

Abstract
Background
Parasitic plants rely on their host to cover their nutritional requirements either for their entire life or a smaller part of it. Depending on the level of parasitism, a proportional reduction on the plastid genome has been found. However, knowledge on gene loss and evolution of the mitogenome of parasitic plants is only available for four hemiparasitic Viscum species (Viscaceae), which lack many of the mitochondrial genes, while the remaining genes exhibit very fast molecular evolution rates. In this study, we include another genus, Phoradendron, from the Viscaceae, as well as 10 other hemiparasitic or holoparasitic taxa from across the phylogeny of the angiosperms to investigate how fast molecular evolution works on their mitogenomes, and the extent of gene loss.
Results
Our observations from Viscum were replicated in Phoradendron liga, whereas the remaining parasitic plants in the study have a complete set of the core mitochondrial genes and exhibit moderate or only slightly raised substitution rates compared to most autotrophic taxa, without any statistically significant difference between the different groups (autotrophs, hemiparasites and holoparasites). Additionally, further evidence is provided for the placement of Balanophoraceae within the order Santalales, while the exact placement of Cynomoriaceae still remains elusive.
Conclusions
We examine the mitochondrial gene content of 11 hemiparasitic and holoparasitic plants and confirm previous observations in Viscaceae. We show that the remaining parasitic plants do not have significantly higher substitution rates than autotrophic plants in their mitochondrial genes. We provide further evidence for the placement of Balanophoraceae in the Santalales.
Electronic supplementary material
The online version of this article (10.1186/s12862-019-1401-8) contains supplementary material, which is available to authorized users.
Keywords: Phylogeny, Parasitic plants, Substitution rates, Evolution, Parasitism, Balanophoraceae, Viscaceae, Mitogenome
Background
Parasitic plants comprise around 1% of all angiosperm species (http://parasiticplants.siu.edu/ParPlantNumbers.pdf). Depending on their photosynthetic ability, they are categorized as hemiparasites (facultative or obligatory) and holoparasites. Current phylogenetic evidence suggests that parasitism has evolved independently at least 12 times [1, 2], offering an excellent framework for studies of correlation between parasitism and genomic evolution. While a series of papers have recently focused on the correlation between levels of parasitism and loss of key photosynthesis genes in the plastome [3–5] and an instance of a possible complete loss of the plastome has also been reported in the holoparasite Rafflesia lagascae Blanco [6], comparatively little attention has been given to mitochondrial evolution.
The plastome of a typical, autotrophic angiosperm is circular, of a rather uniform size, and has a relatively conserved structure, with two unique regions separated by two inverted repeats, as well as a conserved gene content of ca. 40 protein coding genes, four rRNA genes and approximately 20 tRNA genes [7–9]. However, plastomes of the numerous parasitic angiosperms that have been sequenced show a pronounced variation both in size, structure and gene content. Not unexpectedly, there is a clear correlation between the type of parasitism and the level of plastome reduction and modification, e.g. plastomes of the holoparasitic members of Orobanchaceae [10], Cuscuta L. [3, 4], Pilostyles Guil. [11], Rafflesia lagascae [6], Hydnora visseri Bolin, E.Maass & Musselman [12], and others, are much more reduced and modified than plastomes of hemiparasitic members of Orobanchaceae [5] and Viscaceae [13]. Similar evolutionary plastome reductions have been described in achlorophyllous mycoheterotrophic plants, which have either functionally or completely lost most or all genes involved in photosynthesis, e.g., Neottia nidus-avis (L.) Rich. [14], Corallorhiza striata Lindl. [15], Petrosavia stellaris Becc. [16], and several members of Ericaceae [17]. In hemiparasitic plants, capable of photosynthesis, major plastome reductions would not be expected, but nevertheless a recent study of the Santalales, and specifically on the hemiparasitic genera Osyris L. and Viscum showed that their plastomes are reduced by 10–22% compared to Vitis [13] the closest autotrophic relative sequenced.
Evolution of the mitochondrial genomes, henceforth mitogenomes, which are involved in basic cell metabolism, was not expected to be influenced by parasitism. Mitochondria are essential producers of the energy (ATP) of the cell and harbour key metabolic genes that control respiration, stress responses, and additionally defence against microorganisms and aspects of programmed cell apoptosis. Compared to plant mitochondrial genomes, animal mitochondrial genomes are in general fairly small in size (ca. 16 kb long), uniform in structure (circular) and gene content, encoding 22 tRNA genes, two rRNA genes and usually 13 protein coding genes of the respiratory chain complexes I: nad1, nad2, nad3, nad4, nad4L, nad5, nad6, III: cob, IV: cox1, cox2, cox3, and V: atp6, and atp8 [18]. In contrast, plant mitogenomes exhibit profound differences: Their size is 10 to almost 1000 times larger than their animal counterparts, ranging from less than 200 kb to some 11.3 mb as in the case of Silene conica L. [19]. This has been partly attributed to the presence of larger introns, of more repeated sequences, and stretches of DNA transferred either from the nuclear and chloroplast genome, or from bacteria and viruses that have infected the plant [19, 20]. Electron microscopy and DNA sequencing has revealed that plant mitogenomes may be circular, linear, branching and comprising one or more molecules, as is the cases in e.g. Silene conica [19], Oryza sativa L. [21] and Brassica napus L. [22]. Regarding their standard gene content, plant mitogenomes contain all the genes present in the animal mitogenomes, with additional subunits in respiratory complex I: nad7, nad9, V: atp1, atp4, atp9, as well as in complex II: sdh3, sdh4. In addition to the genes of the respiratory chain, plant mitogenomes contain a maturase-related protein gene (matR), genes encoding for proteins of the small and large subunit of the ribosomes (rps and rpl genes) and the genes ccmB, ccmC, ccmFc and ccmFn that are involved in the cytochrome C biogenesis pathway. All genes from complexes I, III, IV, and V, which are considered as the core mitochondrial genes, are present in all hitherto sequenced autotrophic land plants [23], while the remaining genes comprise a variable part of the plant mitogenome. This was also observed in the holoparasitic plants Rafflesia lagascae [6] and Cynomorium coccineum [24], and similar results have been reported for three hemiparasitic and three holoparasitic Orobanchaceae, all of which possessed a complete core mitochondrial gene set [25]. However, recent studies on four species of Viscum showed a surprising reduction in gene content compared to all other known angiosperm mitogenomes [26, 27]. This observation prompts the obvious question, whether a loss of genes in the mitogenome similar to that observed in the Viscaceae is found in other parasitic lineages as well.
In addition to extensive gene loss, a very high level of sequence divergence of the remaining key genes was also observed in the four species of Viscum [26, 27]. Especially the genes in respiratory complex V, atp1, apt6, atp8, and atp9, have very high substitution rates compared to other plants. However, a few genes, e.g. matR in V. album L., retain a low substitution rate. In general, mitochondrial genomes have low substitution rates, lower than what is found in the chloroplast genomes, and 10–20 times lower compared to the plant nuclear genomes [28]. Often, higher substitution rates in the mitogenome are accompanied by higher substitution rates in the other two plant genomes, and are inferred to be a characteristic of the species that exhibits them, e.g. in certain species of Plantago L. [29]. However, it has been shown later that certain plant genera, like Pelargonium L. H. Bailey, have high substitution rates only in their mitogenomes [30]. Comparison of nuclear small-subunit rDNA from partially autotrophic, chlorophyllous hemiparasites, and achlorophyllous holoparasites showed that the latter have much higher substitution rates than the former [31]. Higher chloroplast substitution rates have been suggested to be correlated with species richness, as in the case of the Proteaceae [32]. Early theoretical models have suggested that parasites should evolve much faster than their hosts as a response to the host-parasite arms race [33]. A comparison of parasitic plants to their closest non-parasitic relatives showed that the first tend to have higher substitution rates across all three genomes [34]. Thus, in this study we also want to test how general the significantly higher substitution rates seen in the mitochondrial genes of Viscum are in parasitic plants and whether higher substitution rates are correlated to levels of parasitism or is clade specific.
Materials and methods
Plant material
Plant material was obtained from Herbarium C and the silica gel collection of the Natural History Museum of Denmark, University of Copenhagen, Denmark. To the best of our knowledge all plants have been legally collected. Details on the plants examined in the study (including authors) are listed in Table 1. The vouchers are all deposited in Herbarium C. Author names are according to the International Plant Names Index (www.ipni.org).
Table 1.
