

Abstract
Millions of Americans now entering midlife and old age were exposed to high levels of lead, a neurotoxin, as children. Evidence from animal-model and human observational studies suggest that childhood lead exposure may raise the risk of adult neurodegenerative disease, particularly dementia, through a variety of possible mechanisms including epigenetic modification, delayed cardiovascular and kidney disease, direct degenerative CNS injury from lead remobilized from bone, and lowered neural and cognitive reserve. Within the next ten years, the generation of children with the highest historical lead exposures, those born in the 1960s, 1970s, and 1980s, will begin to enter the age at which dementia symptoms tend to emerge. Many will also enter the age in which lead stored in the skeleton may be remobilized at greater rates, particularly for women entering menopause and men and women experiencing osteoporosis. Should childhood lead exposure prove pro-degenerative, the next twenty years will provide the last opportunities for possible early intervention to forestall greater degenerative disease burden across the aging lead-exposed population. More evidence is needed now to characterize the nature and magnitude of the degenerative risks facing adults exposed to lead as children and to identify interventions to limit long-term harm.
Keywords: Aging, development, epigenetics, lead, neurodegeneration
INTRODUCTION
When do neurodegenerative diseases, like Alzheimer’s (AD) and Parkinson’s (PD), begin? The answer may be: at conception. As with many age-related conditions, neurodegenerative diseases are increasingly considered to result from an array of insults and risk factors operating differentially across the lifespan, with early life, pre- and post-natal emerging as a critical window for the development of risk [1–3]. Following a Developmental Origins of Health and Disease (DOHaD) theoretical approach [4], some emerging theories now view abnormal age-related degeneration as the delayed consequence of disrupted neural development [3, 5, 6].
Although diseases like AD and PD appear to be fundamentally multifactorial in their etiology and mixed in their pathology [7]—with many possible roads leading to the same dysfunctional outcome—the identification of common early-life risk factors holds significant promise for early intervention with at-risk individuals and for primary prevention to reduce risks for future generations. With an aging global population, even small reductions in risk could significantly lower the future worldwide burden of neurodegenerative disease [8].
For millions of Americans now entering midlife and older age, childhood exposure to lead may be one profound, ubiquitous risk factor for age-related neurodegenerative disease.
Until the early-1990s, lead was ever-present in American communities, with lead use in paints, pipes, and gasoline resulting in high lead exposures across the population. Lead was first added to gasoline in 1921 and, until phase-downs began in the mid-1970s, its use increased exponentially [9]. In 1976, when the first America-wide lead-level surveillance began, the average American’s blood-lead level was three times higher than the current reference value for clinical attention [10]. Individuals, and particularly children, living in highly urban areas, beside busy roads, or near lead-emitting industries had the highest exposures [10–13].
Lead, a heavy metal able to substitute for calcium in the body, is a potent neurotoxin. Though its harm for the developing child’s brain is by now wellknown [14], the risk that lead poses for the exposed child later in life is still an active area of research [15]. Accumulating evidence from experimental and observational studies now suggests that childhood lead exposures may result in lasting neural, epigenetic, and behavioral changes not seen in exposed adults—changes that, together, may significantly alter exposed-children’s risks for neurodegenerative disease in old age.
American children born in the 1960s, 1970s, and early 1980s experienced, en mass, lead exposures of a magnitude not seen before or since [9, 13, 16]. Within the next ten years, the oldest of these children will enter the age at which degenerative disease symptoms tend to emerge. By then it will be too late to intervene.
This review article summaries the existing evidence linking lead exposure in childhood to increased neurodegenerative disease risk in adulthood. It considers the possible mechanisms by which increased risk could be conferred, including biological, behavioral, and epigenetic paths, and it articulates the many gaps in knowledge that additional research should fill while considering possible implications for public health and policy.
EVIDENCE LINKING CHILDHOOD LEAD EXPOSURE TO DEGENERATIVE BRAIN DISEASE
Research studies on the potential neurodegenerative consequences of lead exposure have primarily focused on risks for the two most common neurodegenerative diseases, AD and PD. Box 1 describes the general pathological hallmarks of AD and PD. Although these specific pathology syndromes result in selective degradation of different brain networks [17], there is considerable pathological overlap among them [18] and among related diseases (e.g., Lewy-body dementia), with co-morbid or so-called “mixed pathologies” accounting for most cases, particularly of diagnosed dementias [7, 19, 20]. Lead exposure may be a common, or non-specific, risk factor for these disorders and, notably, has been linked to other degenerative diseases not reviewed here, particularly amyotrophic lateral sclerosis [21, 22].
Box 1. The general hallmarks of Alzheimer’s disease (AD) and Parkinson’s disease (PD).
AD is associated with progressive atrophy in cortical and hippocampal brain areas and with the formation of amyloid-β plaques and neurofibrillary tangles between neurons. Plaques and tangles result from abnormal accumulation of amyloid-β protein precursor (AβPP) and microtubule-associated protein tau respectively.
PD is associated with progressive loss of dopaminergic neurons in the substantia nigra of the mid-brain and with the presence of Lewy body protein clumps between neurons, resulting from abnormal accumulation of the α-synuclein protein, among others [229].
Both AD and PD are age-related, with age being their most significant risk factor. Idiopathic AD and PD, the most common forms of each disease, have environmental exposures consistently implicated in their etiology, with increased disease risk linked to exposure to head trauma [23–25], air pollution [26–28], metals [29–31], pesticides [32], and chronic stress [33].
No prospective longitudinal study has yet followed a group of lead-exposed children into old age. The best evidence linking early-life lead exposure and late-life neurodegeneration currently arrives from experimental studies using cellular and animal models, which allow for examination of full-lifespan outcomes. These studies suggest a clear link between early-life lead exposure and late-life brain disease. Observational studies in lead-exposed workers and community-dwelling elderly have, meanwhile, provided consistent evidence that lead’s neurotoxic activities can induce changes in the brain that are pro-degenerative and that increase neurodegenerative risk years after exposure. Recently, a handful of limited follow-up studies in lead-exposed children in young adulthood and at midlife provide some additional suggestion of an exposure-outcome link. These three sources of evidence—cellular and animal model studies, studies of adults exposed to lead, and longitudinal studies of exposed children—are reviewed in order.
