

ABSTRACT
Background
The putative functional variant −265T>C (rs5082) within the APOA2 promoter has shown consistent interactions with saturated fatty acid (SFA) intake to influence the risk of obesity.
Objective
The aim of this study was to implement an integrative approach to characterize the molecular basis of this interaction.
Design
We conducted an epigenome-wide scan on 80 participants carrying either the rs5082 CC or TT genotypes and consuming either a low-SFA (<22 g/d) or high-SFA diet (≥22 g/d), matched for age, sex, BMI, and diabetes status in the Boston Puerto Rican Health Study (BPRHS). We then validated the findings in selected participants in the Genetics of Lipid Lowering Drugs and Diet Network (GOLDN) Study (n = 379) and the Framingham Heart Study (FHS) (n = 243). Transcription and metabolomics analyses were conducted to determine the relation between epigenetic status, APOA2 mRNA expression, and blood metabolites.
Results
In the BPRHS, we identified methylation site cg04436964 as exhibiting significant differences between CC and TT participants consuming a high-SFA diet, but not among those consuming low-SFA. Similar results were observed in the GOLDN Study and the FHS. Additionally, in the FHS, cg04436964 methylation was negatively correlated with APOA2 expression in the blood of participants consuming a high-SFA diet. Furthermore, when consuming a high-SFA diet, CC carriers had lower APOA2 expression than those with the TT genotype. Lastly, metabolomic analysis identified 4 pathways as overrepresented by metabolite differences between CC and TT genotypes with high-SFA intake, including tryptophan and branched-chain amino acid (BCAA) pathways. Interestingly, these pathways were linked to rs5082-specific cg04436964 methylation differences in high-SFA consumers.
Conclusions
The epigenetic status of the APOA2 regulatory region is associated with SFA intake and APOA2 −265T>C genotype, promoting an APOA2 expression difference between APOA2 genotypes on a high-SFA diet, and modulating BCAA and tryptophan metabolic pathways. These findings identify potential mechanisms by which this highly reproducible gene-diet interaction influences obesity risk, and contribute new insights to ongoing investigations of the relation between SFA and human health.
This study was registered at clinicaltrials.gov as NCT03452787.
Keywords: APOA2, epigenomics, metabolomics, transcription, gene-diet interaction, obesity, tryptophan metabolism, branched-chain amino acid metabolism, satiety
INTRODUCTION
A common genetic variant within the promoter of apolipoprotein A-II gene (APOA2), −265T >C (rs5082), with a frequency of the minor C allele ranging from 0.2 to 0.4, depending on the population, directs APOA2 expression in vitro (1). Genetic studies in humans have shown associations with obesity and postprandial metabolism of triglyceride-rich lipoprotein (1, 2). While this single nucleotide polymorphism (SNP) was not associated with obesity in genome-wide studies, we have demonstrated that this variant interacts with SFA intake on risk of obesity in several populations in the United States, Europe, and Southeast Asia (3–7). Individuals with the CC genotype have an elevated obesity risk when consuming a diet high in SFA (i.e., ≥22 g/d) (3, 6), compared with T allele carriers. In addition, CC carriers tend to consume more food, and particularly more high-fat and high-protein foods (7). Despite these consistent observations, the molecular mechanisms underlying this gene-by-diet interaction on obesity remain unknown.
Epigenetic status reflects an individual's profile acquired in response to environmental exposures (8). Early life nutrition, adult lifestyle habits, and other environmental factors can “program” disease risk in later life (9). Gene-diet interactions may reflect, in part, the effects of diet on epigenetic status. Epigenetic changes in promoter and enhancer regions can differentially affect mRNA expression of target genes, modifying their expression and, thus, regulation of specific metabolic pathways, which can be detected through metabolomic analysis (10).
Importantly, APOA2 CC homozygotes exhibited an increased risk of obesity only when consuming a high-SFA diet, but they have no increased risk of obesity when consuming a low-SFA diet (4–8). This observation suggests that higher SFA intake occurs first, followed by APOA2 genotype–associated obesity. Thus, we hypothesized that the genotype-dependent response to a high-SFA diet could involve epigenetic changes, leading to altered APOA2 expression and metabolic changes in energy metabolism and homeostasis. In this study, we examined the correlation between epigenome-wide variation and APOA2 genotype, conditional on dietary SFA intake, in selected participants of the Boston Puerto Rican Health Study (BPRHS), and then validated our findings in 2 additional populations, the Framingham Heart Study (FHS) and the Genetics of Lipid Lowering Drugs and Diet Network (GOLDN) Study. In the FHS, we further examined how the identified methylation site within the APOA2 region affects APOA2 mRNA expression across different APOA2 genotypes and varying SFA intakes. We then investigated differences in metabolomic profiles between CC and TT genotypes in relation to low- or high-SFA intake in order to understand the relation between epigenetic signatures and metabolic pathways in the context of obesity risk. This study was designed to identify potential mechanisms by which this highly reproducible gene-diet interaction influences obesity risk, and also inform the long-standing debates surrounding saturated fat intake and health outcomes.
METHODS
Boston Puerto Rican Health Study
The BPRHS is a longitudinal cohort study (11). The primary objective is to examine and characterize the relations between nutrition, genetics, and health disparities in Puerto Rican adults. The population comprises 1499 individuals, self-identified as Puerto Ricans, with baseline age ranging from 45 to 75 y (11). To maximize the power to detect the effect of the C allele, we designed the study to focus on the difference between 2 homozygotes (CC compared with TT), which we expected to be maximal. Based on our published observations of the APOA2-SFA interaction, which was consistent in several populations (3–6) and an epigenome-wide association study (12), we determined that a sample size of 20 for each CC and TT genotype with either low- or high-SFA has statistical power of 95% to detect epigenetic signatures at a significance of α = 1.1 × 10−7 (1% methylation difference between CC and TT genotype and an SE of 0.2%) of the APOA2-SFA interaction on obesity. Within this study (see Supplemental Figure 1), 40 participants with CC genotype at APOA2 −265T>C were selected, with 20 reporting a low-SFA intake (<22 g/d) and 20 reporting a high-SFA intake (≥22 g/d) (3, 6). By matching age, sex, SFA intake, type 2 diabetes status, and BMI for the 40 participants with CC genotype, the second set of 40 participants with TT genotype at APOA2 −265T>C was selected from the same population, with priority given to participants who were matched most closely for age, sex, SFA intake, and type 2 diabetes (Table 1). The Institutional Review Board at Tufts University approved the study protocol. All participants provided written consent for participation in the study.
Table 1.
Characteristics of participants in 3 populations according to CC and TT genotypes of APOA2 –265T >C1
| TT | CC | |||
|---|---|---|---|---|
| Low-SFA intake | High-SFA intake | Low-SFA intake | High-SFA intake | |
| BPRHS | ||||
| n = 80 | 20 | 20 | 20 | 20 |
| Women, % | 50 | 50 | 50 | 50 |
| Age, y | 59.5 ± 6.8 | 59.6 ± 6.8 | 59.5 ± 6.7 | 58.6 ± 7.6 |
| Drinker, % | 30 | 60 | 35 | 60 |
| Smoker, % | 10 | 25 | 35 | 30 |
| BMI, kg/m2 | 31.6 ± 5.8 | 31.2 ± 5.6 | 29.4 ± 6.0 | 34.0 ± 5.72 |
| SFA, g/d | 14.0 ± 4.2 | 33.7 ± 8.62 | 14.6 ± 5.0 | 35.1 ± 12.62 |
| Total energy, kcal | 1699 ± 561 | 3000 ± 6302 | 1522 ± 416 | 2932 ± 6912 |
| GOLDN | ||||
| n = 379 | 107 | 165 | 39 | 68 |
| Women, % | 77 | 42 | 85 | 40 |
| Age, y | 46.3 ± 16.8 | 41.5 ± 14.0 | 43.8 ± 15.8 | 43.4 ± 12.7 |
| Drinker, % | 47 | 45 | 64 | 54 |
| Smoker, % | 26 | 26 | 31 | 29 |
| BMI, kg/m2 | 27.9 ± 5.5 | 26.8 ± 5.0 | 27.1 ± 5.4 | 29.1 ± 5.9 |
| SFA, g/d | 15.3 ± 4.5 | 38.0 ± 20.82 | 15.9 ± 4.0 | 41.1 ± 19.52 |
| Total energy, kcal | 1351 ± 383 | 2599 ± 14072 | 1367 ± 402 | 2884 ± 12082 |
| FHS | ||||
| n = 243 | 74 | 96 | 34 | 39 |
| Women, % | 73 | 56 | 71 | 62 |
| Age, y | 65.1 ± 9.3 | 62.1 ± 8.2 | 65.4 ± 9.4 | 64.4 ± 7.7 |
| Drinker, % | 76 | 72 | 91 | 82 |
| Smoker, % | 12 | 4 | 6 | 10 |
| BMI, kg/m2 | 25.3 ± 4.2 | 27.5 ± 5.42 | 25.4 ± 3.7 | 26.7 ± 4.5 |
| SFA, g/d | 16.1 ± 4.2 | 31.6 ± 9.22 | 15.9 ± 4.3 | 30.6 ± 7.62 |
| Total energy, kcal | 1532 ± 349 | 2284 ± 5122 | 1528 ± 337 | 2336 ± 5372 |
1Age, BMI, SFA, total energy are expressed as means ± SDs. APOA2, apolipoprotein A-II; BPRHS, Boston Puerto Rican Health Study; FHS, Framingham Heart Study; GOLDN, Genetics of Lipid Lowering Drugs and Diet Network.
