

Abstract
Protein phosphatase 1 (PP1) is a highly conserved protein phosphatase that performs the majority of serine and threonine (Ser-Thr) dephosphorylation reactions in eukaryotes and opposes the actions of a diverse set of Ser-Thr protein kinases. PP1 gains substrate specificity through binding to a large number (> 200) of regulatory proteins that control PP1 localization, activity, and interactions with substrates. PP1 recognizes the well-characterized RVxF binding motif that is present in many of these regulatory proteins, thus generating a multitude of distinct PP1 holoenzymes. Here we show that a subset of the RVxF binding motifs, in which x is a phosphorylatable amino acid (RV[S/T]F), were phosphorylated specifically during mitosis and that this phosphorylation event abrogated the interaction of PP1 with the regulatory protein. We determined that this phosphorylation was primarily governed by the mitotic protein kinase Aurora B and that high phosphorylation site stoichiometry of these sites maintained phosphorylation of PP1 substrates during mitosis by disrupting the assembly of PP1 holoenzymes. We generated an antibody that recognizes the phosphorylated form of the RV[S/T]F motif (RVp[S/T]F) and used it to identify known PP1 regulatory proteins (KNL1, CDCA2, and RIF1) as well as multiple proteins that could potentially act as PP1 binding partners (UBR5, ASPM, SEH1, and ELYS) governed by this mechanism. Taken together, the work presented here suggests a general regulatory mechanism by which the coordinated activities of Aurora B and PP1 control mitotic progression.
INTRODUCTION
In mitosis, a cell segregates its duplicated genome into two daughter cells. Reversible protein phosphorylation is an important regulatory mechanism in this process. The opposing activities of protein kinases and protein phosphatases are responsible for establishing the phosphorylation stoichiometry, the relative amounts of phosphorylated and unphosphorylated forms, of mitotic regulatory proteins. Imbalance in either activity can lead to mitotic defects and genomic instability (1).
Entry into mitosis is initiated by the activation of cyclin-dependent kinase 1 (CDK1) (2). CDK1 phosphorylates and activates multiple mitotic substrates including the mitotic protein kinases Aurora A and B and Polo-like kinase 1 (PLK1). The activation of kinases in the beginning of mitosis results in a dramatic net increase in protein phosphorylation during the early phases of mitosis (3–5). However, to exit mitosis and achieve the lower amount of mitotic protein phosphorylation that is characteristic of interphase, it is necessary for mitotic phosphoproteins to be dephosphorylated in a timely manner (6).
In vertebrates, most of the dephosphorylation reactions during mitotic exit have been attributed to the phosphoprotein phosphatase (PPP) family of serine-threonine phosphatases, in particular PP1 and PP2A (7, 8). Activation of CDK1 in the G2 phase has been reported to inhibit the activities of these major mitotic protein phosphatases (9). For instance, CDK1 phosphorylation of PP1 at Thr320 inhibits PP1 phosphatase activity during mitosis. After the decline of CDK1 activity in anaphase, PP1 auto-dephosphorylates Thr320 and regains full activity to drive mitotic exit (10).
PP1 is responsible for the majority of dephosphorylation reactions in eukaryotic cells (11). The catalytic subunit of PP1 is a ubiquitous enzyme that interacts with a diverse set of regulatory proteins that not only target the phosphatase to its substrate, but also control its activity (12). More than 200 regulatory proteins of PP1 have been identified, with more predicted to be awaiting identification (13, 14). The binding of the PP1 catalytic subunit to its regulatory proteins to generate PP1 holoenzymes is governed by short sequence motifs (15, 16). Nearly 90% of PP1-binding proteins dock to PP1 through their RVxF motif, [R/K]-X(0,1)-[V/I]-{P}-[F/W], wherein X is any residue and {P} any residue but proline (17–20). RVxF-containing regulatory proteins bind to a hydrophobic region on the catalytic PP1 subunit that is distant from the active site (17, 21). Characterization of the RVxF motif in PP1 interactors reveals an over-representation of serine and threonine at the ‘x’ position, with serine occurring in ~21% and threonine in ~18% of known PP1 binding partners (fig. S1) (13). Furthermore, phosphorylation of the serine or threonine residue within the RVxF motif inhibits their binding to PP1 (22–25).
During mitosis, PP1 is enriched on chromosomes and at centrosomes (26, 27), and multiple PP1-targeting subunits have been identified at kinetochores, the mitotic spindle, centromeres, the spindle midzone, and the nuclear envelope (28–32). One of the key mitotic regulatory proteins of PP1 is the kinetochore protein Kinetochore null 1(KNL1), also known as cancer susceptibility candidate 5 (CASC5 ) (22, 28). KNL1 recruits PP1 to the kinetochores through its RVxF motif in which x is a serine residue. Phosphomimetic mutations of the serine in the PP1-binding RVxF and SILK motifs in KNL1 greatly reduce its binding to PP1 (22, 33). PP1 has been shown to antagonize the activity of Aurora B (34). Indeed, the balance of Aurora B and PP1 activities is essential for establishing chromosome bi-orientation and for regulating the spindle-assembly checkpoint (SAC) (35, 36). During early mitosis, Aurora B localizes to centromeres and chromosomes before re-localizing to the spindle midzone during mitotic exit. Incorrect microtubule-kinetochore attachments or the lack of tension at kinetochores leads to phosphorylation of outer kinetochore proteins by Aurora B and activation of the spindle assembly checkpoint (37). Once bipolar attachments are established, PP1 dephosphorylates Aurora B substrates at kinetochores to silence the SAC and drive mitotic exit (38, 39).
Here we identified phosphorylation of RV[S/T]F motifs in known and previously unidentified PP1 regulatory proteins as a cell cycle–dependent event. We show that Aurora B was the primary kinase responsible for this phosphorylation event during mitosis. We propose that Aurora B–dependent phosphorylation of RV[S/T]F motifs is an important regulatory mechanism that prevents PP1 from interacting with its regulatory proteins during mitosis, thereby affecting phosphorylation of a multitude of PP1 substrates and controlling the balance of Aurora B kinase and PP1 phosphatase activities.
RESULTS
PP1 preferentially binds non-phosphorylated RV[S/T]F motifs in vitro
It was previously shown that phosphorylation of the RV[S/T]F motifs in the skeletal muscle glycogen targeting subunit (GM) and KNL1 negatively regulates their interaction with PP1 (22, 23, 40). To determine whether RV[S/T]F phosphorylation is a general mechanism controlling the interaction of PP1 with its regulatory proteins, we examined the binding of PP1 to non-phosphorylated (unmodified) or phosphorylated versions of RV[S/T]F peptides from different PP1 regulatory proteins (table S1). We tested PP1 binding to these peptides using two independent biochemical experiments- overlays using recombinant human PP1 (Fig. 1A) and in vitro peptide pull-downs (Fig. 1B). In PP1 overlays, the RVRW peptide from the protein YLP motif–containing protein 1 (YLPM1) (41) was used as a positive control, and the mutated version RARA, which does not bind PP1 (41), was used as a negative control. Both in overlay as well as in in vitro peptide pull-downs, PP1 exhibited a clear preference for binding to the unmodified versions of all peptides. The phosphorylated form of each peptide was either not bound by PP1 or showed reduced interaction with PP1 compared to the corresponding non-phosphorylated form (Fig. 1, A and B), suggesting that loss of PP1 binding upon phosphorylation of RV[S/T]F motifs is a general phenomenon.
Fig. 1. PP1 preferentially binds nonphosphorylated RV[S/T]F motifs in vitro.
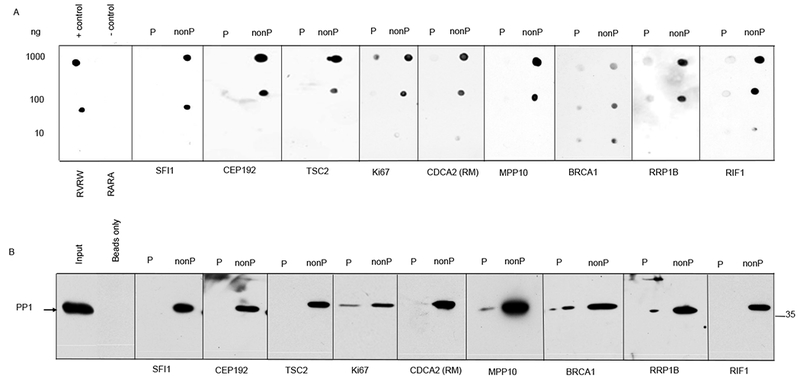
(A) Phosphorylated (P) or nonphosphorylated (nonP) versions of RV[S/T]F-containing peptides from the indicated proteins were spotted onto nitrocellulose membranes in the indicated amounts. The membranes were overlaid with a mixture of recombinant human PP1 and probed with a PP1-specific antibody. A peptide derived from the PP1-binding protein YLPM1 (RVRW) was used as a positive control, and the nonphosphorylable mutant version of this peptide (RARA) as the negative control. (B) Activated 5-Carboxypentyl-Sepharose 4B N-hydroxysuccinimide ester (CH-Sepharose) beads were coupled to the phosphorylated or nonphosphorylated versions of the indicated RV[S/T]F peptides and incubated with clarified HeLa whole cell extracts. After washing, proteins bound to the beads were eluted with SDS sample buffer, run on SDS-PAGE, and immunoblotted for PP1. The position of the 35 kDa mass marker is indicated. Peptides derived from the indicated proteins and sequences are shown in table S1. The data represents one of three independent experiments for both (A) and (B).
