

Abstract
Hyperphosphatemia is common in patients with chronic kidney disease and is increasingly associated with poor clinical outcomes. Current management of hyperphosphatemia with dietary restriction and oral phosphate binders often proves inadequate. Tenapanor, a minimally absorbed, small-molecule inhibitor of the sodium/hydrogen exchanger isoform 3 (NHE3), acts locally in the gastrointestinal tract to inhibit sodium absorption. Because tenapanor also reduces intestinal phosphate absorption, it may have potential as a therapy for hyperphosphatemia. We investigated the mechanism by which tenapanor reduces gastrointestinal phosphate uptake, using in vivo studies in rodents and translational experiments on human small intestinal stem cell–derived enteroid monolayers to model ion transport physiology. We found that tenapanor produces its effect by modulating tight junctions, which increases transepithelial electrical resistance (TEER) and reduces permeability to phosphate, reducing paracellular phosphate absorption. NHE3-deficient monolayers mimicked the phosphate phenotype of tenapanor treatment, and tenapanor did not affect TEER or phosphate flux in the absence of NHE3. Tenapanor also prevents active transcellular phosphate absorption compensation by decreasing the expression of NaPi2b, the major active intestinal phosphate transporter. In healthy human volunteers, tenapanor (15 mg, given twice daily for 4 days) increased stool phosphorus and decreased urinary phosphorus excretion. We determined that tenapanor reduces intestinal phosphate absorption predominantly through reduction of passive paracellular phosphate flux, an effect mediated exclusively via on-target NHE3 inhibition.
INTRODUCTION
Patients with chronic kidney disease (CKD) are unable to maintain fluid and mineral balance. Reduced urinary excretion of phosphate and resultant hyperphosphatemia is associated with multiple complications; thus, addressing this imbalance is increasingly recognized as an important factor for optimizing outcomes in patients with CKD (1). Because intestinal phosphate absorption increases linearly with increasing dietary phosphate intake and does not saturate even at extremely high luminal phosphate concentrations (2–5), phosphate balance is principally maintained through the regulation of urinary phosphate excretion (6, 7). Reabsorption of phosphate in the renal tubule is modulated such that serum phosphate concentrations are maintained within a physiologic range despite considerable variations in daily phosphate intake.
Hyperphosphatemia is a predictable comorbidity in patients with advanced CKD, especially in patients with end-stage renal disease (ESRD) receiving dialysis. This is, at least in part, due to sustained intestinal phosphate absorption in the face of impaired or absent urinary phosphate excretion, which cannot be fully compensated by standard dialysis regimens. Elevated serum phosphate is associated with adverse outcomes in patients with CKD (8), including increased risk of all-cause mortality (9–12), cardiovascular events (13), and CKD progression (14), and is an independent risk factor for left ventricular hypertrophy (15). Furthermore, hyperphosphatemia in CKD is accompanied by increased fibroblast growth factor 23 (FGF-23) concentration and secondary hyperparathyroidism, which contribute to metabolic bone disease, ectopic calcification, renal failure, and progression of cardiovascular disease (16–18).
Oral phosphate binders, together with dietary phosphate restriction, are the primary treatment approaches for patients with ESRD receiving dialysis (1, 19). Restricting dietary phosphate intake can reduce the severity of hyperphosphatemia and secondary hyperparathyroidism, although adherence is typically poor and this diet can result in nutritional deficiencies (20). Poor compliance has also been reported with oral phosphate binders (21). These agents are associated with numerous side effects, such as nausea, vomiting, and constipation; the large amount of binder required to capture dietary phosphate means that treatment is associated with a high pill burden for patients, which can further compromise compliance (22).
Dietary phosphate absorption occurs predominantly in the small intestine by at least two distinct pathways: transcellular and paracellular (23–25). Two families of sodium-dependent phosphate solute carrier (SLC) transporters are responsible for the transport of phosphate into cells: the SLC34 (type II) and SLC20 (type III) families (23). The type II sodium-dependent phosphate transporter 2b (NaPi2b; SLC34A2) mediates the bulk of transcellular phosphate absorption in the intestine (26–28). NaPi2b has a high affinity for phosphate (KM, 10 to 100 μM); thus, it is saturated under most dietary phosphate concentrations (3, 29). The type III sodium-dependent phosphate transporters PiT1 (SLC20A1) and PiT2 (SLC20A2) are also expressed in the intestine, although their contribution to intestinal phosphate absorption is limited, as shown by the minimal transcellular sodium-dependent phosphate uptake remaining in NaPi2b knockout (KO) mice (26, 27, 30, 31).
Paracellular phosphate absorption through tight junction complexes is driven by the electrochemical phosphate gradient. An emerging body of literature demonstrates that tight junctions are dynamically regulated by signal transduction pathways, actively interact with the cytoskeleton, and can display some permeability specificity toward individual or selected ion groups (32, 33). In contrast to NaPi2b, paracellular phosphate absorption does not saturate, increases in a linear fashion with increasing phosphate concentration gradient, and is bidirectional (2, 34). Luminal phosphate concentrations on standard diets have been measured in the millimolar range (3), far in excess of concentrations that saturate NaPi2b and higher than blood phosphate concentrations. Increasing luminal phosphate concentration above NaPi2b saturation facilitates discrimination between these two transport pathways. Thus, paracellular phosphate influx is quantitatively the most important overall mechanism of phosphate absorption under typical conditions of phosphate availability.
Tenapanor is a first-in-class, minimally absorbed, small-molecule inhibitor of the sodium/hydrogen exchanger isoform 3 (NHE3) that acts locally in the gastrointestinal tract. NHE3 was first identified as a drug target in CKD owing to its key role in intestinal sodium and fluid absorption (35). In both healthy rats and human volunteers, tenapanor increases stool sodium and reduces urinary sodium, with minimal systemic drug exposure (35). In nephrectomized rats fed a high-salt diet and exhibiting hypervolemia, cardiac hypertrophy, and arterial stiffening, tenapanor reduced extracellular fluid volume, left ventricular hypertrophy, albuminuria, and blood pressure (35). In addition, tenapanor reduced intestinal absorption of radioactive phosphate, reduced urinary and serum phosphate, and both prevented ectopic calcification and lowered FGF-23 in a rat model of CKD (36).
Here, we delineate the mechanism by which intestinal phosphate absorption is inhibited by tenapanor, investigating its effects on both passive paracellular phosphate absorption and NaPi2b-mediated phosphate transport. Phosphate absorption has been observed along the entire length of the small intestine; however, species-specific regional variations have been described. A direct comparison of segment-specific phosphate absorption in rats and mice, using the in vivo loop technique, revealed it to be quantitatively highest in the duodenum of the rat and in the ileum of the mouse (37). Classic physiology experiments in humans, using the triple-lumen catheter perfusion technique in vivo, showed substantial and nonsaturating phosphate absorption across both the proximal and distal small intestine, although quantitatively higher proximally, more consistent with observations in rats than in mice (2). Therefore, to comprehensively investigate the effect of NHE3 inhibition on phosphate absorption along the length of the small intestine, we used enteroid monolayer cultures (38) derived from human distal duodenum and ileum to capture mechanisms of both proximal and distal absorption in humans and complemented this with experiments in the jejunum of mouse and rat. Mouse ileum, a segment where NaPi2b is highly expressed and plays a dominant role in phosphate absorption, was used to specifically assess the role of NaPi2b in the phosphate-lowering mechanism of tenapanor. We also report the effects of tenapanor on intestinal phosphate absorption in healthy human volunteers.
RESULTS
Tenapanor reduces passive paracellular phosphate absorption
Tenapanor reduces phosphate absorption in vivo
In a rat intestinal loop model, tenapanor reduced radioactive phosphate absorption in the jejunum to an amount similar to that observed in sodium-free conditions (Fig. 1A). The jejunum is a key site of intestinal phosphate absorption in rats (37). Increasing luminal phosphate concentration acutely via oral bolus caused proportionate increases in urinary phosphate excretion, an indirect marker of intestinal phosphate absorption (Fig. 1B). This linear concentration dependence of absorption and failure to saturate, even at high concentrations, is characteristic of passive paracellular absorption. Tenapanor reduced phosphate absorption across all phosphate concentrations, decreasing urinary phosphate excretion even at high phosphate concentrations (Fig. 1B), suggesting that tenapanor reduces paracellular phosphate absorption.
Fig. 1. Effects of tenapanor on phosphate absorption in vivo in rats.

(A) Effect of tenapanor (10 μM) and sodium-free buffer on radioactive phosphate absorption in the rat jejunum in vivo loop model compared with vehicle [dimethyl sulfoxide (DMSO)] (n = 5 to 7 per group). (B) Urinary phosphate excretion 4 hours after an oral (p.o.) bolus of varying phosphate concentrations (0.15 to 1.5 M) in rats pretreated with tenapanor (0.5 mg/kg) or vehicle (acidified water, 0.01% Tween 80) (n = 6 per group). (C) Urinary phosphate excretion in rats at different dietary phosphate intakes at baseline and after 4 days of treatment with tenapanor (0.5 mg/kg) or vehicle (acidified water, 0.01% Tween 80) (n = 7 per group). (D) Urinary phosphate and urinary sodium excretion 4 hours after a fixed quantity (5 g) of high-phosphorus (1.2%) meal in rats treated with tenapanor (0.15 mg/kg) or vehicle (acidified water, 0.01% Tween 80) (n = 8 per group). Cecal (E) sodium delivery, (F) phosphate delivery, (G) water volume, (H) sodium concentration, (I) phosphate concentration, (J) potassium concentration, (K) chloride concentration, (L) calcium concentration, and (M) magnesium concentration in rats fed a fixed quantity of high-phosphorus (1.2%) meal and treated with vehicle (acidified water, 0.01% Tween 80) or tenapanor (0.15 mg/kg) (n = 6 per time point per group). Means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; two-way analysis of variance (ANOVA), with post hoc testing at each time point with Bonferroni’s correction (A to C and E to M); pairwise comparisons by Student’s t test (D).
Chronic increases in dietary phosphate content in rats produced linear and nonsaturating increases in urinary phosphate excretion at baseline, consistent with passive paracellular absorption (Fig. 1C). Tenapanor reduced urinary phosphate excretion after 4 days of administration, suggesting the inhibition of a constant fraction of intestinal phosphate absorption despite increasing dietary phosphate content, indicative of a reduction in paracellular, rather than transcellular, absorption (Fig. 1C).
Paracellular phosphate absorption could be reduced by at least two potential mechanisms as a result of NHE3 inhibition: (i) a general decrease in paracellular water flux and diffusional driving force caused by luminal sodium and water retention, which would be accompanied by minimal change or a decrease in luminal phosphate concentration, or (ii) a decrease in paracellular permeability to phosphate through the tight junction, which would be accompanied by an increase in luminal phosphate concentration. To investigate this, an enteropooling study was performed in healthy rats trained to eat a high-phosphate meal (1.2%), measuring ion mass and ion concentration in the cecum at defined time points. The small intestine is the primary site of phosphate absorption; therefore, the concentration of phosphate and other ions in the cecum reflects the amount not absorbed in the small intestine.
