base changes in tumour DNA have the power to reveal the causes and evolution of cancer. Oncogene 2017;36:158–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Horsfall MJ, Gordon AJ, Burns PA, et al.Mutational specificity of alkylating agents and the influence of DNA repair. Environ Mol Mutagen 1990;15:107–22. [DOI] [PubMed] [Google Scholar]
- 81. Krawczak M, Ball EV, Cooper DN.. Neighboring-nucleotide effects on the rates of germ-line single-base-pair substitution in human genes. Am J Hum Genet 1998;63:474–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Krawczak M, Smith-Sorensen B, Schmidtke J, et al.Somatic spectrum of cancer-associated single basepair substitutions in the TP53 gene is determined mainly by endogenous mechanisms of mutation and by selection. Hum Mutat 1995;5:48–57. [DOI] [PubMed] [Google Scholar]
- 83. Rogozin IB, Babenko VN, Milanesi L, et al.Computational analysis of mutation spectra. Brief Bioinform 2003;4:210–27. [DOI] [PubMed] [Google Scholar]
- 84. Rogozin IB, Pavlov YI.. Theoretical analysis of mutation hotspots and their DNA sequence context specificity. Mutat Res 2003;544:65–85. [DOI] [PubMed] [Google Scholar]
- 85. Zavolan M, Kepler TB.. Statistical inference of sequence-dependent mutation rates. Curr Opin Genet Dev 2001;11:612–15. [DOI] [PubMed] [Google Scholar]
- 86. Matsuda T, Bebenek K, Masutani C, et al.Error rate and specificity of human and murine DNA polymerase eta. J Mol Biol 2001;312:335–46. [DOI] [PubMed] [Google Scholar]
- 87. Kondrashov AS, Rogozin IB.. Context of deletions and insertions in human coding sequences. Hum Mutat 2004;23:177–85. [DOI] [PubMed] [Google Scholar]
- 88. Pham P, Bransteitter R, Petruska J, et al.Processive AID-catalysed cytosine deamination on single-stranded DNA simulates somatic hypermutation. Nature 2003;424:103–7. [DOI] [PubMed] [Google Scholar]
- 89. Topal MD, Eadie JS, Conrad M.. O6-methylguanine mutation and repair is nonuniform. Selection for DNA most interactive with O6-methylguanine. J Biol Chem 1986;261:9879–85. [PubMed] [Google Scholar]
- 90. Day WH, McMorris FR.. Critical comparison of consensus methods for molecular sequences. Nucleic Acids Res 1992;20:1093–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Day WH, McMorris FR.. Threshold consensus methods for molecular sequences. J Theor Biol 1992;159:481–9. [DOI] [PubMed] [Google Scholar]
- 92. Malyarchuk BA, Rogozin IB, Berikov VB, et al.Analysis of phylogenetically reconstructed mutational spectra in human mitochondrial DNA control region. Hum Genet 2002;111:46–53. [DOI] [PubMed] [Google Scholar]
- 93. Stormo GD, Schneider TD, Gold L.. Quantitative analysis of the relationship between nucleotide sequence and functional activity. Nucleic Acids Res 1986;14:6661–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Berikov VB, Rogozin IB.. Regression trees for analysis of mutational spectra in nucleotide sequences. Bioinformatics 1999;15:553–62. [DOI] [PubMed] [Google Scholar]
- 95. Brunet JP, Tamayo P, Golub TR, et al.Metagenes and molecular pattern discovery using matrix factorization. Proc Natl Acad Sci USA 2004;101:4164–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Tan VY, Fevotte C.. Automatic relevance determination in nonnegative matrix factorization with the beta-divergence. IEEE Trans Pattern Anal Mach Intell 2013;35:1592–605. [DOI] [PubMed] [Google Scholar]
- 97. Lee DD, Seung HS.. Learning the parts of objects by non-negative matrix factorization. Nature 1999;401:788–91. [DOI] [PubMed] [Google Scholar]
- 98. Temiz NA, Donohue DE, Bacolla A, et al.The somatic autosomal mutation matrix in cancer genomes. Hum Genet 2015;134:851–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Gehring JS, Fischer B, Lawrence M, et al.SomaticSignatures: inferring mutational signatures from single-nucleotide variants. Bioinformatics 2015;31:3673–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Rosenthal R, McGranahan N, Herrero J, et al.DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol 2016;17:31.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Ardin M, Cahais V, Castells X, et al.MutSpec: a galaxy toolbox for streamlined analyses of somatic mutation spectra in human and mouse cancer genomes. BMC Bioinformatics 2016;17:170.