RV[S/T]F motifs in PP1 binding proteins are phosphorylated in cells during mitosis
To determine whether phosphorylation of RV[S/T]F PP1-binding motifs occurs in cells and in a manner that depends on the cell cycle, we raised antibodies that specifically recognize the phosphorylated forms of the RV[S/T]F motifs (RVp[S/T]F motifs) of several PP1-binding proteins, including Rap1-interacting factor 1 (Rif1), Centrosomal protein of 192 kDa (CEP192), Suppressor of fermentation-induced loss of stress resistance 1 (SFI1), breast cancer type 1 susceptibility protein (BRCA1), and Cell division cycle associated 2 (CDCA2; also called Repoman, RM).
To validate these antibodies, we preformed dot blots using different amounts of phosphorylated and non-phosphorylated versions of the antibody epitopes (fig. S2A). Although the phosphorylated forms of the peptides were readily recognized by the antibodies, the non-phosphorylated (unmodified) versions, even at the highest amount (1000 ng), were not. Next, we used these antibodies to investigate the phosphorylation status of RV[S/T]F motifs in RIF1, CEP192, SFI1, BRCA1, and CDCA2 in mitosis and during cell cycle progression. HeLa cells were synchronized in G1/S using a thymidine block, released, and then captured in mitosis with nocodazole, which arrests the cells in prometaphase. The nocodazole was washed out, and the cells were collected at different time points from 0.5 h to 22 h following release from mitotic blockade. We observed increased phosphorylation of RV[S/T]F motifs of each of the five proteins 30 minutes after nocodazole washout (Fig. 2A). Cell-cycle synchronization was confirmed by immunoblotting with the mitotic marker Cyclin B1 (Fig. 2A) as well as FACS analysis (Fig. 2B). This increase in phosphorylation in mitotic cells was also observed by immunofluorescence staining using antibodies specific for the phosphorylated RVxF epitopes, including phosphorylated CEP192, phosphorylated SFI1, and phosphorylated BRCA1 (fig. S2B, fig. S2C and fig. S2D).
Fig. 2. RV[S/T]F motifs are phosphorylated in cells during mitosis.
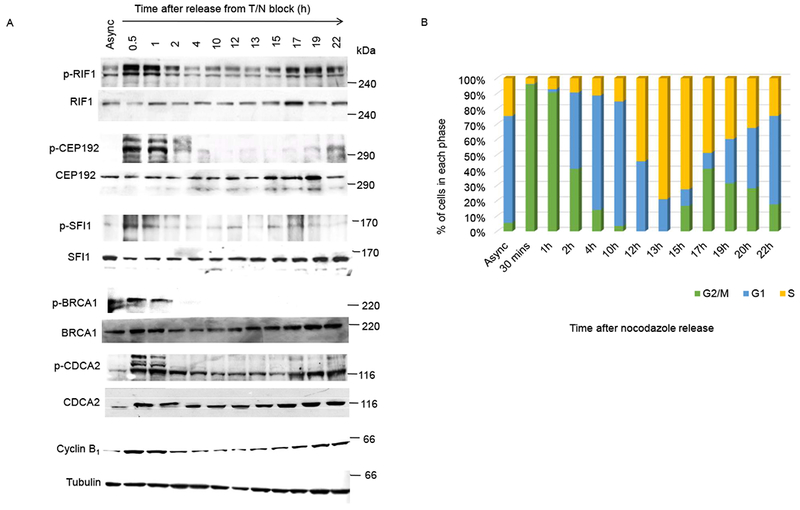
(A) HeLa cells were synchronized using thymidine-nocodazole (T/N) block and released in fresh media thereafter. Samples were collected at the indicated time points, immunoblotted for cyclin B1, and analyzed for phosphorylation within the RV[S/T]F motifs of the indicated proteins using in-house generated phosphoepitope-specific antibodies. Async, asynchronized cells. Antibodies are characterized in fig. S2, and peptide sequences are shown in table S1. Samples were collected three times with the same synchronization technique, and the data represents one of three independent experiments. Equal loading was confirmed immunoblotting for tubulin. Mass markers are indicated in kDa. (B) Quantification of cells in each phase of the cell cycle at the indicated time points following synchronization as determined by FACS analysis.
Global analysis of RV[S/T]F motifs shows increased phosphorylation during mitosis
To globally monitor phosphorylation of the ([R/K]-X(0,1)-[V/I]-[S/T]-[F/W]) consensus motif (referred to as the RV[S/T]F motif henceforth) in PP1 binding partners, we generated an antibody specific for RVp[S/T]F, the phosphorylated form of the RV[S/T]F motif present in multiple PP1 binding proteins in the human proteome. We characterized the phospho-specificity and motif selectivity of this antibody on dot blots using different amounts of phosphorylated and non-phosphorylated (unmodified) versions of peptides corresponding to a common version of the RV[S/T]F and RVp[S/T]F motifs of known PP1-interacting proteins, including RIF1 (RVSF and RVpSF), peptides with mutations in RIF1 RVSF motif (KVSW, KVpSW, RISF, RIpSF, RITW, and RIpTW), Ki67 (RVSF and RVpSF), CDCA2 (RVTF and RVpTF), RRP1B (KVTF and KVpTF), BRCA1 (KVTF and KVpTF), and TSC2 (RSVSW and RSVpSW) (28, 42, 43) (fig. S3A). This confirmed that the RVp[S/T]F antibody is phospho-specific and recognizes to some degree related phosphorylated RV[S/T]F motifs in vitro. The phospho-specificity of the antibody was also confirmed by competition with the phosphopeptide (RRVpSFADK), which was used for generating the antibody (fig. S3B). To determine the phosphorylation status of RV[S/T]F motifs in cells, we performed immunoblotting of extracts from HeLa cells synchronized in mitosis as described above. We found a marked increse in phosphorylation of RV[S/T]F motifs in different proteins during mitosis (Fig. 3A).
Fig. 3. Global analysis of RV[S/T]F motif phosphorylation during mitosis.

(A) The phosphoepitope-specific antibody generated using the peptide RRVpSFADK recognizes several proteins in immunoblots of HeLa cell lysates (upper panel). Lysates from synchronized HeLa cells at the indicated times following release from T/N block were probed with this RVp[S/T]F (p-RV[S/T]F)-specific antibody to monitor changes in the phosphorylation status of this motif during the cell cycle. Phosphorylation of PP1 at Thr320 (p-Thr320) was also assessed at these time points. Equal loading was confirmed by immunoblotting for tubulin. Mass markers are indicated in kDa. Three replicates of the p-RV[S/T]F immunoblot were quantified using ImageJ software and normalized to the mitotic (0.5 h) sample (lower panel). Error bars represent mean ± SD. (B) Immunofluorescence staining of HeLa cells showing DAPI, tubulin, and p-RV[S/T]F at different phases of the cell cycle. >50 cells were imaged for each condition in four independent experiments. Scale bar, 20μm.
Phosphorylation of PP1 Thr320 by CDK1 during mitotic entry inactivates the enzyme until anaphase when PP1 auto-dephosphorylates Thr320 and fully activates itself (10, 44). This dephosphorylation occurs within 2 hours of nocodazole release, which corresponds with the observed decrease in phosphorylation of the RV[S/T]F motifs (Fig. 3A). This suggests that in addition to the direct inhibition of the PP1 catalytic subunit by CDK1 phosphorylation, the catalytic activity of PP1 might also be inhibited by phosphorylation within the RV[S/T]F motifs of PP1 regulatory proteins (the regulatory subunits of the PP1 holoenzyme) during mitosis.
The global increase in phosphorylation of RV[S/T]F motifs was also confirmed by immunofluorescence staining using the RVp[S/T]F-specific antibody (Fig. 3B). Compared to interphase cells, metaphase cells showed a dramatic increase in the phosphorylation of RV[S/T]F motifs in the cytoplasm, spindle poles, and along the mitotic spindle. This phosphorylation remains at spindle poles and the central spindle in anaphase and the midbody and telophasic bridge during cytokinesis (Fig. 3B).
Phosphorylation of the RV[S/T]F motifs depends on Aurora B kinase
Next, we wanted to identify the protein kinase(s) responsible for phosphorylation of the RV[S/T]F motifs during mitosis. To do so, we treated HeLa cells with small molecule inhibitors of the mitotic kinases Aurora A, Aurora B, PLK1, and CDK1, which are activated at mitotic entry and play a role in controlling mitotic progression (4, 6), and monitored the phosphorylation of RV[S/T]F motifs. At low concentration (1μM), the small molecular inhibitor MLN8054 targets Aurora A, but at high concentration (5μM) targets both Aurora and Aurora B (45) Treatment with a low concentration of MLN8054 had only a modest effect on RV[S/T]F phosphorylation, but treatment with a high concentration markedly reduced RV[S/T]F phosphorylation (Fig. 4A), suggesting the effect of MLN8054 treatment at high concentration is likely due to inhibition of Aurora B. This is supported by the observation that specific inhibition of Aurora B by ZM447439 or hesperadin showed a marked decrease in the phosphorylation of RV[S/T]F motifs (Fig. 4A). The RV[S/T]F motif is reminiscent of the basophilic consensus motif of Aurora A and Aurora B (4, 46), supporting our observation that Aurora B participates in RV[S/T]F phosphorylation and suggesting that Aurora B may directly phosphorylate RV[S/T]F motifs. The effectiveness and specificity of each inhibitor was determined by investigating the phosphorylation status of known substrates of each protein kinase by western blotting (Fig. 4B) of HeLa cell extracts. Treatment with the PLK1 inhibitor BI2536 or the CDK1 inhibitor Flavopiridol caused only a modest decline in the phosphorylation of the RV[S/T]F motifs in the substrates. Inhibition of CDK1 forces cells out of mitosis, as indicated by the decrease in the mitotic marker H3 phosphorylated on Ser10 (Fig. 4B). To confirm that the RV[S/T]F phosphorylation was Aurora B–dependent, mitotic cells treated with different kinase inhibitors were fixed and stained with the RVp[S/T]F antibody (Fig. 4C). This showed a marked decrease in phosphorylation of the RV[S/T]F motifs upon treatment with Aurora B inhibitor (hesperadin) in cells (Fig. 4C). Mitotic cells treated with CDK1 inhibitor did not show any decrease in phosphorylated RV[S/T]F by immunofluorescence (Fig. 4C).