Parasitic plants examined in this study
| # | Project ID | Family | Species | Collector | Date of collection | Area of collection | Type of parasitism |
|---|---|---|---|---|---|---|---|
| 1 | C3122 | Boraginaceae |
Pholisma sonorae
(Torr. ex A. Gray) Yatsk |
R. Thome 52,167 | 1978 | USA, California, Algodones dunes | Holoparasitic |
| 2 | C3123 | Lauraceae |
Cassytha pubescens
Schltdl. |
Whibleg 9833 | 1985 | Australia, South Australia | Hemiparasitic |
| 3 | C3124 | Krameriaceae |
Krameria lanceolata
Torr. |
Museelman 4841 | 1975 | USA, Florida | Hemiparasitic |
| 4 | C314 | Balanophoraceae |
Langsdorffia hypogaea
Mart. |
P. Barbour 2646 | 1978 | Peru, Province Bagua, 12 km E of da Peca | Holoparasitic |
| 5 | C1935 | Orobanchaceae | Lathraea squamaria L. | G. Petersen & O. Seberg | 25/04/05 | Denmark, Virum, Geels Skov | Hemiparasitic |
| 6 | C3089 | Orobanchaceae | Lathraea cladestina L. | H. Æ. Pedersen 811 | 16/05/14 | Spain, Alava, Done Bikendi Havana | Hemiparasitic |
| 7 | C3127 | Loranthaceae | Loranthus europaeus Jacq. | Carlos Reif | 22/02/15 | Czeck Republic, Kutna Hora | Hemiparasitic |
| 8 | C3151 | Cynomoriaceae | Cynomorium coccineum L. | F. N. Rasmussen | 01/05/15 | Portugal | Holoparasitic |
| 9 | C3152 | Cytinaceae | Cytinus hypocistis L. ssp. clusii Nyman | F. N. Rasmussen | 05/05/15 | Portugal, Algarve, 13.2 km NE of Albufeira | Holoparasitic |
| 10 | C3158 | Cytinaceae | Cytinus hypocistis L. ssp. hypocistis | G. Petersen | 30/05/15 | Italy, Liguria | Holoparasitic |
| 11 | C2482 | Viscaceae | Phoradendron liga Eichler | O. Seberg & G. Petersen | 02/11/08 | Argentina, Salta | Hemiparasitic |
DNA extraction
DNA was extracted using two different protocols depending on the type of the sample; one for the herbarium samples and one for the silica gel samples. DNA was extracted from the herbarium samples in the clean lab of the Natural History Museum of Denmark, University of Copenhagen, Denmark, using the ancient plant DNA extraction protocol by Wales et al. [35]. DNA was extracted from the silica gel samples using the DNA Plant Minikit (Qiagen) according to the manufacturer’s instructions, with the addition of a Proteinase K treatment step, for 2 h at 65 °C, immediately after the bead-beating step (Qiashredder). Details on tissue type, protocol, cleaning procedures and mass used for DNA extraction are given in the Additional file 1: Table S1.
Library preparation
The DNA extracted from the herbarium samples was already broken down into very small fragments, whereas the DNA from the silica gels samples was sheared using a Bioruptor (Diagenode). 100 μl dilutions of 10 ng/μl DNA concentration were sheared using the following conditions: 5 cycles of 15 s ON, 90 s OFF. The sheared DNA was then run on a 2% agarose gel with a 100 bp DNA ladder (Thermo Scientific), to check the size variation of the DNA fragments. For all samples we obtained DNA fragments in the range of 200–600 bp.
For the resulting 11 samples, the NEBNext DNA Sample Prep Master Mix Set 2 (New England Biolabs, Ipswich, Massachusetts) using blunt-end adapters specified by Meyer and Kircher [36] was used to prepare the fragments for inserting the adapter sequences and unique barcodes. In order to determine the number of cycles needed for each library, qPCRs were performed on 1:40 dilutions of each DNA sample, using the SYBR Green Master Mix 1 (Agilent Technologies) on an Mx3500p qPCR machine (Agilent Technologies). For each sample a common forward primer and a unique, custom, reverse index primer was used in PCRs. Libraries were amplified 10–14 cycles in a total volume of 100 μL, containing 5 U AmpliTaq Gold polymerase (Applied Biosystems, Foster City, CA), 1× AmpliTaq Gold buffer, 2.5 mM MgCl2, 0.4 mg/mL bovine serum albumin (BSA), 0.2 mM of each dNTP, 0.2 μM IS4 forward primer, 0.2 μM indexed reverse primer, and 20 μL DNA library template. Following amplification, libraries were purified using a QIAquick PCR Purification kit (Qiagen, Hilden, Germany), according to manufacturer’s instructions. DNA was eluted in 32 μL EB buffer and the column was incubated for 10 min at 37 °C prior to centrifugation. The libraries were first quantified on a Qubit 2.0 (Life Technologies, Carlsbad, CA) using a dsDNA high sensitivity assay, and then run on a TapeStation 2200 using the high sensitivity tapes (Agilent, Santa Clara, CA) to determine the average insert size and molarity of each library. Then two sample pools were created, one for the samples from the herbarium and another for the samples from silica gel.
Next generation sequencing
The sequences were obtained using Illumina sequencing on a Hiseq 2500 at the National High-Throughput DNA Sequencing Centre of the University of Copenhagen, Denmark. Details on the run, chemistry, number of raw reads obtained and number of reads that passed quality control are given in Table 2. Sequences for Phoradendron liga were obtained from a previous investigation, where 150 bp pair end sequencing was used. For the silica gel samples 100 bp pair end sequencing was chosen, while for the herbarium samples 100 bp single end sequencing is the preferred method as the DNA is already degraded rendering pair end sequencing sometimes inefficient.
Table 2.
Sequencing information
| # | Project ID | Species | Sequencing platform | Sequencing chemistry | Raw reads | QC Trimmed reads | Mitochondrial gene coverage |
|---|---|---|---|---|---|---|---|
| 1 | C3122 | Pholisma sonorae | Illumina HiSeq 2500 | 100 bp Single End | 14,993,887 | 14,838,255 | 7X |
| 2 | C3123 | Cassytha pubescens | Illumina HiSeq 2500 | 100 bp Single End | 129,061,347 | 127,650,167 | 81X |
| 3 | C3124 | Krameria lanceolata | Illumina HiSeq 2500 | 100 bp Single End | 14,392,427 | 14,102,890 | 7X |
| 4 | C314 | Langsdorffia hypogaea | Illumina HiSeq 2500 | 100 bp Single End | 8,584,595 | 8,118,411 | 31X |
| 5 | C1935 | Lathraea squamaria | Illumina HiSeq 2500 | 100 bp Pair End | 78,319,916 | 44,057,801 | 495X |
| 6 | C3089 | Lathraea clandestina | Illumina HiSeq 2500 | 100 bp Pair End | 54,446,884 | 31,074,675 | 92X |
| 7 | C3127 | Loranthus europaeus | Illumina HiSeq 2500 | 100 bp Pair End | 100,902,100 | 57,189,199 | 386X |
| 8 | C3151 | Cynomorium coccineum | Illumina HiSeq 2500 | 100 bp Pair End | 138,075,206 | 49,400,497 | 30X |
| 9 | C3152 | Cytinus hypocistis ssp. clusii | Illumina HiSeq 2500 | 100 bp Pair End | 138,075,206 | 66,503,764 | 901X |
| 10 | C3158 | Cytinus hypocistis ssp. hypocistis | Illumina HiSeq 2500 | 100 bp Pair End | 78,272,446 | 29,053,324 | 330X |
| 11 | C2482 | Phoradendron liga | Illumina HiSeq 2500 | 150 bp Pair End | 29,638,962 | 23,696,088 | 40X |
NGS data analysis
The obtained raw reads were trimmed for quality, adapters and unidentified nucleotides (Ns) using Adapter Removal [37]. The reads that passed the quality control were then imported into Geneious version R9.0.5 (Biomatters Ltd.). The reads from each taxon were mapped to the mitochondrial gene sets of 5 autotrophic taxa spread across the angiosperm phylogeny ( Brassica carinata L.A. Braun NC_016120 [38], Vitis vinifera L. NC_0122119, Liriodendron tulipifera L. NC_021152, Salvia miltiorrhiza NC_023209 and Glycine max NC_020455). The Geneious mapper was used with the following settings: Iterate 5 times, medium/low sensitivity, fast. Consensus sequences were then created using a strict criterion of 75% read agreement, calling gaps when the coverage was below 10x, using the Geneious consensus sequence tool. Additionaly, to recover divergent mitochondrial genes that would not be identified by mapping-to-reference runs, we employed de novo assemblies using SPAdes [39] with default settings. The resulting contigs were annotated as previously to identify those of mitochondrial origin and mapping-to-reference runs were employed as described above to check for mean coverage to use as proxy to identify more mitochondrial contigs which lacked annotations. Using relaxed search criteria on a local BLAST database comprised of the mitochondrial genes of Brassica carinata, Viscum album and Viscum scurruloideum, the presence/absence of mitochondrial genes was tested on the resulting contigs.
