

Abstract
The analysis of hybridization and gene flow among closely related taxa is a common goal for researchers studying speciation and phylogeography. Many methods for hybridization detection use simple site pattern frequencies from observed genomic data and compare them to null models that predict an absence of gene flow. The theory underlying the detection of hybridization using these site pattern probabilities exploits the relationship between the coalescent process for gene trees within population trees and the process of mutation along the branches of the gene trees. For certain models, site patterns are predicted to occur in equal frequency (i.e., their difference is 0), producing a set of functions called phylogenetic invariants. In this article, we introduce HyDe, a software package for detecting hybridization using phylogenetic invariants arising under the coalescent model with hybridization. HyDe is written in Python and can be used interactively or through the command line using pre-packaged scripts. We demonstrate the use of HyDe on simulated data, as well as on two empirical data sets from the literature. We focus in particular on identifying individual hybrids within population samples and on distinguishing between hybrid speciation and gene flow. HyDe is freely available as an open source Python package under the GNU GPL v3 on both GitHub (https://github.com/pblischak/HyDe) and the Python Package Index (PyPI: https://pypi.python.org/pypi/phyde).
Keywords: ABBA-BABA, coalescence, gene flow, hybridization, phylogenetic invariants
It is increasingly recognized that a strictly bifurcating model of population descent is inadequate to describe the evolutionary history of many species and is particularly common among plants, as well as some groups of animals (Mallet 2005; Baack and Rieseberg 2007; Mallet 2007). Hybridization and gene flow are processes that obscure this simple model, but separating the signal of admixture from other sources of incongruence, such as incomplete lineage sorting (ILS), is especially difficult (Maddison 1997). Early methods for detecting hybridization in the presence of ILS used estimated gene trees to detect deviations from the coalescent model under the expectation of a bifurcating species tree (Joly et al. 2009; Kubatko 2009; Meng and Kubatko 2009; Gerard et al. 2011). This work was later extended to include searches over network space to infer species networks with reticulate edges (Yu et al. 2011, 2012, 2013, 2014; Solís-Lumis and Ané 2016). Because these network inference methods do not always scale to large numbers of species, other approaches for detecting hybridization that use genome-wide single nucleotide polymorphism (SNP) data to test for admixture on rooted, four- or five-taxon trees have often been employed (“ABBA-BABA”-like methods; Green et al. 2010; Durand et al. 2011; Patterson et al. 2012; Martin et al. 2014; Eaton and Ree 2013; Pease and Hahn 2015). A common feature of these genome-wide methods for hybridization detection is their use of site patterns to test for deviations from the expected frequency under a neutral coalescent model with no introgression (Green et al. 2010; Durand et al. 2011).
The theoretical underpinnings of these site pattern-based inference methods originate from the study of invariants: functions of site pattern probabilities that contain information about the underlying relationships of the sampled species. For example, the D-statistic (Patterson et al. 2012) is based on invariants and uses the site patterns ABBA and BABA, which should occur in equal frequency in the absence of admixture. The earliest use of invariants to infer phylogenies was a pair of articles from Cavender and Felsenstein (1987) and Lake (1987), who separately derived functions to determine the correct topology for an unrooted quartet using binary and nucleotide data, respectively. Recent applications of phylogenetic invariants include proving identifiability (Chifman and Kubatko 2015), coalescent-based species tree inference (SVDquartets; Chifman and Kubatko 2014), sliding-window analyses of phylogenetic bipartitions (SplitSup; Allman et al. 2016), and the detection of hybridization (Green et al. 2010; Durand et al. 2011; Kubatko and Chifman 2015). The increased interest in methods using invariants is concomitant with the ability to collect genomic sequence data, allowing for accurate estimates of site pattern frequencies. Furthermore, because these methods are based on site pattern frequencies, they offer computationally tractable approaches for analyzing genome-scale data sets.
In this article, we introduce HyDe, a Python package for the detection of hybridization using phylogenetic invariants. HyDe automates the detection of hybridization across large numbers of species and can conduct hypothesis tests at both the population and individual levels. A particular advantage of HyDe over other methods is its ability to assess individual-level variation in the amount of hybridization using per-individual hypothesis tests and individual bootstrap resampling in putative hybrid populations. The programming philosophy behind HyDe is to provide a low-level library of data structures and methods for computing site pattern probabilities and conducting hypothesis tests, along with a core set of Python scripts that use this library to automatically parse and analyze genomic data. This setup allows our software to be easily extended as new methods based on site pattern probabilities are developed. We describe the available features of the software in detail below and demonstrate the use of HyDe on both simulated and empirical data sets. To facilitate the use of HyDe for other researchers, we have provided all of the code for processing, plotting, and interpreting the results of these analyses.
DESCRIPTION
Model Background
In this section, we provide a brief overview of the model used by HyDe for detecting hybridization. The theory behind the model, the derivation of the test statistics and corresponding asymptotic distributions, as well as assessments of statistical power and performance for different simulation settings can be found in Kubatko and Chifman (2015).
Consider a rooted, four-taxon network consisting of an outgroup and a triplet of ingroup populations: two parental populations (P1 and P2) and a hybrid population (Hyb) that is a mixture of P1 and P2 (Fig. 1). Under this model, gene trees arise within the parental population trees following the coalescent process (Kingman 1982), where the hybrid population is either (1) sister to P1 with probability 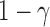 or (2) sister to P2 with probability
or (2) sister to P2 with probability 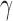 (Meng and Kubatko 2009). Mutations are then placed on these gene trees according to the standard Markov models of nucleotide substitution [e.g., JC69 (Jukes and Cantor 1969), HKY85 (Hasegawa et al. 1985), GTR (Tavaré 1986)]. Given one sampled individual within each population, we can describe a probability distribution on the possible site patterns observed at the tips of the gene trees:
(Meng and Kubatko 2009). Mutations are then placed on these gene trees according to the standard Markov models of nucleotide substitution [e.g., JC69 (Jukes and Cantor 1969), HKY85 (Hasegawa et al. 1985), GTR (Tavaré 1986)]. Given one sampled individual within each population, we can describe a probability distribution on the possible site patterns observed at the tips of the gene trees: 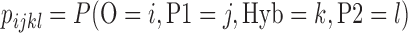 , with
, with 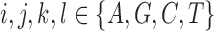 . Using nucleotide sequence data, we can also estimate these probabilities using the observed site patterns to get
. Using nucleotide sequence data, we can also estimate these probabilities using the observed site patterns to get 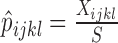 , where
, where 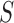 is the total number of observed sites and
is the total number of observed sites and 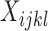 is the number of times that site pattern
is the number of times that site pattern 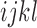 was observed. Implicit in this model is the assumption that each site evolves along its own gene tree, making unlinked sites the most appropriate input data. However, when the number of genes is large, it has been shown through simulations and empirical analyses that multilocus data provide a good approximation for the model and can be used to calculate site pattern probabilities (Chifman and Kubatko 2014; Tian and Kubatko 2017).
was observed. Implicit in this model is the assumption that each site evolves along its own gene tree, making unlinked sites the most appropriate input data. However, when the number of genes is large, it has been shown through simulations and empirical analyses that multilocus data provide a good approximation for the model and can be used to calculate site pattern probabilities (Chifman and Kubatko 2014; Tian and Kubatko 2017).