Fig. 4. Effects of mitotic kinase inhibition on phosphorylation of PP1-binding RV[S/T]F motifs.

(A) Following mitotic arrest and treatment with the proteasome inhibitor MG132, HeLa cells were treated with the indicated kinase inhibitors The cells were harvested and the lysates separated by SDS-PAGE and immunoblotted for phosphorylated RV[S/T]F (p-RV[S/T]F, upper panel). The blot was quantified using ImageJ software and normalized to the mitotic sample (lower panel). MLN8054, Aurora A and B inhibitor; ZM447439 and hesperadin, Aurora B inhibitors; BI2536, PLK1 inhibitor; Flavopiridol, CDK1 inhibitor. N=3. (B) To demonstrate the effectiveness and specificity of each kinase inhibitor, the same samples from panel (A) were immunoblotted to show the indicated phosphorylated proteins. H3 is phosphorylated on Ser10 by Aurora B;Aurora A is autophosphorylated on Thr288; Aurora B is autophosphorylated on Thr232; PP1 is phosphorylated on Thr320 by CDK1 (arrow); TCTP is phosphorylated on Ser46 by PLK1. N=3. (C) HeLa cells were treated with inhibitors of Aurora B (hesperadin), Aurora A (Aurora A inhibitor I), CDK1 (roscovitine), or PLK1 (BI2536) for 2 h before immunostaining for tubulin and phosphorylated RV[S/T]F (p- RV[S/T]F). Nuclei are indicated with DAPI. Control cells were not treated with any inhibitor. >50 cells were imaged for each condition in three independent experiments. Scale bar, 20μm.
Furthermore, treating cells the Aurora B inhibitor hesperadin caused a time-dependent reduction in the phosphorylation of RV[S/T]F-containing proteins in cells released from nocodazole arrest (Fig. 5A). Immunofluorescence of cells treated with Aurora B kinase inhibitor was also used to confirm results from western blotting. The inhibition of Aurora B activity was confirmed by the specific lack of phosphorylation of its substrate H3 at Ser10 (Fig 4B and5B) (47). Immunofluorescence staining of Aurora B inhibitor treated cells revealed a strong reduction of phosphorylated RV[S/T]F staining compared to control cells (Fig. 5C), as quantified in Fig. 5D.
Fig. 5. Aurora B phosphorylates RV[S/T]F motifs during mitosis.
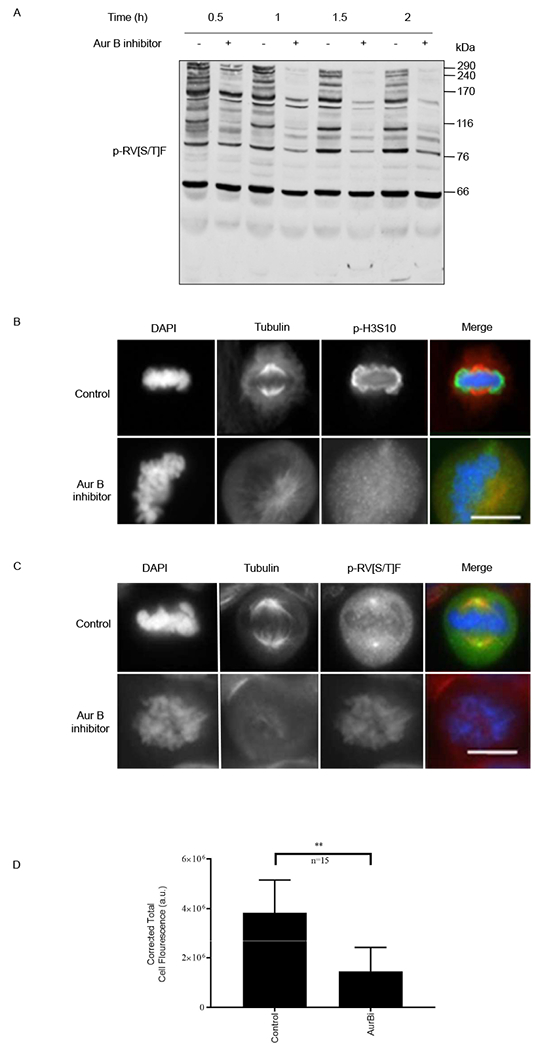
(A) HeLa cells were arrested in mitosis with nocodazole, treated with either DMSO (−) or the Aurora B inhibitor hesperadin (+) in the presence of the proteasome inhibitor MG132, harvested, and immunoblotted for phosphorylated RV[S/T]F motifs (p-RV[S/T]F). Mass markers are indicated in kDa. (B) and (C) Immunofluorescence showing nuclei (DAPI), tubulin, and either H3 phosphorylated on Ser10 (p-H3S10, B) or p-RV[S/T]F (C) in mitotically-arrested HeLa cells treated with the Aurora B inhibitor hesperadin. >15 cells were imaged for three independent replicates. (D) Difference in RVp[S/T]F staining was quantified for each condition, untreated (Control) and Aurora B inhibitor–treated (AurBi) by calculating the total cell fluorescence, using ImageJ. Error bars represent mean ± SD. **p<0.01, paired Student’s t test (n=15). Scale bar, 20 μm.
Proteomics analysis reveals new candidates phosphorylated by Aurora B on RV[S/T]F motifs.
To identify proteins recognized by the antibody specific for RVp[S/T]F we performed immunoprecipitations with the antibody or control IgG from mitotically-arrested HeLa cells (fig. S4A). This showed a marked enrichment of proteins in the RVp[S/T]F immunoprecipitations compared to control IgG immunoprecipitations in mitotic cells. To determine the identity of these proteins, we precipitated them using trichloroacetic acid and analyzed them by liquid chromatography–coupled tandem mass spectroscopy (LC-MS/MS) (table S2). In total, we analyzed five independent control IgG and five independent RVp[S/T]F immunoprecipitations. The degree of confidence for RVp[S/T]F–specific binding was assessed by a computational tool, significance analysis of interactome (SAINT) (48, 49) (table S3). Using this approach, we identified 421 proteins that specifically bound to RVp[S/T]F antibody (table S2 and S3).
Next, we investigated whether these proteins bound to the antibody recognizing RVp[S/T]F following Aurora B inhibition. We compared the binding of proteins to the RVp[S/T]F-specific antibody in immunoprecipitates from untreated cells to that of proteins from cells treated with the Aurora B inhibitor hesperadin by label-free intensity-based absolute quantification (iBAQ) (50) (table S2). This revealed that 227 (~54%) of the 421 proteins that bound to the antibody in the absence of Aurora B exhibited no or significantly reduced binding to antibody (p-value < 0.05) upon Aurora B inhibition (table S2). To determine the functional profile of the proteins specifically enriched in the RVp[S/T]F immunoprecipitates, we performed gene ontology (GO) analysis for these proteins. GO analysis showed an enrichment of proteins involved in mitosis, spindle organization and assembly, and chromosome segregation (table S4). Furthermore, many of the proteins are localized to cellular structures such as the spindle, kinetochore, chromosome, and midbody (table S5). This shows these RV[S/T]F-containing proteins may potentially regulate the binding of PP1 and the antagonistic activities of PP1 and Aurora B at these structures in mitosis.
Out of the 421 proteins that specifically bound to the antibody recognizing RVp[S/T]F, 37 contained the RV[S/T]F motif and were phosphorylated within this motif during mitosis (table S6). Many of the other proteins specifically bound to the antibody are predicted to be in complex with proteins that contain the RV[S/T]F motif but are not phosphorylated during mitosis. Approximately 68% (25) of the RV[S/T]F-containing proteins failed to bind or exhibited significantly reduced binding to the RVp[S/T]F antibody when the cells were treated with the Aurora B inhibitor hesperadin (table S6). The protein KNL1, a PP1 binding protein that associates with PP1 in a manner that depends on its phosphorylation by Aurora B (22), was identified in our study along with other cell cycle regulatory proteins including RIF1,which controls DNA replication by targeting PP1 to the chromatin (51), CDCA2, which recruits PP1 to anaphase chromosomes to maintain chromosome architecture (52), E3 ubiquitin-protein ligase UBR5 (UBR5), which promotes metaphase to anaphase transition through mediating protein turnover (53), Abnormal spindle-like microcephaly-associated protein (ASPM), which controls spindle positioning and orientation (54), Nucleoporin SEH1 (SEH1), which regulates chromosome segregation through controlling Aurora B localization at centromeres(55), and Embryonic large molecule derived from yolk sac (ELYS), which recruits PP1 to kinetochores and chromosomes during mitotic exit(56).