In this enteropooling model, tenapanor reduced urinary phosphate and sodium excretion after the high-phosphate meal (Fig. 1D) and increased sodium (Fig. 1E) and phosphate (Fig. 1F) delivery to the cecum, confirming decreased absorption of these ions in the small intestine. After vehicle treatment, very little phosphate was delivered to the cecum, even in the face of a high-phosphate load, indicating a high capacity for phosphate absorption in the small intestine (Fig. 1F). Luminal sodium retention observed with tenapanor treatment was accompanied by increased luminal water volume (Fig. 1G) and luminal sodium concentration (Fig. 1H). In addition, the inhibition of intestinal phosphate absorption that resulted from tenapanor treatment was accompanied by an increase in luminal phosphate concentration (Fig. 1I). This suggests that, rather than reducing the driving force, tenapanor decreases paracellular permeability to phosphate. However, reduced water absorption may also contribute to decreased phosphate absorption via decreased paracellular solvent drag.
Potassium is reported to be exclusively absorbed in the small intestine via passive paracellular flux (39). In contrast to phosphate, luminal potassium concentration in the enteropooling model was decreased by tenapanor (Fig. 1J), consistent with greater solute dilution secondary to luminal water retention because of decreased transcellular sodium absorption, relative to a reduction in paracellular potassium flux. Combined, these findings illustrate the in vivo selectivity of tenapanor for phosphate relative to potassium.
Cecal chloride concentrations in the enteropooling model were low, indicative of high-capacity chloride absorption in the small intestine against the prevailing chemical gradient opposed by high plasma chloride concentrations (98 to 106 mM), and consistent with a prominent role of active transcellular chloride transport (Fig. 1K). Tenapanor increased the luminal chloride concentration compared with vehicle control; however, the maximal increase in chloride concentration was modest (1.6-fold) compared with the maximal tenapanor-induced increases in phosphate concentration (5.7-fold), demonstrating a preferential effect of tenapanor on phosphate relative to chloride. Given the low cecal concentration of chloride relative to plasma chloride concentrations, which would be insufficient to support paracellular chloride absorption, the modest increase in cecal chloride observed with tenapanor treatment is likely the result of a disruption in electroneutral transcellular sodium chloride absorption, which would be a direct result of NHE3 inhibition, rather than reduced paracellular chloride absorption.
The concentrations of divalent cations calcium (Fig. 1L) and magnesium (Fig. 1M) in the cecum were also low and comparable to their respective plasma concentrations, thus indicating efficient small intestinal absorption of these cations. Tenapanor did not significantly (P > 0.05) affect cecal concentrations of calcium or magnesium, demonstrating specificity toward inhibition of phosphate absorption relative to divalent cation absorption.
Tenapanor inhibits paracellular phosphate flux in an intestinal epithelial cellular model
Intestinal epithelial stem cells from human or mouse gastrointestinal biopsies cultured as monolayers allow for monitoring of ion transport across the intestinal epithelium (38). The enteroid monolayer contains the diversity of intestinal epithelial cell lineages, models the specific gene expression patterns of each individual intestinal segment, expresses the appropriate endogenous ion transporters (for example, NHE3 and NaPi2b) in a segment-specific manner, polarizes to form tight junctions with segment-specific expression of claudins and other tight junction proteins, and generates the expected negative luminal electrical potential observed in vivo. The differentiated enteroid monolayer therefore enables the study of transcellular and paracellular phosphate absorption.
NHE3 is highly expressed along the length of the gastrointestinal tract (40) and is endogenously expressed in each segment of the intestinal monolayer model, as shown in representative images from the ileum (fig. S1A). Similar to the in vivo situation, most cells in the monolayer are absorptive cells and are polarized, expressing NHE3 only at the apical surface (fig. S1B). Proton secretion coupled to sodium absorption by NHE3 results in acidification of the apical media of the monolayer. This was inhibited by tenapanor, monitored by the color change of pH-sensitive phenol red in the apical media (fig. S1C). Concentration-response studies measuring apical pH using 2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein, acetoxymethyl ester (BCECF-AM) dye, where a decrease in the fluorescence emission ratio reflects a decrease in pH, showed that tenapanor inhibited apical acid secretion by NHE3 with a half-maximal inhibitory concentration (IC50) of 2 and 6 nM in human and mouse ileum monolayers, respectively (fig. S1D). This is consistent with the potency of tenapanor against NHE3 in primary target assays (35). In human ileum and duodenum cell monolayer cultures, tenapanor caused a concentration-dependent inhibition of the NHE3-mediated recovery of intracellular pH (pHi) after acid loading (IC50: ileum, 13 nM; duodenum, 9 nM) (fig. S1, E and F). On the basis of the potency of NHE3 inhibition in the intestinal monolayer culture system, subsequent experiments investigating effects on phosphate absorption used a tenapanor concentration of 1 μM to ensure complete NHE3 inhibition.
Assessment of apical-to-basolateral phosphate flux in duodenum cell monolayer cultures derived from two healthy human donors showed significant (P < 0.0001), linear (R2 = 0.989 for donor 1, R2 = 0.998 for donor 2), and nonsaturating increases in phosphate flux with increasing apical phosphate concentrations (Fig. 2A). Under these conditions, NaPi2b would be saturated at even the lowest phosphate concentration. This indicates that passive paracellular absorption is the primary route of phosphate absorption in the human duodenum model, consistent with the absence of phosphate transporter expression in the duodenum epithelial cell monolayer culture and the lack of effect of a NaPi2b inhibitor (41) on duodenal phosphate absorption in the monolayer (fig. S1G).
Fig. 2. Effects of phosphate concentration gradient and tenapanor on phosphate absorption in human duodenum monolayer cultures.

(A) Correlation between phosphate flux and initial apical phosphate concentration in human duodenum monolayer cultures from two separate donors (1, 2) after overnight incubation. The reproducibility of phosphate absorption was assessed in experiments from two separate passages for one donor (n = 4 per group). (B) Basolateral phosphate concentration, phosphate flux, and (D) transepithelial electrical resistance (TEER) at different apical phosphate concentrations (1 to 30 mM) in human duodenum monolayer cultures after 4 hours of treatment with tenapanor (1 μM) or vehicle (DMSO) with an initial starting basolateral phosphate concentration of 0 mM (n = 4 to 16 per group). (E) Apical phosphate retention, (F) apical phosphate concentration, (G) basolateral phosphate concentration, and (H) apical fluid volume at different initial apical phosphate concentrations (1 to 30 mM) in human duodenum monolayer cultures after overnight treatment with tenapanor (1 μM) or vehicle (DMSO) with an initial starting basolateral phosphate concentration of 1 mM; data from the donor used to test reproducibility in (A). (I) Effect of tenapanor (1 μM) or vehicle (DMSO) on bidirectional phosphate flux at varying phosphate concentration gradients (1 to 10 mM) in human duodenum monolayer cultures (n = 4 per group). Means ± SEM. *P < 0.05, ***P < 0.001, ****P < 0.0001 versus vehicle; two-way ANOVA, with post hoc testing at each concentration with Bonferroni’s correction (B to I).
The effects of apical tenapanor administration on phosphate flux were evaluated by measuring changes in basolateral phosphate concentration after a 4-hour treatment period in duodenum cell monolayer cultures, varying the apical phosphate concentration (with initial basolateral phosphate concentration fixed at 0 mM). Phosphate absorption was strongly dependent on the apical-basolateral phosphate concentration gradient, and basolateral phosphate concentration (Fig. 2B) and phosphate flux (Fig. 2C) were lower with tenapanor treatment above 1 mM apical phosphate. Tenapanor also increased TEER compared with vehicle (Fig. 2D) after a 4-hour incubation in human duodenum monolayer cultures, consistent with a decrease in paracellular permeability. Treatment with tenapanor did not affect the potential difference across the monolayer compared with vehicle control (Table 1), indicating that the electrical driving force for phosphate flux is not affected by tenapanor.
Table 1. Transepithelial potential difference across human duodenum monolayer cultures.
Monolayers were treated for 4 hours with vehicle (DMSO) or tenapanor (1 µM) (n = 4 per group). Transepithelial potential difference measured as apical relative to basolateral. Means ± SEM. Pairwise comparisons by Student’s t test (P > 0.05).
| Vehicle | Tenapanor (1 µM) |
|---|---|
| −5.5 ± 0.3 mV | −5.3 ± 0.26 mV |
Extending the treatment period of the duodenum monolayers to overnight allowed for measurable apical-to-basolateral water flux across a range of initial apical phosphate concentrations (with initial basolateral phosphate concentration set at 1 mM). Overnight treatment with tenapanor inhibited phosphate absorption across the range of phosphate concentrations, providing a chemical phosphate gradient as indicated by a higher retention of phosphate in the apical compartment compared with vehicle (Fig. 2E). Consistent with decreased phosphate permeability, the retention of phosphate in the apical chamber produced by tenapanor was accompanied by higher apical phosphate concentrations compared with vehicle (Fig. 2F). Conversely, basolateral phosphate concentrations were reduced with tenapanor exposure (Fig. 2G). NHE3 inhibition by tenapanor decreased apical-to-basolateral water absorption (Fig. 2H) as a result of decreased transcellular sodium flux. Similar results were observed in human ileum monolayers (fig. S2), showing that tenapanor also inhibits phosphate absorption in the distal small intestine of humans.
Paracellular transport is also characterized by bidirectional flux based on the prevailing electrochemical gradient. Consistent with phosphate moving across human duodenum monolayer cultures through the paracellular pathway, we demonstrated basolateral-to-apical phosphate flux that was linearly dependent on the transepithelial phosphate concentration gradient, which is nonsaturating through a 10 mM phosphate gradient (Fig. 2I). The magnitude of phosphate flux at a given phosphate concentration gradient was similar in both directions, although appeared to be slightly reduced in the basolateral-to-apical direction, which is likely a result of the unfavorable electrical gradient to reverse phosphate flux generated by the negative luminal potential established across the monolayer (−5.5 ± 0.3 mV relative to basolateral). Apically applied tenapanor reduced basolateral-to-apical phosphate flux at all phosphate concentrations tested, comparable to the effect on apical-to-basolateral phosphate absorption (Fig. 2I). Combined, these results suggest that tenapanor inhibits phosphate flux by decreasing paracellular permeability, consistent with the in vivo enteropooling results.
Tenapanor increases TEER when there is a favorable gradient for NHE3-mediated proton efflux
Proton secretion by NHE3 gradually acidifies the apical media in the intestinal epithelial cell monolayer cultures, which results in an unfavorable proton gradient for NHE3-mediated transport. The reduction in NHE3-mediated proton efflux in the setting of an elevated luminal proton concentration has been described previously in the renal tubule (42). To initiate phosphate absorption experiments, acidified cell culture media were replaced with media of a defined starting phosphate concentration and a neutral apical pH. The change to neutral apical media immediately restored the proton gradient, enabling NHE3-mediated proton efflux and resulting in a large and sustained reduction in TEER from baseline in the duodenum (Fig. 3A) and ileum (Fig. 3B) monolayer cultures. This effect was blocked by tenapanor (Fig. 3, A and B). This shows that TEER is elevated when NHE3 is inhibited by tenapanor or when NHE3-mediated proton efflux is reduced by an unfavorable proton gradient, both of which result in intracellular proton accumulation.
Fig. 3. TEER and pHi in human intestinal cell monolayer cultures.