Fig. 1.

Illustration of the model for detecting hybridization using HyDe. The hybrid population (Hyb) is modeled as a mixture between two parental populations, P1 (1-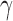 ) and P2 (
) and P2 (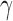 ).
).
These site pattern probabilities form the basis of the test for hybridization that is implemented in HyDe, as well as several other methods. As we noted before, the D-statistic (Patterson et al. 2012) is based on invariants and uses the site pattern probabilities 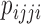 and
and 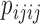 to test for hybridization assuming a null model of no admixture:
to test for hybridization assuming a null model of no admixture:
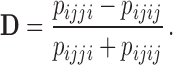 |
(1) |
When admixture is absent, the expected value of 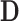 is 0, indicating that
is 0, indicating that 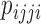 and
and 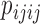 should occur in equal frequency (i.e., they are invariant).
should occur in equal frequency (i.e., they are invariant).
In a similar way, Kubatko and Chifman (2015) presented several different invariants-based test statistics that detect hybridization using linear phylogenetic invariants. Among the invariants that they tested, the ratio of 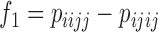 and
and 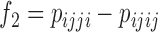 provided the most statistical power and is used to form the test statistic that is used by HyDe to detect hybridization. When the model holds, it can be shown that
provided the most statistical power and is used to form the test statistic that is used by HyDe to detect hybridization. When the model holds, it can be shown that
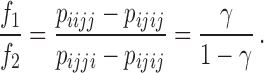 |
(2) |
Deviation of the observed frequencies from this expectation is used to form a test statistic that asymptotically follows a standard Normal distribution, allowing formal hypothesis tests to be conducted (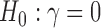 vs.
vs. 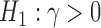 ). Furthermore, because the ratio of
). Furthermore, because the ratio of 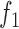 and
and 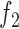 is a function of
is a function of 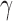 , we can estimate the amount of admixture directly from the observed site pattern frequencies:
, we can estimate the amount of admixture directly from the observed site pattern frequencies:
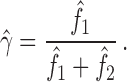 |
(3) |
Software Features
In this section, we outline the main features of the HyDe software. Previously, Kubatko and Chifman (2015) used C code to test their new methods and distributed it in accordance with standard publishing practices. However, the code was only intended to evaluate performance for their method of hybridization detection, and not as a “distribution-level” piece of software. The new Python package that we present here serves that purpose, and provides additional functionality to accommodate more complex hybridization scenarios and sampling designs.
Multiple individuals per population.
We have modified the calculation of the site pattern probabilities originally presented in Kubatko and Chifman (2015) to accommodate multiple individuals per population, rather than testing each sampled individual separately. We do this by considering all permutations of the individuals in the four populations involved in the hypothesis test. For example, if there are 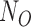 individuals in the outgroup,
individuals in the outgroup, 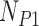 individuals in parental population one,
individuals in parental population one, 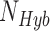 individuals in the putative hybrid population, and
individuals in the putative hybrid population, and 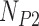 individuals in parental population two, then a total of
individuals in parental population two, then a total of 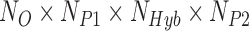 quartets are used to calculate the site patterns for the hypothesis test. Including multiple individuals per population increases the sample size used to calculate the site pattern probabilities and can therefore lead to more accurate detection of hybridization.
quartets are used to calculate the site patterns for the hypothesis test. Including multiple individuals per population increases the sample size used to calculate the site pattern probabilities and can therefore lead to more accurate detection of hybridization.
Identifying individual hybrids.
An underlying assumption of the coalescent with hybridization is that all the individuals in the hybrid population are admixed (i.e., hybrid speciation; Meng and Kubatko 2009). However, when gene flow, rather than hybrid speciation, is responsible for the introgression of genetic material into the admixed population, it is possible that not all individuals will contain these introgressed alleles. When hybridization detection is conducted on populations with a mix of hybrids and non-hybrids, it is possible for invariant-based tests to report significant results, even though the underlying assumption of uniform admixture is violated. To help with the detection of non-uniform introgression into the hybrid population, we have included two methods in HyDe that aim to detect variation in the amount of hybridization in the individuals sampled.
If not all of the individuals in a putative hybrid population are admixed, bootstrap resampling can reveal heterogeneity in the process of introgression when more or fewer hybrid individuals are included in each replicate. For example, if we are testing four diploid individuals in a putative hybrid and only two of them are 50:50 hybrids (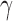 = 0.5 for each), then the value of gamma for the whole population will be
= 0.5 for each), then the value of gamma for the whole population will be 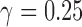 . When we resample individuals with replacement and recalculate
. When we resample individuals with replacement and recalculate 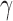 , we will get different values depending on how many times the hybrids are resampled (0 times:
, we will get different values depending on how many times the hybrids are resampled (0 times: 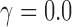 ; 1 time:
; 1 time: 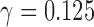 ; 2 times:
; 2 times: 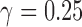 ; 3 times:
; 3 times: 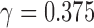 ; 4 times:
; 4 times: 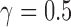 ). Because the process of hybridization is not uniform, the value of
). Because the process of hybridization is not uniform, the value of 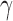 jumps between different values. This can also be visualized by plotting the distribution of
jumps between different values. This can also be visualized by plotting the distribution of 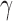 across the bootstrap replicates (e.g., Fig. 2).
across the bootstrap replicates (e.g., Fig. 2).
We have also implemented methods that test all individuals in a putative hybrid separately while treating the parents and outgroup as populations. This approach allows hypothesis tests and estimates of 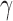 to be calculated for every individual to see if it is a hybrid. A caveat with testing each individual is that the number of sites sampled must be enough to have statistical power to detect that hybridization has occurred (Kubatko and Chifman 2015).
to be calculated for every individual to see if it is a hybrid. A caveat with testing each individual is that the number of sites sampled must be enough to have statistical power to detect that hybridization has occurred (Kubatko and Chifman 2015).
Ambiguity codes and missing data.
To allow more data to be used, and to account for the potentially large amounts of missing data that are common in high throughput sequencing data sets, we have implemented approaches that integrate over missing or ambiguous nucleotides by considering the possible resolutions of the observed bases into site patterns. We note that this approach is also appropriate for high coverage data sets when an individual is heterozygous at a particular locus, but a consensus sequence with an ambiguity code is used to represent the different bases of the heterozygous genotype. As an example, consider the site pattern AGRG. There are two possible resolutions for the ambiguity code R: AGGG and AGAG. To account for this, we add 0.5 to the site pattern counts 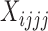 and
and 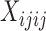 . In general, for any site pattern containing ambiguous or missing bases (but not gaps), we find all possible resolutions and add one divided by the total number of resolutions to the corresponding site pattern counts.