Phylogenetic analysis and statistics
For the phylogenetic analysis, a custom database comprising all mitochondrial gene sequences of the 11 taxa analyzed in this study and of 27 non-parasitic taxa (Fig. 1; Additional file 2: Table S4), which represent all major orders in the angiosperm phylogeny, was created in Geneious. Our emphasis was to have at least one non-parasitic plant as closely related as possible to each of the parasitic plants. Each gene set was aligned using MAFFT v7.222 [40] under the following options: Algorithm FFT-NS-i × 1000, scoring matrix 200PAM / k = 2 and default gap open penalty and offset value, as implemented in Geneious R9.0.5. The overhangs of each alignment were then trimmed using the implemented function in Geneious (Tools- > Mask alignment- > More than 70% gaps), so that all sequences from each gene have the same length. The alignments of the core genes were then concatenated into the following groups of genes: 1) all_atp_genes, which includes atp1, atp4, atp6, apt8, and atp9, 2) all_ccm_genes, which includes ccmB, ccmC, ccmFc, and ccmFn, and 3) all_nad_genes, which includes nad1, nad2, nad3, nad4, nad4L, nad5, nad6, nad7, and nad9. Regarding the variably occurring ribosomal protein genes (rpl and rps) alignments were made for rpl2, rpl5, and rpl16, and rps3 as the remaining genes were frequently missing from some of the 38 taxa. Finally, a concatenation of the mitochondrial core gene alignments, including rps3, rpl2, rpl5, and rpl16 gave the MT_genes alignment with a length of 40,091 positions (29,036 positions without gaps).
Fig. 1.

Protein-gene content of 38 angiosperms, including the 11 parasitic plants analysed in this study. Dark grey boxes indicate presence of a gene, light grey boxes indicate pseudogenes or partial genes, and white boxes indicate absence of the gene. Taxa in green show hemiparasitic and taxa in light red show holoparasitic plants
In order to expand the taxonomic sampling size of our analysis we merged part of our data matrix with the one used by Qiu et al. [41], which contains mitochondrial gene sequences from 380 taxa, but only four mitochondrial genes, namely atp1, rps3, matR and nad5. Thus, we only used these four sequences from our smaller data set as well. The concatenated matrix from the Qiu et al. data set were separated into the four genes mentioned above, re-aligned with the sequences of our data set using MAFFT v.7222 with the same options as previously, the overhangs were trimmed and the sequences were concatenated. The final data set included 418 terminals and had a total length of 9076 positions.
Phylogenetic trees were constructed for 1) each mitochondrial gene (analyses 1 and 2) each concatenated group of aligned genes (analyses 2 and 3) for the concatenated alignment that includes all mitochondrial genes (MT_genes alignment) (analyses 3 and 4) for the concatenated sequences of the larger data set of the 418 terminals (analysis 4). All trees were visually inspected for significant topological conflicts. Phylogenetic analyses were performed in RAxML [42] using the GTR-Gamma substitution model, utilizing the Rapid Bootstrapping and search for best ML-scoring tree algorithm with 1000 bootstrap replicates, and using either Amborella Baill. (analyses 1–3) or Cycas L. plus Zamia L. (analysis 4) as the outgroup. The resulting tree files with bootstrap values were imported into FigTree (http:// tree.bio.ed.ac.uk/software/figtree/) for visualization.
Statistical analyses of the total number of substitutions on the phylogenetic trees were based on all mitochondrial gene sequences (analysis 3) and the large data set (analysis 4) was done in R (http://www.R-project.org), using the MASS [43], APE [44], Faraway [45], and Agricolae [46] libraries (Additional file 3: Code C1). The tree files in Newick format were used as input in both cases. One-way analysis of variance (1-way ANOVA) was performed grouping the plants into 1) four groups: autotrophic, hemiparasitic, holoparasitic plants and Viscaceae, and 2) three groups: autotrophic, hemiparasitic (including the Viscaceae), and holoparasitic plants. The significance of the analysis was verified by running Tukey HSD (Honestly Significant Difference) tests and the significant threshold value was p < 0.1.
Results
Mitochondrial gene content in parasitic plants
The vast majority of all hemiparasitic and holoparasitic plants analyzed in our study possess an almost complete mitochondrial core gene complement, where all genes from complex I (nad1, nad2, nad3, nad4, nad4L, nad5, nad6, nad7 and nad9), complex III (cob), complex IV (cox1, cox2 and cox3) and complex V (atp1, atp4, atp6, atp8 and atp9) are present, along with the maturase-related protein gene (matR) and the genes ccmB, ccmC, ccmFc and ccmFn that are involved in the cytochrome C biogenesis pathway (Fig. 1).
Krameria lanceolata appears to be the only species to possess only a partial, not functional nad4L gene (197 nucleotides, 65% of the complete Brassica carinata nad4L gene, missing part of the 5′ sequence). With regards to complex II (sdh3, sdh4), there is not a distinct pattern of presence/absence. Hence, Cassytha pubescens harbours both genes; Cynomorium coccineum and Langsdorffia hypogaea only possess a partial sdh4 gene, Lathraea clandestina and L. squamaria have a complete sdh4 gene; while Loranthus europaeus, Krameria lanceolata, Cytinus hypocistis ssp. clusii, C. hypocistis ssp. hypocistis and Pholisma sonorae have lost both. The three rRNA genes appear to be present in all taxa in the analysis, except for Phoradendron, while the variable genes encoding for proteins of the small and large ribosomal subunits are absent in a random pattern. Hence, several parasitic taxa possess 5–6 out of the maximally 15 rpl and rps genes in addition to 1–3 pseudogenes, while others have 14 ribosomal protein genes, namely Cassytha pubescens and Cynomorium coccineum, both only lacking the rps2 gene. This gene appears to be missing from all parasitic taxa of this data set, and also from many autotrophic plants. For the remaining genes encoding for proteins of the two ribosomal subunits in the 12 parasites sequenced here, rpl2 is present in six taxa, rpl5 in nine, rpl10 in seven, while rpl16 in just one; rps2 is absent from all, while the rest of the rps genes vary from 5 to 6 to 10 per parasitic plant, without a clear pattern of loss.
A remarkable exception is the mitogenome of Phoradendron liga, which like species of Viscum lacks all nad genes, but also ccmB, matR, and possibly atp4 and rrn26. Many ribosomal protein genes are also lost. The core mitochondrial genes show great divergence compared to “normal” plant mitochondrial genes. Hence, these genes show highest similarity to the respective ones from Viscum album (NC_029039), with atp1 showing 81% identity to V. album and less than 75% to other taxa and atp9 showing 92% identity to V. album when running BLASTN search. The remaining genes exhibit similarities ranging from 80 to 86%, to a variety of distantly related taxa, while the general identity between the respective genes of different taxa is higher than 92% (data not shown).
Phylogenetic analysis
The data set used in analysis 3 consisting of the total aligned and concatenated mitochondrial genes of just 38 taxa results in a tree with most species in their expected phylogenetic position (Fig. 2). Thus, Lathraea clandestina and L. squamaria cluster next to Erythranthe guttata (DC.) G. L. Nesom and Salvia miltiorrhiza Bunge in the order Lamiales, and Pholisma sonorae (Boraginales) is also placed within the Lamiids. Loranthus europaeus and Langsdorffia hypogaea cluster with Osyris alba forming the Santalales and Phoradendron liga clusters with high support (BS = 100%) in the Viscaceae, however the clade is not placed within the Santalales. The two Cytinus hypocistis subspecies cluster with Gossypium hirsutum L. in the order Malvales. Krameria lanceolata (Zygophyllales) is placed as the sister to Cynomorium coccineum (Saxifragales) but the clade has low support (BS = 72%). Cassytha pubescens (Laurales) branches off relatively near the root of the tree, but is not placed as the sister to the other representative of Laurales, Liriodendron tulipifera.
Fig. 2.

Phylogenetic tree of the 38 angiosperms included in the study. Branch length shows substitution rates, while bootstraps support values are shown on the base of each branch. Taxa in green show hemiparasitic and taxa in red show holoparasitic plants
Expanding the analysis to a total of 418 terminals, while reducing the number of mitochondrial genes to four (analysis 4), (Additional file 4: Figure S1), most of the apparent misplacements seen in the smaller data set are corrected due to the denser taxon sampling, hence Cassytha pubescens is placed within the Laurales as sister to Laurus L. (BS = 67%), Krameria lanceolata clusters with Guaiacum L. (Zygophyllales) (BS = 100%), close to other representatives of the Fabiids, while Pholisma sonorae is placed within Boraginales as closely related to Ehretia P. Browne, Hydrophyllum L. and Borago L. (BS = 100%). Similarly, Lathraea cladestina and L. squamaria are well supported within the Lamiales as sister to Erythranthe guttata (BS = 99%). Both Langsdorffia hypogaeae (Balanophoraceae) and Osyris alba (Santalaceae) are placed within the Santalales, the latter as the well-supported sister to Santalum L. (BS = 96%). Additionally, Loranthus europaeus (Loranthaceae) is placed in the Santalales as a strongly supported sister to Gaiadendron G. Don (Loranthaceae) (BS = 98%). Phoradendron liga and the remaining Viscaceae form a strongly supported clade (BS = 100%), however still not included in Santalales, but placed within a well-supported clade including the Ericales (BS = 94%). Cynomorium coccineum (Cynomoriaceae) is places as the sister to a clade corresponding to the superasterids, but the relationship is unsupported.