Effects of tenapanor (1 μM) versus vehicle (DMSO) on TEER after the change from acidic (pH 6.0) apical media at baseline to fresh neutral pH apical media to restore the gradient for NHE3-mediated proton efflux in (A) human duodenum and (B) human ileum cell monolayer cultures (n = 3 per group). (C) TEER after the change from acidic (pH 6.0) apical media at baseline to fresh apical media at different pH in human duodenum monolayer cultures (n = 3 per group). (c) Effect of tenapanor (1 μM) on TEER at varying apical media pH in human duodenum monolayer cultures normalized to equivalent pH level treated with vehicle (DMSO) (n = 3 per group). Recovery of pHi, initiated by the addition of a sodium-containing media and measured using pH-sensitive BCECF-AM dye after acid loading in sodium-free media in (E) human duodenum and (F) human ileum monolayer cells at different apical pH in the presence of tenapanor (1 μM) or vehicle (DMSO). RLU, relative luminescence units. (G) Concentration-response effect of tenapanor on rate of the recovery from intracellular acidification at neutral and acidic apical pH in human ileum monolayer cultures. (H) Change in pHi, measured using BCECF-AM dye, over time after the change to acidic, neutral, or alkaline pH apical media in human ileum monolayer cultures. Effect of tenapanor (1 μM) versus vehicle (DMSO) on change in (I) pHi and (J) TEER over time in human ileum monolayer cultures (n = 3 to 6 per group). Means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 versus vehicle (A, B, and I), versus pH 7.3 (C and D), or versus pH 6.0 (H); two-way ANOVA, with post hoc testing at each concentration with Bonferroni’s correction (A to D, H, and I); non-linear regression analysis, log (inhibitor) versus response (three parameters) (G).
Luminal pH modulates pHi, NHE3-mediated proton efflux, TEER, and phosphate absorption
In human duodenum monolayers, apical pH had a strong influence on TEER. Replacing acidified apical media (pH 6.0) with fresh media produced similar reductions in TEER from baseline under neutral (pH 7.0) and alkaline (pH 8.0) conditions but caused an increase in TEER when acidic media (pH 5.5) were introduced (Fig. 3C). Fresh media, pH-matched to the apical media being replaced (pH 6.0), had no effect on TEER (Fig. 3C). Compared with the TEER of the equivalent pH control, tenapanor increased TEER at neutral and alkaline luminal pH, but not under acidic conditions, when NHE3-mediated proton efflux was unfavorable and TEER was already high (Fig. 3D).
The rate and magnitude of recovery of pHi after acid loading (a direct measure of NHE3-mediated proton efflux in the enterocyte) were reduced with decreasing luminal pH in both duodenum and ileum monolayer cultures (Fig. 3, E and F), and tenapanor reduced pHi recovery at each of the luminal pH values tested. Although NHE3-mediated proton efflux was reduced at pH 6.0 compared with pH 7.0, the potency of tenapanor in inhibiting NHE3 was unaffected by extracellular pH [IC50: 10 ± 1.9 nM (pH 6.0) and 9.5 ± 2.0 nM (pH 7.0); P = 0.87] (Fig. 3G).
In human ileum monolayer cultures, changes in luminal pH were rapidly accompanied by changes in pHi and TEER, within 1 min after manipulation of the apical media pH (Fig. 3, H to J). A rapid reduction in pHi was observed at luminal pH 6.0. A higher pHi was seen when neutral (pH 7.0) or alkaline (pH 8.0) media were added, although there was little difference between pH 7.0 and pH 8.0 (Fig. 3H), similar to the effects of luminal pH on TEER. At neutral apical pH (when NHE3 is effectively effluxing protons), tenapanor reduced pHi to a similar extent observed with pH 6.0 apical media, with effects observed within 1 min (the first time point technically possible to measure) of initiating tenapanor treatment (Fig. 3I). In monolayer cultures mounted in Ussing chambers, which permit dynamic and continuous TEER measurements, tenapanor application resulted in a near-immediate increase in TEER compared with vehicle control (Fig. 3J). Therefore, inhibition of NHE3, by tenapanor or decreased NHE3-mediated proton efflux as a result of acidic luminal media, results in a rapid (<1 min) reduction in pHi and a correspondingly rapid increase in TEER.
Phosphate flux in ileum monolayer cultures was measured during overnight exposure to acidic (pH 6.0), neutral (pH 7.0), and alkaline (pH 8.0) apical media, with and without tenapanor. There was a strong, positive, and significant relationship between apical pH and phosphate flux at initial apical phosphate concentrations of both 10 mM (R2 = 0.94, P < 0.0001) and 30 mM (R2 = 0.92, P < 0.0001) (Fig. 4A). Tenapanor reduced phosphate absorption across the pH range of 6.0 to 8.0, with the reduction in phosphate flux being higher at pH 7.0 and 8.0 compared with pH 6.0, but no difference between pH 7.0 and pH 8.0 (Fig. 4B).
Fig. 4. Effect of apical pH on phosphate flux and effect of tenapanor on phosphate flux at different apical pH in ileum monolayers and jejunum tissue ex vivo.
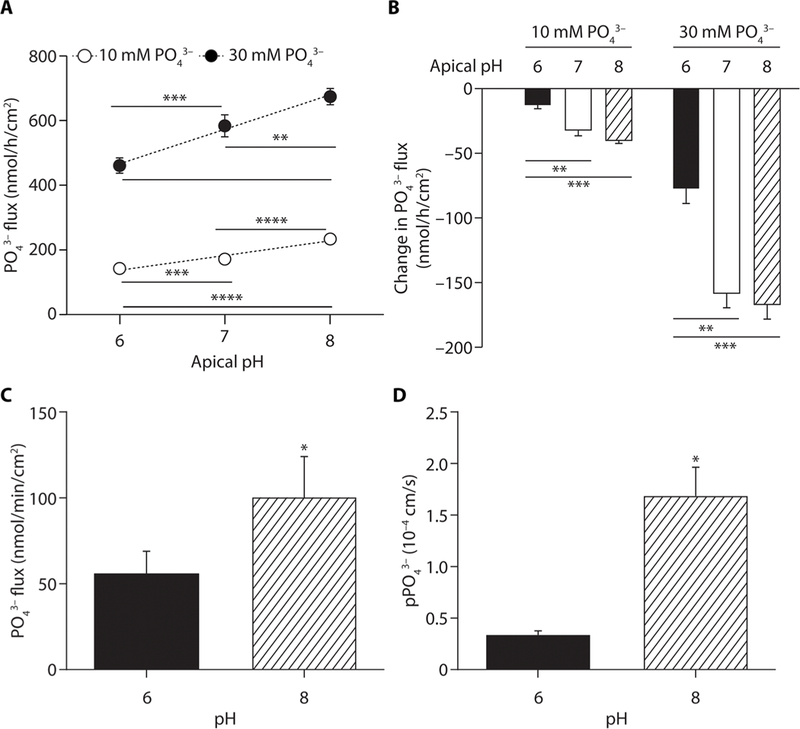
Effect of varying apical pH on basolateral phosphate flux in human ileum epithelial cell monolayer cultures after an overnight incubation at an initial apical phosphate concentration of 10 or 30 mM and an initial basolateral phosphate concentration of 0 mM, with (A) vehicle (DMSO) or (B) tenapanor (1 μM; the effect of tenapanor is represented as change from vehicle). (C) Phosphate flux measured with radioactive tracer and (D) paracellular phosphate permeability (pPO43−) measured by biionic dilution potential in mouse jejunum strips at pH 6.0 and 8.0 (n = 4 to 6 per group). Means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; one-way ANOVA, with post hoc testing at each pH with Bonferroni’s correction (A and B); pairwise comparisons by Student’s t test (C and D).
To confirm that the results obtained in enteroid monolayers translate to intestinal tissue, radioactive tracer (phosphate flux) and biionic dilution potential (paracellular phosphate permeability) were measured in mouse jejunum mounted in Ussing chambers at pH 6.0. and 8.0. Consistent with the human duodenum monolayer results, both phosphate flux (Fig. 4C) and permeability (Fig. 4D) were increased at pH 8.0 compared with pH 6.0. Combined, these results suggest increased phosphate permeability at pH 8.0 in both human duodenum monolayer cultures and mouse jejunum— conditions where the predominant phosphate species is HPO42−.
Tenapanor decreases paracellular permeability as measured by dilution and biionic potentials
Paracellular sodium and chloride permeability and paracellular phosphate permeability were determined from sodium chloride dilution potentials and phosphate biionic potentials, respectively, in both human duodenum monolayer cultures and mouse jejunum mounted in Ussing chambers at pH 8.0. Tenapanor increased TEER (Fig. 5A) and decreased permeability to sodium (Fig. 5B) and chloride (Fig. 5C) in human duodenum monolayer cultures. Sodium-to-chloride permeability (Fig. 5D) was unchanged, whereas phosphate permeability decreased (Fig. 5E), confirming decreased paracellular permeability to phosphate. A consistent effect of tenapanor on paracellular permeability was observed in mouse jejunum ex vivo (Fig. 5, F to J).
Fig. 5. Effect of tenapanor on paracellular permeability measured by direct biophysical methods and the chemical driving forces for paracellular absorption in vivo in rats.

Effect of tenapanor at pH 8.0 on (A) TEER, (B) sodium permeability (pNa+), (C) chloride permeability (pCl−), (D) sodium-to-chloride permeability (pNa+/pCl−), and (E) phosphate permeability (pPO 3−) in human duodenum monolayer cultures mounted in Ussing chambers. Effect of tenapanor on (F) TEER, (G) pNa+, (H) pCl−, (I) pNa+/pCl−, and (J) pPO43− across mouse jejunum in Ussing chambers. Luminal concentrations of (K) sodium, (L) chloride, and (M) phosphate in the proximal and distal small intestine in untreated rats trained to eat a standardized meal (n = 6 per group). Means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; pairwise comparisons by Student’s t test (A to J); two-way ANOVA, with post hoc testing at each time point with Bonferroni’s correction (K to M).
The preferential inhibition of intestinal phosphate and sodium absorption by tenapanor in vivo, relative to other ions including chloride, is not readily reflected in these direct biophysical measurements of paracellular permeability. It is important to recognize that measurements of paracellular ion permeability made via in vitro dilution potential measurements are not equivalent to in vivo paracellular ion flux, which is the integrated result of paracellular permeability and the prevailing electrochemical gradient. Sodium and chloride permeability was assessed in the Ussing chamber by acutely imposing an ~100 mM chemical gradient to enable dilution potential measurement, a gradient unlikely to be achieved in vivo because of the high plasma concentration of both sodium (135 to 145 mM) and chloride (96 to 106 mM) opposing paracellular absorption from the lumen. To better assess the actual driving force for various ions in vivo, we measured ion concentrations in both the proximal and distal small intestine at defined time points in untreated, healthy rats after ingestion of a high-phosphate (1.2%) meal, which would be typical of processed food. Peak luminal intestinal sodium concentrations were observed in the distal small intestine that approximated normal plasma sodium concentrations (Fig. 5K). Combined with the negative luminal potential, the electrochemical gradient did not provide a strong impetus for paracellular sodium absorption; there could potentially be secretion. Luminal chloride concentrations were highest in the proximal small intestine, but still below normal plasma chloride concentrations, and were significantly decreased in the distal small intestine compared with the proximal small intestine at each time point (Fig. 5L). Even with a favorable electrical gradient for absorption of an anion, paracellular chloride absorption would not be supported in this concentration range. The absence of the requisite electro-chemical gradients to drive substantial paracellular sodium and chloride flux in vivo suggests a prominent role for transcellular absorption of these ions, consistent with the high expression of ion exchangers for both sodium (NHE3) and chloride (SLC26A3 and SLC26A6) in the small intestine (43).