. In general, for any site pattern containing ambiguous or missing bases (but not gaps), we find all possible resolutions and add one divided by the total number of resolutions to the corresponding site pattern counts.
Software Design and Implementation
HyDe is implemented in Python and also uses Cython, a superset of the Python language that allows C/C++ capabilities to be incorporated into Python for better efficiency (Behnel et al. 2010). Installing HyDe requires several external Python modules [numpy, cython, matplotlib, multiprocess, and seaborn]. The goal behind our implementation of HyDe was to provide both pre-packaged scripts to conduct standard hybridization detection analyses, as well as a library of functions and data structures that researchers could use to customize their analyses or implement new tests based on site pattern probabilities. Below, we describe the functionality of both of these interfaces, as well as the input files required to run HyDe.
Input files.
The input files for running HyDe are plain text files containing the DNA sequence data and the map of individuals to populations. The DNA sequence data are expected to be in sequential Phylip format and the population map is a two-column table with individual names in the first column and the name of the population that it belongs to in the second column. The third input file that is required for individual-level testing and bootstrapping (optional for running a normal HyDe analysis) is a three column table of triplets. A hypothesis test is then run for each triplet using the first column as parent one, the second column as the putative hybrid, and the third column as parent two. The outgroup for each test is always the same and is specified separately at the command line. Output files from previous HyDe analyses can also be used to specify which triplets are to be tested.
Command line interface.
The command line interface for HyDe consists of a set of Python scripts that can be used to detect hybridization, filter results, conduct individual bootstrapping, and test for hybridization at the individual level. The three main scripts for detecting hybridization with HyDe provide the majority of the basic functionality that is needed to detect hybridization in an empirical data set and to assess if individuals in putative hybrids are all equally admixed. Each of these scripts also has a multithreaded version for parallelizing hypothesis tests across triplets (in square brackets below).
run_hyde.py [run_hyde_mp.py]: The run_hyde.py script detects hybridization by running a hypothesis test on all possible triplets in a data set in all directions (i.e., a “full” HyDe analysis). There is also an option to supply a list of specific triplets (three column text file) to test. The script outputs two files: one with all of the hypothesis tests that were conducted and one with only those hypothesis tests that were significant at an overall
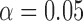 level (after incorporating a Bonferonni correction) with estimates of
level (after incorporating a Bonferonni correction) with estimates of 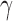 between 0 and 1.
between 0 and 1.individual_hyde.py [individual_hyde_mp.py]: The individual_hyde.py script runs separate hybridization detection analyses on each individual within a putative hybrid lineage. The only additional input needed for the script is a three column list of triplets or a filtered results file from the run_hyde.py script.
bootstrap_hyde.py [bootstrap_hyde_mp.py]: The bootstrap_hyde.py script performs bootstrap resampling of the individuals within a putative hybrid lineage and conducts a hypothesis test for each bootstrap replicate. Similar to the individual_hyde.py script, the script uses as input a table of triplets or a filtered results file from a previous hybridization detection analysis.
The workflow that we envision for these scripts starts with using the run_hyde.py script to test for hybridization on all triplets in all possible directions, and to filter out only those triplets that have significant evidence for hybridization. Then, using the filtered output file, users can either test all of the individuals in each hybrid lineage or perform bootstrap resampling of the individuals using the individual_hyde.py and bootstrap_hyde.py scripts, respectively. Users with specific hypotheses that they want to test can simply create a text file listing the triplets of interest to be run through any of the three scripts.
Python API.
Each of the command line scripts described above makes use of an underlying Python library with built-in data structures for processing the hypothesis tests and bootstrapping analyses conducted by HyDe. This library is exposed through the Python module phyde (Pythonic Hybridization Detection). The main data structures that are part of this module are listed below:
HydeData: The HydeData class is the primary data structure for reading in and storing DNA sequence data and maps assigning individuals to populations. This class also implements three of the main methods for detecting hybridization at the population level [test_triple()] and the individual level [test_individuals()], as well as bootstrapping individuals within populations [bootstrap_triple()] (see Box 1).
HydeResult: The HydeResult class parses the results file output by a hybridization detection analysis and stores the results as a Python dictionary (key-value pair). Values stored by the HydeResult class can be accessed by providing the name of the desired value and the names of the taxa in the triplet of interest as arguments to the variable used when reading in the results. For example, if we read the results of a hybridization detection analysis into a variable named results, we would access the estimated value of
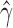 for the triplet (sp1, sp2, sp3) using the following code: results("Gamma", "sp1", "sp2", "sp3")!.
for the triplet (sp1, sp2, sp3) using the following code: results("Gamma", "sp1", "sp2", "sp3")!.Bootstrap: The Bootstrap class is similar to the HydeResult class except that it has built-in methods for parsing the format of the bootstrap output file written by the bootstrap_hyde.py script. Each bootstrap replicate is printed with a single line containing four pound symbols separating each tested triplet (####\n). These results are parsed into a Python dictionary that can be used to access the bootstrap replicates for particular triplets using the variable name, just like the HydeResult class [e.g., bootstraps("Gamma", "sp1", "sp2", "sp3")].
phyde.viz: The viz submodule uses the matplotlib and seaborn libraries to implement basic functions for plotting the distribution of bootstrap replicates for any quantity calculated by the bootstrap_hyde.py script (Z-score, p-value,
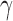 , etc.) for a specified triplet. It does this by taking the name of a Bootstrap object variable, the name of the value to be plotted, and the names of the taxa in the triplet of interest (Supplemental Materials: HyDe_SysBio.ipynb).
, etc.) for a specified triplet. It does this by taking the name of a Bootstrap object variable, the name of the value to be plotted, and the names of the taxa in the triplet of interest (Supplemental Materials: HyDe_SysBio.ipynb).
Box 1.
Example code to run hybridization detection analyses with HyDe using the Python API. The phyde module is loaded using the import command:
# import the phyde module
import phyde
Data are read in and stored using the HydeData class by passing the names of the input file, map file, and outgroup, as well as the number of individuals, the number of populations, and the number of sites.
# read in data using HydeData class
data = phyde.HydeData("data.txt", "map.txt",
"out", 16, 4, 50000)
With the data read in, we can use methods implemented in the HydeData class to test for hybridization at the population level [test_triple(p1, hyb, p2)], at the individual level [test_individuals(p1, hyb, p2)], and can bootstrap resample individuals [bootstrap_triple(p1, hyb, p2, nreps)].