Evidence from cellular and animal studies
Basic cellular and animal studies have, over the last few decades, explicated the toxic actions of lead within the central nervous system (CNS). Lead is a heavy metal that, in the body, is able to substitute for calcium, an element critical to neuronal signaling, neurogenesis, mylenation, synaptic plasticity, and the functioning of glial cells [34, 35]. Lead is believed to pass the blood-brain barrier through calcium channels and, once inside the brain, to enter neurons and glial cells through similar channels [36]. Lead has a half-life in the brain of roughly two years [37].
Inside neurons, lead: suppresses neurotransmitter release, which relies on calcium ions; alters energy metabolism through the inhibition of NMDA-ion channels and the activation of protein kinase C, and; blocks the release of calcium from mitochondria, leading to the formation of reactive oxygen species, mitochondrial “self-destruction,” and apoptosis of the neuron [35]. Lead also leads to neuronal death through enhanced lipid peroxidation and, in glutamate-signaling neurons, excitotoxicity. During neural development lead can disrupt neuron and glial migration, differentiation, and, for neurons, the formation of synapses [35]. Glutamate and dopamine systems appear to be preferentially vulnerable to lead, leading to particular disruptions in hippocampal long-term potentiation and cortical executive-functioning [35]. In mice, lead preferentially accumulates in the hippocampus and cerebral cortex [38], a rough pattern also reported in humans [39].
While most studies of lead toxicity have considered the immediate consequences of exposure, over the past decade a number of studies have considered longer-term consequences, particularly from early-life exposure. This research, conducted primarily in mice and non-human primates, suggests that early-life lead exposure may hold neurodegenerative consequences in old age.
The most compelling evidence of this link is provided by a multi-decadal study of female macaque monkeys differentially exposed to lead acetate in the first year of life. These monkeys, born at the Health Protection Branch of the Canadian government in 1980, were raised for studies on the developmental consequences of lead exposure. Following a number of behavioral experiments across the 1990s, which linked lead to learning and memory deficits [40, 41], the study animals were transferred to the U.S.-National Institute of Environmental Health Sciences (NIEHS), where they were sacrificed, in 2003, in mid-adulthood, at age 23 years. (Macaques are generally considered “old” by age 30; J Harry, personal communication, February 16, 2018). Early blood tests indicated that the lead-exposed monkeys had experienced moderate exposure, with blood-lead levels ranging from 19–26 µg/dL, moderately higher than the U.S. population average in the late 1970 s and well above the current reference level (5 µg/dL). CNS tissue from nine study animals was made available to researchers at the University of Rhode Island for research on long-term lead-related pathology.
Upon examining the primate CNS tissue, investigators observed significantly greater AD-like pathology in the frontal cortex of the lead-exposed monkeys (n = 5) than in the non-lead exposed control monkeys (n = 4), including the presence of diffuse amyloid-β plaques and neurofibrillary tangles [42]. These pathological signs were accompanied by: significantly elevated mRNA levels of amyloid-β-related genes (e.g., amyloid-β protein precursor (AβPP), transcription factor Sp1, and, marginally, BACE1); DNA methylation patterns indicative of enhanced brain aging; and significant elevations in biomarkers of oxidative DNA damage (8-oxo-dG). Greater AD-like hyperphosphorylation of tau and pathological tau deposits were also observed [43].
Unfortunately, these follow-up studies were strictly immunohistochemical (NH Zawia, personal communication, October 12, 2017), and the aged, lead-exposed macaques were not subjected to any formal cognitive or behavioral tests prior to sacrifice, although no gross behavioral abnormalities were noted in any of the macaques’ health records (J. Harry, personal communication, February 16, 2018).
Complementary studies in mice have since provided finer details on the nature and timing of lead’s possible neurodegenerative effects, with at least one study hinting that early-life may represent a critical window for the development of later disease risk. In this study, researchers exposed mice to low levels of lead: a) early in life (within the first month), b) late in life (for three months beginning at midlife), or c) both early and late. In mice exposed early in life, investigators detected AD-like deficits in learning and memory that were accompanied by over-expression of AD-related genes [44] (e.g., genes coding for AβPP), elevated production of AD-related proteins (e.g., tau), and elevated protein phosphorylation at cellular sites matching those seen in brain extracts from AD patients (e.g., serine and threonine sites) [45]. Mice exposed to lead only in midlife displayed no observable impairment in learning or memory ability and, further, evidenced no degenerative epigenetic or morphological brain changes despite having experienced greater cumulative exposure than the early-exposed mice. Notably, the AD-like pathological changes seen in the early-life exposure mice mirrored those seen in the lead-exposed macaques [42, 43]. These findings also match those reported from several other mouse studies [46–49] and from studies in transgenic mice that produce amyloid-β plaques [50].
Collectively, the degenerative pathological outcomes seen in primates and mice exposed to lead early in life suggest that lead exposure may hold long-term neurodegenerative consequences for those individuals exposed in childhood. As mice exposed only at midlife appear to suffer less degenerative pathology, childhood may represent a unique window of vulnerability for the development of disease following lead exposure. As the studies of adults workers and community-dwelling elders reviewed in the next section make clear, however, degenerative disease risk may also increase in those heavily exposed to lead in adulthood or, conversely, those exposed moderately in old age, which may represent a second vulnerable period.
Evidence from adults exposed to lead
Studies of adults exposed to lead through their home environment or through employment in lead-related industries have indicated a link between later-life lead exposure and neurodegenerative disease. Lead is hypothesized to increase disease risk in exposed adults by directly causing inflammatory and pro-degenerative intracellular oxidative damage. It is not yet clear if the mechanisms operating to increase risk following adult exposures are the same as those operating in exposed children. Nevertheless, studies of adults exposed to lead provide confirmation of pro-degenerative neurotoxic activity.