Phosphorylation within RV[S/T]F motifs reduces binding to PP1
To validate the results from the mass spectrometry analysis, we preformed reciprocal immunoprecipitations using antibodies that recognize two known PP1-binding proteins: RIF1 and CDCA2 using protein extracts from HeLa cells cultured in the presence and absence of various mitotic kinase inhibitors. Western blot analysis of RIF1 and CDCA2 immunoprecipitations using antibodies specific for the phosphorylated RVSF of RIF1 or the phosphorylated RVTF of CDCA2 confirmed that Aurora B is required for phosphorylation of these motifs (fig. S4B). These experiments also confirmed that phosphorylation of RIF1 in mitosis negatively regulates binding to PP1. PP1-bound microcystin-Sepharose beads were used to pull down proteins from extracts of asynchronous, mitotic, or Aurora B-inhibited cells, and the elution was analyzed for the presence of RIF1. PP1 readily interacted with RIF1 from asynchronous lysates, but this interaction was significantly reduced in mitotic lysates. Inhibition of Aurora B activity partially restored the interaction in mitotic extracts (Fig. 6A).
Fig. 6. Phosphorylation of the RVxF motif by Aurora B inhibits PP1 binding.
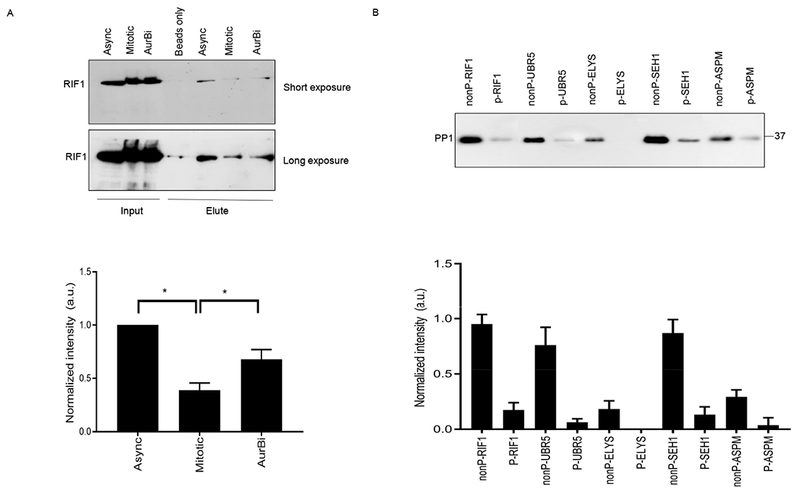
(A) Recombinant human PP1α/β/γ coupled to microcystin-Sepharose beads were used to test in vitro binding between PP1 and proteins from asynchronous (Async), mitotic, or Aurora B–inhibited (AurBi) HeLa extracts. After incubating the PP1-coupled beads with cell lysates, the bound proteins were eluted and tested for the presence of RIF1 by immunoblotting (upper panel). Immunoblots were quantified using ImageJ software and normalized relative to RIF1 binding to PP1-coupled beads in extracts from asynchronous cells. Error bars represent mean ± SD. *p<0.05, paired Student’s t test (n=3 independent experiments) (B) Nonphosphorylated (nonP) and phosphorylated (P) peptides containing the RV[S/T]F motifs from the indicated proteins coupled to activated CH-Sepharose beads were incubated with HeLa cell extracts. After washing, the bound proteins were eluted with SDS sample buffer and immunoblotted for PP1 (upper panel). The sequences of the peptides are shown in table S1. Three replicates of the experiment were quantified and normalized relative to nonP-RIF1 peptide (lower panel). Error bars represent mean ± SD.
To confirm the effect of phosphorylation within RV[S/T]F motifs on PP1 binding, phosphorylated or unmodified versions of RV[S/T]F-containing peptides from proteins identified in our proteomic screen using the Aurora B inhibitor (RIF1, UBR5, ELYS, SEH1 and ASPM), were tested for binding to PP1 in vitro. Whereas the unmodified versions of these peptides bound to PP1, the phosphorylated versions bound considerably less or no PP1 (Fig. 6B), confirming preferential PP1 binding to these peptides in the non-phosphorylated state and supporting the idea that they are true PP1 binders.
DISCUSSION
The substrate specificity of PP1 is controlled by regulatory proteins, most of which utilize the RVxF binding motif for interaction with the phosphatase. Sequence analysis of this motif from known PP1 binding proteins shows an over-representation of either positively charged Arg or Lys residues or phosphorylatable Ser or Thr residues at the ‘x’ position (fig. S1). The positive charge of the Arg or Lys residue helps the regulatory proteins bind tightly to the hydrophobic pocket on the surface of PP1, as in the case of the KVRF docking motif in growth arrest and DNA damage–inducible protein GADD34 for example (57). In contrast, a negative charge introduced by phosphorylation of a Ser or Thr residue at the ‘x’ position provides a regulatory mechanism for the reversible association between the phosphatase and the regulatory protein through the action of protein kinases during specific cellular events. This is consistent with the absence of acidic residues (Asp or Glu) at the ‘x’ position (fig S1). Phosphorylation within or in close proximity to the PP1 docking site of known regulatory proteins including GM and KNL1 blocks their binding to PP1 (22, 23, 40). Here, we confirmed and extended this observation by identifying additional known and previously unidentified PP1-binding proteins that interact with PP1 through an RV[S/T]F motif in a phosphorylation-dependent manner. Furthermore, we provide evidence that this phosphorylation is a cell cycle–dependent event occurring specifically during mitosis (Figs. 2 and 3).
Among the possible mitotic protein kinases, the RV[S/T]F motif most closely fits the Aurora kinase consensus sequence R-X-S/T-Φ, where X is any amino acid and Φ is a hydrophobic amino acid. Indeed, we found that Aurora B, but not Aurora A, phosphorylated the Ser or Thr within the binding motif of PP1 in known and potential regulatory proteins of PP1 (Figs. 4 and 5, table S2). However, not all [R/K][V/I][S/T][F/W] motifs are phosphorylated by Aurora B, therefore other basophilic kinases including protein kinase A (PKA), protein kinase B (PKB), CaMKII, and CHK2, among others, could play a role in the phosphorylation of these motifs in other cellular events or subcellular compartments in which Aurora B is not involved in regulation of PP1 activity. We also observed a modest decrease in RV[S/T]F phosphorylation upon the inhibition of PLK1, Aurora A, and CDK1 (Fig. 4). The PLK1 consensus motif is characterized by acidic amino acids upstream of and hydrophobic amino acids downstream of the phosphorylation site (4, 35). However, in vitro and in vivo analysis of PLK1 substrates have revealed that PLK1 also phosphorylates [S/T]F motifs (4, 58). Thus, it is possible that PLK1 opposes PP1 function by phosphorylating phenylalanine-containing RV[S/T]F motifs. Our results show that among the proteins that were specifically bound to RVp[S/T]F antibody, 68% were lost or reduced upon inhibition of Aurora B (table S2). The other proteins (32%) enriched in RVp[S/T]F immunoprecipitates that do not change upon Aurora B inhibition might be phosphorylated by other kinases, and we believe the localization of the specific proteins containing RV[S/T]F motifs dictates the kinase responsible for their phosphorylation. This has been shown previously in the case of the centrosomal protein CEP192, where the RV[S/T]F motif is targeted by PLK1 (59). The RV[S/T]F motif has almost certainly evolved to regulate PP1 function by controlling its recruitment to binding partners in a phosphorylation-dependent manner in response to specific cellular conditions and at specific cellular locations.
Using affinity enrichment with a RVp[S/T]F antibody and quantitative proteomics, we identified previously unknown phosphorylation sites for the protein kinase Aurora B within the RV[S/T]F motif of 25 proteins: 6 known [RIF1, antigen identified by monoclonal antibody Ki67 (Ki67), CDCA2, KNL1, BRCA1, and ribosomal RNA processing protein 1 homolog B (RRP1B)] and 19 proteins previously unknown to bind PP1. From this latter group of proteins that interact with PP1 in a phosphorylation- and cell cycle–dependent manner, we validated 4 proteins with known links to mitotic progression. Peptides containing RV[S/T]F motifs from these proteins preferentially bound to PP1 when the RV[S/T]F motif was not phosphorylated (Fig. 6). These proteins include the E3 ubiquitin ligase UBR5, the microtubule regulator ASPM, the nucleoporin SEH1, and the mitotic exit PP1 scaffold protein ELYS. UBR5 is an HECT (homology to E6AP C terminus) family E3 ubiquitin ligase that has been reported to facilitate anaphase entry by forming complex with the spindle assembly checkpoint (SAC) proteins BUBR1 and BUB3 and promoting their turnover by ubiquitination (53, 60) (table S2). Phosphorylation of the “KVTF” sequence in UBR5 by Aurora B might promote PP1 dissociation to keep the SAC activated. Once this motif is dephosphorylated, PP1 could be recruited by UBR5 to contribute to silencing the SAC. ASPM localizes to spindle poles during mitosis and is known to be involved in spindle microtubule organization and cytokinesis (61, 62). Phosphorylation within the “KVSF” motif of ASPM during mitosis has been reported in previous large-scale mass spectrometry studies (4, 63). Our data confirms this phosphorylation during mitosis, and we found this phosphorylation event to be primarily governed by Aurora B kinase. The nucleoporin SEH1 is required for the recruitment of the Nup107-160 nucleoporin complex to kinetochores during mitosis and plays a critical role in establishing correct kinetochore-microtubule attachments (64). ELYS has been reported to recruit the Nup107-160 subcomplex to the kinetochores (65), and it has been shown that the Caenorhabditis elegans homolog of ELYS, MEL-28, recruits PP1 specifically at mitotic exit and plays a crucial role during nuclear reassembly (66). Given the role of Aurora B and PP1 during cytokinesis, these potential PP1 binding partners might be crucial for the recruitment of PP1 and regulation of the Aurora B-PP1 axis during cytokinesis and nuclear envelope re-assembly. In budding yeast, RIF1 has been shown to bind PP1, and this interaction has been implicated in replication origin firing (42, 67, 68). Here, we confirmed the RIF1-PP1 interaction in human cells and identified Aurora B–dependent phosphorylation of the RVSF motif of RIF1 during mitosis (fig. S4B). In addition, we also show that phosphorylation within the RVSF of RIF1 reduced PP1 binding, at least in vitro (Fig. 6A).