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Goncearenco A, Rager S, Li M, et al.MutaGene: exploring background mutational processes in cancer and linking them to protein phenotype. Nucliec Acid Res 2017, 10.1093/nar/gkx367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Burns MB, Temiz NA, Harris RS.. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat Genet 2013;45:977–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Smith KS, Yadav VK, Pedersen BS, et al.Signatures of accelerated somatic evolution in gene promoters in multiple cancer types. Nucleic Acids Res 2015;43:5307–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Chan K, Gordenin DA.. Clusters of multiple mutations: incidence and molecular mechanisms. Annu Rev Genet 2015;49:243–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Taylor BJ, Wu YL, Rada C.. Active RNAP pre-initiation sites are highly mutated by cytidine deaminases in yeast, with AID targeting small RNA genes. Elife 2014;3:e03553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Gearhart PJ, Bogenhagen DF.. Clusters of point mutations are found exclusively around rearranged antibody variable genes. Proc Natl Acad Sci USA 1983;80:3439–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Morozov P, Sitnikova T, Churchill G, et al.A new method for characterizing replacement rate variation in molecular sequences. Application of the Fourier and wavelet models to Drosophila and mammalian proteins. Genetics 2000;154:381–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Tang H, Lewontin RC.. Locating regions of differential variability in DNA and protein sequences. Genetics 1999;153:485–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Nik-Zainal S, Alexandrov LB, Wedge DC, et al.Mutational processes molding the genomes of 21 breast cancers. Cell 2012;149:979–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Sakofsky CJ, Roberts SA, Malc E, et al.Break-induced replication is a source of mutation clusters underlying kataegis. Cell Rep 2014;7:1640–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Bacolla A, Jaworski A, Larson JE, et al.Breakpoints of gross deletions coincide with non-B DNA conformations. Proc Natl Acad Sci USA 2004;101:14162–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Bacolla A, Tainer JA, Vasquez KM, et al.Translocation and deletion breakpoints in cancer genomes are associated with potential non-B DNA-forming sequences. Nucleic Acids Res 2016;44:5673–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Rowley JD. Chromosome translocations: dangerous liaisons revisited. Nat Rev Cancer 2001;1:245–50. [DOI] [PubMed] [Google Scholar]
- 115. Green MR, Kihira S, Liu CL, et al.Mutations in early follicular lymphoma progenitors are associated with suppressed antigen presentation. Proc Natl Acad Sci USA 2015;112:E1116–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Sorlie T, Tibshirani R, Parker J, et al.Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci USA 2003;100:8418–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Martin KJ, Kritzman BM, Price LM, et al.Linking gene expression patterns to therapeutic groups in breast cancer. Cancer Res 2000;60:2232–8. [PubMed] [Google Scholar]
- 118. Yu K, Lee CH, Tan PH, et al.Conservation of breast cancer molecular subtypes and transcriptional patterns of tumor progression across distinct ethnic populations. Clin Cancer Res 2004;10:5508–17. [DOI] [PubMed] [Google Scholar]
- 119. Rouzier R, Perou CM, Symmans WF, et al.Breast cancer molecular subtypes respond differently to preoperative chemotherapy. Clin Cancer Res 2005;11:5678–85. [DOI] [PubMed] [Google Scholar]
- 120. Hu Z, Fan C, Oh DS, et al.The molecular portraits of breast tumors are conserved across microarray platforms. BMC Genomics 2006;7:96.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Fumagalli D, Blanchet-Cohen A, Brown D, et al.Transfer of clinically relevant gene expression signatures in breast cancer: from Affymetrix microarray to Illumina RNA-sequencing technology. BMC Genomics 2014;15:1008.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Haibe-Kains B, Desmedt C, Loi S, et al.A three-gene model to robustly identify breast cancer molecular subtypes. J Natl Cancer Inst 2012;104:311–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Sotiriou C, Wirapati P, Loi S, et al.Gene expression profiling in breast cancer: understanding the molecular basis of histologic grade to improve prognosis. J Natl Cancer Inst 2006;98:262–72. [DOI] [PubMed] [Google Scholar]