Substitution rates
Table 3 shows the substitution rates of each taxon for 1) all the mitochondrial genes, and 2) atp1, 3) matR and 4) rps3, respectively. It is evident that in this analysis the mean mitochondrial gene substitution rate is highly influenced by the fast evolving atp1 gene. Butomus umbellatus L. has some of the fastest evolving atp1, matR and rps3 genes, but the presence of the remaining slowly evolving mitochondrial genes lowers its mean substitution rate. The majority of the holoparasitic plants in this analysis tend to have higher substitution rates compared to the autotrophic ones, but Geranium maderense is evolving even faster still. It is notable that Viscum album, albeit having one of the fastest evolving and most divergent atp gene sets, has a slowly evolving matR gene. Among the representatives of Viscaceae analysed here, only V. album has a complete matR gene.
Table 3.
Substitution rates of the atp1, matR, rps3, and all the mitochondrial genes of the 38 taxa of analysis 1 and 3. The taxa are ordered in ascending order based on the total substitution rates of all the mitochondrial genes
| Species | all_mt | atp1 | matR | rps3 |
|---|---|---|---|---|
| Amborella trichopoda | 0.45 | 0.45 | 0.45 | 0.45 |
| Liriodendron tulipifera | 0.460 | 0.463 | 0.464 | 0.465 |
| Cassytha pubescens | 0.484 | 0.511 | 0.468 | 0.478 |
| Vitis vinifera | 0.487 | 0.517 | 0.472 | 0.477 |
| Phoenix dactylifera | 0.491 | 0.477 | 0.479 | 0.486 |
| Citrullus lanatus | 0.500 | 0.516 | 0.496 | 0.493 |
| Ricinus communis | 0.502 | 0.516 | 0.490 | 0.488 |
| Malus x domestica | 0.503 | 0.534 | 0.490 | 0.499 |
| Nicotiana tabacum | 0.508 | 0.535 | 0.497 | 0.490 |
| Glycine max | 0.511 | 0.556 | 0.492 | 0.499 |
| Gossypium hirsutum | 0.511 | 0.531 | 0.486 | 0.510 |
| Pholisma sonorae | 0.511 | 0.538 | 0.495 | 0.520 |
| Osyris alba | 0.513 | 0.529 | 0.477 | 0.483 |
| Krammeria lanceolata | 0.513 | 0.542 | 0.496 | 0.500 |
| Vaccinium macrocarpon | 0.517 | 0.573 | 0.500 | – |
| Erythranthe guttata | 0.519 | 0.596 | 0.495 | – |
| Cucurbita pepo | 0.520 | 0.538 | 0.525 | 0.514 |
| Cynomorium coccineum | 0.521 | 0.547 | 0.500 | 0.531 |
| Lathraea squamaria | 0.521 | 0.599 | 0.491 | 0.507 |
| Lathraea clandestina | 0.522 | 0.594 | 0.491 | 0.508 |
| Asclepias syriaca | 0.522 | 0.569 | 0.502 | 0.502 |
| Brassica carinata | 0.525 | 0.560 | 0.507 | 0.537 |
| Populus tremula | 0.525 | 0.648 | 0.503 | 0.537 |
| Aegilops speltoides | 0.529 | 0.519 | 0.512 | 0.527 |
| Helianthus annuus | 0.529 | 0.533 | 0.492 | – |
| Daucus carota | 0.532 | 0.541 | 0.489 | 0.528 |
| Butomus umbellatus | 0.547 | 0.713 | 0.550 | 0.597 |
| Salvia miltiorrhiza | 0.548 | 0.598 | 0.493 | 0.511 |
| Cytinus hypocistis ssp. clusii | 0.553 | 0.554 | 0.520 | 0.648 |
| Cytinus hypocistis ssp. hypocystis | 0.561 | 0.583 | 0.534 | 0.577 |
| Langsdorffia hypogaea | 0.569 | 0.579 | 0.600 | 0.568 |
| Loranthus europaeus | 0.569 | 0.583 | 0.534 | 0.580 |
| Geranium maderense | 0.593 | 0.614 | 0.524 | 0.880 |
| Viscum album | 1.199 | 1.377 | 0.492 | 1.345 |
| Viscum minimum | 1.203 | 1.382 | – | – |
| Viscum scurruloideum | 1.210 | 1.381 | – | 1.347 |
| Viscum crassulae | 1.255 | 1.383 | – | – |
| Phoradendron liga | 1.621 | 1.681 | – | – |
| Mean | 0.622 | 0.669 | 0.500 | 0.581 |
| Median | 0.521 | 0.554 | 0.495 | 0.511 |
Figure 3 shows the substitution rates of all mitochondrial genes of the 38 taxa used in Table 3. Black lines depict the mean substitution rates of each mitogenome, with regards to its coding sequences, only. As the outgroup, Amborella has a fixed value. This peculiarity is due to a bug in RaXML as has been addressed by the developer of the program, yet since all distances are calculated from the root of the tree, and since Amborella is not the focus of this study, this bug is insignificant to the downstream analysis of the results. Phoradendron liga also has a fixed value due to the lack of the matR and rps3 genes used in the analysis, while its atp1 and all_mt_genes rates are quite similar. It is evident that there are no great differences between the taxa, irrespective of whether they are autotrophic, hemiparasitic or holoparasitic. A profound exception is, as shown earlier [26, 27], the Viscaceae, with Phoradendron liga showing the same pattern as the Viscum spp. The few sequences obtained from this hemiparasite show very low similarity to the homologous sequences of other plant species, thus for atp1 the degree of similarity varies from 49 to 73%, for atp9 from 37% to 82%, for the cox genes from 62 to 71% and for rps12 from 27 to 41%. The boxplot also demonstrates that there are a few autotrophic plants, which show relatively higher substitution rates than the parasitic plants.
Fig. 3.

Barplot with the substitution rates of the mitochondrial genes of the 38 taxa in analysis 1. Taxa in green show hemiparasitic and taxa in red show holoparasitic plants
Expanding the analysis (analysis 4) reveals that across the angiosperms, most taxa show mean substitution rates in the vicinity of 0.65 to 0.75 substitutions per site (Supplementary Material, Figure S1). However, Acorus L., Geranium L., Hydrolea L. and others also exhibit high substitution rates, similar to those found in Viscaceae.
How significant is the difference seen in the Viscaceae, compared to the other taxa? Additional file 5: Table S3 shows the results of the 1-way ANOVA in the small data set, where the samples are put into four categories: autotrophic, hemiparasitic, holoparasitic plants and the Viscaceae. First, it shows once again that atp1 (Additional file 5: Table S3a) evolves faster compared to matR, rps3, and nad5, with mean substitution rates for the 3 groups (autotrophic, hemiparasitic, holoparasitic) ranging from 0.53 to 0.58 (excluding the Viscaceae), followed by matR (Additional file 5: Table S3b), with mean substitution rates ranging between 0.49 and 0.52, and rps3 (Additional file 5: Table S3c), with mean substitution rates ranging between 0.47 and 0.56. The mean substitutions per site for all the mitochondrial genes shown in the four groups are 0.51, 0.51, 0.55 and 1.3 for the autotrophic, hemiparasitic, holoparasitic plants and Viscaceae, respectively (Additional file 5: Table S3d). The Tukey HSD test reveals no statistically significant difference between the autotrophic, hemiparasitic and holoparasitic plants. On the other hand, Viscaceae show statistically significant difference compared to the other three groups (p < 0.01). Similarly, when Viscaceae are included in the hemiparasitic group (data not shown), they still show significant differences compared to the remaining two groups (p < 0.01), which can be attributed to the very small sample size and homogeneity of the group (nine taxa).
Performing the same analyses using the expanded Qui et al. data set, we once again see that Viscaceae have significantly higher substitution rates than the autotrophic, hemiparasitic and holoparasitic plants (p < 0.1) (Fig. 4). However, comparing the individual taxa to each other, Viscum album is not statistically different from non-parasitic taxa such as Acorus, Geranium, Podocarpus Pers. and others (Additional file 6: Table S2). Viscum album is currently the only representative of Viscaceae that still possesses a matR gene, and the low substitution rate of this gene lower the total substitution rate of the mitogenome of the species.
Fig. 4.