Adult lead exposure and PD risk
A number of studies have reported increased rates of PD in adults exposed to lead occupationally. In one U.S. case-control study with elderly participants, individuals with the highest quartile of lifetime lead exposure, calculated from bone and blood-lead measures and from analysis of probable occupational exposures, were found to have a two-fold greater risk of PD than those in the lowest quartile of lifetime exposure [51]. A different case control study using U.S. elders from a general population and bone-lead measurements of cumulative lead exposure reported a three-fold greater risk for those in the highest quartile of exposure compared to those in the first [52]. Occupational studies reliant on self-report measures of lead exposure, determined through interviews about occupation and work tasks, have generally failed to find associations between lead and PD [53–55], although at least one such study reported considerably elevated risk of PD (OR = 5.24) in workers estimated to have been exposed to lead and additional metals (e.g., lead and copper or lead and iron) for greater than 20 years [53]. Prospective studies linking industry lead emissions data to PD risk at U.S. census tract [56] and county levels [57] have also failed to identify PD-lead associations, suggesting that environmental-level exposures, which tend to be lower than occupational-levels, may not alter risk or, conversely, that measures of industrial lead emissions may be poor proxies for actual lead exposure.
Adult lead exposure and AD risk
Unlike with PD, no studies have yet examined AD risk following adult lead exposure. Numerous cross-sectional studies in older adults have, however, reported between-individual cognitive deficits associated with lead exposure [58–63], and several retrospective and prospective longitudinal studies have reported within-individual cognitive decline following lead exposure [64–68]. Collectively, these studies examined lead-outcome associations in both occupationally and environmentally exposed individuals and in socioeconomically and ethnically diverse populations. In one representative study, of healthy elderly men from a general U.S. population (the VA Normative Aging Study, VA-NAS), one interquartile range of higher cumulative lead exposure associated with cognitive deficits akin to aging the brain five additional years [65]. A 2007 review of the evidence collected to that point concluded that there was “moderate evidence” of a likely causal relationship between adult lead exposure and cognitive decline [64].
Cognitive deficits relative to peers and cognitive decline measured across many years represent profound risk factors for AD [69–71], with the premorbid disease phase proceeded by several years of “progressively accelerating” cognitive decline [69]. Cognitive deficits associated with lead exposure consistently appear in AD-related domains, including verbal and visual memory, attention and general executive functioning, and in domains related to motor ability, manual dexterity, and visuospatial ability [64]. Studies are now being planned to more directly evaluate AD diagnosis and symptom severity risk following adult lead exposure (MG Weisskopf, personal communication, October 12, 2017).
Although adult lead exposure appears to confer risk for cognitive decline, a risk factor for AD, it is not clear to what extent studies of lead exposed adults can generalize to lead exposed children. One thing these studies do suggest, however, is that lead doses received in the past, sometimes decades in the past, may lead to delayed or progressive effects in adults [64]. Indeed, in both longitudinal and cross-sectional studies, measures of past lead exposure (typically taken through non-invasive cortical bone K-shell X-ray fluorescence measurements) were better predictors of cognitive impairment or decline than measures of recent exposure in all study subjects except those still experiencing high exposures through work [64]. A 2006 MRI follow-up in one of these groups that was found to have cognitive decline (former organolead workers) reported that past lead exposure associated with the prevalence of white matter lesions and region-specific brain atrophy 18 years after the cessation of exposure. The authors hypothesized that the pattern of brain degeneration detected was “a consequence of progressive changes” following lead exposure [72].
Progressive degenerative changes following adult lead exposure have been detected as gross abnormalities in brain morphometry (e.g., white matter lesions and regional atrophy) [72], but also as more subtle alterations in brain health, which appear to be AD-like. In particular, adult lead exposure has been connected to abnormal ratios of brain metabolites in the hippocampus. A small (n = 31) magnetic resonance spectroscopy (MRS) follow-up of the healthy, elderly members of the VA-NAS with the highest and lowest rates of cumulative lead exposure reported brain metabolite ratios in the hippocampus of the most lead-exposed subjects that are typically seen in the preclinical phases of AD, notably an increase in the ratio of myinsitol-to-creatine [73]. Two MRS studies of heavily-exposed middle-aged Chinese and Taiwanese workers in lead-related industries have reported similar findings, with higher exposure workers displaying greater AD-like abnormalities in brain metabolites in the hippocampus [74, 75]. These metabolic abnormalities are believed to signal the beginning of the hippocampal neuron loss and gliosis associated with AD [73, 76]. MRI measures in the lead-exposed Chinese workers confirmed that these abnormalities were accompanied by hippocampal atrophy [75], a common biomarker for the mild cognitive impairment that precedes AD diagnosis [77, 78]. High brain-lead concentrations have, additionally, been reported following autopsy of lead-exposed Japanese workers with dementia and diffuse neurofibrillary tangles with calcification [79].
Evidence from children exposed to lead
The earliest evidence linking childhood lead exposure to neurodegeneration comes from a 1931 case of a lead poisoned Cincinnati child [80]. Stricken with lead-related encephalopathy after ingesting leaded paint at age two, the child experienced progressive mental deterioration until his death, at age 44, of pneumonia. Brain autopsy following death revealed expansive atrophy associated with AD, in cortical, temporal and hippocampal brain areas, with neural tissue in these regions clouded by neurofibrillary tangles that, even then, were considered hallmarks of AD. At that time at least two other cases of individuals highly exposed to lead “from a very early age” were found, after death, to have experienced AD-type pathology [81]. Following these cases, researchers at Indiana University observed the formation of neurofibrillary tangles in cortical and hippocampal neurons in rabbits within hours of injecting the animals with tetraethyllead [81].