- 124. Wallden B, Storhoff J, Nielsen T, et al.Development and verification of the PAM50-based Prosigna breast cancer gene signature assay. BMC Med Genomics 2015;8:54.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Yu K, Sang QA, Lung PY, et al.Personalized chemotherapy selection for breast cancer using gene expression profiles. Sci Rep 2017;7:43294.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Cooper DN, Bacolla A, Ferec C, et al.On the sequence-directed nature of human gene mutation: the role of genomic architecture and the local DNA sequence environment in mediating gene mutations underlying human inherited disease. Hum Mutat 2011;32:1075–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Jones PA, Baylin SB.. The fundamental role of epigenetic events in cancer. Nat Rev Genet 2002;3:415–28. [DOI] [PubMed] [Google Scholar]
- 128. Toyota M, Ahuja N, Ohe-Toyota M, et al.CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci USA 1999;96:8681–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Hinoue T, Weisenberger DJ, Lange CP, et al.Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res 2012;22:271–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Witte T, Plass C, Gerhauser C.. Pan-cancer patterns of DNA methylation. Genome Med 2014;6:66.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Sanchez-Vega F, Gotea V, Margolin G, et al.Pan-cancer stratification of solid human epithelial tumors and cancer cell lines reveals commonalities and tissue-specific features of the CpG island methylator phenotype. Epigenetics Chromatin 2015;8:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Stratton MR, Campbell PJ, Futreal PA.. The cancer genome. Nature 2009;458:719–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Bozic I, Antal T, Ohtsuki H, et al.Accumulation of driver and passenger mutations during tumor progression. Proc Natl Acad Sci USA 2010;107:18545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Bozic I, Gerold JM, Nowak MA.. Quantifying clonal and subclonal passenger mutations in cancer evolution. PLoS Comput Biol 2016;12:e1004731.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. McFarland CD, Korolev KS, Kryukov GV, et al.Impact of deleterious passenger mutations on cancer progression. Proc Natl Acad Sci USA 2013;110:2910–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. McFarland CD, Mirny LA, Korolev KS.. Tug-of-war between driver and passenger mutations in cancer and other adaptive processes. Proc Natl Acad Sci USA 2014;111:15138–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Chen J, Sun M, Shen B.. Deciphering oncogenic drivers: from single genes to integrated pathways. Brief Bioinform 2015;16:413–28. [DOI] [PubMed] [Google Scholar]
- 138. Nussinov R, Tsai CJ.. ′Latent drivers′ expand the cancer mutational landscape. Curr Opin Struct Biol 2015;32:25–32. [DOI] [PubMed] [Google Scholar]
- 139. Vogelstein B, Papadopoulos N, Velculescu VE, et al.Cancer genome landscapes. Science 2013;339:1546–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Stehr H, Jang SH, Duarte JM, et al.The structural impact of cancer-associated missense mutations in oncogenes and tumor suppressors. Mol Cancer 2011;10:54.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Molina-Vila MA, Nabau-Moreto N, Tornador C, et al.Activating mutations cluster in the “molecular brake” regions of protein kinases and do not associate with conserved or catalytic residues. Hum Mutat 2014;35:318–28. [DOI] [PubMed] [Google Scholar]
- 142. Tamborero D, Gonzalez-Perez A, Lopez-Bigas N.. OncodriveCLUST: exploiting the positional clustering of somatic mutations to identify cancer genes. Bioinformatics 2013;29:2238–44. [DOI] [PubMed] [Google Scholar]
