

Abstract
Several mechanisms of resistance have been identified, underscoring the complex nature of estrogen receptor (ER) signaling and the many connections between this pathway and other essential signaling pathways in breast cancer cells. Many therapeutic targets of cell signaling and cell cycle pathways have met success with endocrine therapy and remain an ongoing area of investigation. This review focuses on two major pathways that have recently emerged as important opportunities for therapeutic intervention in endocrine resistant breast tumors: PI3K/AKT/mTOR cell signaling and cyclinD1/cyclin-dependent kinase 4/6 cell cycle pathways. Additionally, we highlight individual and combination strategies in current clinical trials that target these pathways and others under investigation for the treatment of ER positive breast cancer.
Introduction
Endocrine therapy has dramatically improved survival in breast cancer patients over the past several decades, however resistance to these therapies remains one of the major causes of breast cancer mortality today [1]. Late recurrence and death from estrogen receptor positive (ER+) breast cancer can occur for at least 20 years after the original diagnosis even after 5 years of adjuvant endocrine therapy [2●●]. Identifying mechanisms of resistance and strategies by which to combat these mechanisms is paramount to patient survival.
Several mechanisms of resistance to endocrine therapies have been identified, many centered around the structure, activation, and complex functions of ER, as well as cross-talk between the estrogen signaling network and other cellular pathways. The major form of ER in breast cancer is ERα, encoded by ESR1, and the major function of ER is as a transcription factor controlling genes associated with cell survival and proliferation [3]. ER function is influenced by circulating estrogens and related molecules, giving ER-targeted therapies their success. Post-translational modifications also influence function, localization, and interaction with other regulators. In addition to ERα, there also exists transcription factor ERβ, encoded by ESR2, as well as alternatively spliced and truncated variants of ESR1/ERα [4]. The biology of ER is complex and how breast tumors can gain ER function then maintain this despite ER inhibition is not well understood. Thus, the mechanism of action of various endocrine therapies is complicated, varies, and remains an active area of investigation.
Known mechanisms of resistance to hormone therapies are complex and include epigenetic regulation of ESR1 expression [5,6], ESR1 mutations [7–12], alternative splicing events [3], ESR1 truncation and fusion events [13], post-translational modifications [14,15], alterations in the hormone binding domain [7,16], alter-native recruitment sites within the genome [17], differential recruitment of coregulators [18], feedback loops by ER target genes on expression/activity of ER [19●], downstream actions of ER target genes on growth factor pathways and other signaling networks [20,21●●], influences of the tumor microenvironment [22], and many others (Figure 1). The details of these mechanisms are beyond the scope of this review but have been thoroughly described by others [23,24]. The complexity of ER function in tumor cells underscores the heterogeneity of breast cancer biology and demonstrates a necessity for continued basic research and clinical demonstration to effectively target the pathways essential to tumor cell survival.
Figure 1.
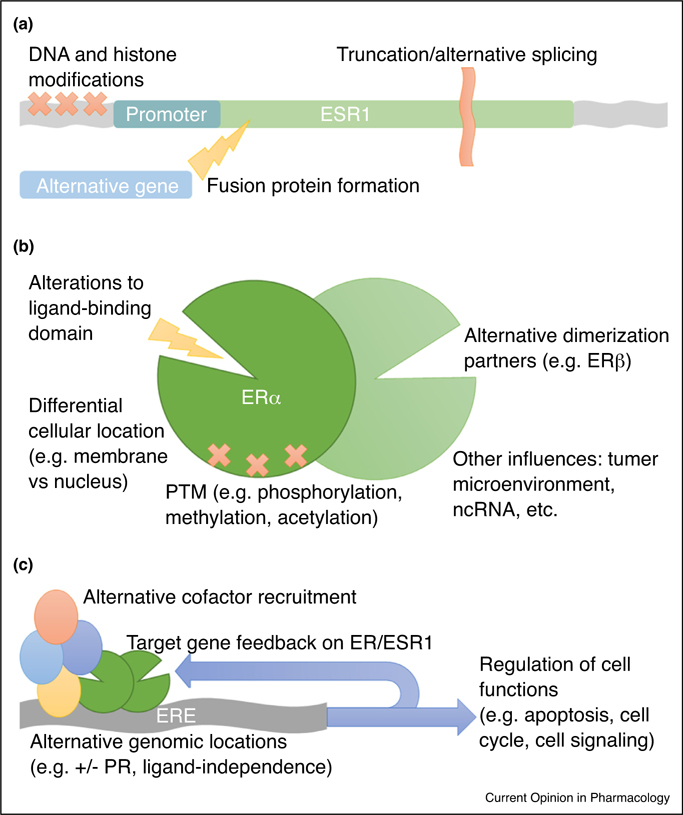
Diagrammatic representation of known mechanisms of resistance to endocrine therapy. Mechanisms of resistance to endocrine therapy are known to occur at several levels. (a) At the level of ESR1, modifications include epigenetic modification of histone proteins and DNA to alter ESR1 transcription, truncation, alternative splicing events, and fusion events with alternative DNA sequences to produce variations of the ER protein. (b) The events outlined above can result in alterations to the ligand-binding domain and differential cellular localization of the receptor, altering function. Additionally, homo-dimerization or heterodimerization can occur as well as various post-translational modifications (PTM), dependent upon alterations to the proteome of tumor cells and possibly other factors, such as tumor microenvironment and non-coding RNA molecules, resulting in modifications to ER function and target gene selection. (c) Activated ER binds estrogen response elements (ERE) to dictate target genes for transcriptional activation, however alternate recruitment sites are well document and largely dependent upon recruitment of specific cofactors and tethering proteins, such as progesterone receptor (PR). Differential recruitment of cofactors can also lead to repression of transcription rather than activation, modifying the downstream outcome of ER activation. Target genes can create feedback loops to modify ESR1/ER expression and behavior, including cross-talk with other signaling pathways in the cell. Finally, all of the events outlined above converge on regulation of essential cellular functions, therefore identifying the many opportunities for development of endocrine therapy resistance has profound effects on clinical control of tumorigenesis.