2Significant differences by t test between high- and low-SFA in participants with CC or TT genotype.
Genetics of Lipid Lowering Drugs and Diet Network Study
The GOLDN Study is a constituent of the Family Heart Study within the NIH National Heart, Lung and Blood Institute (7). Participants (n = 1327), aged ≥18 y, from US families of European descent, were recruited at 2 field centers: Minneapolis, MN and Salt Lake City, UT. The main goal of the GOLDN Study was to identify genetic factors that determine individual responses to high-fat meals and 3-wk treatment with fenofibrate, a triglyceride-lowering medication (12). Institutional Review Boards at Tufts University, the University of Minnesota, University of Utah, and the University of Alabama at Birmingham approved the study protocol. In the current study (see Supplemental Figure 2), 107 participants with CC genotype and 272 with TT genotype for APOA2 −265T>C, who were not taking medication for hypertension, dyslipidemia, or diabetes, were selected to validate the findings from the BPRHS. Participants of each genotype were further divided into 2 subgroups, based on low-SFA (<22 g/d) and high-SFA intake (≥22 g/d) (Table 1) (3, 6).
Framingham Heart Study
Established in 1948, the FHS is a free-living multiple generation study of participants recruited in and near Framingham, MA (13). The Offspring Cohort started in 1971 with the enrollment of 5124 participants recruited from the children and spouses of the original FHS (14). In-person physical and clinical examinations occurred every 4–8 y. In exam 8, 2005–2008, blood samples were collected, and 2741 samples were available for DNA methylome and transcriptome analysis (15). In this study (see Supplemental Figure 3), to validate the findings from the BPRHS, we include 73 unrelated participants with CC genotype and 170 with TT genotype at APOA2 −265T >C who did not take medication for hypertension, dyslipidemia, or diabetes. Participants of each genotype were further divided into 2 subgroups based on SFA intake: low, <22 g/d; high, ≥22 g/d (Table 1) (3, 6).
Assessment of dietary intake
In the BPRHS, diet was assessed using a specially designed and validated food frequency questionnaire (16), whereas the Diet History Questionnaire was administrated in the GOLDN Study (17) and the Willett semi-quantitative food frequency questionnaire was applied in exam 8 of the FHS (18). The dietary intake for each population (16–18) was estimated based on the Harvard University food composition database, the US Department of Agriculture database, and the Minnesota Nutrient System.
DNA extraction
In the BPRHS and the FHS, genomic DNA for methylation analysis was extracted from buffy coats using the QIAamp DNA Blood mini kit or Gentra Puregene DNA extraction kit (Qiagen) (15). In the GOLDN Study, genomic DNA was isolated from CD4+ T cells from frozen buffy coat samples using DNeasy kits (Qiagen) (12).
Methylome analysis
Genome-wide DNA methylation of isolated DNA samples in the BPRHS and the GOLDN Study was quantified using Illumina Infinium human methylation 450K arrays (Illumina), as described previously (15, 19). The methylation signal at each methylation site was estimated as a β score, the proportion of the total methylation-specific signal, and the detection P value was the probability that the total intensity for a given probe fell within the background signal intensity. Methylation sites were excluded from further analysis if one of the following criteria was met: 1) detection P > 0.01 and 1.5% of samples have missing data, or 2) >10% of samples lack sufficient intensity (12). The methylation signal β scores were further adjusted for batch effects across samples and normalized using the ComBat function in the ChAMP R package (20). To account for heterogeneity of different cell types across all samples, principal components were calculated, based on the β scores of all autosomal CpG sites that passed quality control, using the prcomp function in R (v12.12.1). The first 4 principal components were used in all subsequent statistical analyses.
We acquired the FHS DNA methylome data through dbGaP, accession #phg000492.v2, where the methylation statuses of 2741 participants were measured at exam 8, using Illumina Infinium human methylation 450K arrays (15). Methylation signals were processed and normalized as for the BPRHS and the GOLDN Study.
Transcription analysis
We obtained FHS transcriptome data from dbGaP under accession #phe00002.v6. Transcriptome analysis was conducted in exam 8 using the Affymetrix Human Exon 1.0 ST array with mRNA from whole blood samples collected from 725 Offspring Cohort participants after overnight fasting (21). The quality control and normalization of the initial gene expression data have been published elsewhere (22, 23). In this study, we extracted gene expression data for 201 of 243 participants with CC or TT genotypes who were not taking medication for hypertension, dyslipidemia, or diabetes. Furthermore, to determine if the identified methylation site (i.e., cg04436964) was correlated with gene expression of APOA2 or nearby genes, we focused analysis on the expression of those 10 genes that passed quality control (QC) of the 31 genes that map within 50 kb upstream or downstream of the identified methylation site. Genes present on the array but not passing QC can be considered as unresponsive to methylation at cg04436964.
Metabolomic profile
Metabolic profiling of plasma samples from those 80 participants of the BPRHS for whom the methylome analysis was performed was conducted by Metabolon, Inc. (24). Briefly, 80 plasma samples were shipped on dry ice to Metabolon, and stored at −80°C until analysis. After proteins were removed with methanol, metabolomic analysis was performed using ultrahigh-performance liquid chromatography-tandem mass spectroscopy. With reference to a library of over 4500 purified standards for retention time/index, mass-to-charge ratio, and chromatographic data, individual metabolites were identified and quantified by estimating the AUC of the peaks. A total of 808 metabolites that passed QC were identified and assigned to pathway groups.
Statistical analysis
A schematic of the data analysis with sample size information is depicted in Figure 1.
Figure 1.

Analysis scheme. Experiments, described in the leftmost column, proceeded from top to bottom. Cohort source and sample sizes for the various genotypes at APOA2 −265T >C under conditions of low or high SFA intake are given. For all analyses involving the BPRHS cohort, data from the same participants were used, and from the same data collection period. APOA2, apolipoprotein A-II; BPRHS, Boston Puerto Rican Health Study; FHS, Framingham Heart Study; GOLDN, Genetics of Lipid Lowering Drugs and Diet Network.
Epigenome-wide analysis of APOA2 genotype, by high- and low-SFA intake, in the BPRHS
To identify epigenetic sites that differed between APOA2 −265T>C CC and TT genotypes, we conducted an epigenome-wide association study in 80 participants selected according to their genotype using the GEM package R tool suite, with methylation signals as dependent variables with the G-model (detecting methylation markers associated with genotype, i.e., methylation quantitative trait loci) (25), adjusting for medication for hypertension, lipid-lowering, cell-type heterogeneity, and population structure. Population structure was adjusted by principle component analysis (PCA), with 50,704 selected SNPs genotyped using Affymetrix Axiom World LA Arrays, designed especially for Hispanic populations, and probe sets to genotype 817,810 SNPs. The 50,704 SNPs were selected based on call rate >97%, MAF ≥5%, pairwise linkage disequilibrium R2 ≤ 0.1, and P value of the Hardy-Weinberg Equilibrium ≥10−6. Using PCA implemented in SVS (GoldenHelix Inc.), one eigenvalue was selected to represent population structure, based on the Scree plot. This PCA1 principle component variable was used in all regression models to adjust for population structure. Total energy intake was not included as a covariate in the linear regression model as APOA2 −265T >C is known to be associated with total energy intake (3). The Bonferroni correction with an epigenome-wide significance of P = 1.1 × 10−7 was used to adjust for multiple comparisons (12).