Few cases of childhood lead poisoning have since been followed-up in the literature, although one group of lead-poisoned children treated at Boston’s Children Hospital in the 1940s were given cognitive tests in middle age (n = 33, Mean age = 55 years). Evidence of age-related degeneration was not noted at that time, but significant, widespread cognitive deficits relative to matched controls were detected, 50 years after “cessation of exposure” [82].
While cases of lead poisoning can describe the most extreme long-term consequences of early-life lead exposure, the best evidence for the risks facing the millions of Americans exposed environmentally to lead as children in the 1960s, 1970s, and 1980s will come from follow-up studies in general population samples of children born in those years. The few such studies conducted to date suggest that the neurologic harm and cognitive deficits associated with childhood lead exposure persist into adulthood and middle age and are accompanied by changes in brain morphometry, white matter integrity, and metabolism. These studies are summarized in Table 1.
Table 1.
Adult follow-up studies in lead-exposed general-population child cohorts
| Cohort | Age at exposure test | N at most recent relevant follow-up | Average age at most recent relevant follow-up | Estimated cohort age in 2018 | Relevant lead-outcome associations reported |
|---|---|---|---|---|---|
| Massachusetts school cohort | 7.3 years | 132 | 18.4 | 45 years | Neurobehavioral deficits [136]. |
| Impaired academic performance [136]. | |||||
| Cincinnati Lead Study cohort | Prenatal and every 6 months to age 6.5 | 159 | 20.8 | 36 years | Decreased brain volume, particularly in frontal gray matter and anterior cingulate cortex [126]. |
| White matter diffusion abnormalities across brain [137]. | |||||
| Altered metabolism in several gray and white matter regions [203]. | |||||
| Port Pirie Study cohort | Prenatal and 0.5, 1.25, 2, 3, 4, 5, 6, 7, 11–13 years | 402 | 26.9 | 38 years | Mental health problems: likelihood of specific phobia; anxiety problems; depressive symptoms [204]. |
| Boston prospective cohort | Prenatal and 0.5, 1, 1.5, 2, 4.75, and 10 years | 43, 55 | 29 years | 38 years | Lower cognitive function (IQ score) (n = 43) [135]. |
| Altered expression of genes related to amyloid-β production (n = 55) [83]. | |||||
| Lower plasma amyloid-β42 concentrations (n = 55) [83]. | |||||
| Dunedin Study cohort | 11 years | 565 | 38 | 46 years | Cognitive deficits relative to peers/Cognitive decline relative to pre-exposure self [84]. |
| Lower socioeconomic status than peers/Downward social mobility relative to parents [84]. |
Note. The Bunker Hill cohort, a group of children accidentally exposed to high levels of lead in 1974, was not included in this table, as most cohort children did not have blood-lead levels measured. Cohort members did receive neurobehavioral follow-up in young adulthood, however, and were found to suffer from impairment in peripheral nerve and general cognitive function when compared to similar individuals with no accidental exposure [205]. Other lead-tested child cohorts, including those from Cleveland [206], Rochester [207, 208], Mexico [209, 210], and Yugoslavia [211] do not appear to have received follow-up in adulthood.
It is not yet clear if the functional, structural, and metabolic abnormalities found in adults exposed to lead as children are indicative of increased risk for neurodegenerative disease, although the evidence from animal studies reviewed earlier suggest that they may be. Notably, in one group of lead exposed children (n = 55) who had their blood assayed at age 29, childhood lead exposure was linked to altered expression of genes related to the production of amyloid-β, the hallmark AD protein [83]. Study members with greater childhood lead exposure also had lower plasma amyloid-β42 levels, a phenomenon often, though not always, seen in AD patients [83]. This year the oldest age follow-up in lead exposed children, in a population-representative cohort of New Zealanders born in 1972–1973, reported evidence of cognitive deficits in middle-aged adults exposed to lead as children and, further, of cognitive decline across the 30 years preceding follow-up [84]. As noted earlier in this review, cognitive deficits relative to peers and cognitive decline measured across many years represent risk factors for AD [69–71], although these are generally used as risk predictors in older populations (e.g., those >65 years old).
POTENTIAL MECHANISMS OF DISEASE RISK
As reviewed above, lead exposure has been linked to brain abnormalities, cognitive decline, and increased degenerative disease risk long after the cessation of exposure, in mice, non-human primates, and humans. Two classes of potential mechanisms of action are considered here:
Those that increase disease pathology, wherein early-life lead exposure drives changes in brain health that lead directly to increased pathology in adulthood (e.g., loss of synapses, neuronal death, etc.). The best evidence for such a direct lead-disease link involves early-life lead exposures altering epigenetic regulation to drive degenerative protein pathology. Adult brain pathology resulting from lead-induced cardiovascular and kidney disease and from circulating lead remobilized from bone during menopause and osteoporosis also represent potential, but understudied, direct disease pathways.
Those that increase susceptibility to disease pathology, wherein early-life lead exposure drives changes in brain health that lead indirectly to increased clinical outcomes in adulthood (e.g., functional impairment, clinical symptoms, etc.) by making the brain less likely to maintain function in the face of normal age-related decline or other unrelated pathology. Such indirect mechanisms include altered brain reserve through early neuronal loss (structural susceptibility) and altered cognitive reserve through early deficits in intellectual and self-regulatory ability (functional susceptibility).
Generally speaking, too little evidence exists to determine which of these potential mechanisms, if any, are primarily responsible for altering cognitive outcomes or disease risks in exposed individuals. Some mechanisms, particularly altered epigenetic regulation, have received considerable attention while others, including cardiovascular and kidney disease, remain largely uninvestigated. These potential disease pathways are discussed, with suggestions offered for future research.