Much literature exists detailing the mechanisms of resistance. This review highlights two major pathways that have recently emerged as important opportunities for therapeutic intervention in endocrine resistant breast tumors: the phosphatidylinositol 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) cell signaling pathway and the cyclin D1/cyclin-dependent kinase (CDK)4/6 cell cycle pathway. Inhibitors to these pathways have been developed, assessed in preclinical studies, and investigated in multiple clinical trials, each with marked benefit toward improving survival but also with specific challenges and limitations discussed below.
PI3K/AKT/mTOR pathway
The PI3K/AKT/mTOR pathway is essential for cell growth and survival, protein synthesis, and glucose metabolism. It is dysregulated in many tumor types, prompting investigation of inhibition of this pathway in many cancer models, including the triple negative subtype of breast cancer [25]. Cross-talk between this pathway and estrogen-mediated signaling is known [26,27], prompting investigation of inhibitors targeting this pathway in ER+ breast tumors. Genomic alterations in PIK3CA, the gene encoding the catalytic subunit of PI3K, are common in ER+ tumors [28●,29,30]. It is thought that suppression of the PI3K/AKT/mTOR pathway can lead to increased activity of ER, facilitating resistance, and therefore many groups have used preclinical models to elucidate the mechanisms of PI3K/AKT/mTOR pathway inhibitors and the influence these compounds have on estrogen-mediated signaling [19●,21●●,25,31–33].
Evidence suggests that, while inhibition of the PI3K/AKT/mTOR pathway at all levels results in reduced cell proliferation and survival, the complexity of this signaling pathway ensures that compensatory mechanisms are activated that confer resistance to single inhibitors [32,33]. For example, inhibition of mTOR results in activation of AKT as well as extracellular signal-regulated kinase (ERK), leading to increased signaling through alternative branches of these pathways [19●,21●●]. Similarly, inhibition of PI3K activates upstream tyrosine kinases, allowing cells to escape the inhibitory effects on cell proliferation [21●●]. AKT, after activation by PI3K, can phosphorylate ER at S167, resulting in ligand-independent activation of ER-mediated transcription [34,35]. Ribas and colleagues demonstrated that inhibition of AKT with AZD5363 re-sensitizes cells to tamoxifen, acts synergistically with fulvestrant, and prevents emergence of hormone-independent cells in vivo. However, in their model, AKT inhibition activated positive feedback loops driven by MYC, resulting in increased gene expression of human epidermal receptor growth factor 2 and 3 (ERBB2-–ERBB3), ERK5, and insulin-like growth factor 1 (IGF1) [19●]. These observations have led to investigation of the use of multiple inhibitors simultaneously and development of novel inhibitors based on alternative iterations of PI3K ligand binding pockets generated by mutations in PIK3CA. Cross-talk between the PI3K/AKT/mTOR and ER pathways has immense clinical relevance, but further investigation is required to fully understand the interaction between these pathways. We are not aware of active trials combing a mTOR inhibitor and AKT inhibitor but await the results of combined mTOR inhibitors with PI3K inhibitors.
Several inhibitors of the PI3K/AKT/mTOR pathway have been tested in clinical trials in ER+ tumors in conjunction with various endocrine therapies. In 2012, the mTOR inhibitor everolimus was approved by the Food and Drug Administration (FDA) for the treatment of postmenopausal women with advanced ER+/HER2 breast cancer in combination with exemestane. The approval was based on a randomized double-blind multicenter trial (BOLERO-2), where the combination significantly improved disease free survival (DFS) compared to exemestane alone (7.8 months versus 3.2 months) [36]. Recently, the results from a phase 3 trial involving buparlisib, a pan-PI3K inhibitor targeting all four isoforms of class I PI3K (α, β, δ, and γ), were presented and published [37]. Although the efficacy data of buparlisib supports the use of PI3K inhibitors plus endocrine therapy in patients with PIK3CA mutations, the safety profile of buparlisib plus fulvestrant did not support its further development in that setting. Pan-PI3K inhibitors such as buparlisib might be limited by adverse events arising from a broad spectrum of off-target effects [38]. PI3Kα-selective inhibitors are currently under clinical evaluation in combination with endocrine therapy in patients with ER+, HER2-negative breast cancer. Table 1 summarizes the ongoing clinical trials targeting PI3K/AKT/mTOR.