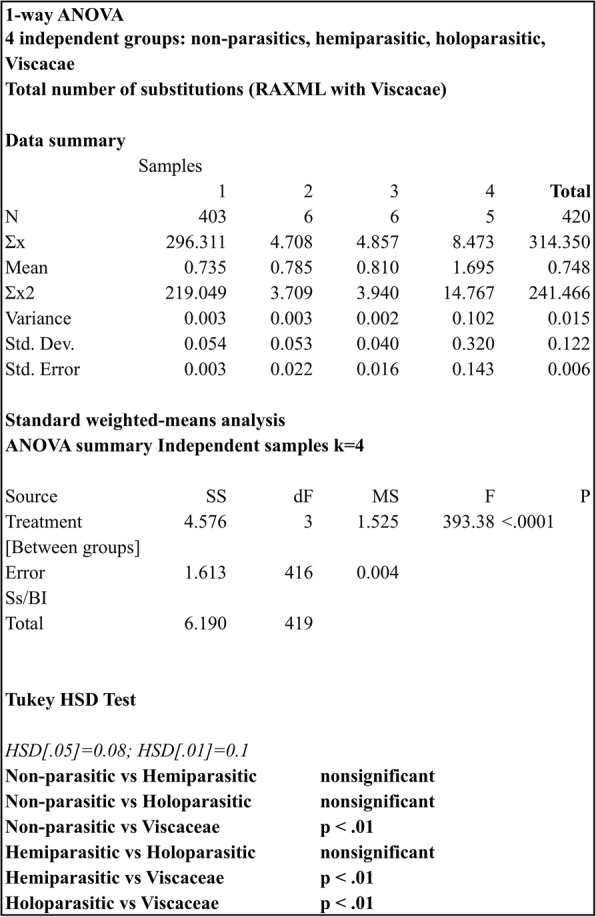
Analysis of variance of the substitution rates in the large data set of the mitochondrial genes (analysis 4)
Discussion
Mitochondrial gene loss in most parasitic plants is confined to the variable part of the mitogenome
Nearly all angiosperm mitogenomes assembled thus far have a full core gene complement, comprising of 24 genes. Exceptions are genera like Vigna Savi, which is missing the cox3 gene [20], and Silene L., which is missing the ccmFc gene [19]. The latter, despite having one of the largest plant mitogenomes known (11.3 mb), has also lost all but one or two of the variable genes (four rpl, 11 rps and two sdh genes). Recently it was found that Viscum possesses one of the smallest and most dramatically reduced plant mitogenomes investigated so far [26, 27] .
In the present investigation, we sequenced five hemiparasitic and seven holoparasitic plants and extracted all known plant mitochondrial genes. Most of them possessed a full mitochondrial core gene complement. One exception is the hemiparasite Krameria lanceolata, which possesses only a partial and most likely unfunctional nad4L gene. This is the first time that a functional loss of a nad gene not accompanied by the loss of any other core mitochondrial gene has been observed in a plant mitogenome, other than in Viscum. The function of the nad4L gene product in complex I (mitochondrial NADH dehydrogenase) is also covered by the nad4 gene product, which in the case of Krameria lanceolata is intact.
The other exception is Phoradendron liga, another hemiparasitic plant belonging to Viscaceae. Like Viscum spp., P. liga is a stem parasite and as in Viscum a massive gene loss is observed. Using relaxed criteria for gene recognition, Skippington et al. [47] proposed that some of the genes (e.g., atp4, atp8, ccmFc), which were reported missing in V. album, are actually present although very highly divergent. Using the same criteria, presence of additional genes may be proposed in the two other species of Viscum previously investigated [26]. Whether these highly divergent sequences do represent homologous and functional genes need further investigation. Irrespective of the precise number of genes present in members of the Viscaceae, massive reduction in gene content remains a fact and a unique feature of the Viscaceae. The closely related hemiparasites Osyris alba (root parasite, Santalaceae) and Loranthus europaeus (stem parasite, Loranthaceae) both possess all core mitochondrial genes. Thus, at this point there is no evidence of a correlation between level of parasitism and gene contents in the mitogenomes; rather it seems to be highly clade specific. This contrasts to the correlation observed between parasitism and gene loss found in the plastomes of parasitic plants [10, 12, 13].
When it comes to the variable mitochondrial genes there is no clear pattern of gene loss in the parasitic plants. Phoradendron has lost most ribosomal protein genes like Viscum [26, 27]. For the remaining parasitic plants, six to 14 of the 15 ribosomal protein genes are present. This is not surprising; it has been observed that ribosomal protein genes can be transcribed in the nuclear genome and transferred to the mitochondria [47].
The massive mitochondrial gene loss observed in the Viscaceae [26, 27] remains enigmatic. The possibility that genes are not truly missing from the mitogenome but just unobserved is negligible. For Phoradendron and all other taxa investigated in this study, sequencing was done in sufficient depth to capture genes both from the mitogenome and the plastome. However, sequencing depth is not sufficient to reliably detect nuclear genes, thus it remains a possibility that genes missing from the mitogenome could be transferred to the nucleus. Due to the huge size of the Viscum nuclear genome (201Gb [48]) it is not a realistic option to increase sequencing depth in order to try to recovered any nuclear copies of the mitochondrial genes. Other methods, e.g., transcriptomics may be more relevant (see below). Thus, the crucial question of how the four Viscum species and P. liga produce the energy they require in order to sustain life remains, as does the possibility that they have developed a novel mechanism to do so [26].
A eukaryote completely lacking mitochondria was recently characterised by Karnkowska et al. [49]. Thus, an oxymonad species, Monocercomonoides sp., has replaced part of the respiratory processes facilitated by mitochondrial genes with others that are suggested to have emerged through lateral gene transfer from bacteria. Also in Archezoa, double membrane-bound, mitochondrion-related organelles were found to harbour genes so modified, that their homology to the standard mitochondrial genes could not be established [50]. Monocercomonoides sp. appears to produce ATP through an extended anaerobic glycolysis pathway, coupled with fermentation. However, this scenario seems unrealistic for an organism that actively produces oxygen and has a functional plastome and a photosynthetic apparatus [13]. A few unicellular organisms, e.g., the fungus Rozella [51], which have lost the entire complex I genes, use alternative pathways that do not require pumping of protons, but this leads to the production of less ATP in total.
To investigate how Viscum and Phoradendron survive with their reduced mitogenomes, especially before they establish contact with their hosts (something that may take months in e.g. Viscum album), it would be worthwhile to try to capture active mitochondrial RNA molecules using probes that target standard mitochondrial genes as well as the ones identified in the previous studies [26, 27] by transcriptomics. In this manner, it will be possible to verify the presence or absence of the ‘normal’ genes, which may have been missed in the whole genome sequencing approach used here and at the same time also check if the very divergent mitochondrial genes found are active or not. Furthermore, for many of the ‘normal’ genes the 3-dimensional structure of their products is known enabling the targeting of probes to RNA sequences of highly conserved regions.
The evolutionary pathway resulting in the massive mitochondrial gene loss observed in Viscum and Phoradendron also needs further studies. The Viscaceae is composed entirely of hemiparasitic stem parasites and includes no less than ~ 465 species in six genera, amounting to approximately 20% of all parasitic plants in the Santalales [52], [www.parasiticplants.siu.edu/ListParasites.htm]. Thus, data from additional species and genera of Viscaceae are strongly needed along with data from the closest relatives to Viscaceae, i.e. Amphorogynaceae [2, 53].
Substitution rates of mitochondrial genes in parasitic plants are not significantly different compared to autotrophic, non-parasitic plants, except for the Viscaceae
In this study, we also wanted to test the hypothesis that parasitic plants have higher substitution rates than autotrophic plants. Early theoretical models have suggested that parasites should evolve much faster than their hosts as a response to the arms race between host and parasite [33]. The highly increased substitution rates of the remaining mitochondrial genes previously shown in Viscum [26, 27] are shared and even exceeded by Phoradendron, but not by the other representatives of Santalales (Fig. 3). Based on the small data set of 38 taxa it is evident that the holoparasites Cytinus hypocistis ssp. hypocistis and C. hypocistis ssp. clusii (Cytinaceae) do indeed have higher substitution rates than their closest autotrophic relative included Gossypium hirsutum (Malvaceae) (Fig. 3). The same trend has been observed in their chloroplast genes [54]. However, in other supposedly more distantly related taxa, e.g. Geranium maderense (Geraniaceae), we observe notably faster substitution rates than in Cytinus. Pholisma sonorae (Boraginaceae) evolves slightly faster than Nicotiana tabacum L. (Solanaceae), but slower than other representatives of the Lamiids, like Erythranthe guttata (Phrymaceae) (Fig. 2). Lathraea clandestine and L. squamaria (Orobanchaceae) both show elevated substitution rates compared to E. guttata, but the non-parasite, Salvia miltiorrhiza (Lamiaceae), evolves faster still. The hemiparasitic Cassytha pubescens (Lauraceae), Osyris alba (Santalaceae), and Krameria lanceolata (Krameriaceae) all have similar substitution rates, in the same range as many autotrophic plants. However, within the Santalales, both the holoparasitic Langsdorffia hypogaea (Balanophoraceae) and the hemiparasitic Loranthus europaeus (Loranthaceae) have higher substitution rates compared to the closely related hemiparasite Osyris alba (Santalaceae), although far from the level observed in Viscaceae, where Phoradendron liga has the fastest substitution rae of all taxa in the data set.