Mechanisms that may increase disease pathology
Epigenetic modification
Much of the recent attention to lead’s potential role as a driver of degenerative brain disease has focused on lead’s role as a potential modifier of gene regulation in exposed individuals [31, 85]. In particular, early-life lead exposure has, in mice, been linked to perturbed regulation and expression of a number of genes related to neuronal development and the neural/glial response to stressors like metals and pathogens. Many of these genes have been implicated in degenerative disease pathology, including those that code for the expression of serine/threonine protein phosphatases [86] (implicated in tau pathology), AβPP [87], and the beta-secretase enzyme [88] (implicated in amyloid-β pathology). Lead appears to alter gene expression primarily by decreasing DNA methytransferase activity in effected cells [87].
In all, roughly 150 genes appear to be differentially expressed in mice exposed to lead early in life [49]. Some of these genes, like those coding for AβPP, are overexpressed in old age relative to healthy controls, leading to pathological protein accumulations [89]. Other genes, like those coding for neprilysin, which removes amyloid-β from the brain, are under-expressed in old age relative to healthy controls, leading to ineffective responses to accumulating proteins [89].
Notably, lead exposure appears to alter cellular epigenetic processing predominantly when exposure occurs early in life. In their study of brain changes in mice following lead exposure at different ages, investigators from the University of Rhode Island found no significant epigenetic or pathologic abnormalities in mice exposed to lead at midlife, while significant alterations had been observed, in both tau and amyloid-β relevant pathways, in adult mice exposed to lead in the first month of life [44, 45]. This general trend, of later-life dysfunction only following early-life exposure, has been reported in other studies of lead-exposed mice using different exposure protocols to examine other degenerative outcomes, like oxidative stress [48, 49].
Of particular relevance to the aging U.S. population, recent evidence suggests that epigenetic changes associated with lead exposure can lay dormant until old age, at least in mice. In a mouse study of “life-time” AβPP gene expression following early-life lead exposure, for example, a transient peak in AβPP gene expression in the mouse cortex immediately after lead exposure was followed by months of normal gene expression levels [88]. These levels abruptly increased again toward the end of the study-mice lifespans (Fig. 1) and were accompanied by accumulation of AβPP and amyloid-β in effected cortex tissue [88]. In this study, as in previous ones that tested the effects of early versus late-life lead exposures, only mice exposed early in life evidenced significant gene expression or protein level changes.
Fig. 1.
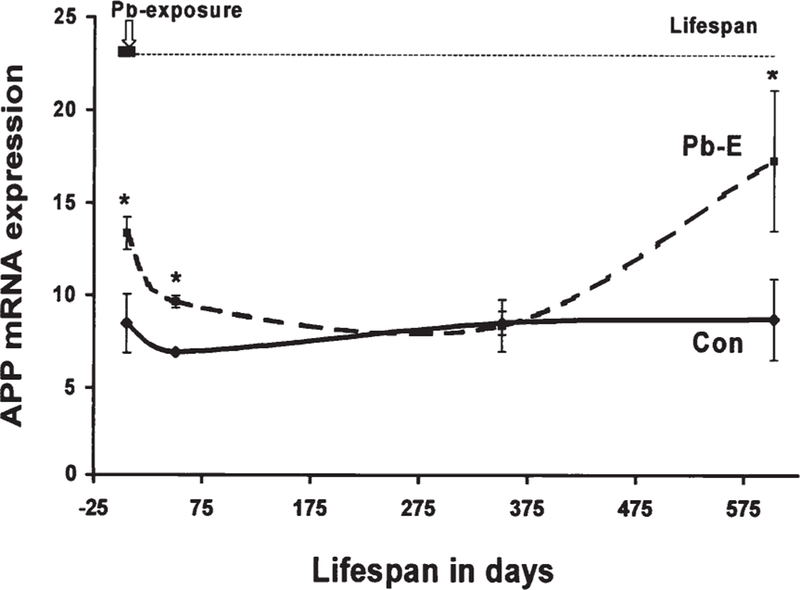
Early-life lead exposure results in delayed gene expression changes in mice. This figure shows the level of amyloid-β protein (APP) mRNA expression in cortex tissue in mice exposed to lead early in life (Pb-E) and in control mice (Con) across the life span. Reproduced from Basha et al. [88], The Journal of Neuroscience.
In general, early lead exposure appears to repress gene expression later in life [49]. Interestingly, most of the genes repressed after early lead exposure are typically upregulated during normal aging. Such genes, which code for DNA repair enzymes [48], immune response to pathogens, cell metabolism, and metal binding [49], are suspected to be involved in compensatory or reactive responses to “stressors acting on the aging brain” [49]. In this way, early life lead exposure may both exacerbate pathologic processes (e.g., those leading to protein accumulation and oxidative damage) and undermine the brain’s ability to cope or respond to these pathological processes. Again, thus far such epigenetic effects have primarily been observed in mice exposed to lead in early life, and not in those exposed in adulthood [48, 49, 88, 90].
These animal findings have largely yet to be replicated in humans; however, at least one analysis of gene expression in a small group of lead-tested children (n = 39) followed-up at age 29 reported significant associations between prenatal lead exposure and the differential expression, three decades later, of a number of genes related to neural development and amyloid-β production and deposition (e.g., ADAM9, RTN4, LRPAP1) [83]. Study members with higher prenatal lead exposure also had lower levels of amyloid-β protein in their blood plasma, a phenomenon thought to reflect greater compartmentalization of amyloid-β in the brain. (AD patients also tend to have lower amyloid-β plasma levels than healthy controls) [91, 92]. These findings represent important extensions of the findings from animal studies and warrant replication in larger cohorts.
Two recent reviews provide longer summaries of the epigenetic mechanisms linking lead exposure to neurodegenerative disease [31, 85]. Both conclude that epigenetic modification may be a primary mechanism by which early-life lead exposure may exert neurodegenerative effects.