Validation in the GOLDN Study and the FHS
To validate the initial findings from the BPRHS, we conducted a similar epigenome-wide association study in selected datasets from the GOLDN Study (see Supplemental Figure 2) and from the FHS (see Supplemental Figure 3). In the GOLDN Study, 271 participants with TT genotype and 107 participants of CC genotype who did not use medication for hypertension, dyslipidemia, or diabetes were included in the validation analysis (Table 1 and Supplemental Figure 2). To identify methylation quantitative trait loci, a linear Gmodel was implemented with methylation measures as dependent variables and APOA2 genotype as a predictor using the GEM package R tool suite (25), adjusting for age, sex, center, cell-type heterogeneity, and family relationship. In the FHS, 171 participants with TT genotype and 73 with CC genotype, who did not take medication for hypertension, dyslipidemia, or diabetes, were included for validation (Table 1 and Supplemental Figure 3). A similar linear G-model was implemented using the GEM package R tool suite (25), controlling for cell-type heterogeneity, age, and sex. All analyses were conducted using R3.3.2, and the results were reviewed and formatted using the genome tool of SVS 8.7.
Meta-analysis
A meta-analysis of the results from the 3 populations was conducted using the meta R package (https://cran.r-project.org/web/packages/meta/index.html). A comparison of allelic effect (β) of APOA2 −265T >C from the meta-analysis between low- and high-SFA intake was conducted with SAS 9.4 (SAS Inc.) using a t test.
Associations between epigenetic variants and APOA2 mRNA expression in the FHS
To determine whether identified methylation sites in the APOA2 region were associated with APOA2 mRNA expression in whole blood, we examined the correlation between the identified methylation site (cg04436964, which showed differential association with APOA2 genotype and differed between low- and high-SFA intake) and FHS exam 8 expression data for genes located within a 50-kb region of the methylation site. Gene expression measures were treated as dependent variables, and methylation sites and APOA2 genotype were tested as predictors, controlling for age, sex, and cell-type heterogeneity. These analyses were conducted using a linear regression model with SAS 9.4 or SVS 8.7.
Metabolic pathway enrichment analysis
All detected metabolites (n = 808) were organized into metabolic pathways based on the annotation database of Metabolon Inc. Only pathways that contained ≥3 detected metabolites were included in the pathway analysis. For each metabolite, comparisons between CC and TT genotypes were conducted for high- or low-SFA intake. These comparisons were conducted using ANCOVA contrast in a 2-way ANOVA model, which included the fixed effects and interaction terms of genotype and SFA, adjusting for BMI. The mean square error for the overall model was used in the calculation of the individual contrasts. The number of metabolites that differ between CC and TT genotypes at P ≤ 0.05 was counted within each pathway. Metabolic pathway enrichment between CC and TT genotypes was determined by z score calculation for each pathway, separately for low- and high-SFA intake, as: Z score = [r – n (R/N)]/√{n (R/N)[1 – (R/N)][1 – (n – 1)/(N – 1)]} (26), where N = 635 is the total number of metabolites detected in the metabolomic profiling that were included in the 63 pathways, R is the total number of metabolites significantly different between CC and TT genotypes within low- or high-SFA intake, n is the total number of metabolites measured in a specific pathway, and r is the number of metabolites significantly different between CC and TT genotypes within a specific pathway. P values of Z scores were derived assuming a normal distribution and 2-sided, and were then corrected for multiple testing by the Bonferroni test (P = 0.05/63, 0.0008).
Linking the epigenomic and metabolomic signatures
To uncover the connection between epigenomic status and metabolic network, we conducted metabolic network enrichment analysis in relation to the identified methylation site cg04436964, which differed between CC and TT genotypes with a high-SFA intake. We performed an association analysis between cg04436964 and all metabolites using a linear regression model, adjusting for age, sex, population structure, and cell-type heterogeneity. The cg04436964-associated (at P ≤ 0.05) metabolites were evaluated for overlap with those that differed (at P ≤ 0.05) between CC and TT genotypes under high-SFA intake. The number of metabolites that met both criteria (associated with cg04436964 at P ≤ 0.05 and differed between CC and TT genotypes under high-SFA intake) were counted within each pathway. z scores and P values were calculated as above for each of 63 pathways. The P values were corrected for multiple testing using a Bonferroni test. Missing data were excluded from all the analyses.
RESULTS
Epigenome-wide association analysis of APOA2 stratified by SFA intake in the BPRHS
The general characteristics of 2 groups of selected participants with CC genotypes (n = 40) and low- (<22 g/d) or high-SFA intake (≥22 g/d) (3, 6) and 2 groups of participants with TT genotypes (n = 40) matched to the CC group by SFA intake, age, and sex are shown in Table 1. While the men:women ratio and age are almost identical among the 4 groups, the mean BMI of the CC participants with high-SFA was significantly higher than that of the low-SFA intake group. In addition, SFA and total energy intake were significantly greater in the high-SFA groups than in the low-SFA groups regardless of APOA2 genotype. These differences are expected, as the participants were selected unbiasedly to represent APOA2 genotype and SFA intake on obesity (3).
To identify the epigenomic signatures of APOA2 by SFA interaction, we conducted an epigenome-wide scan to compare the methylation status of 20 participants with CC genotype and 20 participants with TT genotypes each stratified by low- or high-SFA intake (Figure 1). The Manhattan plots and QQ plots of all methylation sites are depicted in Supplemental Figures 4A and B, and 5A and B. For participants with high-SFA intake (Supplemental Figure 4B), one CpG site, cg04436964, with methylation ranging from 0.77 (77%) to 0.96 (96%), attained genome-wide significance at P = 4.35 × 10−8 between CC and TT genotypes (Table 2). This CpG maps ∼25.7 kb from the APOA2 transcription start site (TSS) and ∼25.9 kb from −265T >C. The physical map of the 50-kb region centered on APOA2 −265T >C (Figure 2B) indicates the single methylation site that displayed significant differences between CC and TT genotypes. One copy of the C allele is associated with a 3.5% increase in methylation at cg04436964 under a high-SFA diet, assuming an additive model for the epigenetic effect. For participants with low-SFA intake (Figure 2A, Table 2, Supplemental Figures 4A and 5A), however, methylation at no other CpG site differed significantly between CC and TT genotypes after correction for multiple testing (P > 1.1 × 10−7). As obesity may influence epigenetic status, we controlled for BMI and observed that the associations remained consistent. Furthermore, a comparison of methylation under conditions of high- and low-SFA intake within each genotype, CC or TT, indicated that no methylation site showed an epigenome-wide significant difference (between low- and high-SFA) within genotype (P > 1.1 × 10−7, data not shown).
Table 2.
CpG sites that show significant difference in methylation status between APOA2 genotypes according to saturated fat intake in 3 populations1
| CC vs. TT | ||||||||
|---|---|---|---|---|---|---|---|---|
| Low-SFA | High-SFA | |||||||
| CpG site | Chr:Position2 | n | β (SE) | P value3 | n | β (SE) | P value3 | |
| BPRHS | cg04436964 | 1:161,167,745 | 20 | 0.031 (0.006) | 1.22 × 10−6 | 20 | 0.035 (0.005) | 4.35 × 10−8* |
| cg24429974 | 1:161,195,180 | 20 | 0.010 (0.006) | 3.77 × 10−3 | 20 | 0.009 (0.008) | 0.255 | |
| cg24847046 | 1:161,135,079 | 20 | −0.009 (0.006) | 0.185 | 20 | −0.017 (0.006) | 0.011 | |
| GOLDN | cg04436964 | 1:161,167,745 | 146 | 0.009 (0.002) | 1.59 × 10−3 | 233 | 0.013 (0.002) | 1.02 × 10−10* |
| cg24429974 | 1:161,195,180 | 146 | 0.009 (0.002) | 1.51 × 10−4 | 233 | 0.011 (0.002) | 1.84 × 10−10* | |
| cg24847046 | 1:161,135,079 | 146 | −0.004 (0.003) | 0.106 | 233 | −0.007 (0.002) | 2.51 × 10−4 | |
| FHS | cg04436964 | 1:161,167,745 | 108 | 0.031 (0.004) | 5.23 × 10−12* | 135 | 0.040 (0.003) | 1.21 × 10−25* |
| cg24429974 | 1:161,195,180 | 108 | 0.022 (0.003) | 1.63 × 10−10* | 135 | 0.021 (0.003) | 3.33 × 10−12* | |
| cg24847046 | 1:161,135,079 | 108 | −0.013 (0.003) | 3.38 × 10−6 | 135 | −0.018 (0.003) | 2.70 × 10−8* | |
1*P value ≤1.1 × 10−7. APOA2, apolipoprotein A-II; BPRHS, Boston Puerto Rican Health Study; FHS, Framingham Heart Study; GOLDN, Genetics of Lipid Lowering Drugs and Diet Network.