Table 1.
Summary of ongoing clinical trials involving PI3K/AKT/mTOR in ER+ breast cancer
| Target | Compound | NCT number | Population |
|---|---|---|---|
| PI3K | AZD5363, Paclitaxel | NCT01625286 | ER+ MBC |
| BKM120 or BYL719 and Capecitabine | NCT01300962 | ||
| BYL719, Letrozole or Exemestane | NCT01870505 | ||
| BYL719, Letrozole | NCT01791478 | ||
| GDC-0032, Letrozole or Fulvestrant | NCT01296555 | ||
| Taselisib, Fulvestrant | NCT02340221 | ||
| Alpelisib, Fulvestrant | NCT02437318 | ||
| Alpelisib, hormone therapy | NCT03056755 | PIK3CA-mutant | |
| BYL719, Fulvestrant | NCT01219699 | ||
| AZD8186, Docetaxel | NCT03218826 | PTEN or PIK3CB mutant | |
| Taselisib, Enzalutamide | NCT02457910 | Androgen Receptor+ MBC | |
| mTOR | Everolimus to adjuvant hormone therapy | NCT01805271 | Adjuvant ER+ |
| Everolimus, standard hormone therapy | NCT01805271 | ||
| TAK-228 then Letrozole | NCT02619669 | ||
| TAK-228, Tamoxifen | NCT02988986 | ER+ MBC | |
| Everolimus, Letrozole | NCT01698918 | ||
| AZD2014, Fulvestrant | NCT02216786 | ||
| Everolimus, Exemestane | NCT01783444 | ||
| Everolimus, Tamoxifen | NCT01298713 | ||
| mTOR, IGF1R | Ridaforolimus, Dalotuzumab, Exemestane | NCT01605396 | ER+ MBC |
| AKT | AZD5363 | NCT03310541 | AKT-mutant |
| AZD5363, Fulvestrant | NCT01992952 | ER+ MBC | |
| AZD5363 | NCT01226316 | MBC | |
| MSC2363318A, with Trastuzumab or Tamoxifen | NCT01971515 | ||
| AKT, PD-L1, MEK, VEGF | Ipatasertib, Atezolizumab, Cobimetinib, Bevacizumab | NCT03395899 | Neoadjuvant ER+ |
| PI3K, mTOR, CDK | Ribociclib, Everolimus, Exemestane | NCT01857193 | ER+ MBC |
| Gedatolisib, Fulvestrant, Palbociclib, Goserelin | NCT02626507 | ||
| PI3K, CDK | Ribociclib, Fulvestrant and BYL719 or BKM120 | NCT02088684 | |
| Ribociclib, Letrozole, BYL719 | NCT01872260 | ||
| Ribociclib, Tamoxifen, Letrozole, Anastrozole, Goserelin | NCT02278120 | ||
| mTOR, CDK | Ribociclib, Everolimus, Exemestane | NCT02732119 | |
| Hormone therapy, hormone therapy with Everolimus, or hormone therapy with CDK4/6 inhibition | NCT02753686 |
MBC, metastatic breast cancer; IGF1R, Insulin Growth Factor 1 Receptor; PD-L1, programmed death-ligand 1; MEK, Mitogen-activated protein kinase kinase. Table updated from clinicaltrials.gov on March 11, 2018.
CyclinD1/CDK4/6 pathway
Cell cycle progression is regulated by many proteins, including the cyclins and cyclin-dependent kinases (CDK). Cyclin D1 binds to CDK4/6 to regulate progression through the G1 phase of the cell cycle and is a known downstream target of the PI3K/AKT/mTOR pathway [39]. CDK4/6 has also been shown to crosstalk with the ER signaling pathway [40]. Vora and colleagues showed that insensitivity to PI3K inhibitors is evident by persistent RB phosphorylation and can be effectively overcome by combining a CDK inhibitor with a PI3K inhibitor [40]. Since tumorigenesis relies heavily on unchecked progression through the cell cycle, the cyclinD1/CDK4/6 pathway has emerged as a desirable target in the treatment of many cancers, including breast [41–43]. Originally, inhibitors such as palbociclib were hypothesized to be most efficacious in the triple negative subtype of breast cancer, but early studies identified ER expression as highly correlated with response to palbociclib [44]. Over the past 3 years, multiple large-scale clinical trials have demonstrated that inhibiting CDK4/6 leads to significant clinical benefit when combined with standard endocrine therapies in metastatic ER+, HER2-negative breast cancer [45–52]. Currently CDK4/6 inhibitors, including palbociclib, ribociclib and abemaciclib, are being increasingly employed in clinical practice combined with endocrine therapy. Also, abemaciclib was approved as a monotherapy in ER+, HER2-negative metastatic breast cancer whose disease progressed during or after endocrine therapy [50]. However, CDK4/6 inhibitors typically induce tumor stabilization with only modest increased rates of tumor shrinkage and their cytostatic effects are limited by primary and acquired resistance. Primary and acquired resistance to CDK4/6 inhibitors mediated by loss of the RB1 gene has been demonstrated in preclinical models [53,54●●,55]. In a recent study, the emergence of acquired resistance to palbociclib or ribociclib with the concurrent development of multiple de novo somatic RB1 mutations was shown in metastatic ER+ patients [56]. Table 2 summarizes the ongoing clinical trials with CDK4/6 inhibitors.