# test for hybridization in the "sp2" population
res1 = data.test_triple("sp1", "sp2", "sp3")
# test all individuals in the "sp2" population
res2 = data.test_individuals("sp1", "sp2", "sp3")
# bootstrap individuals in the "sp2" population 200 times
res3 = data.bootstrap_triple("sp1", "sp2", "sp3", 200)
BENCHMARKS
All code used to complete the simulations and example analyses are available as a Jupyter Notebook on Dryad (http://dx.doi.org/10.5061/dryad.372sq). We have also included all output files needed to reproduce the tables and figures. Note that in this section we use the names out, sp1, sp2, and sp3 for the names of the populations, rather than O, P1, Hyb, and P2. We do this to denote that these taxa have not yet been tested for hybridization, whereas the model is explicitly constructed to contain a hybrid species and its parents.
Non-Uniform Hybridization Simulations
To demonstrate the use of the methods in HyDe that are designed to identify individual hybrids, we set up a simulated example for four taxa (out, sp1, sp2, sp3) that intentionally violated the assumption of hybrid speciation by including a single hybrid individual in the population being tested for admixture (sp2). We simulated gene trees using the program ms (Hudson 2002) with population branch lengths of 1.0 coalescent unit assuming a model of coalescent independent sites (i.e., one gene tree per site; Kubatko and Chifman 2015) for 10,000, 50,000, 100,000, 250,000, and 500,000 sites. The parental populations were simulated with five individuals each and the outgroup had only one individual. The “admixed” population (sp2) had four non-admixed individuals that were most closely related to parental population sp1, and a single hybrid individual that was a 50:50 (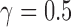 ) mix between sp1 and sp3. A single DNA base was simulated on each gene tree for the different numbers of sites using the program Seq-Gen (Rambaut and Grassly 1997). We rescaled gene tree branch lengths from coalescent units to mutation units using a population scaled mutation rate (
) mix between sp1 and sp3. A single DNA base was simulated on each gene tree for the different numbers of sites using the program Seq-Gen (Rambaut and Grassly 1997). We rescaled gene tree branch lengths from coalescent units to mutation units using a population scaled mutation rate (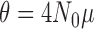 ) of 0.01 per site and used a GTR+Gamma model of nucleotide substitution with three rate categories:
) of 0.01 per site and used a GTR+Gamma model of nucleotide substitution with three rate categories:
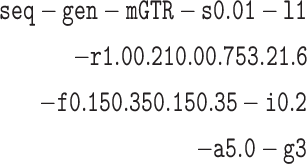 |
Output from Seq-Gen was formatted for input to HyDe using a Python script (seqgen2hyde.py; available on Dryad). Hybridization detection was completed for the simulated data sets with HyDe v0.4.1 using the run_hyde.py, bootstrap_hyde.py (500 bootstrap replicates), and individual_hyde.py scripts. Output files were processed and plotted in Python v2.7 using the phyde, matplotlib, and seaborn modules.
Results.
Testing for hybridization at the population level using the run_hyde.py script produced a test statistic indicating that there was significant admixture in the sp2 population at the 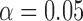 level when the number of sites was at least 50,000 (Table 1). The estimated values of
level when the number of sites was at least 50,000 (Table 1). The estimated values of 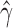 for these tests were close to 0.9, which is inconsistent with the data that we simulated. The value of
for these tests were close to 0.9, which is inconsistent with the data that we simulated. The value of 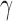 that we would expect is 0.1. However, because this is an explicit violation of the coalescent model with hybridization, the formula for the estimation of
that we would expect is 0.1. However, because this is an explicit violation of the coalescent model with hybridization, the formula for the estimation of 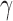 is no longer valid.
is no longer valid.
Table 1.
Results of the population-level hybridization detection analyses for the non-uniform hybridization simulation using the run_hyde.py script
| # sites | Z-score | P-value |
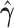
|
|---|---|---|---|
10,000
|
1.100 | 0.136 | 0.896 |
| 50,000 | 2.234 | 0.013 | 0.897 |
| 100,000 | 3.274 |
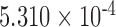
|
0.900 |
| 250,000 | 5.420 |
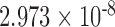
|
0.896 |
| 500,000 | 6.808 |
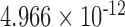
|
0.905 |
Note that the result for the simulation with 10,000 sites was not significant at the 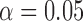 level (marked with
level (marked with  ).
).
If we did not already know that not all of the individuals were hybrids, we would mistakenly infer that the sp2 population has 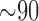 % of its genetic material inherited from population sp3. However, when we test each individual using the individual_hyde.py script, we correctly infer that only one of the individuals is admixed (individual ‘i10’,
% of its genetic material inherited from population sp3. However, when we test each individual using the individual_hyde.py script, we correctly infer that only one of the individuals is admixed (individual ‘i10’, 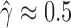 ) and that the rest of the individuals are not hybrids (Table 2). Bootstrap resampling of the individuals in population sp2 also indicates that hybridization is not uniform. Figure 2 shows the distribution of
) and that the rest of the individuals are not hybrids (Table 2). Bootstrap resampling of the individuals in population sp2 also indicates that hybridization is not uniform. Figure 2 shows the distribution of 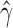 across all 500 bootstrap replicates and demonstrates that the value of
across all 500 bootstrap replicates and demonstrates that the value of 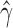 jumps between different values depending on the number of times the hybrid individual is resampled.
jumps between different values depending on the number of times the hybrid individual is resampled.
Table 2.
Results of the individual-level hybridization detection analyses for the non-uniform hybridization simulation using the individual_hyde.py script
| 10,000 | 50,000 | 100,000 | 250,000 | 500,000 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Individual | Z-score | P-value |
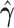
|
Z-score | P-value |
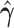
|
Z-score | P-value |
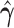
|
Z-score | P-value |
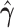
|
Z-score | P-value |
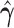
|
| i6 | 0.408 | 0.342 | n.s. | 0.054 | 0.478 | n.s. | 0.049 | 0.480 | n.s. | 0.387 | 0.349 | n.s. |
 0.015 0.015 |
0.506 | n.s. |
| i7 | 0.241 | 0.405 | n.s. |
 0.295 0.295 |
0.616 | n.s. |
 0.289 0.289 |
0.614 | n.s. | 0.322 | 0.374 | n.s. | 0.012 | 0.496 | n.s. |
| i8 | 1.128 | 0.130 | n.s. |
 0.994 0.994 |
0.840 | n.s. | 0.106 | 0.458 | n.s. | 0.773 | 0.220 | n.s. |
 0.275 0.275 |
0.609 | n.s. |
| i9 | 0.711 | 0.239 | n.s. |
 0.445 0.445 |
0.672 | n.s. | 0.554 | 0.290 | n.s. | 0.734 | 0.231 | n.s. |
 0.130 0.130 |
0.552 | n.s. |
| i10 | 2.267 | 0.012 | 0.562 | 6.464 |
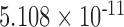
|
0.450 | 9.011 |
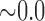
|
0.497 | 14.015 |
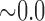
|
0.497 | 20.114 |
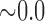
|
0.508 |
n.s. = not significant. Individual ‘i10’ was the only hybrid in the population and was simulated with 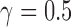 .