- 143. Meyer MJ, Lapcevic R, Romero AE, et al.Mutation3D: cancer gene prediction through atomic clustering of coding variants in the structural proteome. Hum Mutat 2016;37:447–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Nishi H, Tyagi M, Teng S, et al.Cancer missense mutations alter binding properties of proteins and their interaction networks. PLoS One 2013;8:e66273.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Vazquez M, Valencia A, Pons T.. Structure-PPi: a module for the annotation of cancer-related single-nucleotide variants at protein-protein interfaces. Bioinformatics 2015;31:2397–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Hashimoto K, Rogozin IB, Panchenko AR.. Oncogenic potential is related to activating effect of cancer single and double somatic mutations in receptor tyrosine kinases. Hum Mutat 2012;33:1566–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Li M, Kales SC, Ma K, et al.Balancing protein stability and activity in cancer: a new approach for identifying driver mutations affecting CBL ubiquitin ligase activation. Cancer Res 2016;76:561–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Miosge LA, Field MA, Sontani Y, et al.Comparison of predicted and actual consequences of missense mutations. Proc Natl Acad Sci USA 2015;112:E5189–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Babenko VN, Basu MK, Kondrashov FA, et al.Signs of positive selection of somatic mutations in human cancers detected by EST sequence analysis. BMC Cancer 2006;6:36.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Glazko GV, Babenko VN, Koonin EV, et al.Mutational hotspots in the TP53 gene and, possibly, other tumor suppressors evolve by positive selection. Biol Direct 2006;1:4.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Futreal PA, Coin L, Marshall M, et al.A census of human cancer genes. Nat Rev Cancer 2004;4:177–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Santarius T, Shipley J, Brewer D, et al.A census of amplified and overexpressed human cancer genes. Nat Rev Cancer 2010;10:59–64. [DOI] [PubMed] [Google Scholar]
- 153. Poliakov E, Managadze D, Rogozin IB.. Generalized portrait of cancer metabolic pathways inferred from a list of genes overexpressed in cancer. Genet Res Int 2014;2014:646193.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Lawrence MS, Stojanov P, Mermel CH, et al.Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014;505:495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Lawrence MS, Stojanov P, Polak P, et al.Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013;499:214–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. de Bruin EC, McGranahan N, Mitter R, et al.Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science 2014;346:251–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Williams MJ, Werner B, Barnes CP, et al.Identification of neutral tumor evolution across cancer types. Nat Genet 2016;48:238–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Sottoriva A, Kang H, Ma Z, et al.A big bang model of human colorectal tumor growth. Nat Genet 2015;47:209–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Uchi R, Takahashi Y, Niida A, et al.Integrated multiregional analysis proposing a new model of colorectal cancer evolution. PLoS Genet 2016;12:e1005778.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Ling S, Hu Z, Yang Z, et al.Extremely high genetic diversity in a single tumor points to prevalence of non-Darwinian cell evolution. Proc Natl Acad Sci USA 2015;112:E6496–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Gao R, Davis A, McDonald TO, et al.Punctuated copy number evolution and clonal stasis in triple-negative breast cancer. Nat Genet 2016;48:1119–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Markowetz F. A saltationist theory of cancer evolution. Nat Genet 2016;48:1102–3. [DOI] [PubMed] [Google Scholar]
- 163. Cross W, Graham TA, Wright NA.. New paradigms in clonal evolution: punctuated equilibrium in cancer. J Pathol 2016;240:126–36. [DOI] [PubMed] [Google Scholar]
- 164. Bettegowda C, Sausen M, Leary RJ, et al.Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med 2014;6:224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Aravanis AM, Lee M, Klausner RD.. Next-generation sequencing of circulating tumor DNA for early cancer detection. Cell 2017;168:571–4. [DOI] [PubMed] [Google Scholar]