Collectively our data reveal that, during mitosis, in addition to inhibitory phosphorylation of PP1 catalytic subunits by CDK1 (10), PP1 function is controlled through a mechanism that relies on PP1-binding regulatory proteins. This dual inhibition mechanism provides on the one hand a fail-safe to limit PP1 activity during mitosis, while on the other hand allowing for more specific control of the phosphorylation status of select substrates. We propose that the RV[S/T]F motifs phosphorylated by Aurora B during mitosis are dephosphorylated by a yet unknown mitotic phosphatase. After this dephosphorylation event, PP1 can be recruited to its regulatory proteins, which can be themselves PP1 substrates, or target the catalytic subunit to multiple other mitotic phosphorylated substrates (Fig. 7), thus contributing to the global dephosphorylation events that drive mitotic exit. In conclusion, we have identified a previous unknown regulatory mechanism that coordinates the activities of the key mitotic enzymes PP1 and Aurora B for maintaining the kinase-phosphatase balance during mitosis. This balance between the counteracting activities of protein kinases and protein phosphatases is essential to maintain genomic integrity and cell survival.
Fig. 7. Proposed model of PP1 regulation during the cell cycle.
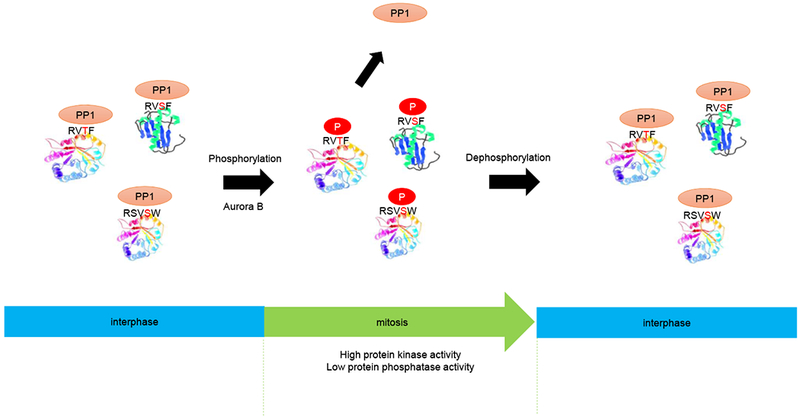
PPI is the catalytic subunit of the PPI holoenzyme, which includes both PP1 and any one of various regulatory proteins that bind to PP1 and control its catalytic activity. PP1 binds to RV[S/T]F motifs in the regulatory proteins during interphase. Upon activation of Aurora B during mitosis, these regulatory proteins are phosphorylated within the RV[S/T]F motifs. This phosphorylation leads to the dissociation of PP1 from the regulatory proteins thereby interfering with targeting of the phosphatase to its substrates. At the end of mitosis, these motifs are dephosphorylated, which regenerates the docking site for PP1, enabling the holoenzyme to target phosphorylated substrates for their dephosphorylation at mitotic exit.
MATERIALS AND METHODS
Cell culture and synchronization
Mycoplasma-free HeLa cells were obtained from ATCC and cultured in DMEM, supplemented with 10% FBS and 100 U ml−1 penicillin-streptomycin. For cell cycle analysis, cells were arrested with 2 mM thymidine for 17 h followed by a 7 h release. Following release, cells were blocked in prometaphase with 40 ng ml−1 nocodazole. After 9 h arrest, the cells were released into fresh media and harvested at different time points within a 24 h cycle. A part of each sample was fixed in ethanol and stained with propidium iodide for FACS analysis performed at the Flow Cytometry Facility at the University of Calgary.
Antibodies and reagents
The following commercial antibodies were used in this study at the indicated concentrations: PP1 (Santa Cruz, 1:500), RIF1 (Bethyl antibodies, 1:500), SFI1 (Proteintech Group Inc., 1:250), BRCA1 (Cell Signaling, 1:1,000), CDCA2 (Repoman) (Abcam, 1:1,000), Cyclin B1 (Santa Cruz, 1:500), tubulin (Sigma-Aldrich, 1:1,000), H3 pSer10 (Millipore (Upstate), 1:1,1000), p-Aurora A/B/C (Cell Signaling, 1:500), PP1 pThr320 (Cell Signaling, 1:1000), TCTP pSer46 (Cell Signaling, 1:1000), and actin (Cell Signaling, 1:5000). HRP-conjugated goat anti-Mouse and goat anti-Rabbit were obtained from Pierce and Thermo Fisher Scientific and were used at 1:5,000. The following peptides were used in this study, either unmodified or phosphorylated within RV[S/T]F motif: ZAP (GKKRVRWADLE), ZAPRARA (GKKRARAADLE), RIF1 (KRRVSFADK), CEP192 (SEKHVTFENHK), SFI1 (SRKVTFEGPK), CDCA2/Repoman (KRKRVTFGED), TSC2 (QLHRSVSWADSAK), RRP1B (SSKKVTFGLN), MPP10 (KESLKRVTFAL), ORC2 (KTPQKSVSFSLK), BRCA1 (QSPKVTFECEQK), Ki67 (KRRRVSFGGH), UBR5 (VHRVKVTFKDEK), ELYS (RLKETRISFVEEK), SEH1 (KQVWRVSWNIT), ASPM (SANVSKVSFNEK). The peptides were synthesized with 98% purity at GL Biochem (Shanghai) Ltd. The consensus RV[S/T]F logo from known PP1 interactors was generated using WebLogo (69). For the generation of phospho-specific antibodies, the RVp[S/T]F peptides derived from the proteins were coupled to keyhole limpet haemocyanin (KLH) (Imject Maleimide-activated KLH kit, Pierce) and bovine serum albumin (BSA) before injecting them into rabbits. In-house generated antibodies were affinity purified on phospho-peptide columns and used in Western blots at the following concentrations: phosphoRIF1 (2 μg ml−1), phosphoCEP192 (2 μg ml−1), phosphoSFI1 (1 μg ml−1), phosphoCDCA2 (RM) (5 μg ml−1), phosphoBRCA1 (5 μg ml−1). 25 mM NaF and 5 μg ml−1 of the respective dephospho-peptides were added to all the phospho-epitope specific antibody solutions.
PP1 binding assays
For the peptide pull-downs, the RV[S/T]F–containing peptides (either unmodified or phosphorylated within the RV[S/T]F motif) derived from different proteins were coupled to activated CNBr-Sepharose beads (GE Healthcare) using the manufacturer’s protocol. Peptide-coupled beads were incubated with HeLa whole cell extracts for 3 h at 4°C. The beads were washed with Tris buffer (pH 7.5) plus 500 mM NaCl and 0.5% NP-40 and the bound proteins were eluted and analyzed for the presence of PP1 by immunoblotting. For PP1 overlays, recombinant PP1 was expressed in E. coli BL21 and affinity purified using microcystin-Sepharose beads as described previously (70). RV[S/T]F-containing peptides were coupled to BSA using glutaraldehyde (G5882, Sigma-Aldrich) as the coupling agent. These peptides were spotted onto nitrocellulose membrane in different amounts and overlaid with a mixture of recombinant human PP1(α/β/γ) (1 μg ml−1). Phosphatase inhibitors (25mM NaF and 0.5 μM microcystin-LR) were included at all steps. PP1 binding was analyzed by immunoblotting.
Kinase inhibitor assays
HeLa cells were treated with 2 mM thymidine for 17 h, released for 7 h followed by treatment with 100 ng ml−1 nocodazole or 100 nM taxol for 15 h for inducing prometaphase arrest. MLN8054 (a gift from S. Gerber, Dartmouth, used at 1 μM to inhibit Aurora A and at 5 μM to inhibit both Aurora A and Aurora B), Aurora A inhibitor I (100nM, Selleckchem), ZM447439 (5μM, Tocris), hesperadin (100nM, Selleckchem), BI2536 (100nM, Selleckchem), flavopiridol (2 μM, Selleckchem), or roscovitine (50 μM, Millipore) were added for 45 mins. The cells were treated with MG132 (10 μM, Millipore) for 30 mins prior to adding the kinase inhibitors.
Immunoprecipitation
Nocodazole-treated (100 ng ml−1) HeLa mitotic cells with or without 1 h hesperadin treatment (100 nM) were collected by mitotic shake-off and released for 30 mins in the presence or absence of hesperadin. The cells were lysed using nucleosome preparation buffer (10 mM HEPES pH 7.9, 10 mM KCl, 1 mM CaCl2, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 0.1% Triton X-100 with 100 U ml−1 micrococcal nuclease and 100 U ml−1 TurboNuclease). After 10 min incubation at 37°C, an equal volume of high-salt buffer (600 mM NaCl with 2% Triton X-100) was added followed by sonication and clearing of extract at 10,000 rpm for 10 mins. All the buffers were supplemented with protease inhibitors (EDTA-free protease inhibitor cocktail tablets, S8830, Sigma Aldrich) and phosphatase inhibitors (25 mM NaF, 0.5 μM MC-LR). The protein concentration was determined using Bradford reagent and BSA as standard. Prior to the IP, lysate was pre-cleared with CL-4B Sepharose beads (1/10th lysate volume).