.
Fig. 2.
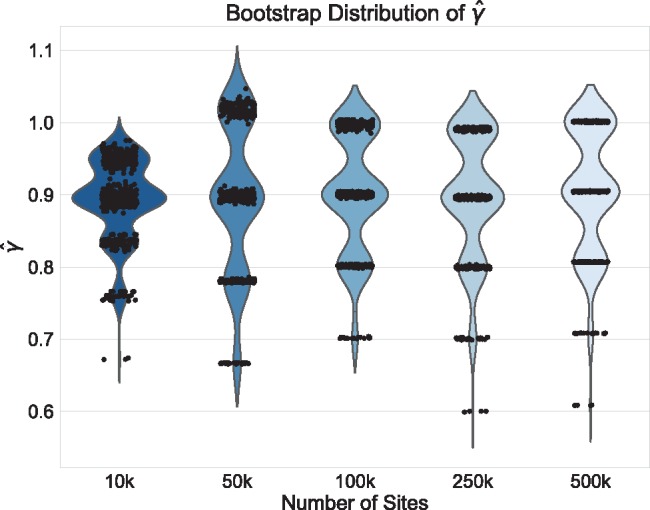
Violin plots of the distribution of 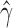 across 500 bootstrap replicates for 10,000, 50,000, 100,000, 250,000, and 500,000 sites for the non-uniform hybridization simulations. The black dots in each violin plot represent the actual values of
across 500 bootstrap replicates for 10,000, 50,000, 100,000, 250,000, and 500,000 sites for the non-uniform hybridization simulations. The black dots in each violin plot represent the actual values of 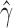 that were estimated by each replicate (black dots are jittered). The pattern of jumping between distinct values of
that were estimated by each replicate (black dots are jittered). The pattern of jumping between distinct values of 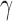 with no estimates in between are a strong indication of non-uniform admixture.
with no estimates in between are a strong indication of non-uniform admixture.
Validating Population-Level Hybridization Detection
To contrast with the previous simulation, and to validate hybridization at the population level, we repeated the same procedure as above for simulating DNA sequence data for four taxa, but this time all individuals in the sp2 population were 10% admixed. This maintains the overall level of 10% admixture in the population, but now it is distributed evenly among the individuals. We then ran a hybridization detection analysis on these simulated data using the run_hyde.py, bootstrap_hyde.py (500 replicates), and individual_hyde.py scripts.
Results.
At the population level, the analysis with run_hyde.py detected significant hybridization (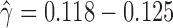 ) in all cases except for the simulation with 10,000 sites (Table 3). For this scenario, there was not enough data to detect the hybridization at the
) in all cases except for the simulation with 10,000 sites (Table 3). For this scenario, there was not enough data to detect the hybridization at the 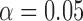 level. Analyzing each individual in the sp2 population correctly detected hybridization in all individuals, with estimates of
level. Analyzing each individual in the sp2 population correctly detected hybridization in all individuals, with estimates of 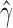 slightly over the original level of 10% admixture (Table 4). Bootstrap resampling of the individuals in the sp2 population also demonstrated that hybridization was uniform, as can be seen by the unimodal distribution of
slightly over the original level of 10% admixture (Table 4). Bootstrap resampling of the individuals in the sp2 population also demonstrated that hybridization was uniform, as can be seen by the unimodal distribution of 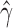 values across bootstrap replicates in Fig. 3.
values across bootstrap replicates in Fig. 3.
Table 3.
Results of the population-level hybridization detection analyses for the 10% admixture simulation using the run_hyde.py script
| # sites | Z-score | P-value |
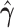
|
|---|---|---|---|
10,000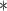
|
0.071 | 0.472 | 0.008 |
| 50,000 | 2.363 | 0.009 | 0.125 |
| 100,000 | 3.677 |
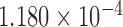
|
0.125 |
| 250,000 | 5.796 |
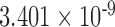
|
0.119 |
| 500,000 | 8.425 |
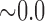
|
0.118 |
Note that the result for the simulation with 10,000 sites was not significant at the 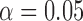 level (marked with
level (marked with 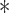 ).
).
Table 4.
Results of the individual-level hybridization detection analyses for the 10% admixture simulation using the individual_hyde.py script
| 10 000 | 50 000 | 100 000 | 250 000 | 500 000 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Individual | Z-score | P-value |
|
Z-score | P-value |
|
Z-score | P-value |
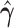
|
Z-score | P-value |
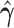
|
Z-score | P-value |
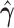
|
|
| i6 | 0.092 | 0.463 | n.s. | 2.141 | 0.016 | 0.117 | 3.967 |
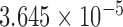
|
0.133 | 5.545 |
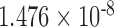
|
0.116 | 8.151 |
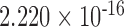
|
0.116 | |
| i7 | 0.089 | 0.464 | n.s. | 2.542 | 0.006 | 0.132 | 4.397 |
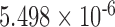
|
0.144 | 6.795 |
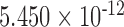
|
0.136 | 8.092 |
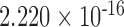
|
0.115 | |
| i8 | 0.2457 | 0.399 | n.s. | 2.155 | 0.016 | 0.113 | 3.326 |
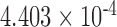
|
0.117 | 5.421 |
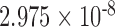
|
0.110 | 8.682 |
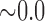
|
0.122 | |
| i9 | – | – | – | 2.663 | 0.004 | 0.132 | 2.458 | 0.007 | 0.091 | 5.538 |
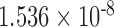
|
0.115 | 8.637 |
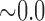
|
0.119 | |
| i10 | – | – | – | 2.315 | 0.010 | 0.128 | 4.217 |
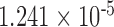
|
0.135 | 5.664 |
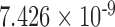
|
0.118 | 8.563 |
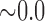
|
0.119 | |
n.s. = not significant. All individuals were equally admixed with 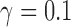 .
.
Fig. 3.
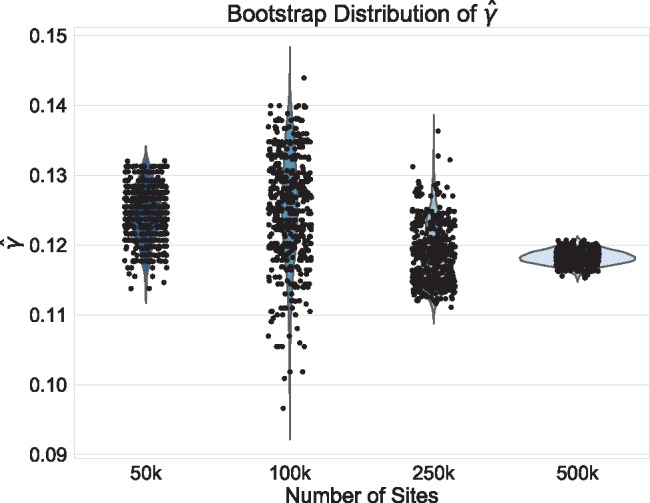
Violin plots of the distribution of 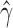 across 500 bootstrap replicates for 50,000, 100,000, 250,000, and 500,000 sites for the uniform hybridization simulations. The black dots in each violin plot represent the actual values of
across 500 bootstrap replicates for 50,000, 100,000, 250,000, and 500,000 sites for the uniform hybridization simulations. The black dots in each violin plot represent the actual values of 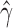 that were estimated by each replicate (black dots are jittered). The distribution of the estimates across bootstrap samples indicates that hybridization is uniform. The analyses with 10,000 sites did not have enough statistical power to detect hybridization and were therefore left out of the plot.
that were estimated by each replicate (black dots are jittered). The distribution of the estimates across bootstrap samples indicates that hybridization is uniform. The analyses with 10,000 sites did not have enough statistical power to detect hybridization and were therefore left out of the plot.