A previous comparison of parasitic plants with their closest autotrophic relatives showed that the parasites tended to have higher substitution rates across all three genomes [34]. The study compared the substitution rates of four mitochondrial genes (matR, atp1, nad1 and cox1) from 12 parasitic lineages with representatives of the assumed most closely related, available autotrophic lineages, and using pairwise comparisons of the sequences. However, this approach has several pitfalls. Firstly, the phylogenetic position of certain parasitic taxa is ambiguously resolved and the choice of close relatives for comparison thus not necessarily ideal. Further, results can be biased by the exact choice of close relatives from groups where substitution rates vary considerably even among autotrophic members.The Wilcoxon signed-rank test only evaluates pairwise comparisons of two related or matched samples and gives no information as whether the finding is significant for the species, family or group level.
To overcome some of these problems we inferred the phylogenetic placement of all parasitic taxa analysed and calculated substitution rates from the root of the tree, based on the out-group taxa. Furthermore, we performed a one-way analysis of variance on the taxa and groups of taxa as well. The most substantial result is that the Viscaceae, including Phoradendron liga, Viscum album, V. crassulae, V. minimum and V. scurruloideum, have significantly higher substitution rates of all holoparasitic, hemiparasitic, and autotrophic plants in the 38 taxa data set. There were no statistically significant differences between any of the remaining hemiparasitic plants compared to either the holoparasitic or the autotrophic ones. Several hemiparasitic and holoparasitic plants evolve faster than their closest autotrophic relatives, yet without statically significantly differences. As shown earlier by Bromham et al. [34] Krameria lanceolata evolves faster than Guaiacaum, however, the difference in mean substitution rates is insignificant (Additional file 6: Table S2). In the Lauraceae, Cassytha pubescens has a higher substitution rate than its autotrophic closest relatives, while Cytinus hypocistis ssp. hypocistis and C. hypocistis ssp. clusii evolve much faster than their closest relatives (Brassicaceae). The same is observed for Langsdorffia hypogaea and Loranthus europaeus. On the other hand, Osyris alba, the two Lathraea species, as well as Pholisma sonorae exhibit slower molecular evolution rates than the closely related autotrophs. Thus, there is no evidence that parasitic plants necessarily evolve faster than their closest autotrophic relatives.
An obvious pitfall of the limited taxon sampling in the small dataset of 38 taxa is that we unintentionally may make comparison with either a particular fast or slowly evolving relative, hence biasing the results. To overcome this problem we expanded our data set to a total of 418 taxa but only four genes making use of the data matrix by Qiu et al. [41] (including a few duplicates as a means of verification of similar sequences used in the analysis). Running a group- or clade-specific (Viscaceae, holoparasitic, hemiparasitic, autotrophic plants) one-way analysis of variance of all the taxa of the large data set we again found the Viscaceae to have a significantly faster substitution rate than any other group of holoparasitic, hemiparasitic, and autotrophic taxa. However, running the same analysis this time comparing each parasitic taxon to the rest and not grouping them into categories we made some unexpected observations. While Phoradendron and three of the Viscum species still had significantly higher substitution rates, V. album did not (Additional file 6: Table S2). The explanation for this seems to be the difference in gene content of the five Viscaceae species. All species possess the fast evolving atp1 gene, all lack nad5, V. album and V. scurruloideum possess rps3, but only V. album possess matR, which in this species has a low substitution rate. Thus, the presence of matR lowers the overall substitution rate to a similar level as observed in several autotrophic taxa like Acorus, Hydrolea and Geranium. Similarly, the remaining holoparasitic and hemiparasitic taxa of the analysis exhibit mean evolution rates that fall within the same range as the autotrophic taxa. Thus, there is no statistically significant difference between the remaining taxa, equal to what we observed within the group-specific one-way analysis of variance. Even parasitic plants that have eliminated photosynthesis and lost most or all of their plastid genes still maintain their mitochondrial genes with only minor alterations.
Considering the suggestion that parasites should evolve much faster than their hosts as a response to an arms race between host and parasite [33], a comparison of substitution rates between hosts and parasites would appear relevant. However, many parasitic plants have a worldwide distribution and parasitize on a variety of hosts belonging to different species, genera, or even families (e.g., Viscum album is known to parasite 367 hosts – including species, subspecies and varieties, 97 genera and 46 families [55]). Moreover, in certain cases – especially among root parasites – it can be exceedingly difficult to locate the host of the parasite with certainty. Thus, the remark based on what is observed from bacteria in regards to arms race between the parasite and the host cannot easily be tested in plants. However, the global analysis using as many taxa as possible as conducted here does not support the postulate with regards to mitochondrial gene evolution.
Some Phylogenetic Observations
Holoparasites very often have a strongly reduced and modified morphology, which makes them very difficult to place in a phylogenetic context [56]. Additionally they usually have a strongly reduced and divergent plastome that have made it equally difficult to place them in a molecular phylogenetic context as most major phylogenies are based on plastome genes [57, 58] or whole plastomes [59]. The current phylogenetic analyes based only on mitochondrial genes, may thus provide further evidence on the relationships of these parasitic taxa.
Here, we find Langsdorffia embedded in the Santalales (Fig. 2; Additional file 4: Figure S1). This is consistent with the recent phylogenetic analysis of the Santalales, where three plastid genes (matK, rbcL and accD), one mitochondrial gene (matR), and three nuclear loci (SSU rDNA, LSU rDNA and rpb2) were used to demonstrate the placement of the Balanophoraceae within the order [2]. Su et al. [2] further observed that the Balanophoraceae is non-monophyletic consisting of two clades with different relationships. The genus Langsdorffia was not included in their analysis, but due to the much less dense taxon sampling of Santalales in our analyses and some differences in tree topology, we cannot place Langsdorffia in either of the clades. However, we do provide independent evidence for the inclusion of Balanophoraceae in Santalales.
In the present study, we have performed two widely different phylogenetic analyses: one with a shallow taxon sampling, but massive data sampling (38 taxa, 40,091 characters), and one with very dense taxon sampling, but modest data sampling (418 taxa, 9076 characters). Compared to the general conception of angiosperms phylogeny as represented by APG IV [60], the latter data set results in a considerably more accurate phylogenetic tree. This observation is in full accordance with the general importance of density in taxon sampling as discussed by e.g. Heath et al. [61], and Nabhan and Sarkar [62].
However, while some of the parasitic taxa, which seem misplaced in the analysis using few taxa (i.e. Cassytha, Krameria), are recovered in expected positions when using many taxa, some remain in unexpected positions. Cynomorium, which most likely belongs to the Saxifragales, is placed as sister to a clade including all superasterids, however this position lacks bootstrap support. Further, we recover the Viscaceae clade, which would also be expected to be included in the Santalales, within a rather strongly supported Ericales clade (Additional file 4: Figure S1). This position is surprising since the phylogenetic inclusion of Viscaceae in Santalales has been demonstrated repeatedly and never questioned (e.g. [2, 53]). However, Skippington et al. [47] recently suggested that the matR gene present only in V. album, but not in any other Viscaceae, had been required through horizontal gene transfer from a member of the Ericales. Although the evidence is circumstantial this could potentially explain the position seen even here in our four-gene analysis, since the only other sequence data which are included for the Viscaceae taxa are the highly divergent atp1 sequences.
Conclusions
In this project, we sequenced and analysed 11 parasitic plants spread throughout the angiosperm phylogeny. We show that the Viscaceae are truly unique among parasitic plants both in regards to their mitochondrial gene content and their mitochondrial gene evolution, having the most reduced and fastest evolving mitogenomes to date. We also show that the mitogenomes of parasitic plants in general do not evolve faster than that of their autotrophic relatives. There are several autotrophic taxa that have significantly higher evolutionary rates in their mitochondrial genes than the parasites. Comparison of the substitution rates of the mitogenomes of holoparasitic, hemiparasitic, and autotrophic plants shows no significant difference. This goes hand in hand with the fundamental role of the mitogenome in respiration, a feature of high importance, commonly shared across all plant taxa. Lastly, we pinpointed the phylogenetic position of Langsdorffia hypogaea (Balanophoraceae) in the Santalales.
Additional files
Table S1. Tissue type, protocol, cleaning procedures and mass used for DNA extraction of the 11 samples analysed in this study. (DOCX 57 kb)
Table S4. GeneBank Accession Numbers of the non-parasitic taxa used in the small dataset of 38 taxa. (XLS 104 kb)
Code C1: R code used for the analyses of variance in the small and large data set. (DOCX 12 kb)
Figure S1. Phylogenetic tree of the 418 taxa included in the study. Branch length shows substitution rates, while bootstraps support values are shown on the base of each branch. (PDF 682 kb)
Table S3. ANOVA of the substitution rates in the data set of the 38 taxa for the: a) atp1 gene, b) matR gene, c) rsp3 gene and d) all_mt_genes. (PDF 114 kb)
Table S2. ANOVA of the substitution rates in the data set of 418 taxa using 4 mitochondrial genes (analysis 4). (XLSX 41 kb)
Acknowledgements
The authors are grateful to Associate Professor Yin-Long Qiu, University of Michigan, for giving us access to the four mitochondrial gene matrix [41]. Special thanks to Charlotte Hansen for capable laboratory assistance and Nathan Wales for providing the aDNA protocol and assisting with the DNA extraction process and library preparation of the herbarium samples. Lastly the authors thank the staff at the National High Throughput DNA Sequencing Centre of the University of Copenhagen for Next Generation Sequencing.