In contrast, low plasma phosphate concentrations, high luminal phosphate concentrations, and a negative luminal potential difference across the intestinal epithelium provide a strong electrochemical gradient for paracellular luminal phosphate absorption (Fig. 5M). The electrical gradient across the intestinal epithelium is lumen-negative, and luminal phosphate is pre-dominantly a divalent anion, magnifying the driving force for paracellular phosphate fourfold relative to chloride. Combined with the absence of a low-affinity, high-capacity transcellular phosphate transporter, at high luminal concentrations, phosphate appears to be dependent on paracellular flux for absorption.
NHE3-independent changes in pHi modulate TEER
The effects of intracellular acidification, occurring independent of changes in NHE3 activity, on TEER were evaluated using the cell monolayer culture system. The ionophore nigericin reduced pHi compared with control (fig. S3A) and produced a similar increase in TEER as was observed with tenapanor (fig. S3B). Similarly, the compounds N5,N6-bis(2-fluorophenyl)[1,2,5]oxadiazolo [3,4-b] pyrazine-5,6-diamine (BAM15) and mesoxalonitrile 4-trifluoromethoxy-phenylhydrazone (FCCP), which decrease pHi by uncoupling oxidative phosphorylation in mitochondria (fig. S3C), increased TEER compared with control (fig. S3D). These results show that pHi is a direct regulator of TEER, independent of NHE3 activity.
Tenapanor requires NHE3 to inhibit paracellular phosphate absorption
Clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9)–mediated gene editing was used to produce human ileum epithelial stem cell clones with nucleotide insertions and deletions (Fig. 6A), which resulted in a complete loss of NHE3 expression and function (Fig. 6, B to E). NHE3 KO ileum monolayers were otherwise morphologically indistinguishable from control monolayers and had similar baseline TEER values. In monolayer culture studies, the NHE3 KO clones showed the same phenotype as tenapanor-treated cells. After overnight incubation, NHE3 KO monolayers showed reduced absorption of water (Fig. 6F), sodium (Fig. 6G), and phosphate (Fig. 6H), as indicated by their apical retention compared with control monolayers. NHE3 KO cells had reduced apical proton secretion and increased apical media pH compared with the control monolayers (Fig. 6I). With the restoration of neutral apical media and, hence, NHE3-mediated proton secretion in control cells, there was a marked decrease in TEER from baseline in control cultures; however, no change in TEER was observed in the NHE3 KO cells (Fig. 6J).
Fig. 6. Paracellular phosphate absorption in NHE3 KO human ileum monolayer cultures.

(A) DNA sequencing showing CRISPR/Cas9-mediated gene editing of NHE3 resulting in nucleotide deletions and insertions. The single-guide RNA (sgRNA) targeting human NHE3 genomic DNA exon 2 (sgNHE3–13) is shown in red, and the protospacer adjacent motif (PAM) region is shown in blue. The NHE3 KO line (sgNHE3–13 clone 45) contains two different types of mutations (Mut 1 and Mut 2), shown in gray boxes. (A) Western blot showing NHE3 and β-actin (ACTB) protein expression in control and NHE3-edited cells and (C) apical media showing acidification (yellow) or absence of acidification (pink) in control and NHE3-edited cells. Recovery from intracellular acidification after acid loading in the presence of tenapanor or vehicle (DMSO) in (D) control (nontargeting sgControl clone 4) and (E) NHE3 KO (sgNHE3–13 clone 45) human ileum epithelial cell clones. (F) Apical volume, (G) apical sodium, (H) apical phosphate, and (I) apical pH in control and NHE3 KO human ileum monolayers after overnight incubation (n = 6 per group). (J) TEER after the change from acidic (pH 6.0) apical media at baseline to fresh neutral pH apical media to restore the gradient for NHE3-mediated proton efflux in control and NHE3 KO monolayers (n = 4 per group). (K) Change in apical-to-basolateral phosphate absorption and (L) change in apical phosphate concentration with tenapanor (1 μM), normalized to the effect of vehicle (DMSO) in NHE3 KO and control monolayers (n = 4 per group). (M) Change in TEER with tenapanor (1 μM) relative to vehicle (DMSO) after the change to neutral pH apical media in control and NHE3 KO monolayers (n = 4 per group). Means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; pairwise comparisons by Student’s t test (F to I, K, and L); two-way ANOVA, with post hoc testing at each time point with Bonferroni’s correction (J and M).
Consistent with earlier results, tenapanor inhibited apical-to-basolateral phosphate absorption in ileum monolayer control cells (Fig. 6K); this was accompanied by an increased apical phosphate concentration compared with control (Fig. 6L). By contrast, tenapanor had little effect on phosphate absorption (Fig. 6K) or apical phosphate concentration (Fig. 6L) in NHE3 KO cells. Similarly, tenapanor administration increased TEER relative to vehicle only in control cells and had no effect on TEER in NHE3 KO cells (Fig. 6M). These findings indicate that the effects of tenapanor on phosphate absorption, paracellular phosphate permeability, and TEER are mediated via on-target NHE3 inhibition. Similar results were obtained with and without tenapanor in NHE3 KO human duodenum monolayers (fig. S4).
Tenapanor affects 24-hour urinary ion excretion and NaPi2b
Tenapanor reduces urinary sodium and phosphate but not chloride or potassium excretion in rats
Tenapanor decreased urinary sodium (fig. S5A) and phosphate (fig. S5B) excretion but had no effect on urinary chloride (fig. S5C) or potassium (fig. S5D) excretion in healthy rats dosed for 14 days. Tenapanor did not affect plasma sodium or phosphate concentration (fig. S5E), because renal clearances of both phosphate and sodium were appropriately decreased relative to control (fig. S5F). Tenapanor had minimal effect on phosphate-regulating hormones (FGF-23, parathyroid hormone, and vitamin D) in healthy rats receiving a regular phosphate diet [fig. S5G, although tenapanor does reduce FGF-23 concentration in animal models of CKD (36)].
The effect of tenapanor on urinary sodium excretion was maximal on the first few days of treatment (fig. S5A) and remained reduced compared with vehicle for the duration of treatment. However, the reduction was attenuated in magnitude from treatment day 5, suggesting the engagement of a compensatory response to inhibition of intestinal sodium absorption. To investigate this, the expression of transcellular sodium transporters was measured by RNA sequencing (RNA-seq) along the length of the gastrointestinal tract. In tenapanor-treated rats, NHE3 expression was increased in the jejunum, ileum, and proximal colon (fig. S6A), and epithelial sodium channel γ subunit (ENaCγ) expression was increased in the distal colon (fig. S6B), indicating an attempt to increase intestinal sodium absorption. Because NHE3 will remain inhibited in the continued presence of tenapanor, it is likely that increased colonic sodium absorption via up-regulated ENaC is responsible for the attenuation of the reduction of urinary sodium excretion over time. To assess whether the lack of effect of tenapanor on urinary chloride excretion (fig. S5C) required a similar compensatory response as observed for sodium, we also measured the expression of the most prominent chloride exchangers and transporters along the length of the gastrointestinal tract. The expression of chloride exchangers SLC26A3 (fig. S6C), SLC26A6 (fig. S6D), and chloride channel CFTR (cystic fibrosis transmembrane conductance regulator) (fig. S6E) was unchanged by tenapanor treatment. The maintenance of normal chloride balance in the absence of a transcriptional response to up-regulate chloride exchangers and transporters, in contrast to the effect on sodium transporters and channels, is consistent with minimal impact of tenapanor on intestinal chloride absorption.
Tenapanor produces a modest decrease in NaPi2b expression
Expression of mRNA of the active phosphate transporter NaPi2b was modestly (about 30%) but statistically significantly (P < 0.01) lower in rat distal jejunum and ileum after 14 days of tenapanor treatment (Fig. 7A). In addition, immunohistochemistry using an antibody specific to NaPi2b showed a modest decrease in staining intensity in the jejunum after tenapanor treatment in a separate study (Fig. 7B). Transcellular phosphate transport was assessed using intestinal brush border membrane vesicles (BBMVs) isolated after in vivo tenapanor administration in rats. This transport system is predominantly driven by NaPi2b. Tenapanor had little effect on transcellular phosphate uptake, including sodium-dependent phosphate absorption, in either duodenum or jejunum BBMVs (fig. S7, A and B), indicating minimal functional effect of tenapanor on NaPi2b-mediated phosphate absorption. Combined, these findings indicate that NaPi2b expression and, consequently, transcellular phosphate absorption do not increase to compensate for the decrease in passive paracellular phosphate absorption produced by tenapanor. In addition, tenapanor did not affect sodium-dependent glucose absorption in duodenum BBMVs (fig. S7C).
Fig. 7. Effects of tenapanor on NaPi2b expression in rats and NaPi2b activity in mouse ileum monolayer cultures and in vivo in mice.
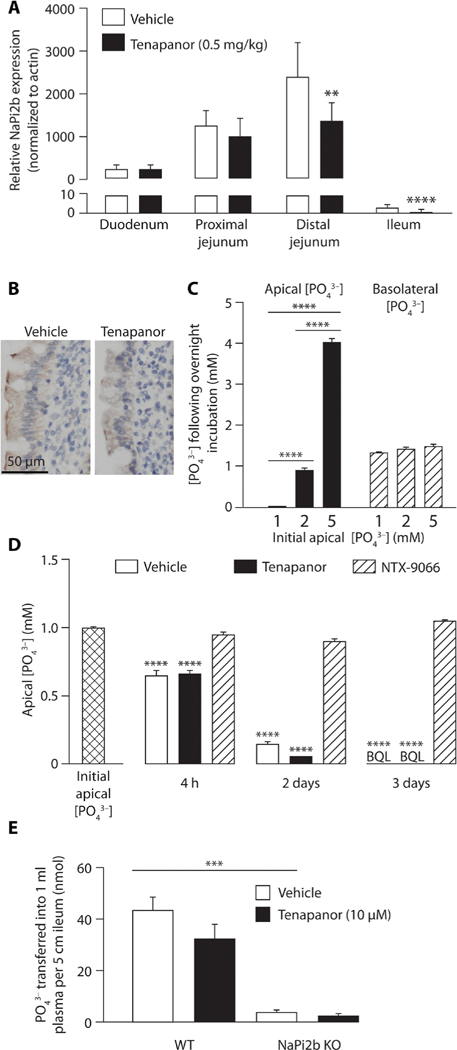
(A) NaPi2b mRNA expression in different intestinal segments after 14 days of tenapanor (0.5 mg/kg) or vehicle (acidified water, 0.01% Tween 80) treatment in rats (n = 8 per group). (B) Immunohistochemistry showing NaPi2b expression in rat jejunum after in vivo treatment with tenapanor or vehicle. (C) Apical and basolateral phosphate concentrations after overnight incubation at different initial apical phosphate concentrations (1 to 5 mM) in mouse ileum monolayer cultures with initial basolateral phosphate concentration of 1 mM (n = 6 per group). (D) Apical phosphate concentrations after a 4-hour, 2-day, or 3-day incubation with tenapanor (1 μM), NTX-9066 (NaPi2b inhibitor; 1 μM), or vehicle (DMSO) in mouse ileum monolayer cultures (n = 6 per group). BQL, below quantification limit. (E) Phosphate absorption with tenapanor (10 μM) versus vehicle in NaPi2b KO and control [wild-type (WT)] mouse ileum in an in vivo ileum loop model (n = 4 to 5 per group). Means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; two-way ANOVA, with post hoc testing at each segment with Bonferroni’s correction (A); one-way ANOVA, with post hoc testing at each concentration (C) or time point (D) with Bonferroni’s correction.