Adult mobilization of sequestered lead in bone
Adult mobilization of lead sequestered in bone during childhood could also potentially drive adult disease pathology, although this mechanism has received little research attention. Only 1% of the body burden of lead is accounted for by lead in the blood. In children, the skeleton is estimated to contain 70% of the lead stored in the body; in adults the skeleton contains roughly 95% [93–95]. While the elimination half-time of lead in the blood is approximately one month, lead may be stored in bone for decades [95]. Within the bone, lead is “essentially inert” [94], but resorption, or “turnover,” of bone tissue can re-mobilize stored lead to form a novel, endogenous source of exposure for other organ systems [95]. Evidence suggests that lead remobilization from bone can lead to significant availability of lead to sensitive organs, like the brain, during high-turnover events, including pregnancy, nursing, peri and post-menopause, and osteoporosis.
No studies have yet evaluated whether childhood lead exposures may lead to adult neurodegenerative disease through the pathway of bone re-mobilization –but evidence on the nature and magnitude of such remobilization suggests that this is a plausible mechanism linking childhood exposure to adult disease, one which would benefit from increased investigation. Box 2 summarizes what is known about the nature and magnitude of harm posed by childhood lead recirculated in adulthood. For now we may conclude: first, that lead stored in bone can be mobilized and re-circulated decades after the cessation of exposure; and, second, that the greater the magnitude of the initial exposure, the greater the subsequent exposure [94–98].
Box 2. How lead absorbed in childhood recirculates later in life.
Lead is incorporated into bone during bone formation and remains sequestered until “turnover,” or resorption, of the bone tissue [94]. In young children bone turnover rates are high, and lead accumulation is limited. As children age, however, bone formation slows, and from roughly age 11 onward lead may begin to accumulate [230]. Though lead stored in the body’s soft, “trabecular” bone, which contains the marrow, tends to be released over the course of several years, lead stored in the more rigid “cortical” bone, which supports the body, can have an elimination half-time of anywhere from 5 to 50 years [94]. Greater initial lead exposures appear to result in larger half-times for the stored lead and, indeed, greater levels of lead remobilization [94, 95].
Bone loss begins, in humans, within the third decade of life, when bone resoprtion rates exceed formation rates [231]. From this time until death, the average woman will lose 35% of her cortical bone and 50% of her trabecular bone; the average man will lose over 25% of his cortical bone and 37% of his trabecular bone [231]. For women, bone loss occurs most acutely during pregnancy and lactation, when calcium is needed for the developing offspring, and during perimenopause (beginning roughly in the 4th decade of life) and menopause. For men, bone loss appears to be relatively constant, with some acceleration in the 5th decade of life onward in those who will develop osteoporosis [231–233].
For pregnant and nursing women and middle-aged and elderly men and women, bone resorption may be a “potentially important” source of lead dose in adulthood [95–97, 234–236]. For example, in a 1988 comparison of 2,981 women at various stages of pre and post-menopause in the second National Health and Nutrition Examination Survey (NHANES-II), post-menopausal women had, on average, blood-lead levels 2.56 µg/dL higher than pre-menopausal women, after controlling for potential cohort effects and other covariates, like race and socioeconomic status [96]. (The mean blood-lead level for pre-menopausal women in the study was 11.63 µg/dL). In a small (n = 15) study of pregnant Australian women, published in 1999, blood-lead levels increased between 10% and 50% (Mean = 25%) during pregnancy and between 30% and 95% (Mean = 65%) in the post-partum period [237]. For the most highly exposed subject, an “extra” 10.1 µg of lead was released into the blood per day over the gestation and postpartum periods. In a separate case study of a pregnant Australian women with high childhood lead exposures, pregnancy actually appeared to trigger acute lead poisoning thirty years, in her case, after the cessation of lead exposure [190, 191].
Though it is clear that bone-lead mobilization can be significant, it is difficult, methodologically, to determine the extent to which adult lead burdens arising from resorbed bone represent lead accumulated in childhood rather than more recently. Retrospective studies using teeth extracted for dental necessity may shed light on this issue, as core tooth enamel provides a record of lead exposure at the time of tooth formation (childhood) that preserves the ratio of lead isotopes present at the time for later comparison with isotope ratios of lead circulating in the blood in adulthood [16].
At least one creative study, from California, has used stable lead isotope analysis to examine the potential contribution of early-life lead exposure to later-life blood-lead levels. This study examined lead in blood and in removed bones from older male and female hip and knee joint replacement patients (n = 5, age range 52–75 years) to determine the percent of lead in the patient’s blood that was “old” lead accumulated decades earlier and mobilized from the skeleton [98]. The authors reported that between 40% and 70% of the lead circulating in patient blood (the percent varied by patient) derived from bone-lead stores that resembled the isotopic composition of environmental lead in California from several decades prior [98].
Cardiovascular/systemic organ disease
A final mechanism through which childhood lead exposure may be a direct driver of degenerative brain disease (i.e., may drive actual brain pathology) may be through the disruption of other, non-CNS organ systems that, in turn, help determine the integrity of the aging brain. No studies have yet examined this mechanistic pathway between early-life lead exposure and later-life cognitive decline and degeneration directly. However, lead is known to disrupt organ function and health in a number of systems integral to brain health, including the cardiovascular and renal systems, which suggests that this mechanism may be worth greater research attention.
Chronic lead exposure, even at low levels (<5 µg/dL), is considered to cause hypertension [99], and adult occupational and residential exposure has been linked to increased rates of coronary heart disease, peripheral arterial disease, alterations in cardiac rhythm, elevations in blood homocysteine levels, and ischemic heart disease [100, 101]. In one VA-NAS sample, for example, the risk of ischemic heart disease was five times greater in individuals in the highest tertile of bone lead levels compared to those in the lowest [101]. Experimental studies suggest that this pathology is induced through oxidative stress, inhibited endothelial repair, and impaired angiogenesis [102].