Table 2.
Ongoing clinical trials involving CDK inhibitors in ER+ breast cancer
| Target | Compound | NCT number | Population |
|---|---|---|---|
| CDK | Ribociclib, Hormone therapy | NCT03285412 | Adjuvant ER+ |
| Ribociclib, Hormone therapy | NCT03078751 | ||
| Ribociclib, Hormone therapy | NCT03081234 | ||
| Palbociclib, Anastrozole, Goserelin | NCT01723774 | ||
| Abemaciclib | NCT02831530 | ||
| Chemotherapy or Ribociclib and Letrozole | NCT03283384 | Neoadjuvant ER+ | |
| Fulvestrant with or without Palbociclib | NCT03447132 | ||
| Paclitaxel, Tamoxifen + Palbociclib, Aromatase Inhibitor | NCT02603679 | ||
| + Palbociclib, Goserelin + Aromatase Inhibitor + Palbociclib | |||
| Palbociclib, Hormone therapy | NCT01864746 | ER+ post-neoadjuvant | |
| Ribociclib, Letrozole | NCT03439046 | ER+ MBC | |
| Ribociclib, Letrozole | NCT01958021 | ||
| Ribociclib, Letrozole, Goserelin | NCT02941926 | ||
| Ribociclib, Fulvestrant | NCT02422615 | ||
| Endocrine therapy plus CDK 4/6 | NCT03425838 | ||
| Ribociclib, Letrozole, Goserelin | NCT03096847 | ||
| Ribociclib or Palbociclib, Fulvestrant | NCT02632045 | ||
| Palbociclib, Letrozole | NCT02499146 | ||
| Palbociclib, hormone therapy | NCT02040857 | ||
| G1T38 with Fulvestrant | NCT02983071 | ||
| Letrozole alone or with Palbociclib | NCT00721409 | ||
| Palbociclib, hormone therapy | NCT03184090 | ||
| Palbociclib, Letrozole | NCT01740427 | ||
| Palbociclib, Exemestane, Goserelin | NCT02917005 | ||
| Palbociclib, Letrozole | NCT02297438 | ||
| Palbociclib | NCT03159195 | ||
| Palbociclib, Tamoxifen | NCT02668666 | ||
| Aromatase inhibitor, Palbociclib | NCT03439735 | ||
| Palbociclib and aromatase inhibitor or Palbociclib and Fulvestrant | NCT03220178 | ||
| Exemestane plus Goserelin with Palbociclib versus Capecitabine | NCT02592746 | ||
| Abemaciclib, Fulvestrant | NCT02107703 | ||
| Abemaciclib | NCT02763566 | ||
| Palbociclib, Letrozole | NCT01684215 | ||
| CDK, HER2 | Palbociclib, Letrozole, Trastuzumab, Goserelin | NCT02907918 | Neoadjuvant ER+/HER2+ |
| Ribociclib with Trastuzumab or Trastuzumab emtansine | NCT02657343 | ER+/HER2+ MBC | |
| Tucatinib in combination with Palbociclib and Letrozole | NCT03054363 | ||
| CDK, PD-1 | Ribociclib, PDR001, Fulvestrant | NCT03294694 | ER+ MBC |
| CDK, IGF | Xentuzumab, Abemaciclib, Fulvestrant | NCT03099174 | ER+ MBC refractory to CDK4/6 |
| CDK, FGFR | Erdafitinib, Palbociclib, Fulvestrant | NCT03238196 | FGFR-amplified ER+ |
MBC, metastatic breast cancer; PD-1, programmed cell death protein 1; IGF, Insulin Growth Factor; FGFR, Fibroblast Growth Factor Receptor. Table updated from clinicaltrials.gov on March 11, 2018.
Conclusions
Resistance to endocrine therapy remains the most significant challenge in treatment of ER+ breast tumors. Despite this resistance, ER signaling remains biologically significant in these cells, and full understanding of the complex signaling pathways responsible for tumor progression remain to be elucidated. There is a lack of evidence informing optimal sequencing of available therapies in the treatment of advanced ER+ breast cancer [57●]. Currently, the working hypothesis within the field is that multiple signaling pathways must converge on essential biological functions such as cell cycle progression, cell survival, and estrogen-independent ER-mediated transcription, such that effective blockade of tumor progression will only be achieved by combination therapies that affect all compensatory mechanisms and alternative pathways. Breast cancer is a highly heterogeneous disease with novel essential factors constantly being discovered at the preclinical level. This is seen here by combination studies that include hormone therapy with addition of inhibitors targeting RAS/rapidly accelerated fibrosarcoma (RAF)/mitogen-activated protein kinase (MAPKinase), MAPK kinase (MEK), Vascular Endothelial Growth Factor (VEGF), other growth factor receptor inhibitors (e.g. IGF, Insulin Growth Factor; FGFR, Fibroblast Growth Factor Receptor) [23], and other immunotherapy inhibitors [58]. Additionally, more potent and bioavailable selective estrogen receptor degraders (SERD) are being tested to overcome resistance induced by ESR1 mutations [10]. Advances in genomic and other technologies that allow deeper understanding of individual tumors and further investigation into the cross-talk between these signaling pathways have provided a plethora of data that require continued mining, both in the preclinical and clinical settings, to personalize therapeutic regimens for each patient.