Tenapanor has minimal effect on NaPi2b activity in cellular and in vivo studies
Mouse ileum demonstrates high NaPi2b expression (26). In mouse ileum monolayers, evaluation of phosphate absorption at increasing initial apical phosphate concentrations (1 to 5 mM) with a fixed initial basolateral phosphate concentration (1 mM) showed similar decreases in apical phosphate concentration after overnight incubation, irrespective of initial starting concentration (Fig. 7C); phosphate was transported in the absence of a concentration gradient, consistent with transcellular flux. Final basolateral phosphate concentration was also similar despite the different starting apical phosphate concentrations (Fig. 7C), showing that phosphate absorption in the mouse ileum was minimally affected by the phosphate concentration gradient. This is a typical characteristic of high-affinity active transport that saturates at low phosphate concentrations of less than 1 mM, and is consistent with the known characteristics of NaPi2b.
In the absence of an apical-to-basolateral phosphate gradient, incubation of mouse ileum monolayers with NTX-9066 (41), a potent NaPi2b inhibitor (1 μM for 4 hours, 2 days, or 3 days), completely blocked apical-to-basolateral phosphate absorption at all time points (Fig. 7D). By contrast, phosphate absorption in the mouse ileum monolayer was unaffected by tenapanor (1 μM) compared with vehicle at all time points measured (Fig. 7D). These findings indicate that phosphate absorption in the mouse ileum monolayer cultures is active, saturates at low luminal phosphate concentrations (imparting a relatively low capacity of phosphate absorption), and is mediated exclusively by NaPi2b; they also indicate that tenapanor does not affect NaPi2b-mediated phosphate absorption.
The effects of tenapanor were also evaluated in the in vivo loop model (ileum of wild-type and NaPi2b KO mice) (26). Almost all (90%) phosphate absorption in the mouse ileum was mediated by NaPi2b (Fig. 7E), similar to the mouse ileum monolayer (Fig. 7D). Tenapanor produced a small, nonsignificant (P > 0.05) decrease in phosphate absorption in both wild-type and NaPi2b KO mouse ileum (Fig. 7E). This further suggests that the phosphate-lowering effect of tenapanor is not dependent on NaPi2b.
Tenapanor does not affect tight junction protein localization and trafficking
There was no obvious change in the localization of tight junction proteins zona occludens-1, occludin, claudin 7, or claudin 3 assessed after 30, 60, or 120 min of tenapanor treatment in the human ileum monolayer model (fig. S8, A to C). Because the panel of validated tight junction protein antibodies for immunocytochemistry is limited, we further evaluated the involvement of protein trafficking in the tenapanor-induced TEER effect to determine whether protein trafficking from the tight junction is responsible. Pitstop 2 and dynasore (inhibitors of clathrin and dynamin, respectively) inhibited endocytosis in the ileum monolayer by preventing carbachol-induced NHE3 internalization (fig. S8D). However, neither Pitstop 2 (fig. S8E) nor dynasore (fig. S8F) blocked tenapanor-induced increases in TEER compared with control, indicating that endocytosis is not required for this effect of tenapanor. The rapid increase in TEER without obvious tight junction protein trafficking involvement might be consistent with the inhibition of NHE3 by tenapanor producing a pH-sensitive conformational change in the tight junction to decrease paracellular phosphate permeability. Effects of pH on TEER have been previously reported (44).
Tenapanor does not affect paracellular macromolecule absorption
Although ions and small macromolecules can traverse cell monolayers via paracellular pathways, it has been proposed that ions and macromolecules are absorbed via distinct pathways (45). Mannitol, a small (molecular weight, 182) hydrophilic molecule thought to be exclusively absorbed via the paracellular route in vivo, was used as a marker of paracellular macromolecule absorption. Tenapanor inhibited 33P absorption in rats at both 0.5 and 10 mg/kg (Fig. 8A) but had no effect on 3H-mannitol absorption (Fig. 8B), suggesting that tenapanor is selective for the paracellular ion absorption pathway over the macromolecule pathway. We also specifically tested the effect of tenapanor on intestinal glucose absorption, because NHE3 activity has previously been proposed to be coupled to paracellular glucose absorption (46). Unabsorbed glucose remaining in the luminal contents from the entire small intestine was measured at fixed time points after initiation of a standardized meal consumed over 4 hours in trained rats pretreated with tenapanor or vehicle. Dietary glucose was efficiently absorbed in the small intestine, and this was unaffected by tenapanor treatment (Fig. 8C).
Fig. 8. Effects of tenapanor on paracellular macromolecule absorption in rats and on phosphate, sodium, and potassium absorption in healthy human volunteers.

(A) Radioactive phosphate and (B) radioactive mannitol absorption in vivo in rats after treatment with vehicle (acidified water, 0.01% Tween 80) or different doses of tenapanor. dpm, disintegrations per minute; AUC, area under the curve. (C) Small intestinal glucose content after initiation of a standardized meal in rats pretreated with vehicle or tenapanor (0.15 mg/kg) or vehicle (acidified water, 0.01% Tween 80) (n = 4 to 7 per group). (D) Stool and (E) urinary phosphorus, (F) urinary sodium, and (G) urinary potassium excretion at baseline (day −1) and after 4 days of tenapanor treatment [15 mg twice daily (b.i.d.)] in healthy volunteers. Means ± SEM (A to C), means ± SD (D to G). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; two-way ANOVA, with post hoc testing at time point with Bonferroni’s correction (A to C); pairwise comparisons by Student’s t test (D to G).
Tenapanor reduces phosphate absorption in healthy human volunteers
In a phase 1 study conducted in healthy human volunteers, treatment with tenapanor (15 mg twice daily) for 4 days significantly (P < 0.05) increased mean daily stool phosphorus excretion from baseline (Fig. 8D), which was accompanied by a comparable and significant (P < 0.001) decrease in mean daily urinary phosphorus excretion (Fig. 8E). These results confirm that the ability of tenapanor to reduce gastrointestinal phosphate absorption in rats translates successfully into humans. Tenapanor also significantly (P < 0.001) reduced mean daily urinary sodium excretion (Fig. 8F) but had no effect on urinary potassium excretion (Fig. 8G), consistent with its effects in rats.
DISCUSSION
Current medical management of hyperphosphatemia with dietary phosphate restriction and oral phosphate binders often proves inadequate to consistently achieve serum phosphate concentration within the target range (22, 47–49). Additional strategies to optimize serum phosphate control are required because hyperphosphatemia is associated with poor clinical outcomes (8–14). Tenapanor is an investigational, small-molecule agent with a different mechanism of action from phosphate binders and has the potential to address unmet needs in hyperphosphatemia therapy. Our investigations provide evidence for the mechanism by which tenapanor, an NHE3 inhibitor, reduces intestinal phosphate absorption. In vivo studies in rodents, combined with translational experiments conducted on human intestinal stem cell–derived enteroid monolayers to model intestinal ion transport physiology, indicate that the predominant mechanism by which tenapanor produces its effect is the reduction of passive paracellular phosphate absorption, retaining activity even at high luminal phosphate concentrations.
Our findings demonstrate that tenapanor decreases the paracellular absorption of phosphate by modulating tight junctions to increase TEER, thereby reducing paracellular phosphate permeability. Experiments using monolayers of NHE3 KO cells showed that the effect of tenapanor on TEER and paracellular phosphate permeability is mediated exclusively via on-target inhibition of NHE3 and is likely the result of decreased pHi, a consequence of inhibition of proton secretion by NHE3. The onset of action to increase TEER and reduce paracellular phosphate permeability is near-immediate after NHE3 inhibition by tenapanor. Previous studies confirm that NHE3 inhibition or deletion is associated with increased TEER (46, 50, 51), although an effect on phosphate absorption has not previously been described. Reduced urinary phosphate excretion has been reported in NHE3 KO mice (52), consistent with our finding of an NHE3-mediated reduction in intestinal phosphate absorption.
Our studies have also shown that tenapanor is not a direct inhibitor of the most important active phosphate transporter, NaPi2b, although repeat administration of tenapanor in vivo does reduce the expression of NaPi2b at the transcriptional level. The contribution of reduced NaPi2b expression to the overall phosphate-lowering effect of tenapanor is likely minor; however, it does indicate that tenapanor prevents increased active transcellular phosphate uptake from compensating for reduced paracellular phosphate absorption. In contrast, the commonly used phosphate binder sevelamer increases NaPi2b expression in mice (28), leading to enhanced transcellular phosphate absorption, which has been proposed to limit the phosphate-lowering efficacy of sevelamer (53).
The effects of tenapanor on overall ion balance in preclinical models and in humans appear to be specific for sodium and phosphate. We have previously reported that tenapanor does not affect the overall balance of other ions in healthy humans and rats (35). Here, we show that tenapanor reduced urinary phosphate excretion in humans, accompanied by increased stool phosphate, but had no effect on urinary potassium excretion. We previously reported that tenapanor had no effect on stool potassium excretion in healthy humans (35), confirming the lack of effect of tenapanor on intestinal potassium absorption, an ion reliant on paracellular absorption in the small intestine. Enteropooling studies measuring luminal ion concentrations in rats confirm the preferential inhibition of phosphate absorption by tenapanor relative to chloride and potassium as well as the divalent cations, calcium and magnesium, which were unaffected by tenapanor. We have previously shown that urinary calcium excretion is unaffected by tenapanor in healthy humans, consistent with this result in rats (35).
Although tight junction permeability can display some specificity toward individual or selected ion groups (32, 33), the in vivo phosphate preference was not readily reflected in direct biophysical measurements of paracellular permeability, suggesting that tight junction ion specificity is not the basis of tenapanor’s in vivo phenotypic selectivity. The reliance of phosphate on paracellular absorption likely explains the observed preferential effect of tenapanor on phosphate absorption. This is a consequence of the driving force for phosphate flux, which itself is a result of a negative luminal potential applied to a divalent anion and low plasma phosphate concentrations relative to high luminal concentrations, and the absence of a low-affinity, high-capacity transcellular phosphate transporter. An examination of tenapanor’s effect on chloride, an anion presumably with similar inherent paracellular permeability characteristics to phosphate, highlights the in vivo specificity of tenapanor. Paracellular ion flux is an integrated outcome determined by paracellular permeability and the prevailing electrochemical gradient. Chloride permeability was reduced by tenapanor when measured by acutely imposing a nearly 100 mM chemical gradient to capture dilution potentials in Ussing chamber experiments ex vivo. However, measured luminal chloride concentrations in vivo were insufficient to support paracellular chloride absorption against normal plasma chloride concentrations, which are higher, indicating a prominent role of active transcellular chloride flux. Consequently, tenapanor had minimal overall impact on chloride such that 24-hour urinary chloride excretion was unchanged in the absence of a transcriptional response to up-regulate chloride exchangers, transporters, or channels.