2The physical position of CpG sites was based on the genome build 37.
3The threshold of epigenome-wide significance is P value ≤1.1 × 10−7.
Figure 2.

Close-up of the 81-kb genomic region surrounding the APOA2 −265T >C site (rs5082, the orange dot) of an epigenome-wide association study with APOA2 −265T >C in the BPRHS, GRCh37/hg19 approximate coordinates: chr1:161,134,000–1,612,215,000. (A) Under low saturated fat intake or <22 g/d (low-SFA), B) under high saturated fat intake or ≥ 22 g/d (high-SFA). The vertical axis displays the –log10(P value) of association, whereas the horizontal axis displays the physical position of CpG sites and genes (genome build 37). Each blue dot depicts one CpG site. The dashed line indicates the threshold of epigenome-wide significance at P = 1.1 × 10−7. ADAMTS4, a disintegrin and metalloproteinase with thrombospondin motifs 4; APOA2, apolipoprotein A-II; BPRHS, Boston Puerto Rican Health Study; B4GALT3, Beta-1,4-Galactosyltransferase 3; FCER1G, Fc fragment of IgE receptor Ig; NDUFS2, NADH:ubiquinone oxidoreductase core subunit S2; NR1I3, nuclear receptor subfamily 1 group I member 3; PPOX, protoporphyrinogen oxidase; TOMM40L, translocase of outer mitochondrial membrane 40 like.
Validation of epigenetic signature of interaction between APOA2 and SFA intake
To validate the initial finding that cg04436964 methylation status correlates with APOA2 −265T>C genotype when SFA intake is high in the BPRHS, we conducted in the GOLDN Study an epigenome-wide association study similar to the BPRHS. The general characteristics of 379 participants of either CC (n = 107) or TT (n = 272) genotype from the GOLDN Study are listed in Table 1, excluding those taking medication for hypertension, diabetes, or dyslipidemia. Similar to the BPRHS, in the GOLDN Study, participants’ methylation at cg04436964 showed a significant difference between CC and TT genotypes with high-SFA intake at P = 1.02 × 10−10 (Table 2). In addition, cg24429974, 1.5 kb distal to −265T >C, also exhibited a significant genotype-specific methylation difference at P = 1.84 × 10−10 in participants with high-SFA intake, and the significance of the results remained after adjustment for BMI. These significant associations between methylation sites and APOA2 genotype are depicted for the 80 kb region around APOA2 −265T >C in Supplemental Figure 6B. One copy of the C allele reflects an increase in methylation of 1.3% at cg04436964 with high-SFA intake (Table 2), assuming an epigenetic effect of an additive model. However, in GOLDN Study participants with low-SFA intake (<22 g/d) (3, 6), these 2 methylation sites (cg04436964 and cg24429974) did not reach epigenome-wide significance between CC and TT genotypes (Supplemental Figure 6A, β = 0.009 and 0.009, P = 1.59 × 10−3 and 1.51 × 10−4, respectively).
To strengthen the validation, we conducted an additional epigenome-wide association study with CC and TT participants in the FHS at exam 8 who were not taking medication for hypertension, diabetes, or dyslipidemia. The general characteristics of the participants are given in Table 1. Similarly, methylation site cg04436964 showed greater significant differences between CC and TT genotypes in participants with high-SFA intake than with low-SFA intake (Supplemental Figure 7, β = 0.040 vs. 0.031, P = 1.21 × 10−25 vs. 5.23 × 10−12, Table 2). In comparison with cg04436964, cg24429974 showed a similar but weaker genotype-related difference between FHS participants with low-and high-SFA intake (Supplemental Figure 7, β = 0.021 vs. 0.022, P = 3.33 × 10−12 vs. 1.63 × 10−10, Table 2). In addition, cg24847046, 58.6 kb from rs5028, showed a significant association with APOA2 genotype in participants with low- or high-SFA intake (Supplemental Figure 7, β = −0.013 vs. −0.018, P = 3.38 × 10−6 vs. 2.70 × 10−8, Table 2). Even after adjustment for BMI, the findings remained consistent in all 3 populations. Together, each of the associations described in 3 distinct populations for cg04436964 are directionally identical, reach epigenome-wide significance thresholds, are dependent on SFA intake and rs5028 genotype, and share robustness with the gene-diet interaction described for this variant.
Meta-analysis of methylation differences between CC and TT genotypes
To combine the results on these populations, we conducted a meta-analysis with a fixed-effect model and found that methylation site cg04436964 showed a greater difference between CC and TT genotypes in high-SFA intake (Figure 3B, β = 0.023, 95% CI: 0.020, 0.026, P = 9.22 × 10−47) than low-SFA intake (Figure 3A, β = 0.015, 95% CI: 0.012, 0.018, P = 5.15 × 10−18). For cg24429974, the difference in methylation status between CC and TT genotypes was less apparent between high- (Figure 3D, β = 0.014, 95% CI: 0.011, 0.017, P = 5.76 × 10−17) and low-SFA intake (Figure 3C, β = 0.013, 95% CI: 0.010, 0.016, P = 1.59 × 10−15). Similarly, the difference in methylation site cg2487046 between CC and TT genotypes was less obvious between high- (Figure 3F, β = −0.011, 95% CI: −0.014, −0.008, P = 1.29 × 10−11) and low-SFA intake (Figure 3E, β = −0.009, 95% CI: −0.013, −0.005, P = 1.87 × 10−5).
Figure 3.

Forest plots of meta-analysis of methylation differences at 3 methylation sites over 3 populations (BPRHS, GOLDN, and FHS) between CC and TT genotypes according to low- and high-SFA intake. The summary of statistics (β, SE, 95% CI, P value) of the meta-analysis are listed on each panel: (A) cg04436964 at low-SFA intake; (B) cg04436964 at high-SFA intake; (C) cg24429674 at low-SFA intake; (D) cg24429674 at high-SFA intake; (E) cg24847046 at low-SFA intake; (F) cg24847046 at high-SFA intake. BPRHS, Boston Puerto Rican Health Study; FHS, Framingham Heart Study; GOLDN, Genetics of Lipid Lowering Drugs and Diet Network.
To determine differential methylation associated with both SFA intake and −265T>C genotype, we compared the difference in the allelic effect of the C allele on 3 methylation sites between low- and high-SFA intake. Using a t test, we found that participants with the CC genotype and high-SFA intake had 0.8% greater methylation at cg04436964, on average, when compared with participants with the TT genotype and low-SFA intake (P = 0.041). For cg24429974 and cg24847046, however, the difference between high- and low-SFA intake was not statistically significant (0.1% and −0.2%, P = 0.565 and 0.337, respectively).
Annotation of CpG cg04436964
Data available for human genome build 37 at genome.ucsc.edu indicate that cg04436964 is a solitary CpG, with the nearest CpG island annotated approximately 4.1 kb distant. This information is corroborated by the absence of correlation in methylation status between this CpG and other regional CpGs (data not shown). The cg04436964 CpG appears to be just within or on the edge of an enhancer region, as determined by querying adipose, kidney, and liver cell data at the WashU Epigenome Browser (27). This enhancer is not observed in all assays or cell types, but data indicate overlap with binding sites for RXRA and ZBTB7A, both transcription factors, with roles in the epigenetics of obesity and the regulation of genes highly expressed in adipose, respectively (28–30). However, data are lacking as to whether this region functions as an enhancer under conditions driven by the diet, either habitual or acute (e.g., an intervention). The −265T>C and cg04436964 site are ∼26 kb apart. The SNP maps with the APOA2 proximal promoter region, and there is some evidence to indicate that the CpG is within an enhancer (27). Thus, it is plausible to hypothesize that elevated intake of SFA serves to promote a looping of the DNA between these 2 regions, providing a means by which they can interact functionally. This interaction would have a stronger effect on APOA2 expression only with the CC genotype at rs5082 and under conditions of high intake of SFA.