Acknowledgements
A. Giordano is supported in part by research funding,Hollings Cancer Center Paul Calabresi Clinical Oncology Training Program Plan 5K12CA157688 at the Medical University of South Carolina.
Footnotes
Conflict of interest statement
Nothing declared.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
● of special interest
●● of outstanding interest
- 1.Early Breast Cancer Trialists’ Collaborative G: Aromatase inhibitors versus tamoxifen in early breast cancer: patient-level meta-analysis of the randomised trials. Lancet 2015, 386:1341–1352. [DOI] [PubMed] [Google Scholar]
- 2. ●●.Pan H, Gray R, Braybrooke J, Davies C, Taylor C, McGale P, Peto R, Pritchard KI, Bergh J, Dowsett M, Hayes DF et al. : 20-year risks of breast-cancer recurrence after stopping endocrine therapy at 5 years. N Engl J Med 2017, 377:1836–1846.A meta-analysis combining individual patient data from 88 trials in the Early Breast Cancer Trialists’ Collaborative Group (EBCTCG) database of randomized trials.
- 3.Wang ZY, Yin L : Estrogen receptor alpha-36 (ER-alpha36): a new player in human breast cancer. Mol Cell Endocrinol 2015, 418(Pt 3):193–206. [DOI] [PubMed] [Google Scholar]
- 4.Ma R, Karthik GM, Lovrot J, Haglund F, Rosin G, Katchy A, Zhang X, Viberg L, Frisell J, Williams C, Linder S et al. : Estrogen receptor beta as a therapeutic target in breast cancer stem cells. J Natl Cancer Inst 2017, 109:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Marchi T, Liu NQ, Stingl C, Timmermans MA, Smid M, Look MP, Tjoa M, Braakman RB, Opdam M, Linn SC, Sweep FC et al. : 4-protein signature predicting tamoxifen treatment outcome in recurrent breast cancer. Mol Oncol 2016, 10:24–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang J, Zhou C, Jiang H, Liang L, Shi W, Zhang Q, Sun P, Xiang R, Wang Y, Yang S: Zeb1 induces ER-alpha promoter hypermethylation and confers antiestrogen resistance in breast cancer. Cell Death Dis 2017, 8:e2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Toy W, Shen Y, Won H, Green B, Sakr RA, Will M, Li Z, Gala K, Fanning S, King TA, Hudis C et al. : ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet 2013, 45:1439–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jeselsohn R, Buchwalter G, De Angelis C, Brown M, Schiff R: ESR1 mutations — a mechanism for acquired endocrine resistance in breast cancer. Nat Rev Clin Oncol 2015, 12:573–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fribbens C, O’Leary B, Kilburn L, Hrebien S, Garcia-Murillas I, Beaney M, Cristofanilli M, Andre F, Loi S, Loibl S, Jiang J et al. : Plasma ESR1 mutations and the treatment of estrogen receptor-positive advanced breast cancer. J Clin Oncol 2016, 34:2961–2968. [DOI] [PubMed] [Google Scholar]
- 10.Bihani T, Patel HK, Arlt H, Tao N, Jiang H, Brown JL, Purandare DM, Hattersley G, Garner F: Elacestrant (RAD1901), a selective estrogen receptor degrader (SERD), has antitumor activity in multiple ER+ breast cancer patient-derived xenograft models. Clin Cancer Res 2017, 23:4793–4804. [DOI] [PubMed] [Google Scholar]
- 11.Toy W, Weir H, Razavi P, Lawson M, Goeppert AU, Mazzola AM, Smith A, Wilson J, Morrow C, Wong WL, De Stanchina E et al. : Activating ESR1 mutations differentially affect the efficacy of ER antagonists. Cancer Discov 2017, 7:277–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takeshita T, Yamamoto Y, Yamamoto-Ibusuki M, Tomiguchi M, Sueta A, Murakami K, Omoto Y, Iwase H: Analysis of ESR1 and PIK3CA mutations in plasma cell-free DNA from ER-positive breast cancer patients. Oncotarget 2017, 8:52142–52155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hartmaier RJ, Trabucco SE, Priedigkeit N, Chung JH, Parachoniak CA, Vanden Borre P, Morley S, Rosenzweig M, Gay LM, Goldberg ME, Suh J et al. : Recurrent hyperactive ESR1 fusion proteins in endocrine therapy resistant breast cancer. Ann Oncol 2018. [DOI] [PMC free article] [PubMed]
- 14.Zhang X, Tanaka K, Yan J, Li J, Peng D, Jiang Y, Yang Z, Barton MC, Wen H, Shi X: Regulation of estrogen receptor alpha by histone methyltransferase SMYD2-mediated protein methylation. Proc Natl Acad Sci U S A 2013, 110:17284–17289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Piggott L, da Silva AM, Robinson T, Santiago-Gomez A, Simoes BM, Becker M, Fichtner I, Andera L, Piva M, Vivanco MD, Morris C et al. : Acquired resistance of ER-positive breast cancer to endocrine treatment confers an adaptive sensitivity to trail through post-translational downregulation of c-FLIP. Clin Cancer Res 2018. [DOI] [PubMed]