For immunoprecipitations, the phosphoRV[S/T]F IgG and the control IgG were coupled to protein A-Sepharose beads using dimethyl pimelimidate before incubation with the cell extracts. The beads were washed 3 times with high-salt wash buffer (400 mM NaCl with 0.5% NP-40 in PBS) followed by 2 washes with low-salt wash buffer (150 mM NaCl with 0.2% NP-40 in PBS). The beads were washed two times with PBS before release with the elution buffer (1% SDS, 15% glycerol, 50 mM Tris-HCl, pH 8.7 and 150 mM NaCl).
Mass spectrometry analysis
Immunoprecipitations were TCA precipitated and digested in solution with trypsin in 50 mM ammonium bicarbonate. Reactions were quenched by the addition of 50% acetonitrile/5% formic acid and dried. Peptides were analyzed on a Q-Exactive Plus mass spectrometer (Thermo Scientific) equipped with an Easy-nLC 1000 (Thermo Scientific) as previously reported (8). Peptides were resuspended in 5% methanol / 1% formic acid and loaded on to a trap column (1 cm length, 100 μm inner diameter, ReproSil, C18 AQ 5 μm 120 Å pore (Dr. Maisch, Ammerbuch, Germany)) vented to waste via a micro-tee and eluted across a fritless analytical resolving column (35 cm length, 100 μm inner diameter, ReproSil, C18 AQ 3 μm 120 Å pore) pulled in-house (Sutter P-2000, Sutter Instruments, San Francisco, CA) with a 60 minute gradient of 5-30% LC-MS buffer B (LC-MS buffer A: 0.0625% formic acid, 3% ACN; LC-MS buffer B: 0.0625% formic acid, 95% ACN). The Q-Exactive Plus was set to perform an Orbitrap MS1 scan (R=70K; AGC target = 3e6) from 350 – 1500 Thomson, followed by HCD MS2 spectra on the 10 most abundant precursor ions detected by Orbitrap scanning (R=17.5K; AGC target = 1e5; max ion time = 75 ms) before repeating the cycle. Precursor ions were isolated for HCD by quadrupole isolation at width = 0.8 Thomson and HCD fragmentation at 26 normalized collision energy (NCE). Charge state 2, 3 and 4 ions were selected for MS2. Precursor ions were added to a dynamic exclusion list +/− 20 ppm for 20 seconds. The resulting data files were searched using Comet (release version 2014.01) in high resolution mode (71) against a target-decoy (reversed) (72) version of the human proteome sequence database (UniProt; downloaded 2/2013, , 40482 entries of forward and reverse protein sequences) with a precursor mass tolerance of +/− 1 Da and a fragment ion mass tolerance of 0.02 Da, and requiring fully tryptic peptides with up to three miscleavages. Carbamidomethylcysteine was enabled as a fixed modification and oxidized methionine as variable modification. The resulting peptide spectral matches were filtered to < 1% false discovery rate (FDR) based on reverse-hit counting (73). Peptide quantification was performed using MassChroQ (74). Protein quantification was performed by iBAQ (50).
Data analysis
Total peptide counts from -RVp[S/T]F and control immunoprecipitations from mitotically-arrested HeLa cells were input into SAINT (49) using the CRAPome interface (48). For a protein to be considered specific to the RVp[S/T]F immunoprecipitations, we required the interaction to have an AvgP score in the SAINT analysis of 0.9 or above in at least two of the five replicates. Changes in RVp[S/T]F binding upon Aurora B kinase inhibitor addition were determined by Student’s t test. Proteins not quantified in immunoprecipitations upon Aurora kinase B inhibitor addition were called “lost”; proteins quantified in four or five out of five replicates of control immunoprecipitations and only one out of five replicates of immunoprecipitations upon Aurora kinase inhibitor addition (p-value: nd) were called “lost”; proteins quantified in two or three out of five replicates of control immunoprecipitations and only one out of five replicates of immunoprecipitations upon Aurora kinase inhibitor addition (p-value: nd) with a decrease of 2-fold ore more were called “lost”; proteins quantified in immunoprecipitations in the presence or absence of Aurora kinase inhibitor with a p-value < 0.05 and a fold-change decrease of 2-fold or more were called “reduced”; protein quantified in immunoprecipitations in the presence or absence of Aurora B inhibitor with a p-value > 0.05 or p-value: nd were called “change not significant”. GO annotations were performed in Cytoscape using BiNGO to test for ontology enrichment. To determine significance of enrichment of terms a Bonferroni corrected p-value cutoff of 0.05 was used.
Immunofluorescence staining and microscopy
For immunofluorescence studies, HeLa cells were grown on poly-lysine coated coverslips, fixed with 3.7% formaldehyde, permeabilized with 0.5% Triton X-100 and blocked in 1% BSA/PBS. The coverslips were then incubated with primary antibodies for 2 h followed by Alexa Fluor 488-conjugated goat anti-rabbit and Alexa Fluor 594-conjugated goat anti-mouse secondary antibodies (Molecular Probes, Thermo Fisher Scientific) for 1 h. Nuclei were counterstained with DAPI (Sigma Aldrich, 1μg ml−1) and images acquired using a Leica DMIRE2 microscope as described above. RVp[S/T]F staining in control and Aurora B inhibitor treated cells was quantified using ImageJ as described in (75). Total corrected cellular fluorescence was calculated using (TCCF = integrated density – (area of selected cell × mean fluorescence of background readings), where a neighboring interphase cell was used as the background fluorescence intensity.
Supplementary Material
Table S1. List of peptide sequences used in this study.
Table S2. List of proteins specifically bound to p-RV[S/T]F antibody and their binding behavior upon Aurora B inhibition.
Table S3. Table containing SAINT analysis of the proteins enriched in the p-RV[S/T]F IPs.
Table S4. Table containing Gene Ontology (GO) analysis based on biological processes for proteins enriched in p-RV[S/T]F IPs.
Table S5. Table containing Gene Ontology (GO) analysis based on cellular component localization for proteins enriched in p-RV[S/T]F IPs.
Table S6. List of proteins containing “RV[S/T]F” motifs specifically enriched in the p-RV[S/T]F immunoprecipitation and their Aurora B sensitivity.
Fig. S1. Consensus RV[S/T]F motif in PP1-interacting proteins.
Fig. S2. Validation of phospho-specific antibodies by dot blots and immunofluorescence.
Fig. S3. Validation and characterization of the RVp[S/T]F antibody.
Fig. S4. Phosphorylation within RVS/TF motifs during mitosis.
Acknowledgements:
The authors would like to thank Drs. P. Douglas and S. Lees-Miller for help with microscopy and Dr. S. Gerber for MLN8054 inhibitor.
Funding: Funding for this work was provided by the Cancer Research Society of Canada (to G.M.), Alberta Cancer Foundation, Eyes High International Doctoral Scholarship and Faculty of Graduate Studies Doctoral Scholarship from University of Calgary (to I.N.), American Cancer Society Research Grant (IRG-82-003-30) and the National Institute of General Medical Sciences (R35GM119455) (to A.N.K.).
Footnotes
Competing interests: The authors declare no competing financial interests.
Data and Materials Availability: The MS data have been deposited to ProteomeXchange Consortium (76), http://www.proteomexchange.org, with accession number PXD009369 . All other data needed to evaluate the conclusions are present in the article or in the Supplementary Materials.