BIOLOGICAL EXAMPLES
Heliconius Butterflies
DNA sequence data from Martin et al. (2013) was downloaded for four populations of Heliconius butterflies (248,822,400 sites; available on Dryad). The number of individuals per population was as follows: four individuals of H. melpomene rosina, four individuals of H. melpomene timareta, four individuals of H. cydno, and one individual of H. hecale. We tested the hypothesis that H. cydno is a hybrid between H. melpomene rosina and H. melpomene timareta with H. hecale as an outgroup using the run_hyde.py script. We then conducted bootstrapping (500 replicates) of the H. cydno individuals, as well as testing each individual separately using the bootstrap_hyde.py and individual_hyde.py scripts, respectively. As a last comparison, we reanalyzed the data with the run_hyde.py script, but changed the underlying code so that all missing or ambiguous sites were ignored in order to test the effect of including/excluding such data.
Results.
Significant hybridization was detected in H. cydno at the population level (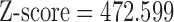 ,
, 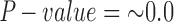 ,
, 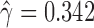 ). Each individual also showed significant levels of hybridization, with
). Each individual also showed significant levels of hybridization, with 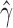 ranging from 0.324 to 0.393, indicating that hybridization in H. cydno is mostly uniform across the individuals sampled. Figure 4 shows a density plot for the estimated values of
ranging from 0.324 to 0.393, indicating that hybridization in H. cydno is mostly uniform across the individuals sampled. Figure 4 shows a density plot for the estimated values of 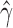 across the 500 bootstrap replicates. Although the distribution does exhibit some amount of jumping between different values they are still between the lower and upper bounds for the individual level estimates of
across the 500 bootstrap replicates. Although the distribution does exhibit some amount of jumping between different values they are still between the lower and upper bounds for the individual level estimates of 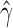 , providing corroborating evidence that all the individuals in the H. cydno population are admixed. When we ignored missing or ambiguous sites, we still detected significant hybridization, but the value of the test statistic was less than half of the value from the original analysis (
, providing corroborating evidence that all the individuals in the H. cydno population are admixed. When we ignored missing or ambiguous sites, we still detected significant hybridization, but the value of the test statistic was less than half of the value from the original analysis (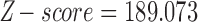 ,
, 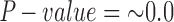 ,
, 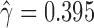 ). This demonstrates the increase in power that can result from including all relevant data in the analysis.
). This demonstrates the increase in power that can result from including all relevant data in the analysis.
Fig. 4.
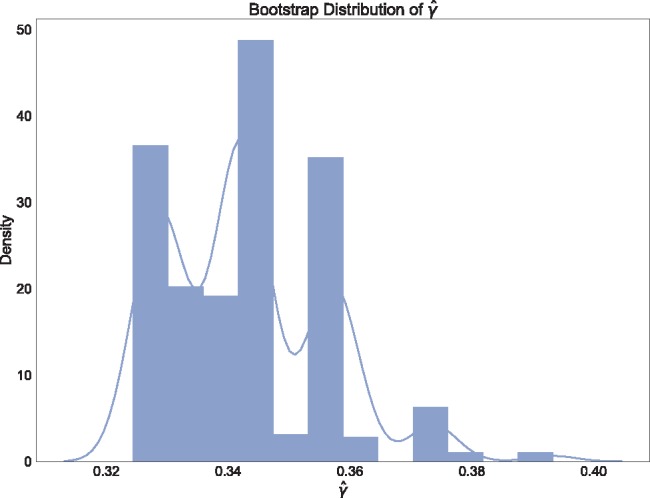
Density plot of the estimated values of 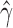 across 500 bootstrap replicates testing for hybridization in Heliconius cydno between H. melpomene rosina and H. melpomene timareta.
across 500 bootstrap replicates testing for hybridization in Heliconius cydno between H. melpomene rosina and H. melpomene timareta.
Swordtail Fish (Xiphophorous)
Transcriptome data from Cui et al. (2013) was obtained from the authors for 26 species of swordtail fish (25 species of Xiphophorous + 1 outgroup from the genus Priapella). The data (3,706,285 sites) were first put in sequential Phylip format for analysis with HyDe. Each taxon was then analyzed at the individual level, resulting in 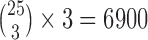 hypothesis tests. Because there were not multiple individuals per taxon, we only analyzed these data using the run_hyde.py script. As with the Heliconius example, we also reanalyzed these data ignoring all sites with missing or ambiguous bases.
hypothesis tests. Because there were not multiple individuals per taxon, we only analyzed these data using the run_hyde.py script. As with the Heliconius example, we also reanalyzed these data ignoring all sites with missing or ambiguous bases.
Results.
Out of 6900 tests, 2199 reported significant levels of hybridization (Bonferroni corrected P-value of 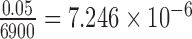 ). When ambiguous or missing bases were ignored, only 867 tests were reported as significant, again demonstrating the increase in power to detect hybridization when including these sites. The distribution of the
). When ambiguous or missing bases were ignored, only 867 tests were reported as significant, again demonstrating the increase in power to detect hybridization when including these sites. The distribution of the 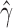 values for the 2199 significant tests is plotted in Fig. 5. This plot shows that most of the admixture occurring in Xiphophorous is at low levels (
values for the 2199 significant tests is plotted in Fig. 5. This plot shows that most of the admixture occurring in Xiphophorous is at low levels (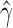 close to either 0.0 or 1.0). However, there are many instances of higher levels of admixture, which matches the findings of Cui et al. (2013) regarding the extensive amount of gene flow and introgression in this group. Solís-Lumis and Ané (2016) also analyzed these data and found evidence for an ancestral hybridization event between X. xiphidium and the northern swordtail clade (denoted NS in Solís-Lumis and Ané (2016), Fig. 10), a pattern we recover as well (259 significant hypothesis tests involving X. xiphidium).
close to either 0.0 or 1.0). However, there are many instances of higher levels of admixture, which matches the findings of Cui et al. (2013) regarding the extensive amount of gene flow and introgression in this group. Solís-Lumis and Ané (2016) also analyzed these data and found evidence for an ancestral hybridization event between X. xiphidium and the northern swordtail clade (denoted NS in Solís-Lumis and Ané (2016), Fig. 10), a pattern we recover as well (259 significant hypothesis tests involving X. xiphidium).