- 16.Harrod A, Fulton J, Nguyen VTM, Periyasamy M, Ramos-Garcia L, Lai CF, Metodieva G, de Giorgio A, Williams RL, Santos DB, Gomez PJ et al. : Genomic modelling of the ESR1 Y537S mutation for evaluating function and new therapeutic approaches for metastatic breast cancer. Oncogene 2017, 36:2286–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abou-Kandil A, Eisa N, Jabareen A, Huleihel M: Differential effects of HTLV-1 tax oncoprotein on the different estrogen-induced-ER alpha-mediated transcriptional activities. Cell Cycle 2016, 15:2626–2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cottu PH: Systemic neoadjuvant therapy of luminal breast cancer in 2016. Bull Cancer 2017, 104:69–78. [DOI] [PubMed] [Google Scholar]
- 19. ●●.Ribas R, Pancholi S, Guest SK, Marangoni E, Gao Q, Thuleau A, Simigdala N, Polanska UM, Campbell H, Rani A, Liccardi G et al. : Akt antagonist AZD5363 influences estrogen receptor function in endocrine-resistant breast cancer and synergizes with fulvestrant (ICI182780) in vivo. Mol Cancer Ther 2015, 14:2035–2048.This study assessed the effects of AKT inhibition in endocrine therapy-resistant cells and how inhibition of this pathway affected ER-mediated signaling.
- 20.Steelman LS, Martelli AM, Cocco L, Libra M, Nicoletti F, Abrams SL, McCubrey JA: The therapeutic potential of mTOR inhibitors in breast cancer. Br J Clin Pharmacol 2016, 82:1189–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. ●●.Bosch A, Li Z, Bergamaschi A, Ellis H, Toska E, Prat A, Tao JJ, Spratt DE, Viola-Villegas NT, Castel, Minuesa G et al. : PI3K inhibition results in enhanced estrogen receptor function and dependence in hormone receptor-positive breast cancer. Sci Transl Med 2015, 7:283ra251.This study demonstrates the cross-talk between PI3K pathway inhibitors and ER-mediated signaling as it relates to resistance and the need to target these pathways simultaneously.
- 22.Andre F, Bachelot T, Campone M, Dalenc F, Perez-Garcia JM, Hurvitz SA, Turner N, Rugo H, Smith JW, Deudon S, Shi M et al. : Targeting FGFR with dovitinib (TKI258): preclinical and clinical data in breast cancer. Clin Cancer Res 2013, 19:3693–3702. [DOI] [PubMed] [Google Scholar]
- 23.Osborne CK, Schiff R: Mechanisms of endocrine resistance in breast cancer. Annu Rev Med 2011, 62:233–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gu G, Dustin D, Fuqua SA: Targeted therapy for breast cancer and molecular mechanisms of resistance to treatment. Curr Opin Pharmacol 2016, 31:97–103. [DOI] [PubMed] [Google Scholar]
- 25.Politz O, Siegel F, Barfacker L, Bomer U, Hagebarth A, Scott WJ, Michels M, Ince S, Neuhaus R, Meyer K, Fernandez-Montalvan AE et al. : Bay 1125976, a selective allosteric AKT1/2 inhibitor, exhibits high efficacy on akt signaling-dependent tumor growth in mouse models. Int J Cancer 2017, 140:449–459. [DOI] [PubMed] [Google Scholar]
- 26.Boulay A, Rudloff J, Ye J, Zumstein-Mecker S, O’Reilly T, Evans DB, Chen S, Lane HA: Dual inhibition of mTOR and estrogen receptor signaling in vitro induces cell death in models of breast cancer. Clin Cancer Res 2005, 11:5319–5328. [DOI] [PubMed] [Google Scholar]
- 27.Miller TW, Hennessy BT, Gonzalez-Angulo AM, Fox EM, Mills GB, Chen H, Higham C, Garcia-Echeverria C, Shyr Y, Arteaga CL: Hyperactivation of phosphatidylinositol-3 kinase promotes escape from hormone dependence in estrogen receptor-positive human breast cancer. J Clin Invest 2010, 120:2406–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. ●.Zardavas D, Te Marvelde L, Milne RL, Fumagalli D, Fountzilas G, Kotoula V, Razis E, Papaxoinis G, Joensuu H, Moynahan ME, Hennessy BT et al. : Tumor PIK3CA genotype and prognosis in early-stage breast cancer: a pooled analysis of individual patient data. J Clin Oncol 2018. JCO2017748301.Clinical study from over 10 000 patients assessing the prognostic value of PIK3CA mutations in different subtypes of breast cancer.