REFERENCES AND NOTES
- 1.Kastan MB, Bartek J, Cell-cycle checkpoints and cancer. Nature 432, 316–323 (2004). [DOI] [PubMed] [Google Scholar]
- 2.Malumbres M, Cyclin-dependent kinases. Genome Biol 15, 122 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dephoure N, Zhou C, Villen J, Beausoleil SA, Bakalarski CE, Elledge SJ, Gygi SP, A quantitative atlas of mitotic phosphorylation. Proc Natl Acad Sci U S A 105, 10762–10767 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kettenbach AN, Schweppe DK, Faherty BK, Pechenick D, Pletnev AA, Gerber SA, Quantitative phosphoproteomics identifies substrates and functional modules of Aurora and Polo-like kinase activities in mitotic cells. Sci Signal 4, rs5 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Olsen JV, Vermeulen M, Santamaria A, Kumar C, Miller ML, Jensen LJ, Gnad F, Cox J, Jensen TS, Nigg EA, Brunak S, Mann M, Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci Signal 3, ra3 (2010). [DOI] [PubMed] [Google Scholar]
- 6.McCloy RA, Parker BL, Rogers S, Chaudhuri R, Gayevskiy V, Hoffman NJ, Ali N, Watkins DN, Daly RJ, James DE, Lorca T, Castro A, Burgess A, Global Phosphoproteomic Mapping of Early Mitotic Exit in Human Cells Identifies Novel Substrate Dephosphorylation Motifs. Mol Cell Proteomics 14, 2194–2212 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barr FA, Elliott PR, Gruneberg U, Protein phosphatases and the regulation of mitosis. J Cell Sci 124, 2323–2334 (2011). [DOI] [PubMed] [Google Scholar]
- 8.Rusin SF, Schlosser KA, Adamo ME, Kettenbach AN, Quantitative phosphoproteomics reveals new roles for the protein phosphatase PP6 in mitotic cells. Sci Signal 8, rs12 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qian J, Winkler C, Bollen M, 4D-networking by mitotic phosphatases. Curr Opin Cell Biol 25, 697–703 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Wu JQ, Guo JY, Tang W, Yang CS, Freel CD, Chen C, Nairn AC, Kornbluth S, PP1-mediated dephosphorylation of phosphoproteins at mitotic exit is controlled by inhibitor-1 and PP1 phosphorylation. Nat Cell Biol 11, 644–651 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moorhead GB, Trinkle-Mulcahy L, Ulke-Lemee A, Emerging roles of nuclear protein phosphatases. Nat Rev Mol Cell Biol 8, 234–244 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Bollen M, Peti W, Ragusa MJ, Beullens M, The extended PP1 toolkit: designed to create specificity. Trends Biochem Sci 35, 450–458 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hendrickx A, Beullens M, Ceulemans H, Den Abt T, Van Eynde A, Nicolaescu E, Lesage B, Bollen M, Docking motif-guided mapping of the interactome of protein phosphatase-1. Chem Biol 16, 365–371 (2009). [DOI] [PubMed] [Google Scholar]
- 14.Heroes E, Lesage B, Gornemann J, Beullens M, Van Meervelt L, Bollen M, The PP1 binding code: a molecular-lego strategy that governs specificity. FEBS J 280, 584–595 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Hurley TD, Yang J, Zhang L, Goodwin KD, Zou Q, Cortese M, Dunker AK, DePaoli-Roach AA, Structural basis for regulation of protein phosphatase 1 by inhibitor-2. J Biol Chem 282, 28874–28883 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Moorhead GB, Trinkle-Mulcahy L, Nimick M, De Wever V, Campbell DG, Gourlay R, Lam YW, Lamond AI, Displacement affinity chromatography of protein phosphatase one (PP1) complexes. BMC Biochem 9, 28 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Egloff MP, Johnson DF, Moorhead G, Cohen PT, Cohen P, Barford D, Structural basis for the recognition of regulatory subunits by the catalytic subunit of protein phosphatase 1. EMBO J 16, 1876–1887 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wakula P, Beullens M, Ceulemans H, Stalmans W, Bollen M, Degeneracy and function of the ubiquitous RVXF motif that mediates binding to protein phosphatase-1. J Biol Chem 278, 18817–18823 (2003). [DOI] [PubMed] [Google Scholar]
- 19.Meiselbach H, Sticht H, Enz R, Structural analysis of the protein phosphatase 1 docking motif: molecular description of binding specificities identifies interacting proteins. Chem Biol 13, 49–59 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Ceulemans H, Bollen M, Functional diversity of protein phosphatase-1, a cellular economizer and reset button. Physiol Rev 84, 1–39 (2004). [DOI] [PubMed] [Google Scholar]
- 21.Terrak M, Kerff F, Langsetmo K, Tao T, Dominguez R, Structural basis of protein phosphatase 1 regulation. Nature 429, 780–784 (2004). [DOI] [PubMed] [Google Scholar]
- 22.Liu D, Vleugel M, Backer CB, Hori T, Fukagawa T, Cheeseman IM, Lampson MA, Regulated targeting of protein phosphatase 1 to the outer kinetochore by KNL1 opposes Aurora B kinase. The Journal of cell biology 188, 809–820 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dent P, Campbell DG, Caudwell FB, Cohen P, Identification of three in vivo phosphorylation sites on the glycogen-binding subunit of protein phosphatase 1 from rabbit skeletal muscle, and their response to adrenaline. FEBS Lett 259, 281–285 (1990). [DOI] [PubMed] [Google Scholar]
- 24.Kim Y, Holland AJ, Lan W, Cleveland DW, Aurora kinases and protein phosphatase 1 mediate chromosome congression through regulation of CENP-E. Cell 142, 444–455 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kumar GS, Gokhan E, De Munter S, Bollen M, Vagnarelli P, Peti W, Page R, The Ki-67 and RepoMan mitotic phosphatases assemble via an identical, yet novel mechanism. eLife 5, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trinkle-Mulcahy L, Andrews PD, Wickramasinghe S, Sleeman J, Prescott A, Lam YW, Lyon C, Swedlow JR, Lamond AI, Time-lapse imaging reveals dynamic relocalization of PP1gamma throughout the mammalian cell cycle. Mol Biol Cell 14, 107–117 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trinkle-Mulcahy L, Chusainow J, Lam YW, Swift S, Lamond A, Visualization of intracellular PP1 targeting through transiently and stably expressed fluorescent protein fusions. Methods Mol Biol 365, 133–154 (2007). [DOI] [PubMed] [Google Scholar]
- 28.Rosenberg JS, Cross FR, Funabiki H, KNL1/Spc105 recruits PP1 to silence the spindle assembly checkpoint. Curr Biol 21, 942–947 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Trinkle-Mulcahy L, Andersen J, Lam YW, Moorhead G, Mann M, Lamond AI, Repo-Man recruits PP1 gamma to chromatin and is essential for cell viability. The Journal of cell biology 172, 679–692 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rodrigues NT, Lekomtsev S, Jananji S, Kriston-Vizi J, Hickson GR, Baum B, Kinetochore-localized PP1-Sds22 couples chromosome segregation to polar relaxation. Nature 524, 489–492 (2015). [DOI] [PubMed] [Google Scholar]
- 31.De Wever V, Nasa I, Chamousset D, Lloyd D, Nimick M, Xu H, Trinkle-Mulcahy L, Moorhead GB, The human mitotic kinesin KIF18A binds protein phosphatase 1 (PP1) through a highly conserved docking motif. Biochem Biophys Res Commun 453, 432–437 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Steen RL, Beullens M, Landsverk HB, Bollen M, Collas P, AKAP149 is a novel PP1 specifier required to maintain nuclear envelope integrity in G1 phase. J Cell Sci 116, 2237–2246 (2003). [DOI] [PubMed] [Google Scholar]
- 33.Welburn JP, Vleugel M, Liu D, Yates JR 3rd, Lampson MA, Fukagawa T, Cheeseman IM, Aurora B phosphorylates spatially distinct targets to differentially regulate the kinetochore-microtubule interface. Mol Cell 38, 383–392 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Francisco L, Wang W, Chan CS, Type 1 protein phosphatase acts in opposition to IpL1 protein kinase in regulating yeast chromosome segregation. Mol Cell Biol 14, 4731–4740 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Emanuele MJ, Lan W, Jwa M, Miller SA, Chan CS, Stukenberg PT, Aurora B kinase and protein phosphatase 1 have opposing roles in modulating kinetochore assembly. The Journal of cell biology 181, 241–254 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meadows JC, Interplay between mitotic kinesins and the Aurora kinase-PP1 (protein phosphatase 1) axis. Biochem Soc Trans 41, 1761–1765 (2013). [DOI] [PubMed] [Google Scholar]
- 37.Lampson MA, Cheeseman IM, Sensing centromere tension: Aurora B and the regulation of kinetochore function. Trends Cell Biol 21, 133–140 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lesage B, Qian J, Bollen M, Spindle checkpoint silencing: PP1 tips the balance. Curr Biol 21, R898–903 (2011). [DOI] [PubMed] [Google Scholar]
- 39.Meadows JC, Shepperd LA, Vanoosthuyse V, Lancaster TC, Sochaj AM, Buttrick GJ, Hardwick KG, Millar JB, Spindle checkpoint silencing requires association of PP1 to both Spc7 and kinesin-8 motors. Developmental cell 20, 739–750 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hubbard MJ, Cohen P, On target with a new mechanism for the regulation of protein phosphorylation. Trends in biochemical sciences 18, 172–177 (1993). [DOI] [PubMed] [Google Scholar]
- 41.Ulke-Lemee A, Trinkle-Mulcahy L, Chaulk S, Bernstein NK, Morrice N, Glover M, Lamond AI, Moorhead GB, The nuclear PP1 interacting protein ZAP3 (ZAP) is a putative nucleoside kinase that complexes with SAM68, CIA, NF110/45, and HNRNP-G. Biochim Biophys Acta 1774, 1339–1350 (2007). [DOI] [PubMed] [Google Scholar]