Further to the assessments of ion flux, tenapanor did not affect the absorption of mannitol tracer, indicating that tenapanor does not affect the paracellular macromolecule absorption pathway under physiological conditions in the small intestine. Moreover, tenapanor had no effect on intestinal glucose absorption. This indicates a low risk of inhibiting the paracellular absorption of dietary macro-molecules and a low risk of drug-drug interactions with the few agents absorbed by this pathway.
A limitation of our study is that the method we used to follow pHi, using a pH-sensitive dye, only measured relative differences between interventions rather than an absolute quantification of pHi. This method was, however, sufficient to follow rapid dynamic pH changes that occurred after treatment with tenapanor and acidic apical media. These measurements are consistent with the ability of tenapanor to modulate tight junction phosphate permeability because of intracellular proton retention, resulting in a rapid decrease in pHi, as the effects of tenapanor on paracellular permeability were mimicked by luminal acidification and other interventions that reduce pHi. Because pHi is tightly regulated within a narrow range to support normal cellular functions, it is likely that the magnitude of the effect of tenapanor on pHi is modest and probably locally confined to NHE3-expressing enterocytes. We have previously reported in healthy volunteers that treatment with tenapanor does not affect serum bicarbonate or urinary pH, suggesting that systemic acid-base balance is not perturbed by this drug (35).
A further limitation to these studies is the unknown identity of the paracellular phosphate pore. The rapid, almost instantaneous increase in TEER with no obvious signaling pathway or tight junction protein trafficking suggests that a direct pH-sensitive conformational change to the tight junction may decrease paracellular phosphate permeability. For example, histidine residues on the intracellular termini of tight junction claudins could be direct pH sensors for the tight junction. Consistent with this possibility, a recently developed trans–tight junction patch clamp technique, applied to measure flux across an individual claudin channel, confirms that modulation of the gating kinetics of tight junction channels is an important mechanism of barrier regulation (54). Unfortunately, the molecular identity of the paracellular phosphate pore has not yet been elucidated, preventing characterization of the specific tight junction proteins involved. Further investigation of the structural and functional response of the tight junction to NHE3 inhibition is therefore required.
Our results are most consistent with pHi mediating the effect of NHE3 inhibition on altered paracellular phosphate absorption. However, our work does not exclude possible effects due to a reduction in intracellular sodium (or resultant potential changes in osmolality and cell volume) or chloride, which may accompany NHE3 inhibition as a result of direct inhibition of transcellular sodium absorption or indirectly through inhibition of electroneutral sodium chloride absorption. Low intracellular sodium concentrations are established and maintained by the basolateral sodium/potassium (Na+/K+) adenosine triphosphatase (ATPase), and the resultant electrochemical sodium gradient provides the driving force for secondary active intestinal transport of solutes coupled to luminal sodium absorption. The Na+/K+ ATPase likely adapts to reduced transcellular sodium influx via NHE3, such as is encountered in periods of fasting, to buffer variations in intracellular sodium and to prevent perturbations in cell osmolality and volume. However, the specific potential contributions of intracellular sodium or chloride concentrations should be the subject of future investigation.
We have previously shown that tenapanor reduced urinary and serum phosphate in a rat model of CKD with vascular calcification (36) and here show that these effects on intestinal phosphate absorption translate to healthy humans. Tenapanor, a minimally absorbed, small-molecule inhibitor of NHE3, effectively increased stool phosphorus excretion and decreased urinary phosphorus excretion in humans when one 15-mg tablet was administered twice daily. Despite currently available treatments, many patients with ESRD fail to maintain target range serum phosphate concentrations (55, 56), with a key contributing factor being poor adherence to phosphate binders as a result of the high pill burden required to effectively capture dietary phosphate (57). Tenapanor has potential to reduce the pill burden for these patients, thereby improving treatment compliance. In addition, tenapanor remains effective in inhibiting intestinal phosphate absorption at high luminal phosphate concentrations. Tenapanor may therefore effectively inhibit intestinal phosphate absorption in the setting of a liberalized phosphate diet. This could contribute an important nutritional benefit to patients with ESRD.
In conclusion, tenapanor reduces intestinal phosphate absorption through the reduction of passive paracellular phosphate influx, quantitatively the most important overall mechanism of intestinal phosphate absorption. Tenapanor modulates tight junctions to increase TEER and reduce paracellular permeability to phosphate; this effect is mediated exclusively via on-target inhibition of NHE3 and is likely the result of decreased pHi due to the inhibition of proton secretion. Tenapanor retains efficacy at high luminal phosphate concentrations, and this translates to reduced intestinal phosphate absorption in healthy humans. We have recently reported that tenapanor significantly reduces serum phosphate and FGF-23 in ESRD patients with hyperphosphatemia receiving dialysis (58, 59); further investigation is ongoing to determine whether tenapanor offers a new approach to serum phosphate control in the clinical setting (60, 61).
MATERIALS AND METHODS
Study design
The primary objective of this work was to elucidate the mechanism by which tenapanor reduces intestinal phosphate absorption, which was explored using a combination of in vivo studies in healthy rodents and translational experiments on human intestinal stem cell– derived enteroid monolayers to model ion transport physiology. Intestinal phosphate absorption was assessed in vivo by measuring luminal and urinary phosphate concentration and excretion under various experimental conditions that manipulated the phosphate concentration gradient. Phosphate flux, TEER, and paracellular ion permeability were measured in human intestinal epithelial stem cell–derived enteroid monolayers under varying phosphate concentration gradients. The animal and cellular monolayer studies were controlled with vehicle control groups. Assignment to treatment or control was randomized. Rigorous blinding of cellular and animal experiments with tenapanor was precluded because of obvious visual pharmacodynamic effects of tenapanor to prevent apical acid secretion in the monolayers and to alter fecal form in rodents; however, the analytical measurements for ion concentration were performed in a blinded manner. The translational effect of tenapanor on intestinal phosphate absorption was evaluated in a single-center, randomized, open-label study in 18 healthy human volunteers [ClinicalTrials.gov, identifier NCT02249936 (62)].
Chemical tools
Pentobarbitone sodium (Pentoject, Animalcare Ltd.), tenapanor {N,N′-(10,17-dioxo-3,6,21,24-tetraoxa-9,11,16,18-tetraazahexacosane-1,26-diyl)bis[3-(6,8-dichloro-2-methyl-1,2,3,4-tetrahydroisoquinolin-4-yl) benzenesulfonamide]} (prepared at Ardelyx), NTX-9066 {4-(3-[4-(2-[3-(N-[(1r,4r)-4-carboxycyclohexyl]-N-ethylsulfamoyl)benzamido]-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxamido)phenyl]propyl) benzoic acid} (prepared at Ardelyx), nigericin (N1495, Life Technologies), BAM15 (5737/10, R&D Systems), FCP (0453/10, R&D Systems), carbachol (1092009, Sigma-Aldrich), Pitstop 2 (ab120687, Abcam), and Dynasore (ab120192, Abcam) were used.
Animal studies
Animal experiments performed in the United States were conducted using experimental protocols approved by the Institutional Animal Care and Use Committee and performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Experiments in the UK were performed in accordance with the UK Animals (Scientific Procedures) Act, 1986, Amendment Regulations 2012, using protocols approved by the University College London (Royal Free Campus) Comparative Biology Unit Animal Welfare and Ethical Review Body Committee. Experiments in Canada were performed in compliance with the animal ethics board at the University of Alberta, Health Sciences Section under approved protocol number AUP00000213. Experiments in Sweden were approved by The Regional Animal Ethics Committee for Experimental Animals, University of Gothenburg.
Intestinal epithelial stem cell monolayer culture model
Intestinal epithelial stem cell monolayers were cultured and differentiated on Transwells as described in detail by Kozuka et al. (38). Human biopsies from which stem cells were sourced were obtained from visibly healthy tissue from male donors according to a protocol approved by the Copernicus Group Institutional Review Board. Experiments were initiated in each differentiated monolayer culture well by washing the apical and basolateral side twice with fresh supplemented basal media and phosphate-free Dulbecco’s modified Eagle’s medium. Compounds were dosed only on the apical side of the monolayer, as detailed in the text; DMSO at an equivalent concentration was used as the control. Phosphate concentration and pH were manipulated as described in the text. Apical and basolateral ion concentrations were measured by ion chromatography, pH was measured using a pH meter, and TEER values were recorded using a volt/ohm meter. TEER results are reported as normalized to baseline. Absolute baseline TEER values are reported in table S1.
Tenapanor in healthy volunteers
Healthy male and female volunteers (aged 19 to 65 years; body mass index, 18 to 29.9 kg/m2) were enrolled in a single-center, randomized, open-label, three-way crossover study to evaluate different tenapanor formulations (ClinicalTrials.gov identifier: NCT02249936). The study protocol was approved by the IntegReview Institutional Review Board. All participants provided written informed consent, and the study was conducted in accordance with the Declaration of Helsinki and the International Conference on Harmonisation and Good Clinical Practice guidelines. Volunteers received a different one of the three tenapanor formulations—HCl capsule, HCl tablet, or free-base tablet—in each treatment period (days 1 to 4, 7 to 10, and 13 to 16), according to the randomization scheme. Tenapanor was administered twice daily before a standardized meal, and 24-hour urine and stool collections were made daily throughout the study period (day −2 to day 17). Data are presented for the tenapanor HCl tablet formulation.
Statistical analysis
The data analyzed were continuous, quantitative, and normally distributed, as assessed by D’Agostino-Pearson omnibus normality test. Between-group differences over time were assessed by a two-way mixed-design ANOVA, and post hoc testing at each time point was performed using Bonferroni’s procedure to correct for multiple comparisons (GraphPad Prism 6). Between-group differences (three or more groups) with only one time point were assessed by one-way ANOVA and post hoc multiple comparisons with Dunnett’s test. Pairwise comparisons were made by Student’s t test. P < 0.05 was considered significant. All results are presented as means ± SE, except the clinical study (Fig. 8, D to G) for which results are means ± SD. Complete individual subject-level data for all experiments are provided in table S2.
Supplementary Material
Fig. S1. NHE3 expression and tenapanor treatment in human and mouse enteroid monolayer cultures.
Fig. S2. Tenapanor inhibits phosphate absorption in human ileum monolayer cultures.
Fig. S3. pHi modulates TEER in monolayer cultures.
Fig. S4. Paracellular phosphate absorption in NHE3 KO human duodenum monolayer cultures.
Fig. S5. Renal and hormonal responses to chronic in vivo tenapanor treatment in healthy rats.
Fig. S6. Changes in intestinal transporter expression in response to chronic in vivo tenapanor treatment in healthy rats.
Fig. S7. Tenapanor has little effect on phosphate or glucose uptake in rat intestinal BBMVs.
Fig. S8. Tenapanor does not alter TEER via redistribution of tight junction proteins.
Table S1. Baseline TEER for all experimental conditions.
www.sciencetranslationalmedicine.org/cgi/content/full/10/456/eaam6474/DC1 Materials and Methods
Table S2. Individual subject-level data for all experiments.
Table S3. Author contributions to the different experiments.