- 29.Kang S, Bader AG, Vogt PK: Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc Natl Acad Sci U S A 2005, 102:802–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Banerji S, Cibulskis K, Rangel-Escareno C, Brown KK, Carter SL, Frederick AM, Lawrence MS, Sivachenko AY, Sougnez C, Zou L, Cortes ML et al. : Sequence analysis of mutations and translocations across breast cancer subtypes. Nature 2012, 486:405–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Estevez LG, Garcia E, Hidalgo M: Inhibiting the PI3K signaling pathway: buparlisib as a new targeted option in breast carcinoma. Clin Transl Oncol 2016, 18:541–549. [DOI] [PubMed] [Google Scholar]
- 32.Choi AR, Kim JH, Woo YH, Cheon JH, Kim HS, Yoon S: Co-treatment of LY294002 or MK-2206 with AZD5363 attenuates AZD5363-induced increase in the level of phosphorylated AKT. Anticancer Res 2016, 36:5849–5858. [DOI] [PubMed] [Google Scholar]
- 33.Lui A, New J, Ogony J, Thomas S, Lewis-Wambi J: Everolimus downregulates estrogen receptor and induces autophagy in aromatase inhibitor-resistant breast cancer cells. BMC Cancer 2016, 16:487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Campbell RA, Bhat-Nakshatri P, Patel NM, Constantinidou D, Ali S, Nakshatri H: Phosphatidylinositol 3-kinase/AKT-mediated activation of estrogen receptor alpha: a new model for anti-estrogen resistance. J Biol Chem 2001, 276:9817–9824. [DOI] [PubMed] [Google Scholar]
- 35.Yamnik RL, Digilova A, Davis DC, Brodt ZN, Murphy CJ, Holz MK: S6 kinase 1 regulates estrogen receptor alpha in control of breast cancer cell proliferation. J Biol Chem 2009, 284:6361–6369. [DOI] [PubMed] [Google Scholar]
- 36.Yardley DA, Noguchi S, Pritchard KI, Burris HA III, Baselga J, Gnant M, Hortobagyi GN, Campone M, Pistilli B, Piccart M, Melichar B et al. : Everolimus plus exemestane in postmenopausal patients with HR(+) breast cancer: BOLERO-2 final progression-free survival analysis. Adv Ther 2013, 30:870–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Di Leo A, Johnston S, Lee KS, Ciruelos E, Lonning PE, Janni W, O’Regan R, Mouret-Reynier MA, Kalev D, Egle D, Csoszi T et al. : Buparlisib plus fulvestrant in postmenopausal women with hormone-receptor-positive, HER2-negative, advanced breast cancer progressing on or after mTOR inhibition (BELLE-3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 2018, 19:87–100. [DOI] [PubMed] [Google Scholar]
- 38.Baselga J, Im SA, Iwata H, Cortes J, De Laurentiis M, Jiang Z, Arteaga CL, Jonat W, Clemons M, Ito Y, Awada A et al. : Buparlisib plus fulvestrant versus placebo plus fulvestrant in postmenopausal, hormone receptor-positive, HER2-negative, advanced breast cancer (BELLE-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 2017, 18:904–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Muise-Helmericks RC, Grimes HL, Bellacosa A, Malstrom SE, Tsichlis PN, Rosen N: Cyclin D expression is controlled post-transcriptionally via a phosphatidylinositol 3-kinase/Akt-dependent pathway. J Biol Chem 1998, 273:29864–29872. [DOI] [PubMed] [Google Scholar]
- 40.Vora SR, Juric D, Kim N, Mino-Kenudson M, Huynh T, Costa C, Lockerman EL, Pollack SF, Liu M, Li X, Lehar J et al. : CDK 4/6 inhibitors sensitize PIK3CA mutant breast cancer to PI3K inhibitors. Cancer Cell 2014, 26:136–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kenny FS, Hui R, Musgrove EA, Gee JM, Blamey RW, Nicholson RI, Sutherland RL, Robertson JF: Overexpression of cyclin D1 messenger RNA predicts for poor prognosis in estrogen receptor-positive breast cancer. Clin Cancer Res 1999, 5:2069–2076. [PubMed] [Google Scholar]
- 42.Kilker RL, Planas-Silva MD: Cyclin D1 is necessary for tamoxifen-induced cell cycle progression in human breast cancer cells. Cancer Res 2006, 66:11478–11484. [DOI] [PubMed] [Google Scholar]
- 43.Wilcken NR, Prall OW, Musgrove EA, Sutherland RL: Inducible overexpression of cyclin D1 in breast cancer cells reverses the growth-inhibitory effects of antiestrogens. Clin Cancer Res 1997, 3:849–854. [PubMed] [Google Scholar]