- 42.Sreesankar E, Bharathi V, Mishra RK, Mishra K, Drosophila Rif1 is an essential gene and controls late developmental events by direct interaction with PP1–87B. Sci Rep 5, 10679 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Takagi M, Nishiyama Y, Taguchi A, Imamoto N, Ki67 antigen contributes to the timely accumulation of protein phosphatase 1gamma on anaphase chromosomes. J Biol Chem 289, 22877–22887 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Helps NR, Luo X, Barker HM, Cohen PT, NIMA-related kinase 2 (Nek2), a cell-cycle-regulated protein kinase localized to centrosomes, is complexed to protein phosphatase 1. Biochem J 349, 509–518 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Manfredi MG, Ecsedy JA, Meetze KA, Balani SK, Burenkova O, Chen W, Galvin KM, Hoar KM, Huck JJ, LeRoy PJ, Ray ET, Sells TB, Stringer B, Stroud SG, Vos TJ, Weatherhead GS, Wysong DR, Zhang M, Bolen JB, Claiborne CF, Antitumor activity of MLN8054, an orally active small-molecule inhibitor of Aurora A kinase. Proceedings of the National Academy of Sciences 104, 4106–4111 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miller ML, Jensen LJ, Diella F, Jørgensen C, Tinti M, Li L, Hsiung M, Parker SA, Bordeaux J, Sicheritz-Ponten T, Olhovsky M, Pasculescu A, Alexander J, Knapp S, Blom N, Bork P, Li S, Cesareni G, Pawson T, Turk BE, Yaffe MB, Brunak S, Linding R, Linear Motif Atlas for Phosphorylation-Dependent Signaling. Science Signaling 1, ra2–ra2 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Giet R, Glover DM, Drosophila aurora B kinase is required for histone H3 phosphorylation and condensin recruitment during chromosome condensation and to organize the central spindle during cytokinesis. The Journal of cell biology 152, 669–682 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mellacheruvu D, Wright Z, Couzens AL, Lambert JP, St-Denis NA, Li T, Miteva YV, Hauri S, Sardiu ME, Low TY, Halim VA, Bagshaw RD, Hubner NC, Al-Hakim A, Bouchard A, Faubert D, Fermin D, Dunham WH, Goudreault M, Lin ZY, Badillo BG, Pawson T, Durocher D, Coulombe B, Aebersold R, Superti-Furga G, Colinge J, Heck AJ, Choi H, Gstaiger M, Mohammed S, Cristea IM, Bennett KL, Washburn MP, Raught B, Ewing RM, Gingras AC, Nesvizhskii AI, The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat Methods 10, 730–736 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Choi H, Larsen B, Lin ZY, Breitkreutz A, Mellacheruvu D, Fermin D, Qin ZS, Tyers M, Gingras AC, Nesvizhskii AI, SAINT: probabilistic scoring of affinity purification-mass spectrometry data. Nat Methods 8, 70–73 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M, Global quantification of mammalian gene expression control. Nature 473, 337–342 (2011). [DOI] [PubMed] [Google Scholar]
- 51.Hiraga S, Alvino GM, Chang F, Lian HY, Sridhar A, Kubota T, Brewer BJ, Weinreich M, Raghuraman MK, Donaldson AD, Rif1 controls DNA replication by directing Protein Phosphatase 1 to reverse Cdc7-mediated phosphorylation of the MCM complex. Genes & development 28, 372–383 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vagnarelli P, Ribeiro S, Sennels L, Sanchez-Pulido L, de Lima Alves F, Verheyen T, Kelly DA, Ponting CP, Rappsilber J, Earnshaw WC, Repo-Man coordinates chromosomal reorganization with nuclear envelope reassembly during mitotic exit. Developmental cell 21, 328–342 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jiang H, He X, Feng D, Zhu X, Zheng Y, RanGTP aids anaphase entry through Ubr5-mediated protein turnover. The Journal of cell biology 211, 7–18 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gai M, Bianchi FT, Vagnoni C, Verni F, Bonaccorsi S, Pasquero S, Berto GE, Sgro F, Chiotto AM, Annaratone L, Sapino A, Bergo A, Landsberger N, Bond J, Huttner WB, Di Cunto F, ASPM and CITK regulate spindle orientation by affecting the dynamics of astral microtubules. EMBO reports 17, 1396–1409 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Platani M, Santarella-Mellwig R, Posch M, Walczak R, Swedlow JR, Mattaj IW, The Nup107–160 nucleoporin complex promotes mitotic events via control of the localization state of the chromosome passenger complex. Mol Biol Cell 20, 5260–5275 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hattersley N, Desai A, The nucleoporin MEL-28/ELYS: A PP1 scaffold during M-phase exit. Cell Cycle 16, 489–490 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Choy MS, Yusoff P, Lee IC, Newton JC, Goh CW, Page R, Shenolikar S, Peti W, Structural and Functional Analysis of the GADD34:PP1 eIF2alpha Phosphatase. Cell Rep 11, 1885–1891 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Santamaria A, Wang B, Elowe S, Malik R, Zhang F, Bauer M, Schmidt A, Silljé HHW, Körner R, Nigg EA, The Plk1-dependent Phosphoproteome of the Early Mitotic Spindle. Molecular & Cellular Proteomics : MCP 10, M110.004457 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nasa I, Trinkle-Mulcahy L, Douglas P, Chaudhuri S, Lees-Miller SP, Lee KS, Moorhead GB, Recruitment of PP1 to the centrosomal scaffold protein CEP192. Biochem Biophys Res Commun 484, 864–870 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Scialpi F, Mellis D, Ditzel M, EDD, a ubiquitin-protein ligase of the N-end rule pathway, associates with spindle assembly checkpoint components and regulates the mitotic response to nocodazole. J Biol Chem 290, 12585–12594 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Higgins J, Midgley C, Bergh AM, Bell SM, Askham JM, Roberts E, Binns RK, Sharif SM, Bennett C, Glover DM, Woods CG, Morrison EE, Bond J, Human ASPM participates in spindle organisation, spindle orientation and cytokinesis. BMC Cell Biol 11, 85 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhong X, Liu L, Zhao A, Pfeifer GP, Xu X, The Abnormal Spindle-like, Microcephaly-associated (ASPM) Gene Encodes a Centrosomal Protein. Cell Cycle 4, 1227–1229 (2005). [DOI] [PubMed] [Google Scholar]
- 63.Nousiainen M, Sillje HH, Sauer G, Nigg EA, Korner R, Phosphoproteome analysis of the human mitotic spindle. Proc Natl Acad Sci U S A 103, 5391–5396 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zuccolo M, Alves A, Galy V, Bolhy S, Formstecher E, Racine V, Sibarita JB, Fukagawa T, Shiekhattar R, Yen T, Doye V, The human Nup107–160 nuclear pore subcomplex contributes to proper kinetochore functions. EMBO J 26, 1853–1864 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rasala BA, Orjalo AV, Shen Z, Briggs S, Forbes DJ, ELYS is a dual nucleoporin/kinetochore protein required for nuclear pore assembly and proper cell division. Proc Natl Acad Sci U S A 103, 17801–17806 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hattersley N, Cheerambathur D, Moyle M, Stefanutti M, Richardson A, Lee KY, Dumont J, Oegema K, Desai A, A Nucleoporin Docks Protein Phosphatase 1 to Direct Meiotic Chromosome Segregation and Nuclear Assembly. Developmental cell 38, 463–477 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mattarocci S, Shyian M, Lemmens L, Damay P, Altintas DM, Shi T, Bartholomew CR, Thoma NH, Hardy CF, Shore D, Rif1 controls DNA replication timing in yeast through the PP1 phosphatase Glc7. Cell Rep 7, 62–69 (2014). [DOI] [PubMed] [Google Scholar]
- 68.Dave A, Cooley C, Garg M, Bianchi A, Protein phosphatase 1 recruitment by Rif1 regulates DNA replication origin firing by counteracting DDK activity. Cell Rep 7, 53–61 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Crooks GE, Hon G, Chandonia JM, Brenner SE, WebLogo: a sequence logo generator. Genome Res 14, 1188–1190 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Moorhead G, MacKintosh RW, Morrice N, Gallagher T, MacKintosh C, Purification of type 1 protein (serine/threonine) phosphatases by microcystin-Sepharose affinity chromatography. FEBS Lett 356, 46–50 (1994). [DOI] [PubMed] [Google Scholar]
- 71.Eng JK, Jahan TA, Hoopmann MR, Comet: an open-source MS/MS sequence database search tool. Proteomics 13, 22–24 (2013). [DOI] [PubMed] [Google Scholar]
- 72.Elias JE, Gygi SP, Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods 4, 207–214 (2007). [DOI] [PubMed] [Google Scholar]
- 73.Elias JE, Gygi SP, Target-decoy search strategy for mass spectrometry-based proteomics. Methods Mol Biol 604, 55–71 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Valot B, Langella O, Nano E, Zivy M, MassChroQ: a versatile tool for mass spectrometry quantification. Proteomics 11, 3572–3577 (2011). [DOI] [PubMed] [Google Scholar]
- 75.McCloy RA, Rogers S, Caldon CE, Lorca T, Castro A, Burgess A, Partial inhibition of Cdk1 in G2 phase overrides the SAC and decouples mitotic events. Cell Cycle 13, 1400–1412 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vizcaino JA, Deutsch EW, Wang R, Csordas A, Reisinger F, Rios D, Dianes JA, Sun Z, Farrah T, Bandeira N, Binz PA, Xenarios I, Eisenacher M, Mayer G, Gatto L, Campos A, Chalkley RJ, Kraus HJ, Albar JP, Martinez-Bartolome S, Apweiler R, Omenn GS, Martens L, Jones AR, Hermjakob H, ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nature biotechnology 32, 223–226 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of peptide sequences used in this study.
Table S2. List of proteins specifically bound to p-RV[S/T]F antibody and their binding behavior upon Aurora B inhibition.
Table S3. Table containing SAINT analysis of the proteins enriched in the p-RV[S/T]F IPs.
Table S4. Table containing Gene Ontology (GO) analysis based on biological processes for proteins enriched in p-RV[S/T]F IPs.
Table S5. Table containing Gene Ontology (GO) analysis based on cellular component localization for proteins enriched in p-RV[S/T]F IPs.
Table S6. List of proteins containing “RV[S/T]F” motifs specifically enriched in the p-RV[S/T]F immunoprecipitation and their Aurora B sensitivity.
Fig. S1. Consensus RV[S/T]F motif in PP1-interacting proteins.
Fig. S2. Validation of phospho-specific antibodies by dot blots and immunofluorescence.
Fig. S3. Validation and characterization of the RVp[S/T]F antibody.
Fig. S4. Phosphorylation within RVS/TF motifs during mitosis.