Fig. 5.
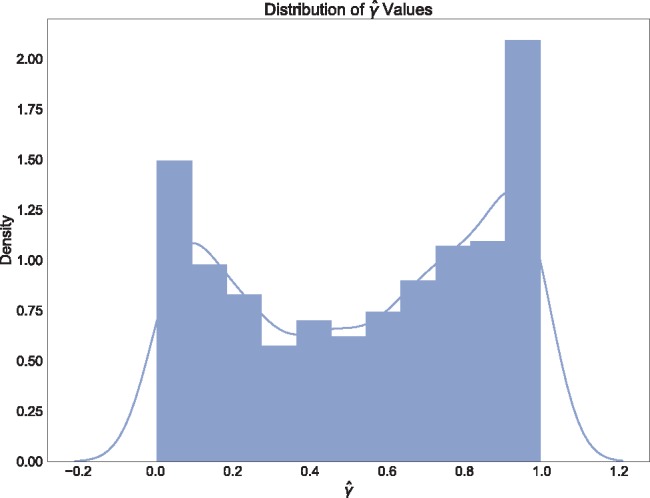
Density plot of the estimated values of  across the 2199 hypothesis tests that detected significant levels of hybridization in Xiphophorous.
across the 2199 hypothesis tests that detected significant levels of hybridization in Xiphophorous.
Timing
We timed all analyses for the Heliconius and swordtail data using the Unix time command. For Heliconius, the analysis using the run_hyde.py script took 11m 49.377s, and the analysis that ignored missing or ambiguous sites took 4m 47.667s. Analyzing the four individuals of H. cydno separately with the individual_hyde.py script took 10m 5.120s. Completing 500 bootstrap replicates for this data set ran the longest of any analysis that we conducted, taking a total of 4105m 33.312s (2.851 days). For the swordtail data, the analysis with the run_hyde.py script took 22m 40.673s, almost twice as long as the analysis for the Heliconius data. This indicates that analyzing more triplets is more computationally intensive than analyzing more sites. When ambiguous or missing sites were ignored, the analysis took 5m 27.889s.
CONCLUSIONS
Hybridization and gene flow are increasingly recognized as important forces in the evolution of taxa across the Tree of Life (Seehausen 2004; Arnold and Kunte 2017; Kagawa and Takimoto 2017). In addition, for groups such as plants, fish, and amphibians, hybridization can also be accompanied by whole genome duplication (allopolyploidy; Otto and Whitton 2000). Kamneva et al. (2017) recently used methods based on the coalescent with hybridization to model haplotypes in allopolyploid strawberries, and we anticipate that approaches using site pattern probabilities should be equally applicable in such situations. Gaining an understanding of when these complex processes are occurring and how they may vary among individuals within a population are important steps to understand the spatial distribution of shared genetic variation among diverging lineages. The methods we have implemented in HyDe to detect hybridization at the population and individual level provide a set of computationally efficient methods for researchers to assess patterns of admixture in natural populations. HyDe also provides an open source Python library for the calculation of site pattern probabilities that can easily be extended to calculate additional statistics that may be developed in the future. As access to genomic data continues to grow, we anticipate that methods such as HyDe that use phylogenetic invariants will play an important role for phylogenomic inferences in non-model species.
AVAILABILITY
HyDe is available as an open source Python package distributed under the GNU General Public License v3 for both Python 2.7 and 3.6. Developmental code can be found on GitHub (https://github.com/pblischak/HyDe) with official code releases uploaded to the Python Package Index (PyPI: https://pypi.python.org/pypi/phyde). Documentation for installing and running HyDe can be found on ReadTheDocs (http://hybridization-detection.readthedocs.io).
SUPPLEMENTARY MATERIALS
Data available from the Dryad Digital Repository: http://dx.doi.org/10.5061/dryad.372sq.
ACKNOWLEDGEMENTS
The authors thank Cécile Ané and Frank Burbink for the invitation to be a part of this special issue on the impact of gene flow and reticulation in phylogenetics, as well as David Posada and three anonymous reviewers for comments that improved the manuscript. We also thank R. Cui, C. Solís-Lemus, and C. Ané for their help with the Xiphophorous data set.
FUNDING
This work was supported in part by grants from the National Science Foundation under the awards DMS-1106706 (J.C. and L.S.K.) and DEB-1455399 (A.D.W. and L.S.K.), and National Institutes of Health Cancer Biology Training Grant T32-CA079448 at Wake Forest School of Medicine (J.C.).
References
- Allman E.S.,, Kubatko L.S.,, Rhodes J.A. 2016. Split scores: a tool to quantify phylogenetic signal in genome-scale data. Syst. Biol. 66:620–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold M.J.,, Kunte K. 2017. Adaptive genetic exchange: a tangled history of admixture and evolutionary innovation. Trends Ecol. Evol. 32:601–611. [DOI] [PubMed] [Google Scholar]
- Baack E.J.,, Rieseberg L.H. 2007. A genomic view of introgression and hybrid speciation. Curr. Opin. Genet. Dev. 17:513–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behnel S.,, Bradshaw R.,, Citro C.,, Dalcin L.,, Seljebotn D.,, Smith K. 2010. Cython: the best of both worlds. Comput. Sci. Eng. 13:31–39. [Google Scholar]
- Cavender J.A.,, Felsenstein J. 1987. Invariants of phylogenies in a simple case with discrete states. J. Classification 4:57–71. [Google Scholar]
- Chifman J.,, Kubatko L.S. 2014. Quartet inference from SNP data under the coalescent model. Bioinformatics 30:3317–3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chifman J.,, Kubatko L.S. 2015. Identifiability of the unrooted species tree topology under the coalescent model with time-reversible substitution processes, site-specific rate variation, and invariable sites. J. Theor. Biol. 374:35–47. [DOI] [PubMed] [Google Scholar]
- Cui R.,, Schumer M.,, Kruesi K.,, Walter R.,, Andolfatto P.,, Rosenthal G.G. 2013. Phylogenomics reveals extensive reticulate evolution in Xiphophorus fishes. Evolution 67:2166–2179. [DOI] [PubMed] [Google Scholar]