Hypertension, cardiovascular disease, and high homocysteine levels all represent profound risk factors for dementia and degenerative brain disease, as thickening cerebral arteries can lead to infarction and poor-perfusion-related hypoxia that, in turn, leads to neuronal death and the up-regulation of pathological protein pathways, including AβPP [103]. Children are known to suffer hypertensive effects of lead exposure [104] but no studies appear to have yet examined adult cardiovascular outcomes in lead-exposed children. A recent mortality linkage study using data from the third National Health and Nutrition Examination Survey (NHANES-III) has, however, performed a roughly 20-year follow-up on a representative cohort of lead-tested U.S. adults (Mean age at baseline = 44.1) [105]. That study reported significantly elevated risk of cardiovascular disease-related (HR1.70) and ischemic heart disease-related (HR2.08) mortality for individuals at the 90th percentile of blood-lead levels at baseline compared to those at the 10th percentile, suggesting that cardiovascular harms from early lead exposure persist over time and may result in premature death. Should children exposed to lead suffer greater rates of hypertension and cardiovascular disease in adulthood, they would also be placed at greater risk of cardiovascular-related degenerative brain pathology.
Lead exposure, even at low levels, is now also considered a “cofactor” in kidney disease, with greater lead dose associated with worse renal function [106]. As with hypertension and cardiovascular disease, chronic kidney dysfunction represents a risk factor for cognitive decline, dementia, and neurodegenerative disease [107, 108]. Part of the effect is likely attributable to kidney disease leading to poor cardiovascular health, but direct brain-kidney interaction has also been proposed, as the consequences of renal dysfunction (e.g., anemia, toxic uremic accumulation, chronic inflammation, acidosis, etc.) can directly impair CNS health [108]. Again, should children exposed to lead suffer greater rates of kidney disease in adulthood, they would be placed at greater risk of degenerative brain pathology. These organ-system-related risks require greater research attention.
Mechanisms that may increase disease susceptibility
Lead harms the developing brain in ways that may increase the risk for neurodegenerative disease later in life by directly causing disease pathology (e.g., epigenetic changes leading to pathological protein accumulation). However, even in the absence of direct lead-triggered disease in adulthood, child-hood lead exposures may increase the susceptibility of the aging brain to lose function in the face of normal age-related decline. Specifically, lead-related losses in brain (structural) and cognitive (functional) reserve may make lead-exposed children more likely to develop diagnosable neurodegenerative disease later in life.
Structural susceptibility
Following a “structural” mechanistic approach to considering lead’s long-term toxic effects, an early loss of functioning neurons and neural networks may later reduce the brain’s capacity to sustain function amid atrophy and neuronal loss associated with normal aging (Fig. 2) [109, 110]. Such a hypothesis was first proposed nearly two decades ago as one explanation for idiopathic PD [111]. Since then, the concept has been expanded to explain the influence of early-life events on the risk for neurodegenerative diseases more broadly [109]. Lead would be pro-degenerative through a structural mechanism by decreasing overall passive “brain reserve” [112].
Fig. 2.

Early brain damage could lead to increased risk of degenerative disease by reducing the brain’s capacity to sustain function in the face of normal age-related neuronal loss. The figure presents a theoretical model of differential onset of Parkinson’s disease (PD) symptoms where the loss of dopamine (DA) producing neurons in the substania nigra proceeds similarly for those with and without developmental damage but the onset of PD symptoms occurs earlier for those with early damage. Reproduced from Landrigan et al. [109], Environmental Health Perspectives.
What is brain reserve – and does lead alter it?
“Reserve” is a concept that seeks to account for individual differences in susceptibility to age-related degenerative disease, particularly for those individuals that maintain high function in the face of advanced pathology [112]. Brain reserve involves the contribution of brain morphology, or structure, to an individual’s tolerance to pathology while cognitive reserve, in contrast, involves the contribution of brain function. Brain reserve is considered to be primarily quantitative and, according to reserve theory, is likely related to the number of neurons and synapses an individual can afford to lose before pathology manifests in symptoms of clinical impairment (Fig. 2). The brain reserve concept is supported by numerous studies reporting lower rates of dementia in individuals with larger premorbid brain size [113–116].
Although dynamic measures of brain structure are now receiving research attention (particularly biomarkers of adult neurogenesis) the static measures of whole brain volume and head circumference (a measure of premorbid brain size) represent the best-studied measures of brain reserve to date [112]. In numerous cross-sectional studies in diverse populations, brain size and head circumference have been linked to the risk of developing AD [113, 117], the age of symptom onset [118], and the severity of symptoms [117, 119]. Brain size has also been linked to the severity of dementia symptoms in PD [120]. The effects are particularly pronounced for those with lower than average brain and head size [121]. In a large cross-sectional study of aging Manhattanites, for example, women in the lowest quintile of head circumference were nearly three times more likely to have AD after adjustments were made for age, education, and ethnicity [113].
The brain accomplishes the majority of its growth (93%) by age 6 [2]. Pre and post-natal lead exposure up to this age has been linked to retarded brain and head growth in a number of studies, in the U.S., Mexico, and Greece [122–125], with an increase in child blood-lead level of 10 µg/dL relating to between 0.33 cm and 0.52 cm smaller head diameter, in both cross-sectional and longitudinal studies. While no studies have yet evaluated the role of diminished brain size in fostering long-term lead-related cognitive impairment or disease, the magnitude of head size deficits seen in lead-exposed children are within the range for elevated dementia risk in later-life. In the cross-sectional study of aging Manhattanites described earlier, the group mean difference in head circumference between elders with AD and those without was 0.57 cm, after adjustments were made for age, education, gender, and ethnicity. In that population, meanwhile, an increase in cross-sectional brain size of one cm2 associated with a delay in AD symptom onset of one third of a year [118].
36 weeks pre-natal gestational age and 12-months post-natal age appear to be critical windows for lead exposure to influence brain and head size development [123]. Lead exposures within this window that decrease brain size will also decrease brain reserve unless neuroplasticity mechanisms somehow compensate for early losses. Few studies have examined brain-size changes over time in lead-exposed children but MRI-based follow-up of the children in the Cincinnati Lead Study at age 20 reported the persistence of lead-related brain alterations, particularly decreased brain volume, decades after lead exposure [126]. This finding suggests that adults exposed to lead as children could have lower brain reserve than less exposed peers, putting them at greater risk of dementia.