CpG methylation and APOA2 genotype are associated with APOA2 mRNA expression, depending on SFA intake
As epigenetic status can affect expression of targeted genes, we determined if the identified methylation site in the APOA2 region was associated with APOA2 mRNA expression. To do this, we examined the correlation between cg04436964 methylation and mRNA expression in whole blood for the 31 protein- and RNA-coding genes located within 50 kb of this CpG site by mining transcriptome data from FHS exam 8. Among the 31 genes, mRNA expression of 10 genes was detected and passed QC (see Supplemental Table 1). For participants with high-SFA intake, cg04436964 methylation exhibited a negative and significant association (Figure 4B, β = −0.891, P = 0.028) with APOA2 mRNA expression. In contrast, those with low-SFA intake, APOA2 mRNA expression was not associated with methylation at cg04436964 (Figure 4A, β = 0.091, P = 0.857). The results remained the same even after adjusting for the additional potential confounder BMI.
Figure 4.
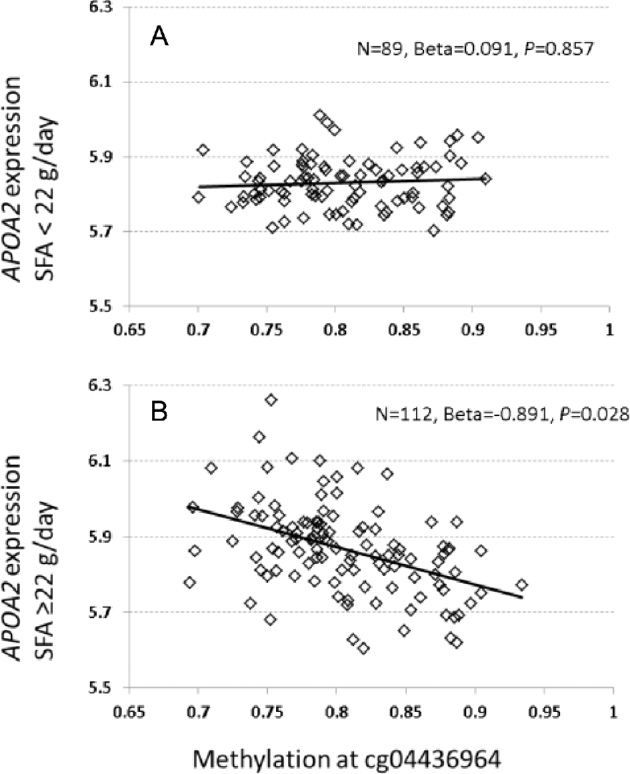
Correlation between APOA2 mRNA expression in whole blood and methylation at cg04436964 in the FHS at exam 8. (A) Under low saturated fat intake or <22 g/d (low-SFA), (B) under high saturated fat intake or ≥22 g/d (high-SFA). The y-axis displays a normalized and log2-transformed signal of APOA2 mRNA expression adjusted for age, gender, and heterogeneity of cell type, whereas the x-axis displays the methylation level of cg04436964. APOA2, apolipoprotein A-II; FHS, Framingham Heart Study.
Further, we observed an association between APOA2 −265T >C genotype and APOA2 mRNA expression that resembled the association which links methylation and expression, in that the 2 associations were limited to participants with high-SFA intake (Figure 5). Among participants with high-SFA intake, we saw that CC homozygotes had significantly lower APOA2 expression than TT homozygotes [mean expression: 5.79 (CC) compared with 5.90 (TT), P = 0.010]. In contrast, among participants with low-SFA intake, there was no significant difference in APOA2 expression between CC and TT genotypes [mean expression: 5.86 (CC) compared with 5.84 (TT), P = 0.670].
Figure 5.
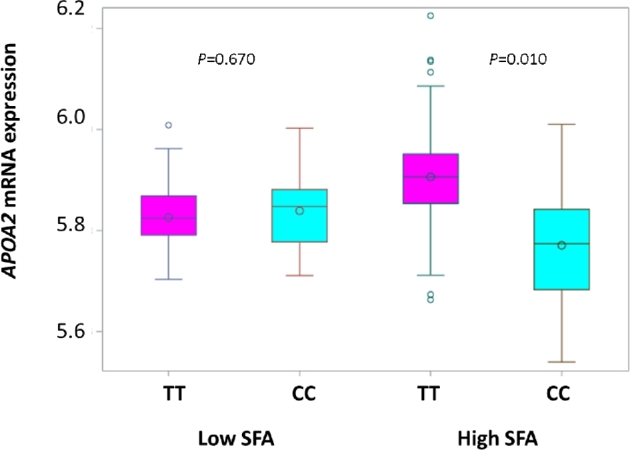
Box plot of APOA2 mRNA expression according to APOA2 –265T >C (rs5082) genotypes in the FHS at exam 8. The cyan box indicates participants with the CC genotype and the magenta box is for participants with the TT genotype, not taking medication for hypertension, dyslipidemia, or diabetes. Inside the box, the empty circle indicates means and the horizontal lines are medians. The bottom and top edges of the box indicate the range of values between the first and third quartiles (the 25th and 75th percentiles), whereas whiskers indicate the data range outside the box. Beyond the whiskers are the outliers. APOA2, apolipoprotein A-II; FHS, Framingham Heart Study.
In participants with a high-SFA diet, there is no correlation between APOA2 expression and expression of NDUFS2 (P = 0.775) or ADAMTS4 (P = 0.699). Hence, under a high-SFA diet, there is no influence of expression of the 2 genes on APOA2 expression. It is possible that the CpG cg04436964 participates in regulating the expression of other genes in tissues other than peripheral blood cells. However, biologically relevant tissues, including liver, subcutaneous and visceral adipose, and kidney, were not readily available for such analysis, especially with regard to APOA2 genotype and saturated fat intake. Lastly, mining Genotype-Tissue Expression Project data (version 7) shows a weak expression quantitative trait locus for −265T>C in liver and subcutaneous adipose for TOMM40L, whose TSS maps about 2.3 kb more distant to cg04436964 than the APOA2 TSS. However, TOMM40L expression in blood did not correlate with cg04436964 methylation (Supplemental Table 1).
High-SFA intake highlights metabolic differences between APOA2 genotypes
To examine the metabolic differences between CC and TT genotypes, metabolic pathway enrichment analysis was performed separately for low- and high-SFA intake datasets, with adjustment for BMI in the analysis. Among 808 metabolites detected in 80 plasma samples from the BPRHS, 635 compounds were grouped into 63 metabolic pathways, each of which contained ≥3 metabolites. Comparing CC to TT carriers on the low-SFA diet, 23 and 26 metabolites showed significant increases and decreases at P ≤ 0.05 (Supplemental Table 2), respectively. Comparing CC to TT carriers under high-SFA conditions, 7 and 64 metabolites displayed significant increases and decreases at P ≤ 0.05 (Supplemental Table 2), respectively. Metabolites that differed between CC and TT genotypes at P ≤ 0.05 were counted within each pathway (Supplemental Table 3). For low-SFA intake, there were 4 pathways with a z score of ≥1.96 (P ≤ 0.05), but none passed significance after correction for multiple testing (P = 0.05/63 = 0.0008 for 63 pathways). For high-SFA intake, 6 pathways had a z score of ≥1.96 (P ≤ 0.05). After correction for multiple testing, 4 pathways remained significantly overrepresented (Supplemental Table 3): leucine, isoleucine, and valine metabolism (P = 2.00 × 10−6), monoacylglycerol metabolism (P = 7.94 × 10−5), tryptophan metabolism (P = 0.0003), and phenylalanine metabolism (P = 0.0005). These 4 pathways highlight important metabolic differences between APOA2 genotypes (CC vs. TT) with a high-SFA intake.