- Durand E.Y.,, Patterson N.,, Reich D.,, Slatkin M. 2011. Testing for ancient admixture between closely related populations. Mol. Biol. Evol. 28:2239–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton D.A.R.,, Ree R.H. 2013. Inferring phylogeny and introgression using RADseq data: an example from the flowering plants (Pedicularis: Orobanchaceae). Syst. Biol. 62:689–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerard D.,, Gibbs H.L.,, Kubatko L.S. 2011. Estimating hybridization in the presence of coalescence using phylogenetic intraspecific sampling. BMC Evol. Biol. 11:291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green R.E.,, Krause, J.,, Briggs A.W.,, Maricic T.,, Stenzel U.,, Kircher M.,, Patterson N.,, Li H.,, Zhai W.,, Fritz M.H.-Y.,, Hansen N.F.,, Durand E.Y.,, Malaspinas A.-S.,, Jensen J.D.,, Marques-Bonet T.,, Alkan C.,, Prüfer K.,, Meyer M.,, Burbano H.A.,, Good J.M.,, Schultz R.,, Aximu-Petri A.,, Butthof A.,, Höber B.,, Höffner B.,, Siegemund M.,, Weihmann A.,, Nusbaum C.,, Lander E.S.,, Russ C.,, Novod N.,, Affourtit J.,, Egholm M.,, Verna C.,, Rudan P.,, Brajkovic D.,, Kucan Ž,, Gušic I.,, Doronichev V.B.,, Golovanova L.V.,, Lalueza-Fox C.,, de la Rasilla M.,, Fortea J.,, Rosas A.,, Schmitz R.W.,, Johnson P.L.F.,, Eichler E.E.,, Falush D.,, Birney E.,, Mullikin J.C.,, Slatkin M.,, Nielsen R.,, Kelso J.,, Lachmann M.,, Reich D.,, Pääbo S. 2010. A draft sequence of the Neandertal genome. Science 328:710–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa M.,, Kishino H.,, Yano T. 1985. Dating of human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 22:160–174. [DOI] [PubMed] [Google Scholar]
- Hudson R.R. 2002. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics 18:337–338. [DOI] [PubMed] [Google Scholar]
- Joly S.,, McLenachan P.A.,, Lockhart P.J. 2009. A statistical approach for distinguishing hybridization and incomplete lineage sorting. Am. Nat. 174:E54–E70. [DOI] [PubMed] [Google Scholar]
- Jukes T.H.,, Cantor C.R. 1969. Evolution of protein molecules. In: Monroe H., editor. Mammalian protein metabolism. New York: Academic Press. p. 21–132. [Google Scholar]
- Kagawa K.,, Takimoto, G. 2017. Hybridization can promote adaptive radiation by means of transgressive segregation. Ecol. Lett. 21:264–274. [DOI] [PubMed] [Google Scholar]
- Kamneva O.K.,, Syring J.,, Liston A.,, Rosenberg N.A. 2017. Evaluating allopolyploid origins in strawberries (fragaria) using haplotypes generated from target capture sequencing. BMC Evol. Biol. 17:180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingman J.F.C. 1982. On the genealogy of large populations. J. Appl. Prob. 19:27–43. [Google Scholar]
- Kubatko L.S. 2009. Identifying hybridization events in the presence of coalescence via model selection. Syst. Biol. 58:478–488. [DOI] [PubMed] [Google Scholar]
- Kubatko L.S.,, Chifman J. 2015. An invariants-based method for hybridization detection from genome-scale sequence data. bioRxiv doi.org/10.1101/034348. [Google Scholar]
- Lake J.A. 1987. A rate-independent technique for analysis of nucleic acid sequences: evolutionary parsimony. Mol. Biol. Evol. 4:167–191. [DOI] [PubMed] [Google Scholar]
- Maddison W.P. 1997. Gene trees in species trees. Syst. Biol. 46:523–536. [Google Scholar]
- Mallet J. 2005. Hybridization as an invasion of the genome. Trends Ecol. Evol. 20:229–237. [DOI] [PubMed] [Google Scholar]
- Mallet J. 2007. Hybrid speciation. Nature 446:279–283. [DOI] [PubMed] [Google Scholar]
- Martin S.H.,, Dasmahapatra K.K.,, Nadeau N.J.,, Salazar C.,, Walters J.R.,, Simpson F.,, Blaxter M.,, Manica A.,, Mallet J.,, Jiggins C.D. 2013. Genome-wide evidence for speciation with gene flow in Heliconius butterflies. Genome Res. 23:1817–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin S.H.,, Davey J.W.,, Jiggins C.D. 2014. Evaluating the use of ABBA-BABA statistics to locate introgressed loci. Mol. Biol. Evol. 32:244–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng C.,, Kubatko L.S. 2009. Detecting hybrid speciation in the presence of incomplete lineage sorting using gene tree incongruence: a model. Theor. Popul. Biol. 75:35–45. [DOI] [PubMed] [Google Scholar]
- Otto S.P.,, Whitton J. 2000. Polyploid incidence and evolution. Ann. Rev. Genet. 34:401–437. [DOI] [PubMed] [Google Scholar]
- Patterson N.,, Moorjani P.,, Luo Y.,, Swapan M.,, Rohland N.,, Zhan Y.,, Genschoreck T.,, Webster T.,, Reich D. 2012. Ancient admixture in human history. Genetics 192:1065–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pease J.B.,, Hahn M.W. 2015. Detection and polarization of introgression in a five-taxon phylogeny. Syst. Biol. 64:651–662. [DOI] [PubMed] [Google Scholar]
- Rambaut A.,, Grassly N.C. 1997. Seq-Gen: an application for the Monte Carlo simulation of DNA sequence evolution along phylogenetic trees. Comput. Appl. Biosci. 13:235–238. [DOI] [PubMed] [Google Scholar]
- Seehausen O. 2004. Hybridization and adaptive radiation. Trends Ecol. Evol. 19:198–207. [DOI] [PubMed] [Google Scholar]
- Solís-Lumis C.,, Ané C. 2016. Inferring phylogenetic networks with maximum pseudolikelihood under incomplete lineage sorting. PLoS Genet. 12:e1005896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavaré S. 1986. Some probabilistic and statistical problems in the analysis of DNA sequences. Lectures on Mathematics in the Life Sciences 17:57–86. [Google Scholar]
- Tian Y.,, Kubatko L.S. 2017. Rooting phylogenetic trees under the coalescent model using site pattern probabilities. BMC Evol. Biol. 17:263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y.,, Barnett R.M.,, Nakhleh L. 2013. Parsimonious inference of hybridization in the presence of incomplete lineage sorting. Syst. Biol. 62:738–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y.,, Degnan J.H.,, Nakhleh L. 2012. The probability of a gene tree topology within a phylogenetic network with applications to hybridization detection. PLoS Genet. 8:e1002660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y.,, Dong J.,, Liu K.J.,, Nakhleh L. 2014. Maximum likelihood inference of reticulate evolutionary histories. Proc. Natl. Acad. Sci. USA 111:16448–16453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y.,, Than C.,, Degnan J.H.,, Nakhleh L. 2011. Coalescent histories on phylogenetic networks and detection of hybridization despite incomplete lineage sorting. Syst. Biol. 60:138–149. [DOI] [PMC free article] [PubMed] [Google Scholar]