Acknowledgments:
We thank C. Plato (Plato BioPharma, Westminster, CO) for execution of the rat phosphate diet study. Technical assistance from C. Ericsson and A.-C. Andréasson (AstraZeneca Gothenburg, Mölndal, Sweden) and U. Schnitzbauer (University of Zurich, Switzerland) is also acknowledged. Editorial support toward the writing of the manuscript was provided by R. Claes of Oxford PharmaGenesis, funded by Ardelyx. Enteroid monolayer experiments were performed by Ardelyx under a license agreement with Hubrecht Organoid Technology providing nonexclusive rights to use patented technology covering enteroid methods.
Funding: This work was funded by Ardelyx, The Kidney Foundation of Canada (KFOC170002 to R.T.A.), and the National Centre for Competence in Research (NCCR) Kidney (to C.A.W.). C.A.W. is supported by the Swiss National Science Foundation. M.D. received funds from Emulate Inc., NIH/National Institute of Diabetes and Digestive and Kidney Diseases (RO1DK02653, R24DK99803, UO1DK10316, and P30DK089501), and NIH/National Institute of Allergy and Infectious Diseases (PO1AI25181).
Footnotes
Competing interests: A.J.K., M. Siegel, Y.H., B.N., J.W., S.K.-M., N.A.M., Q.J., D.P., J.K., P.K., K.K., J.G.L., D.D., D.P.R., and J.S.C. are current or former employees of Ardelyx and have ownership interest in Ardelyx. P.J.G., A.-C.J.-R., D.K., M.B., M. Strömstedt, and T.R.-B. are employees of AstraZeneca and have ownership interest in AstraZeneca. J.M. has a consultancy agreement with Ardelyx and AstraZeneca. R.T.A. has a consultancy agreement with Ardelyx and is the Canada Research Chair in Renal Tubular Epithelial Transport Physiology. Tenapanor is covered by patent WO2010078449 (63). J.G.L. is an inventor on patent WO2014169094A2 assigned to Ardelyx, which is directed to certain aspects of phosphate uptake inhibition by NHE3 inhibitors that are disclosed in this article (64).
Data and materials availability: All data associated with this study are present in the paper or the Supplementary Materials.
REFERENCES AND NOTES
- 1.Kidney Disease: Improving Global Outcomes (KDIGO) CKD-MBD Work Group, KDIGO clinical practice guideline for the diagnosis, evaluation, prevention, and treatment of chronic kidney disease-mineral and bone disorder (CKD-MBD). Kidney Int. Suppl, S1–S130 (2009). [DOI] [PubMed]
- 2.Walton J, Gray TK, Absorption of inorganic phosphate in the human small intestine. Clin. Sci 56, 407–412 (1979). [DOI] [PubMed] [Google Scholar]
- 3.Davis GR, Zerwekh JE, Parker TF, Krejs GJ, Pak CY, Fordtran JS, Absorption of phosphate in the jejunum of patients with chronic renal failure before and after correction of vitamin D deficiency. Gastroenterology 85, 908–916 (1983). [PubMed] [Google Scholar]
- 4.Aloia JF, Yeh JK, Effect of hypophysectomy on intestinal phosphate absorption in rats. Bone 6, 73–77 (1985). [DOI] [PubMed] [Google Scholar]
- 5.Williams KB, DeLuca HF, Characterization of intestinal phosphate absorption using a novel in vivo method. Am. J. Physiol. Endocrinol. Metab 292, E1917–E1921 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Hruska KA, Mathew S, Lund R, Qiu P, Pratt R, Hyperphosphatemia of chronic kidney disease. Kidney Int 74, 148–157 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wagner CA, Hernando N, Forster IC, Biber J, The SLC34 family of sodium-dependent phosphate transporters. Pflugers Arch 466, 139–153 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Toussaint ND, Pedagogos E, Tan S-J, Badve SV, Hawley CM, Perkovic V, Elder GJ, Phosphate in early chronic kidney disease: Associations with clinical outcomes and a target to reduce cardiovascular risk. Nephrology 17, 433–444 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Kestenbaum B, Sampson JN, Rudser KD, Patterson DJ, Seliger SL, Young B, Sherrard DJ, Andress DL, Serum phosphate levels and mortality risk among people with chronic kidney disease. J. Am. Soc. Nephrol 16, 520–528 (2005). [DOI] [PubMed] [Google Scholar]
- 10.Kovesdy CP, Ahmadzadeh S, Anderson JE, Kalantar-Zadeh K, Secondary hyperparathyroidism is associated with higher mortality in men with moderate to severe chronic kidney disease. Kidney Int 73, 1296–1302 (2008). [DOI] [PubMed] [Google Scholar]
- 11.Eddington H, Hoefield R, Sinha S, Chrysochou C, Lane B, Foley RN, Hegarty J, New J, O’Donoghue DJ, Middleton RJ, Kalra PA, Serum phosphate and mortality in patients with chronic kidney disease. Clin. J. Am. Soc. Nephrol 5, 2251–2257 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Palmer SC, Hayen A, Macaskill P, Pellegrini F, Craig JC, Elder GJ, Strippoli GFM, Serum levels of phosphorus, parathyroid hormone, and calcium and risks of death and cardiovascular disease in individuals with chronic kidney disease: A systematic review and meta-analysis. JAMA 305, 1119–1127 (2011). [DOI] [PubMed] [Google Scholar]
- 13.McGovern AP, de Lusignan S, van Vlymen J, Liyanage H, Tomson CR, Gallagher H, Rafiq M, Jones S, Serum phosphate as a risk factor for cardiovascular events in people with and without chronic kidney disease: A large community based cohort study. PLOS ONE 8, e74996 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schwarz S, Trivedi BK, Kalantar-Zadeh K, Kovesdy CP, Association of disorders in mineral metabolism with progression of chronic kidney disease. Clin. J. Am. Soc. Nephrol 1, 825–831 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Zhou C, Wang F, Wang J-W, Zhang L-X, Zhao M-H, Mineral and bone disorder and its association with cardiovascular parameters in Chinese patients with chronic kidney disease. Chin. Med. J 129, 2275–2280 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Collins AJ, Foley RN, Gilbertson DT, Chen S-C, The state of chronic kidney disease, ESRD, and morbidity and mortality in the first year of dialysis. Clin. J. Am. Soc. Nephrol 4 (suppl. 1), S5–S11 (2009). [DOI] [PubMed] [Google Scholar]
- 17.Isakova T, Wahl P, Vargas GS, Gutiérrez OM, Scialla J, Xie H, Appleby D, Nessel L, Bellovich K, Chen J, Hamm L, Gadegbeku C, Horwitz E, Townsend RR, Anderson CAM, Lash JP, Hsu C-Y, Leonard MB, Wolf M, Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int 79, 1370–1378 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Isakova T, Xie H, Yang W, Xie D, Anderson AH, Scialla J, Wahl P, Gutiérrez OM, Steigerwalt S, He J, Schwartz S, Lo J, Ojo A, Sondheimer J, Hsu C-Y, Lash J, Leonard M, Kusek JW, Feldman HI, Wolf M Chronic Renal Insufficiency Cohort (CRIC) Study Group, Fibroblast growth factor 23 and risks of mortality and end-stage renal disease in patients with chronic kidney disease. JAMA 305, 2432–2439 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cannata-Andía JB, Fernández-Martín JL, Locatelli F, London G, Gorriz JL, Floege J, Ketteler M, Ferreira A, Covic A, Rutkowski B, Memmos D, Bos WJ, Teplan V, Nagy J, Tielemans C, Verbeelen D, Goldsmith D, Kramar R, Martin PY, Wüthrich RP, Pavlovic D, Benedik M, Sánchez JE, Martínez-Camblor P, Naves-Díaz M, Carrero JJ, Zoccali C, Use of phosphate-binding agents is associated with a lower risk of mortality. Kidney Int 84, 998–1008 (2013). [DOI] [PubMed] [Google Scholar]
- 20.Sullivan C, Sayre SS, Leon JB, Machekano R, Love TE, Porter D, Marbury M, Sehgal AR, Effect of food additives on hyperphosphatemia among patients with end-stage renal disease: A randomized controlled trial. JAMA 301, 629–635 (2009). [DOI] [PubMed] [Google Scholar]
- 21.Karamanidou C, Clatworthy J, Weinman J, Horne R, A systematic review of the prevalence and determinants of nonadherence to phosphate binding medication in patients with end-stage renal disease. BMC Nephrol 9, 2 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chiu YW, Teitelbaum I, Misra M, de Leon EM, Adzize T, Mehrotra R, Pill burden, adherence, hyperphosphatemia, and quality of life in maintenance dialysis patients. Clin. J. Am. Soc. Nephrol 4, 1089–1096 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sabbagh Y, Giral H, Caldas Y, Levi M, Schiavi SC, Intestinal phosphate transport. Adv. Chronic Kidney Dis 18, 85–90 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marks J, Debnam ES, Unwin RJ, The role of the gastrointestinal tract in phosphate homeostasis in health and chronic kidney disease. Curr. Opin. Nephrol. Hypertens 22, 481–487 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Amanzadeh J, Reilly RF Jr., Hypophosphatemia: An evidence-based approach to its clinical consequences and management. Nat. Clin. Pract. Nephrol 2, 136–148 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Hernando N, Myakala K, Simona F, Knopfel T, Thomas L, Murer H, Wagner CA, Biber J, Intestinal depletion of NaPi-IIb/Slc34a2 in mice: Renal and hormonal adaptation. J. Bone Miner. Res 30, 1925–1937 (2015). [DOI] [PubMed] [Google Scholar]
- 27.Sabbagh Y, O’Brien SP, Song W, Boulanger JH, Stockmann A, Arbeeny C, Schiavi SC, Intestinal npt2b plays a major role in phosphate absorption and homeostasis. J. Am. Soc. Nephrol 20, 2348–2358 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schiavi SC, Tang W, Bracken C, O’Brien SP, Song W, Boulanger J, Ryan S, Phillips L, Liu S, Arbeeny C, Ledbetter S, Sabbagh Y, Npt2b deletion attenuates hyperphosphatemia associated with CKD. J. Am. Soc. Nephrol 23, 1691–1700 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Forster IC, Virkki L, Bossi E, Murer H, Biber J, Electrogenic kinetics of a mammalian intestinal type IIb Na+/Pi cotransporter. J. Membr. Biol 212, 177–190 (2006). [DOI] [PubMed] [Google Scholar]
- 30.Lee GJ, Marks J, Intestinal phosphate transport: A therapeutic target in chronic kidney disease and beyond? Pediatr. Nephrol 30, 363–371 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Marks J, Debnam ES, Unwin RJ, Phosphate homeostasis and the renal-gastrointestinal axis. Am. J. Physiol. Renal Physiol 299, F285–F296 (2010). [DOI] [PubMed] [Google Scholar]
- 32.Günzel D, Fromm M, Claudins and other tight junction proteins. Compr. Physiol 2, 1819–1852 (2012). [DOI] [PubMed] [Google Scholar]
- 33.Günzel D, Yu ASL, Claudins and the modulation of tight junction permeability. Physiol. Rev 93, 525–569 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee DB, Walling MW, Corry DB, Phosphate transport across rat jejunum: Influence of sodium, pH, and 1,25-dihydroxyvitamin D3. Am. J. Physiol 251 (Pt. 1), G90–G95 (1986). [DOI] [PubMed] [Google Scholar]
- 35.Spencer AG, Labonte ED, Rosenbaum DP, Plato CF, Carreras CW, Leadbetter MR, Kozuka K, Kohler J, Koo-McCoy S, He L, Bell N, Tabora J, Joly KM, Navre M, Jacobs JW, Charmot D, Intestinal inhibition of the Na+/H+ exchanger 3 prevents cardiorenal damage in rats and inhibits Na+ uptake in humans. Sci. Transl. Med 6, 227ra236 (2014). [DOI] [PubMed] [Google Scholar]
- 36.Labonté ED, Carreras CW, Leadbetter MR, Kozuka K, Kohler J, Koo-McCoy S, He L, Dy E, Black D, Zhong Z, Langsetmo I, Spencer AG, Bell N, Deshpande D, Navre M, Lewis JG, Jacobs JW, Charmot D, Gastrointestinal inhibition of sodium-hydrogen exchanger 3 reduces phosphorus absorption and protects against vascular calcification in CKD. J. Am. Soc. Nephrol 26, 1138–1149 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marks J, Srai SK, Biber J, Murer H, Unwin RJ, Debnam ES, Intestinal phosphate absorption and the effect of vitamin D: A comparison of rats with mice. Exp. Physiol 91, 531–537 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Kozuka K, He Y, Koo-McCoy S, Kumaraswamy P, Nie B, Shaw K, Chan P, Leadbetter M, He L, Lewis JG, Zhong Z, Charmot D, Balaa M, King AJ, Caldwell JS, Siegel M, Development and characterization of a human and mouse intestinal epithelial cell monolayer platform. Stem Cell Reports 9, 1976–1990 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Agarwal R, Afzalpurkar R, Fordtran JS, Pathophysiology of potassium absorption and secretion by the human intestine. Gastroenterology 107, 548–571 (1994). [DOI] [PubMed] [Google Scholar]
- 40.Orlowski J, Kandasamy RA, Shull GE, Molecular cloning of putative members of the Na/H exchanger gene family. cDNA cloning, deduced amino acid sequence, and mRNA tissue expression of the rat Na/H exchanger NHE-1 and two structurally related proteins. J. Biol. Chem 267, 9331–9339 (1992). [PubMed] [Google Scholar]
- 41.Hachiya S, Miura M, Imamura Y, Kaga D, Sato I, Moritomo H, Kato K, Terai K, Terada Y, Tetrahydrobenzothiophene compound US8729068 B2 (2014).