Linking the epigenomic signature of the APOA2-SFA interaction and the metabolic network
To characterize the connections between the epigenomic signature and the metabolic network, we conducted metabolic pathway enrichment analysis to investigate differential methylation between APOA2 genotypes with high-SFA intake. We examined the correlation between methylation at cg04436964 and 635 metabolites in 63 pathways. The cg04436964-associated (at P ≤ 0.05) metabolites were merged with those that differed (at P ≤ 0.05) between CC and TT genotypes under a high-SFA intake. Eighteen metabolites met both criteria, as their levels in blood demonstrated association with cg04436964 methylation at P ≤ 0.05 and differed between CC and TT genotypes under high-SFA intake (Supplemental Table 4). Enrichment analysis with correction for multiple testing identified 2 pathways as being significantly enriched: tryptophan metabolism (Supplemental Table 4, Z = 5.029, P = 4.92 × 10−7) and leucine, isoleucine, and valine metabolism, also called branched-chain amino acid (BCAA) metabolism (z = 4.048, P = 5.16 × 10−5). In the tryptophan metabolism pathway (Figure 6), 6 of 18 detected metabolites (33%) were correlated with cg04436964 methylation. In CC carriers, 4 of those metabolites, 3-indoxyl sulfate, indolelactate, indoleacetyl glutamine, and xanthurenate, were at lower concentrations than in TT carriers under high-SFA intake.
Figure 6.

CC homozygotes display perturbations in the tryptophan metabolism pathway. The metabolites highlighted in green were significantly (P < 0.05) decreased in CC genotype (cyan box) when compared with TT genotype (magenta box) with high-SFA intake. Box plots of 4 metabolites that met 2 criteria—1) differed between CC and TT genotypes, and 2) was associated with cg04436964—were shown according to APOA2 genotypes and SFA intakes. Inside the box, the empty circle indicates means and the horizontal lines are medians. The bottom and top edges of the box indicate the range of values between the first and third quartiles (the 25th and 75th percentiles), whereas whiskers indicate the data range outside the box. Beyond the whiskers are the outliers.
In the BCAA metabolism pathway, 10 of 25 metabolites (40%) displayed negative correlation with methylation of cg04436964, 4 of which differed between CC and TT genotypes with high-SFA intake: α-hydroxyisocaproate, β-hydroxyisovalerate, 2-methylbutyrylcarnitine, and 3-hydroxy-2-ethylpropionate. Taken together, these observations of altered metabolite levels imply that with high-SFA intake, tryptophan metabolism and BCAA metabolism are reduced to a greater extent in CC carriers than in TT carriers.
DISCUSSION
A high-SFA diet can synergistically augment the risk of obesity for individuals with a particular genotype or portfolio of risk-increasing alleles (31, 32). The mechanisms by which such gene-diet interactions manifest their effects on obesity are not well characterized. In several populations encompassing various ancestries, we have demonstrated a gene-diet interaction whereby APOA2 CC homozygotes at −265T>C exhibited an increased risk of obesity when consuming a diet high in SFA compared with TT carriers (3–7), but they (CC homozygotes) had no increased risk of obesity when consuming a low-SFA diet (4–8). Thus, this replicated interaction between APOA2 genotype and SFA provides a useful platform for dissecting and characterizing the mechanisms of a gene-by-diet interaction on obesity. To achieve this objective, we report methylome, transcription, and metabolomics analyses based on 3 populations. We showed that participants with the CC genotype and high-SFA intake had increased methylation at cg04436964, near the APOA2 gene, compared with TT carriers, echoing the differences in BMI that have been replicated in 6 populations (3–7). Just as BMI did not differ by −265T>C genotype with low-SFA intake, genotype-based differences in methylation were also less apparent when SFA intake was low. The current study broadens our understanding, in that increased methylation at cg04436964 is associated with decreased expression of APOA2, again under high-SFA intake conditions. Both the methylation differences and the altered gene expression reflect the signals of the gene-diet interaction between APOA2 genotype and dietary SFA intake.
A comparison of the metabolomic profiles for CC and TT genotypes in the BPRHS for high-SFA intake, but not for low-SFA intake, identified 4 metabolic pathways that were significantly overrepresented: phenylalanine, monoacylglycerol, tryptophan, and BCAA. These results suggest that key differences between CC and TT carriers with high intake of SFA are explained by the metabolism of lipids and amino acids. We further demonstrated that the correlation between cg04436964 methylation and genotype-specific metabolites was significantly enriched in 2 of these pathways, namely tryptophan metabolism and BCAA metabolism. This indicates that, under conditions of elevated intake of SFA, CC carriers have dysregulated tryptophan metabolism and BCAA pathways to a greater degree than do TT carriers.
In mammals, tryptophan metabolism is a powerful regulator of feeding and satiety (33, 34). Over 95% of tryptophan is metabolized through the kynurenine pathway (35, 36). While plasma serotonin cannot enter the brain, kynurenine easily passes the blood-brain barrier. Thus, plasma kynurenine concentration reliably reflects the status in brain of kynurenic acids or kynurenate (35). Recently, kynurenic acid has been characterized as regulating food-dependent behavioral plasticity in Caenorhabditis elegans via a system whose elements are conserved with mammals (37). Fasting depletes kynurenic acid, and low kynurenic acid activates NMDA-receptor-expressing interneurons to promote food cravings through a neuropeptide-Y-like signaling axis and serotonin release upon feeding (37). In our study, under a high-SFA diet, CC carriers exhibited reduced concentrations of tryptophan metabolites in the kynurenine pathway (Figure 6), including xanthurenate and kynurenic acid, relative to TT carriers. Appetite in CC carriers would then be expected to be greater than in TT carriers, hence CC individuals would consume more food. Indeed, CC carriers, in general, consume more kilocalories than TT carriers (4, 5), which elevates their obesity risk. Furthermore, CC individuals also showed low levels of 3-indoxyl sulfate, indolelactate, and indoleacetyl glutamine, perhaps indicating reduced doleamine-2,3-dioxygenase-independent tryptophan catabolism (35). Lastly, associations between eating behavior and variants in genes encoding monoamine oxidase A and serotonin receptor 2A underscore the role of the tryptophan-serotonin-kynurenine pathway in appetite (38). Thus, altering tryptophan catabolism can lead to an imbalance between energy intake and expenditure via impact on appetite, thereby augmenting the risk of obesity.
In the enriched BCAA metabolism pathway, where metabolites differed between CC and TT genotypes and were associated with cg04436964 methylation and APOA2 expression, CC homozygotes with high-SFA intake uniquely showed a decrease of 4 BCAA catabolites: 3-hydroxy-2-ethylpropionate, α-hydroxyioscaproate, β-hydroxyisovalerate, and 2-methylbutyrylcarnitine. Impaired flux through the BCAA metabolic pathway is a sign of overall metabolic inefficiency. Specifically, with high-fat diets, reduced flux through the BCAA metabolic pathway was associated with obesity onset in mini-pigs (39), hindering BCAA-driven modulation of glucose metabolism in the liver, thereby elevating insulin resistance (40). Furthermore, when the metabolic capacity of the liver is compromised by high-fat feeding, BCAA homeostasis is less stably maintained, and this impairs the liver-skeletal muscle axis of cooperative BCAA catabolism (41). In parallel, defects in BCAA oxidation in muscle serve to disrupt normal lipid metabolism (42), possibly by curbing mTOR-mediated mitochondrial biogenesis (43), which could be of greater effect with high-SFA intakes. Together, the observed changes in metabolites within the Trp/KynA and BCAA pathways, examined in conjunction with reduced succinyl carnitine of the TCA cycle (Supplemental Table 2), serve as dual indicators of altered signaling—appetite/feeding and energy balance—that promote the development of obesity.