Funding
This work was supported by the Danish Council for Independent Research | Natural Sciences (grant number DFF-4002-00505).
Availability of data and materials
The sequences obtained for each taxon were submitted to GenBank under accession numbers: Cassytha pubescens MG233923 – MG233957, Cynomorium coccineum MG234165 – MG234233, Cytinus hypocistis ssp. clusii MG233958 – MG233992, Cytinus hypocistis ssp. hypocistis MG234028 – MG234062, Krameria lanceolata MG234063 – MG234097, Langsdorffia hypogaeae MG234098 – MG234130, Lathraea cladestina MG234131 – MG234164, Lathraea squamaria MG234165 – MG234164, Loranthus europaeus MG233993–234027, Pholisma sonorae MG234234–234268, Phoradendron liga MG234269-MG234275.
Abbreviations
- ANOVA
Analysis of variance
- APG
Angiosperm phylogeny group
- BS
Bootstraps support
- LSU
Large subunit
- mt
Mitochondrial
- SSU
Small subunit
Authors’ contribution
AZ, GP, and OS conceived and designed the study. AZ carried out the laboratory work, data treatment, data analysis, statistical analysis, and drafted the manuscript. GP and OS helped revising the manuscript. All authors read and approved the manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Athanasios Zervas, Email: zervas@snm.ku.dk.
Gitte Petersen, Email: gittep@snm.ku.dk.
Ole Seberg, Email: oles@snm.ku.dk.
References
- 1.Westwood JH, Yoder JI, Timko MP, dePamphilis CW. The evolution of parasitism in plants. Trends Plant Sci. 2010;15:227–235. doi: 10.1016/j.tplants.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 2.Su H, Hu J, Anderson FE, Der JP, Nickrent DL. Phylogenetic relationships of Santalales with insights into the origins of holoparasitic Balanophoraceae. Taxon. 2015;64:491–506. doi: 10.12705/643.2. [DOI] [Google Scholar]
- 3.Funk HT, Berg S, Krupinska K, Maier UG, Krause K. Complete DNA sequences of the plastid genomes of two parasitic flowering plant species, Cuscuta reflexa and Cuscuta gronovii. BMC Plant Biol. 2007;12:1–12. doi: 10.1186/1471-2229-7-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McNeal JR, Kuehl JV, Boore JL, De CW. Complete plastid genome sequences suggest strong selection for retention of photosynthetic genes in the parasitic plant genus Cuscuta. BMC Plant Biol. 2007;22:1–22. doi: 10.1186/1471-2229-7-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wicke S., Muller K. F., de Pamphilis C. W., Quandt D., Wickett N. J., Zhang Y., Renner S. S., Schneeweiss G. M. Mechanisms of Functional and Physical Genome Reduction in Photosynthetic and Nonphotosynthetic Parasitic Plants of the Broomrape Family. The Plant Cell. 2013;25(10):3711–3725. doi: 10.1105/tpc.113.113373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Molina J, Hazzouri KM, Nickrent D, Geisler M, Meyer RS, Pentony MM, et al. Possible loss of the chloroplast genome in the parasitic flowering plant Rafflesia lagascae (Rafflesiaceae) Mol Biol Evol. 2014;31:793–803. doi: 10.1093/molbev/msu051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lagerkvist U. “Two out of three”: an alternative method for codon reading. Proc Natl Acad Sci U S A. 1978;75:1759–1762. doi: 10.1073/pnas.75.4.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palmer JD. Contrasting modes and tempos of genome evolution in land plant organelles. Trends Genet. 1990;6:115–120. doi: 10.1016/0168-9525(90)90125-P. [DOI] [PubMed] [Google Scholar]
- 9.Alkatib S, Fleischmann TT, Scharff LB, Bock R. Evolutionary constraints on the plastid tRNA set decoding methionine and isoleucine. Nucleic Acids Res. 2012;40:6713–6724. doi: 10.1093/nar/gks350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cusimano N, Wicke S. Massive intracellular gene transfer during plastid genome reduction in nongreen Orobanchaceae. New Phytol. 2016;210:680–693. doi: 10.1111/nph.13784. [DOI] [PubMed] [Google Scholar]
- 11.Bellot S, Renner SS. The Plastomes of two species in the Endoparasite genus Pilostyles (Apodanthaceae) each retain just five or six possibly functional genes. Genome Biol Evol. 2016;8:189–201. doi: 10.1093/gbe/evv251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Naumann J, Der JP, Wafula EK, Jones SS, Wagner ST, Honaas A, et al. Detecting and characterizing the highly divergent plastid. Genome Biol Evol. 2015;8:345–363. doi: 10.1093/gbe/evv256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Petersen G, Cuenca A, Seberg O. Plastome evolution in Hemiparasitic mistletoes. Genome Biol Evol. 2015;7:2520–2532. doi: 10.1093/gbe/evv165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Logacheva MD, Schelkunov MI, Penin AA. Sequencing and analysis of plastid genome in Mycoheterotrophic orchid Neottia nidus-avis. Genome Biol Evol. 2011;3:1296–1303. doi: 10.1093/gbe/evr102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barrett CF, Davis JI. The plastid genome of the mycoheterotrophic Corallorhiza striata (Orchidaceae) is in the relatively early stages of degradation. Am J Bot. 2012;99:1513–1523. doi: 10.3732/ajb.1200256. [DOI] [PubMed] [Google Scholar]
- 16.Logacheva MD, Schelkunov MI, Nuraliev MS, Samigullin TH, Penin AA. The plastid genome of Mycoheterotrophic monocot Petrosavia stellaris exhibits both gene losses and multiple rearrangements. Genome Biol Evol. 2014;6:238–246. doi: 10.1093/gbe/evu001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Braukmann T, Sasa S. Plastid genome evolution in mycoheterotrophic Ericaceae. Plant Mol Biol. 2012;79:5–20. doi: 10.1007/s11103-012-9884-3. [DOI] [PubMed] [Google Scholar]
- 18.Boore JL. Animal mitochondrial genomes. Nucleic Acids Res. 1999;27:1767–1780. doi: 10.1093/nar/27.8.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sloan DB, Alverson AJ, Chuckalovcak JP, Wu M, Mccauley DE, Palmer JD, et al. Rapid evolution of enormous , multichromosomal genomes in flowering plant mitochondria with exceptionally high mutation rates. PLoS Biol. 2012;10:e1001241. doi: 10.1371/journal.pbio.1001241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alverson AJ, Zhuo S, Rice DW, Sloan DB, Palmer JD. The mitochondrial genome of the legume Vigna radiata and the analysis of recombination across short mitochondrial repeats. PLoS One. 2011;6:e16404. doi: 10.1371/journal.pone.0016404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Notsu Y, Masood S, Nishikawa T, Kubo N, Akiduki G, Nakazono M, et al. The complete sequence of the rice (Oryza sativa L.) mitochondrial genome: frequent DNA sequence acquisition and loss during the evolution of flowering plants. Mol Gen Genomics. 2002;268:434–445. doi: 10.1007/s00438-002-0767-1. [DOI] [PubMed] [Google Scholar]
- 22.Handa H. The complete nucleotide sequence and RNA editing content of the mitochondrial genome of rapeseed (Brassica napus L.): comparative analysis of the mitochondrial genomes of rapeseed and Arabidopsis thaliana. Nucleic Acids Res. 2003;31:5907–5916. doi: 10.1093/nar/gkg795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adams KL, Palmer JD. Evolution of mitochondrial gene content: gene loss and transfer to the nucleus. Mol Phylogenet Evol. 2003;29:380–395. doi: 10.1016/S1055-7903(03)00194-5. [DOI] [PubMed] [Google Scholar]
- 24.Bellot S, Cusimano N, Luo S, Sun G, Zarre S, Gröger A, et al. Assembled plastid and mitochondrial genomes, as well as nuclear genes, place the parasite family Cynomoriaceae in the Saxifragales. Genome Biol Evol. 2016;8:2214–2230. doi: 10.1093/gbe/evw147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fan W. Comparative evolutionary analysis of Organellar genomic diversity in green plants. 2016. [Google Scholar]