- 44.Finn RS, Aleshin A, Slamon DJ: Targeting the cyclin-dependent kinases (CDK) 4/6 in estrogen receptor-positive breast cancers. Breast Cancer Res 2016, 18:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Finn RS, Martin M, Rugo HS, Jones S, Im SA, Gelmon K, Harbeck N, Lipatov ON, Walshe JM, Moulder S, Gauthier E et al. : Palbociclib and letrozole in advanced breast cancer. N Engl J Med 2016, 375:1925–1936. [DOI] [PubMed] [Google Scholar]
- 46.Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO, Ettl J, Patel R, Pinter T, Schmidt M, Shparyk Y et al. : The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol 2015, 16:25–35. [DOI] [PubMed] [Google Scholar]
- 47.Turner NC, Ro J, Andre F, Loi S, Verma S, Iwata H, Harbeck N, Loibl S, Huang Bartlett C, Zhang K, Giorgetti C et al. : Palbociclib in hormone-receptor-positive advanced breast cancer. N Engl J Med 2015, 373:209–219. [DOI] [PubMed] [Google Scholar]
- 48.Cristofanilli M, Turner NC, Bondarenko I, Ro J, Im SA, Masuda N, Colleoni M, DeMichele A, Loi S, Verma S, Iwata H et al. : Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol 2016, 17:425–439. [DOI] [PubMed] [Google Scholar]
- 49.Hortobagyi GN, Stemmer SM, Burris HA, Yap YS, Sonke GS, Paluch-Shimon S, Campone M, Blackwell KL, Andre F, Winer EP, Janni W et al. : Ribociclib as first-line therapy for HR-positive, advanced breast cancer. N Engl J Med 2016, 375:1738–1748. [DOI] [PubMed] [Google Scholar]
- 50.Dickler MN, Tolaney SM, Rugo HS, Cortes J, Dieras V, Patt D, Wildiers H, Hudis CA, O’Shaughnessy J, Zamora E, Yardley DA et al. : MONARCH 1, a phase II study of abemaciclib, a CDK4 and CDK6 inhibitor, as a single agent, in patients with refractory HR(+)/HER2( ) metastatic breast cancer. Clin Cancer Res 2017, 23:5218–5224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sledge GW Jr, Toi M, Neven P, Sohn J, Inoue K, Pivot X, Burdaeva O, Okera M, Masuda N, Kaufman PA, Koh H et al. : MONARCH 2: Abemaciclib in combination with fulvestrant in women with HR+/HER2 advanced breast cancer who had progressed while receiving endocrine therapy. J Clin Oncol 2017, 35:2875–2884. [DOI] [PubMed] [Google Scholar]
- 52.Goetz MP, Toi M, Campone M, Sohn J, Paluch-Shimon S, Huober J, Park IH, Tredan O, Chen SC, Manso L, Freedman OC et al. : Monarch 3: Abemaciclib as initial therapy for advanced breast cancer. J Clin Oncol 2017, 35:3638–3646. [DOI] [PubMed] [Google Scholar]
- 53.Dean JL, Thangavel C, McClendon AK, Reed CA, Knudsen ES: Therapeutic CDK4/6 inhibition in breast cancer: key mechanisms of response and failure. Oncogene 2010, 29:4018–4032. [DOI] [PubMed] [Google Scholar]
- 54. ●●.Herrera-Abreu MT, Palafox M, Asghar U, Rivas MA, Cutts RJ, Garcia-Murillas I, Pearson A, Guzman M, Rodriguez O, Grueso J, Bellet M et al. : Early adaptation and acquired resistance to CDK4/6 inhibition in estrogen receptor-positive breast cancer. Cancer Res 2016, 76:2301–2313.This original work illustrates convergent mechanisms of early adaptation and acquired resistance to CDK4/6 inhibitors and supports the clinical development of combinations of CDK4/6 and PI3K/mTOR inhibitors in ER + cancers.
- 55.Johnson J, Thijssen B, McDermott U, Garnett M, Wessels LF, Bernards R: Targeting the RB-E2F pathway in breast cancer. Oncogene 2016, 35:4829–4835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Condorelli R, Spring L, O’Shaughnessy J, Lacroix L, Bailleux C, Scott V, Dubois J, Nagy RJ, Lanman RB, Iafrate AJ, Andre F et al. : Polyclonal RB1 mutations and acquired resistance to CDK 4/6 inhibitors in patients with metastatic breast cancer. Ann Oncol 2017. [DOI] [PubMed]
- 57. ●.Pritchard KI, Chia SK, Simmons C, McLeod D, Paterson A, Provencher L, Rayson D: Enhancing endocrine therapy combination strategies for the treatment of postmenopausal HR+/HER2 advanced breast cancer. Oncologist 2017, 22:12–24.This article reviewed phase III clinical trials evaluating endocrine therapy combination therapy in postmenopausal patients with ER+ HER2-nega-tive breast cancer, with the purpose of providing practical clinical guidance.
- 58.Polk A, Svane IM, Andersson M, Nielsen D: Checkpoint inhibitors in breast cancer — current status. Cancer Treat Rev 2018, 63:122–134. [DOI] [PubMed] [Google Scholar]