Our application of omics in 3 populations is a major strength of this study. Beginning with the multiancestral BPRHS, we initially detected methylation differences between APOA2 genotype in participants with high-SFA intake, examining 20 participants of CC genotype and 20 participants of TT genotype. One may argue that the small sample size of the BPRHS discovery cohort limits the power to identify other methylation sites that are associated with the APOA2 genotype differentially under low- and high-SFA intake. However, we have validated our findings by conducting 2 additional epigenome-wide associations in the FHS and GOLDN populations. Three methylation sites—cg04436964, cg24429974, and cg24847046—were identified at the threshold of epigenome-wide significance after correction for multiple testing. Meta-analysis of these results confirmed that only the methylation at cg04436964 differed between APOA2 genotypes and correlated with APOA2 mRNA under high-SFA intake. CC carriers with high-SFA intake tend to have high BMI, so obesity may be contributing to the differences in epigenetic status. Throughout our analyses, we have controlled for BMI in all 3 populations and the results remain consistent. In the FHS, we observed a significant difference in cg04436964 methylation between CC and TT carriers with low-SFA intake, which was not observed in the BPRHS or the GOLDN Study. Detection of the difference in the FHS alone cannot be attributed to greater power stemming from greater sample size because the sample size was greatest in the GOLDN Study. Instead, our ability to detect the difference in the FHS could be explained by the possibility that CC carriers modified SFA intake over time, i.e., some participants might have consumed higher SFA intake early and then changed to a low-SFA diet at a later time point. Indeed, when we examined the SFA intake across 4 examinations—exams 5–8 of the FHS—we found that only 15 CC (6.2%) and 24 TT (9.9%) carriers of 243 participants (without taking medications for lowering lipids, diabetes and hypertension) maintained a high-SFA diet (≥22 g/d) (3, 6), whereas 23 CC (9.5%) and 38 TT (15.6%) carriers maintained a low-SFA diet (<22 g/d) across all 4 exams. An epigenome-wide scan with 23 CC and 38 TT participants with consistent low-SFA intake across all 4 exams indicated that the difference in cg04436964 methylation between CC and TT genotypes did not reach epigenome-wide significance (P = 1.7 × 10−6 > 1.1 × 10−7). On the other hand, for 15 CC and 24 TT participants with consistent high-SFA intake across all 4 exams, the difference in cg04436964 methylation between CC and TT genotypes did reach epigenome-wide significance (P = 4.11 × 10−11). This observation further confirms that the SFA-dependent cg04436964 methylation differs between CC and TT genotypes consistently in all 3 distinct populations.
Some limitations of the current study must be noted. First, the epigenome analysis of the GOLDN Study used CD4 + T cells from a frozen buffy coat, whereas the BPRHS and FHS analyses were conducted using whole-blood buffy coats. This difference could introduce inconsistencies in estimating the population-specific effect sizes associated with methylation. Indeed, the β value for methylation associated with the C-allele was smaller in the GOLDN Study than in the BPRHS and FHS, but the directions were consistent in all cohorts. Second, the dietary patterns and food sources of SFA and their corresponding impacts on health could vary across groups because of regional and cultural differences. In this regard, the BPRHS, which studied a population of Caribbean Hispanic origin, is quite different from the FHS and the GOLDN Study. Population level differences in lifestyle, including diet, could also contribute to epigenetic differences between populations. Lastly, although we employed a targeted metabolomics approach, this is inherently limited to those metabolites that have been identified.
In summary, we applied multiomics approaches to investigate the mechanistic foundations of one of the most consistently replicated gene-diet interactions, that of SFA and the APOA2 genotype. Our findings from 3 populations of diverse ancestries strongly support that the epigenetic status of the APOA2 regulatory region is associated with SFA intake and the APOA2 −265T>C genotype, leading to differential diet-dependent and genotype-dependent APOA2 expression. As appropriate for a gene-diet interaction linked to the disruptive condition of obesity, we also uncovered plausible dysregulation of select metabolic pathways. The epigenetic signature of the gene-diet interaction is correlated with the down-regulation of the kynurenine pathway of tryptophan metabolism and BCAA metabolism, with possible implications for food intake. Collectively, these findings illustrate the effectiveness of multiomic approaches to well-established gene-diet interactions, and contribute novel evidence to ongoing explorations of the impact of saturated fat on human health.
Supplementary Material
Acknowledgements
The authors’ responsibilities were as follows—C-QL and JMO: conceived and designed the study; C-QL, CES, LDP, Y-CL, DA, and DC: acquired the data; C-QL, CES, LDP, MAP, and KLT: analyzed and interpreted the data; JMO and C-QL: performed statistical analysis and drafted the manuscript; C-QL, CES, LDP, JMO, and KLT: critically revised the manuscript for intellectual content; JMO, KLT, and DKA: provided funding and supervision; and all authors: read and approved the final manuscript. The Genotype-Tissue Expression Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by the NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The data used for the analyses described here were obtained from the Genotype-Tissue Expression Portal on 10 January 2018. None of the authors had a conflict of interest. Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the US Department of Agriculture. The USDA is an equal opportunity provider and employer.
Notes
This work was funded by the US Department of Agriculture, under agreement no. 8050-51000-098-00D, by National Heart, Lung, and Blood Institute grants U01-HL072524-04 and R01HL104135, and NIH grants P01 AG023394, P50 HL105185, and R01 AG027087. CES is supported by K08 HL112845. Any opinions, findings, conclusion, or recommendations expressed in this publication are those of the authors and do not necessarily reflect the view of the US Department of Agriculture.
Supplemental Tables 1–4 and Supplemental Figures 1–7 are available from the “Supplementary data” link in the online posting of the article and from the same link in the online table of contents at https://academic.oup.com/ajcn/.
Abbreviations:
- APOA2
apolipoprotein A-II
- BPRHS
Boston Puerto Rican Health Study
- FHS
Framingham Heart Study
- GOLDN
Genetics of Lipid Lowering Drugs and Diet Network
- PCA
principle component analysis
- QC
quality control
- SNP
single nucleotide polymorphism
- TSS
transcription start site
REFERENCES
- 1. van't Hooft FM, Ruotolo G, Boquist S, de Faire U, Eggertsen G, Hamsten A. Human evidence that the apolipoprotein A-II gene is implicated in visceral fat accumulation and metabolism of triglyceride-rich lipoproteins. Circulation 2001;104(11):1223–8. [DOI] [PubMed] [Google Scholar]
- 2. Delgado-Lista J, Perez-Jimenez F, Tanaka T, Pablo PM, Jimenez-Gomez Y, Marin C, Ruano J, Parnell L, Ordovas JM, Lopez-Miranda J. An apolipoprotein A-II polymorphism (−265T/C, rs5082) regulates postprandial response to a saturated fat overload in healthy men. J Nutr 2007;137(9):2024–8. [DOI] [PubMed] [Google Scholar]
- 3. Corella D, Peloso G, Arnett DK, Demissie S, Cupples LA, Tucker K, Lai CQ, Parnell LD, Coltell O, Lee YC et al. APOA2, dietary fat, and body mass index: replication of a gene-diet interaction in 3 independent populations. Arch Intern Med 2009;169(20):1897–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Smith CE, Ordovas JM, Sanchez-Moreno C, Lee YC, Garaulet M. Apolipoprotein A-II polymorphism: relationships to behavioural and hormonal mediators of obesity. Int J Obes 2012;36(1):130–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Smith CE, Tucker KL, Arnett DK, Noel SE, Corella D, Borecki IB, Feitosa MF, Aslibekyan S, Parnell LD, Lai CQ et al. Apolipoprotein A2 polymorphism interacts with intakes of dairy foods to influence body weight in 2 US populations. J Nutr 2013;143(12):1865–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Corella D, Tai ES, Sorli JV, Chew SK, Coltell O, Sotos-Prieto M, Garcia-Rios A, Estruch R, Ordovas JM. Association between the APOA2 promoter polymorphism and body weight in Mediterranean and Asian populations: replication of a gene-saturated fat interaction. Int J Obes 2011;35(5):666–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Corella D, Arnett DK, Tsai MY, Kabagambe EK, Peacock JM, Hixson JE, Straka RJ, Province M, Lai CQ, Parnell LD et al. The-256T >C polymorphism in the apolipoprotein A-II gene promoter is associated with body mass index and food intake in the genetics of lipid lowering drugs and diet network study. Clin Chem 2007;53(6):1144–52. [DOI] [PubMed] [Google Scholar]