- 42.Kinsella JL, Aronson PS, Properties of the Na+-H+ exchanger in renal microvillus membrane vesicles. Am. J. Physiol 238, F461–F469 (1980). [DOI] [PubMed] [Google Scholar]
- 43.Kato A, Romero MF, Regulation of electroneutral NaCl absorption by the small intestine. Annu. Rev. Physiol 73, 261–281 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vastag M, Neuhofer W, Nagel W, Beck FX, Ammonium affects tight junctions and the cytoskeleton in MDCK cells. Pflugers Arch 449, 384–391 (2005). [DOI] [PubMed] [Google Scholar]
- 45.Krug SM, Schulzke JD, Fromm M, Tight junction, selective permeability, and related diseases. Semin. Cell Dev. Biol 36, 166–176 (2014). [DOI] [PubMed] [Google Scholar]
- 46.Turner JR, Black ED, Ward J, Tse C-M, Uchwat FA, Alli HA, Donowitz M, Madara JL, Angle JM, Transepithelial resistance can be regulated by the intestinal brush-border Na+/H+ exchanger NHE3. Am. J. Physiol. Cell Physiol 279, C1918–C1924 (2000). [DOI] [PubMed] [Google Scholar]
- 47.Farrand KF, Copley JB, Heise J, Fridman M, Keith MS, Poole L, Analysis of serum phosphate control and phosphate binder utilization in incident hemodialysis patients. Int. J. Nephrol. Renovasc. Dis 7, 261–269 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fissell RB, Karaboyas A, Bieber BA, Sen A, Li Y, Lopes AA, Akiba T, Bommer J, Ethier J, Jadoul M, Pisoni RL, Robinson BM, Tentori F, Phosphate binder pill burden, patient-reported non-adherence, and mineral bone disorder markers: Findings from the DOPPS. Hemodial. Int 20, 38–49 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang S, Alfieri T, Ramakrishnan K, Braunhofer P, Newsome BA, Serum phosphorus levels and pill burden are inversely associated with adherence in patients on hemodialysis. Nephrol. Dial. Transplant 29, 2092–2099 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gawenis LR, Stien X, Shull GE, Schultheis PJ, Woo AL, Walker NM, Clarke LL, Intestinal NaCl transport in NHE2 and NHE3 knockout mice. Am. J. Physiol. Gastrointest. Liver Physiol 282, G776–G784 (2002). [DOI] [PubMed] [Google Scholar]
- 51.Rievaj J, Pan W, Cordat E, Alexander RT, The Na+/H+ exchanger isoform 3 is required for active paracellular and transcellular Ca2+ transport across murine cecum. Am. J. Physiol. Gastrointest. Liver Physiol 305, G303–G313 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pan W, Borovac J, Spicer Z, Hoenderop JG, Bindels RJ, Shull GE, Doschak MR, Cordat E, Alexander RT, The epithelial sodium/proton exchanger, NHE3, is necessary for renal and intestinal calcium (re)absorption. Am. J. Physiol. Renal Physiol 302, F943–F956 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Komaba H, Fukagawa M, Phosphate—A poison for humans? Kidney Int 90, 753–763 (2016). [DOI] [PubMed] [Google Scholar]
- 54.Weber CR, Liang GH, Wang Y, Das S, Shen L, Yu ASL, Nelson DJ, Turner JR, Claudin-2-dependent paracellular channels are dynamically gated. eLife 4, e09906 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Benner D, Nissenson AR, Van Wyck D, Focused clinical campaign improves mineral and bone disorder outcomes. J. Ren. Care 38, 2–8 (2012). [DOI] [PubMed] [Google Scholar]
- 56.Port FK, Pisoni RL, Bommer J, Locatelli F, Jadoul M, Eknoyan G, Kurokawa K, Canaud BJ, Finley MP, Young EW, Improving outcomes for dialysis patients in the international Dialysis Outcomes and Practice Patterns Study. Clin. J. Am. Soc. Nephrol 1, 246–255 (2006). [DOI] [PubMed] [Google Scholar]
- 57.Joson CG, Henry SL, Kim S, Cheung MY, Parab P, Abcar AC, Jacobsen SJ, Morisky DE, Sim JJ, Patient-reported factors associated with poor phosphorus control in a maintenance hemodialysis population. J. Ren. Nutr 26, 141–148 (2016). [DOI] [PubMed] [Google Scholar]
- 58.Block GA, Rosenbaum DP, Leonsson-Zachrisson M, Astrand M, Johansson S, Knutsson M, Langkilde AM, Chertow GM, Effect of tenapanor on serum phosphate in patients receiving hemodialysis. J. Am. Soc. Nephrol 28, 1933–1942 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Block GA, Rosenbaum DP, Yan A, Greasley PJ, Chertow GM, Wolf M, The effects of tenapanor on serum fibroblast growth factor 23 in patients receiving hemodialysis with hyperphosphatemia. Nephrol. Dial. Transplant (2018). [DOI] [PMC free article] [PubMed]
- 60.A phase 3 study of tenapanor to treat hyperphosphatemia in ESRD patients on dialysis, https://clinicaltrials.gov/ct2/show/NCT03427125 [accessed 25 July 2018].
- 61.An 8-week study to evaluate tenapanor in the treatment of hyperphosphatemia in end-stage renal disease patients on hemodialysis (ESRD-HD), https://clinicaltrials.gov/ct2/show/NCT02675998 [accessed 25 July 2018].
- 62.A phase 1 study to examine the pharmacodynamics of different AZD1722 formulations, https://clinicaltrials.gov/ct2/show/NCT02249936 [accessed 20 December 2016].
- 63.Charmot D, Jacobs JW, Leadbetter MR, Navre M, Carreras C, Bell N, Compounds and methods for inhibiting NHE-mediated antiport in the treatment of disorders associated with fluid retention or salt overload and gastrointestinal tract disorders WO2010078449 A2 (2010).
- 64.Carreras CW, Charmot D, Jacobs JW, Labonte ED, Lewis JG, NHE3-binding compounds and methods for inhibiting phosphate transport WO2014169094A2 (2014).
- 65.Borovac J, Barker RS, Rievaj J, Rasmussen A, Pan W, Wevrick R, Alexander RT, Claudin-4 forms a paracellular barrier, revealing the interdependence of claudin expression in the loose epithelial cell culture model opossum kidney cells. Am. J. Physiol. Cell Physiol 303, C1278–C1291 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kimizuka H, Koketsu K, Ion transport through cell membrane. J. Theor. Biol 6, 290–305 (1964). [DOI] [PubMed] [Google Scholar]
- 67.Günzel D, Stuiver M, Kausalya PJ, Haisch L, Krug SM, Rosenthal R, Meij IC, Hunziker W, Fromm M, Müller D, Claudin-10 exists in six alternatively spliced isoforms that exhibit distinct localization and function. J. Cell Sci 122, 1507–1517 (2009). [DOI] [PubMed] [Google Scholar]
- 68.Plain A, Wulfmeyer VC, Milatz S, Klietz A, Hou J, Bleich M, Himmerkus N, Corticomedullary difference in the effects of dietary Ca2+ on tight junction properties in thick ascending limbs of Henle’s loop. Pflugers Arch 468, 293–303 (2016). [DOI] [PubMed] [Google Scholar]
- 69.Stieger B, Murer H, Heterogeneity of brush-border-membrane vesicles from rat small intestine prepared by a precipitation method using Mg/EGTA. Eur. J. Biochem 135, 95–101 (1983). [DOI] [PubMed] [Google Scholar]
- 70.Bradford MM, A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem 72, 248–254 (1976). [DOI] [PubMed] [Google Scholar]
- 71.Hildmann B, Storelli C, Danisi G, Murer H, Regulation of Na+-Pi cotransport by 1,25-dihydroxyvitamin D3 in rabbit duodenal brush-border membrane. Am. J. Physiol 242, G533–G539 (1982). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. NHE3 expression and tenapanor treatment in human and mouse enteroid monolayer cultures.
Fig. S2. Tenapanor inhibits phosphate absorption in human ileum monolayer cultures.
Fig. S3. pHi modulates TEER in monolayer cultures.
Fig. S4. Paracellular phosphate absorption in NHE3 KO human duodenum monolayer cultures.
Fig. S5. Renal and hormonal responses to chronic in vivo tenapanor treatment in healthy rats.
Fig. S6. Changes in intestinal transporter expression in response to chronic in vivo tenapanor treatment in healthy rats.
Fig. S7. Tenapanor has little effect on phosphate or glucose uptake in rat intestinal BBMVs.
Fig. S8. Tenapanor does not alter TEER via redistribution of tight junction proteins.
Table S1. Baseline TEER for all experimental conditions.
www.sciencetranslationalmedicine.org/cgi/content/full/10/456/eaam6474/DC1 Materials and Methods
Table S2. Individual subject-level data for all experiments.
Table S3. Author contributions to the different experiments.