- 26.Petersen G, Cuenca A, Møller IM, Seberg O. Massive gene loss in mistletoe (Viscum, Viscaceae) mitochondria. Sci Rep. 2015;5:17588. doi: 10.1038/srep17588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Skippington E, Barkman TJ, Rice DW, Palmer JD. Miniaturized mitogenome of the parasitic plant Viscum scurruloideum is extremely divergent and dynamic and has lost all nad genes. Proc Natl Acad Sci U S A. 2015;112:E3515–E3524. doi: 10.1073/pnas.1504491112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wolfe KH, Li W-H, Sharp PM. Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc Natl Acad Sci U S A. 1987;84:9054–9058. doi: 10.1073/pnas.84.24.9054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Palmer JD, Adams KL, Cho Y, Parkinson CL, Qiu YL, Song K. Dynamic evolution of plant mitochondrial genomes: mobile genes and introns and highly variable mutation rates. Proc Natl Acad Sci U S A. 2000;97:6960–6966. doi: 10.1073/pnas.97.13.6960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parkinson CL, Mower JP, Qiu Y, Shirk AJ, Song K, Young ND, et al. Multiple major increases and decreases in mitochondrial substitution rates in the plant family Geraniaceae. BMC Evol Biol. 2005;5:73. doi: 10.1186/1471-2148-5-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nickrent DL, Starr EM. High rates of nucleotide substitution in nuclear small-subunit (18S) rDNA from holoparasitic flowering plants. J Mol Evol. 1994;39:62–70. doi: 10.1007/BF00178250. [DOI] [PubMed] [Google Scholar]
- 32.Duchene D, Bromham L. Rates of molecular evolution and diversification in plants: chloroplast substitution rates correlated with species-richness in the Proteaceae. BMC Evol Biol. 2013;13:65. doi: 10.1186/1471-2148-13-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haraguchi Y, Sasaki A. Host-parasite arms race in mutation modifications: indefinite escalation despite a heavy load? J Theor Biol. 1996;183:121–137. doi: 10.1006/jtbi.1996.9999. [DOI] [PubMed] [Google Scholar]
- 34.Bromham L, Cowman PF, Lanfear R. Parasitic plants have increased rates of molecular evolution across all three genomes. BMC Evol Biol. 2013;13:1–11. doi: 10.1186/1471-2148-13-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wales N, Andersen K, Cappellini E, Ávila-Arcos MC, Gilbert MTP. Optimization of DNA recovery and amplification from non-carbonized archaeobotanical remains. PLoS One. 2014;9:e86827. doi: 10.1371/journal.pone.0086827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meyer M., Kircher M. Illumina Sequencing Library Preparation for Highly Multiplexed Target Capture and Sequencing. Cold Spring Harbor Protocols. 2010;2010(6):pdb.prot5448-pdb.prot5448. doi: 10.1101/pdb.prot5448. [DOI] [PubMed] [Google Scholar]
- 37.Lindgreen S. AdapterRemoval: easy cleaning of next-generation sequencing reads. BMC Res Notes. 2012;5:337. doi: 10.1186/1756-0500-5-337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chang S, Yang T, Du T, Huang Y, Chen J, Yan J, et al. Mitochondrial genome sequencing helps show the evolutionary mechanism of mitochondrial genome formation in Brassica. BMC Genomics. 2011;12:497. doi: 10.1186/1471-2164-12-497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qiu YL, Li LB, Wang B, Xue JY, T a H, Li RQ, et al. Angiosperm phylogeny inferred from sequences of four mitochondrial genes. J Syst Evol. 2010;48:391–425. doi: 10.1111/j.1759-6831.2010.00097.x. [DOI] [Google Scholar]
- 42.Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 43.Venables WN, Ripley BD. Modern Applie statistics with S. 4. New York: Springer; 2002. [Google Scholar]
- 44.Paradis E, Claude J, Strimmer K. APE: analyses of phylogenetics and evolution in R language. Bioinformatics. 2004;20:289–290. doi: 10.1093/bioinformatics/btg412. [DOI] [PubMed] [Google Scholar]
- 45.Faraway J. Extending the linear model with R. 1st ed. Boca Raton: CRC press; 2005.
- 46.de Mendiburu F. A statistical analysis tool for agricultural research. Lima-Peru: National Engineering University (UNI); 2006. [Google Scholar]
- 47.Grohmann L, Brennicke A, Schuster W. The mitochondrial gene encoding ribosomal protein S12 has been translocated to the nuclear genome in Oenothera. Nucleic Acids Res. 1992;20:5641–5646. doi: 10.1093/nar/20.21.5641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zonneveld BJM. New record holders for maximum genome size in eudicots and monocots. J Bot. 2010;2010:1–4. [Google Scholar]
- 49.Karnkowska A, Vacek V, Zubáčová Z, Treitli SC, Petrželková R, Eme L, et al. A eukaryote without a mitochondrial organelle. Curr Biol. 2016;26:1274–1284. doi: 10.1016/j.cub.2016.03.053. [DOI] [PubMed] [Google Scholar]
- 50.Lindmark DG, Muller M. Superoxide dismutase in the anaerobic flagellates, Tritrichomonas foetus and Monocercomonas sp. J Biol Chem. 1974;249:4034–4037. [PubMed] [Google Scholar]
- 51.James TY, Pelin A, Bonen L, Ahrendt S, Sain D, Corradi N, et al. Shared signatures of parasitism and phylogenomics unite cryptomycota and microsporidia. Curr Biol. 2013;23:1548–1553. doi: 10.1016/j.cub.2013.06.057. [DOI] [PubMed] [Google Scholar]
- 52.Mabberley DJ. Mabberley’s plant-book. 3rd ed. Cambridge: Cambridge University press; 2008.
- 53.Nickrent DL, Malécot V, Vidal-russell R, Der JP. A revised classification of Santalales. Taxon. 2010;59:538–558. doi: 10.1002/tax.592019. [DOI] [Google Scholar]
- 54.Roquet C, Coissac E, Cruaud C, Boleda M, Boyer F, Alberti A, et al. Understanding the evolution of holoparasitic plants : the complete plastid genome of the holoparasite Cytinus hypocistis ( Cytinaceae ) Ann Bot. 2016;118:885–896. doi: 10.1093/aob/mcw135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Barney CW, Hawksworth FG, Geils BW. Hosts of Viscum album. For Pathol. 1998;28:187–208. doi: 10.1111/j.1439-0329.1998.tb01249.x. [DOI] [Google Scholar]
- 56.Nickrent DL, Der JP, Anderson FE. Discovery of the photosynthetic relatives of the “Maltese mushroom” Cynomorium. BMC Evol Biol. 2005;5:38. doi: 10.1186/1471-2148-5-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Graham SW, Olmstead RIGO. Utility of 17 chloroplast genes for inferring the phylogeny of the basal angiosperms. Am J Bot. 2000;87:1712–1730. doi: 10.2307/2656749. [DOI] [PubMed] [Google Scholar]
- 58.Dong W, Liu J, Yu J, Wang L, Zhou S. Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS One. 2012;7:1–9. doi: 10.1371/journal.pone.0035071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Martin G, Aury J, Baurens F. The complete chloroplast genome of Banana (Musa acuminata , Zingiberales): insight into plastid monocotyledon evolution. PLoS One. 2013;8:e67350. doi: 10.1371/journal.pone.0067350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chase MW, Christenhusz MJM, Fay MF, Byng JW, Judd WS, Soltis DE, et al. An update of the angiosperm phylogeny group classification for the orders and families of flowering plants: APG IV. Bot J Linn Soc. 2016;181:1–20. doi: 10.1111/boj.12385. [DOI] [Google Scholar]
- 61.Heath TA, Hedtke SM, Hillis DM. Taxon sampling and the accuracy of phylogenetic analyses. J Syst Evol. 2008;46:239–257. [Google Scholar]
- 62.Nabhan AR, Sarkar IN. The impact of taxon sampling on phylogenetic inference: a review of two decades of controversy. Brief Bioinform. 2012;13:122–134. doi: 10.1093/bib/bbr014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Tissue type, protocol, cleaning procedures and mass used for DNA extraction of the 11 samples analysed in this study. (DOCX 57 kb)
Table S4. GeneBank Accession Numbers of the non-parasitic taxa used in the small dataset of 38 taxa. (XLS 104 kb)
Code C1: R code used for the analyses of variance in the small and large data set. (DOCX 12 kb)
Figure S1. Phylogenetic tree of the 418 taxa included in the study. Branch length shows substitution rates, while bootstraps support values are shown on the base of each branch. (PDF 682 kb)
Table S3. ANOVA of the substitution rates in the data set of the 38 taxa for the: a) atp1 gene, b) matR gene, c) rsp3 gene and d) all_mt_genes. (PDF 114 kb)
Table S2. ANOVA of the substitution rates in the data set of 418 taxa using 4 mitochondrial genes (analysis 4). (XLSX 41 kb)
Data Availability Statement
The sequences obtained for each taxon were submitted to GenBank under accession numbers: Cassytha pubescens MG233923 – MG233957, Cynomorium coccineum MG234165 – MG234233, Cytinus hypocistis ssp. clusii MG233958 – MG233992, Cytinus hypocistis ssp. hypocistis MG234028 – MG234062, Krameria lanceolata MG234063 – MG234097, Langsdorffia hypogaeae MG234098 – MG234130, Lathraea cladestina MG234131 – MG234164, Lathraea squamaria MG234165 – MG234164, Loranthus europaeus MG233993–234027, Pholisma sonorae MG234234–234268, Phoradendron liga MG234269-MG234275.