- 8. Ling C, Groop L. Epigenetics: a molecular link between environmental factors and type 2 diabetes. Diabetes 2009;58(12):2718–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vickers MH. Early life nutrition, epigenetics and programming of later life disease. Nutrients 2014;6(6):2165–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cros J, Raffenne J, Couvelard A, Pote N. Tumor heterogeneity in pancreatic adenocarcinoma. Pathobiology 2018;85(1–2):64–71. [DOI] [PubMed] [Google Scholar]
- 11. Tucker KL, Mattei J, Noel SE, Collado BM, Mendez J, Nelson J, Griffith J, Ordovas JM, Falcon LM. The Boston Puerto Rican Health Study, a longitudinal cohort study on health disparities in Puerto Rican adults: challenges and opportunities. BMC Public Health 2010;10:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lai CQ, Wojczynski MK, Parnell LD, Hidalgo BA, Irvin MR, Aslibekyan S, Province MA, Absher DM, Arnett DK, Ordovas JM. Epigenome-wide association study of triglyceride postprandial responses to a high-fat dietary challenge. J Lipid Res 2016;57(12):2200–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dawber TR, Meadors GF, Moore FE. Epidemiological approaches to heart disease: the Framingham Study. Am J Public Health Nations Health 1951;41(3):279–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kannel WB, Feinleib M, Mcnamara PM, Garrison RJ, Castelli WP. Investigation of coronary heart-disease in families—Framingham Offspring Study. Am J Epidemiol 1979;110(3):281–90. [DOI] [PubMed] [Google Scholar]
- 15. Marioni RE, Shah S, McRae AF, Chen BH, Colicino E, Harris SE, Gibson J, Henders AK, Redmond P, Cox SR et al. DNA methylation age of blood predicts all-cause mortality in later life. Genome Biol 2015;16:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tucker KL, Bianchi LA, Maras J, Bermudez OI. Adaptation of a food frequency questionnaire to assess diets of Puerto Rican and non-Hispanic adults. Am J Epidemiol 1998;148(5):507–18. [DOI] [PubMed] [Google Scholar]
- 17. Subar AF, Thompson FE, Kipnis V, Midthune D, Hurwitz P, McNutt S, McIntosh A, Rosenfeld S. Comparative validation of the Block, Willett, and National Cancer Institute food frequency questionnaires—the eating at America's table study. Am J Epidemiol 2001;154(12):1089–99. [DOI] [PubMed] [Google Scholar]
- 18. Rimm EB, Giovannucci EL, Stampfer MJ, Colditz GA, Litin LB, Willett WC. Reproducibility and validity of an expanded self-administered semiquantitative food frequency questionnaire among male health-professionals. Am J Epidemiol 1992;135(10):1114–26. [DOI] [PubMed] [Google Scholar]
- 19. Absher DM, Li XR, Waite LL, Gibson A, Roberts K, Edberg J, Chatham WW, Kimberly RP. Genome-wide DNA methylation analysis of systemic lupus erythematosus reveals persistent hypomethylation of interferon genes and compositional changes to CD4+ T-cell populations. PLoS Genet 2013;9(8):e1003678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Morris TJ, Butcher LM, Feber A, Teschendorff AE, Chakravarthy AR, Wojdacz TK, Beck S. ChAMP: 450k Chip Analysis Methylation Pipeline. Bioinformatics 2014;30(3):428–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McManus DD, Rong J, Huan TX, Lacey S, Tanriverdi K, Munson PJ, Larson MG, Joehanes R, Murthy V, Shah R et al. Messenger RNA and MicroRNA transcriptomic signatures of cardiometabolic risk factors. BMC Genomics 2017;18:139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Katz S, Irizarry RA, Lin X, Tripputi M, Porter MW. A summarization approach for Affymetrix GeneChip data using a reference training set from a large, biologically diverse database. BMC Bioinformatics 2006;7:464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Joehanes R, Ying SX, Huan TX, Johnson AD, Raghavachari N, Wang R, Liu PC, Woodhouse KA, Sen SK, Tanriverdi K et al. Gene expression signatures of coronary heart disease. Arterioscl Throm Vas 2013;33(6):1418–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Evans AM, DeHaven CD, Barrett T, Mitchell M, Milgram E. Integrated, nontargeted ultrahigh performance liquid chromatography/electrospray ionization tandem mass spectrometry platform for the identification and relative quantification of the small-molecule complement of biological systems. Anal Chem 2009;81(16):6656–67. [DOI] [PubMed] [Google Scholar]
- 25. Pan H, Holbrook JD, Karnani N, Kwoh CK. Gene, Environment and Methylation (GEM): a tool suite to efficiently navigate large scale epigenome wide association studies and integrate genotype and interaction between genotype and environment. BMC Bioinformatics 2016;17:299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Doniger SW, Salomonis N, Dahlquist KD, Vranizan K, Lawlor SC, Conklin BR. MAPPFinder: using Gene Ontology and GenMAPP to create a global gene-expression profile from microarray data. Genome Biol 2003;4(1):R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhou X, Li D, Zhang B, Lowdon RF, Rockweiler NB, Sears RL, Madden PA, Smirnov I, Costello JF, Wang T. Epigenomic annotation of genetic variants using the Roadmap Epigenome Browser. Nat Biotechnol 2015;33(4):345–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Godfrey KM, Sheppard A, Gluckman PD, Lillycrop KA, Burdge GC, McLean C, Rodford J, Slater-Jefferies JL, Garratt E, Crozier SR et al. Epigenetic gene promoter methylation at birth is associated with child's later adiposity. Diabetes 2011;60(5):1528–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Day SE, Coletta RL, Kim JY, Campbell LE, Benjamin TR, Roust LR, De Filippis EA, Dinu V, Shaibi GQ, Mandarino LJ et al. Next-generation sequencing methylation profiling of subjects with obesity identifies novel gene changes. Clin Epigenetics 2016;8:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bakhtiarizadeh MR, Moradi-Shahrbabak M, Ebrahimi M, Ebrahimie E. Neural network and SVM classifiers accurately predict lipid binding proteins, irrespective of sequence homology. J Theor Biol 2014;356:213–22. [DOI] [PubMed] [Google Scholar]
- 31. Casas-Agustench P, Arnett DK, Smith CE, Lai CQ, Parnell LD, Borecki IB, Frazier-Wood AC, Allison M, Chen YDI, Taylor KD et al. Saturated fat intake modulates the association between an obesity genetic risk score and body mass index in two US populations. J Acad Nutr Diet 2014;114(12):1954–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ordovas JM, Tai ES. Why study gene-environment interactions? Curr Opin Lipidol 2008;19(2):158–67. [DOI] [PubMed] [Google Scholar]
- 33. Voigt JP, Fink H. Serotonin controlling feeding and satiety. Behav Brain Res 2015;277:14–31. [DOI] [PubMed] [Google Scholar]
- 34. O'Mahony SM, Clarke G, Borre YE, Dinan TG, Cryan JF. Serotonin, tryptophan metabolism and the brain-gut-microbiome axis. Behav Brain Res 2015;277:32–48. [DOI] [PubMed] [Google Scholar]
- 35. Schwarcz R, Bruno JP, Muchowski PJ, Wu HQ. Kynurenines in the mammalian brain: when physiology meets pathology. Nat Rev Neurosci 2012;13(7):465–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Leklem JE. Quantitative aspects of tryptophan metabolism in humans and other species: a review. Am J Clin Nutr 1971;24(6):659–72. [DOI] [PubMed] [Google Scholar]
- 37. Lemieux GA, Cunningham KA, Lin L, Mayer F, Werb Z, Ashrafi K. Kynurenic acid is a nutritional cue that enables behavioral plasticity. Cell 2015;160(1-2):119–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Carr KA, Lin H, Fletcher KD, Sucheston L, Singh PK, Salis RJ, Erbe RW, Faith MS, Allison DB, Stice E et al. Two functional serotonin polymorphisms moderate the effect of food reinforcement on BMI. Behav Neurosci 2013;127(3):387–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Polakof S, Remond D, Bernalier-Donadille A, Rambeau M, Pujos-Guillot E, Comte B, Dardevet D, Savary-Auzeloux I. Metabolic adaptations to HFHS overfeeding: how whole body and tissues postprandial metabolic flexibility adapt in Yucatan mini-pigs. Eur J Nutr 2016;57(1):119–35. [DOI] [PubMed] [Google Scholar]
- 40. Arrieta-Cruz I, Su Y, Gutierrez-Juarez R. Suppression of endogenous glucose production by isoleucine and valine and impact of diet composition. Nutrients 2016;8(2):79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ananieva EA, Van Horn CG, Jones MR, Hutson SM. Liver BCATm transgenic mouse model reveals the important role of the liver in maintaining BCAA homeostasis. J Nutr Biochem 2017;40:132–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lerin C, Goldfine AB, Boes T, Liu M, Kasif S, Dreyfuss JM, De Sousa-Coelho AL, Daher G, Manoli I, Sysol JR et al. Defects in muscle branched-chain amino acid oxidation contribute to impaired lipid metabolism. Mol Metab 2016;5(10):926–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang L, Han J. Branched-chain amino acid transaminase 1 (BCAT1) promotes the growth of breast cancer cells through improving mTOR-mediated mitochondrial biogenesis and function. Biochem Biophys Res Commun 2017;486(2):224–31. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.