Behavioral susceptibility
Following a “behavioral” mechanistic approach to considering lead’s long-term toxic effects, early disruption of the brain’s development may alter exposed children’s cognitive and behavioral development in ways that would later increase the risk of degenerative disease, specifically by reducing their capacity to sustain function amid the atrophy and neuronal loss associated with normal aging [109, 110]. Lead would be pro-degenerative through a behavioral mechanism by decreasing overall active “cognitive reserve” [112], and, possibly, by increasing health risk behaviors.
What is cognitive reserve – and does lead alter it?
The concept of cognitive reserve views brain function during cognition as a factor capable of altering an individual’s tolerance to brain pathology. Under this view, brain-damaged individuals with the same brain size and the same degree of brain pathology may still experience different levels of functional impairment as a result of differences in their brains’ cognitive efficiency, capacity, or flexibility [127]. Such functional reserve is considered modifiable and is believed to reflect the contribution of both genes and life experiences. Unlike brain reserve, cognitive reserve cannot be measured directly, but is instead typically measured through proxies believed to relate to an individual’s cognitive activity, such as intelligence, years of education, and the degree of intellectual complexity required by occupational tasks and leisure pursuits. The concept of cognitive reserve is supported by numerous studies finding lower risks of cognitive decline [115], AD [128–130], and dementia in PD [120] in individuals with higher cognitive reserve as measured by proxies like IQ, education and occupational attainment. The latent cognitive reserve construct has demonstrated both convergent and discriminant validity in multiethnic cohorts [131]. And, finally, adults exposed to lead have been found to suffer fewer cognitive deficits if they have higher cognitive reserve prior to exposure [132].
All evidence suggests that childhood lead exposure lowers cognitive reserve in exposed individuals. Even low-level (<7.5 µg/dL) early-life lead exposure has been linked to significantly lower child intellectual function, attention, focus, emotion regulation ability, and fine motor skills [14]. Young children with higher lead burdens have also been found to display greater hyperactivity, distractibility and antisocial behavior [133, 134]. Intellectual and behavioral deficits in lead-exposed children appear to persist into adulthood [135–137] and, in at least one midlife follow-up study, into middle-age [84].
Regardless of intellectual ability, lead exposed children appear to consistently under-perform in school and, on average, leave school earlier than less exposed peers [14]. This, in turn, may set up lead-exposed children for lower-socioeconomic-status jobs in adulthood with potentially lower intellectual demands [138]. In the one midlife follow-up study of a general population of lead-exposed children, those with higher lead exposures did, in fact, attain slightly lower-status occupations than their less exposed peers and, on average, than their own parents [84]. This outcome was partially but significantly mediated (40% of the effect) by cognitive decline following lead exposure.
If childhood lead exposure did not trigger degenerative brain disease directly, the known intellectual and behavioral consequences of such exposure would nevertheless make exposed individuals less tolerant of later brain pathology. Lowered cognitive reserve suggests that adults exposed to lead as children will be at greater risk of developing dementia regardless of their risk for pathology.
Finally, if lead-related child behavioral dysfunction were to result in the performance of greater health risk behaviors, such as substance abuse, unhealthy eating, or poor sleep hygiene, then lead-exposed individuals may experience greater rates of degenerative disease than their less exposed peers [139–141]. This physical link has not been evaluated, although there is some evidence of increased substance abuse in lead-exposed individuals [142], which is supported by limited experimental studies in mice [143, 144].
IMPLICATIONS FOR PUBLIC HEALTH AND POLICY
The accumulating research findings that suggest that childhood lead exposures may lead, directly or indirectly, to diagnosable neurodegenerative outcomes hold several implications for public health and policy.
Implications for the global burden of disease
Childhood lead exposures from the 1960s, 1970s, and 1980s could plausibly lead to a greater overall incidence of dementia in the U.S. and other developed countries in the coming years. Across the developed world, dementia rates have been in steady decline for at least the past thirty years, a phenomenon partially attributed to greater rates of educational attainment (cognitive reserve) among aging individuals and, additionally, to better treatment of cardiovascular disease, although the precise reasons behind the decline are not known [145–147]. Such declines likely do not, however, reflect the influence of historic child-hood lead exposures, which may have peaked in the U.S. around the mid-1970s [9, 13, 148].
Lead was first added to gasoline to improve engine performance in the early 1920s [9, 149]. From then onwards the use of leaded gasoline increased steadily in the U.S. until the mid 1970s, when the introduction of platinum catalytic converters, which are ruined by lead, triggered a phase-down in leaded gasoline use that was later accelerated in the early 1980s by U.S.-Environmental Protection Agency regulations responding to public health concerns. Although there was no routine population surveillance of American blood-lead levels before 1976, when lead testing was added to the second National Health and Nutrition Examination Survey (NHANES-II), analysis of historical trends in U.S. consumption of lead in gasoline and the levels of lead found in lake sediments, sphagnum moss, and sampled populations’ teeth enamel suggests that individuals now age 65 years and older (those considered in national dementia surveys) likely experienced childhood lead exposures similar to those of children born in the late 1980s, after lead had largely been phased out of use in gasoline [9, 16]. (From 1988–1991, the average blood-lead level for a child under the age of 5 years was 3.75 µg/dL, well below the current reference value) [13]. Americans now in their 40 s and 50 s, meanwhile, born in the 1960s and the 1970s, represent those with the greatest childhood lead exposures. From 1976–1980, the average blood-lead level for a child under the age of 5 years was 16.0 µg/dL, over three times the current reference value [148]. Figure 3 depicts the high, and declining, blood-lead levels recorded across the years of the NHANES-II, 1976–1980.
Fig. 3.
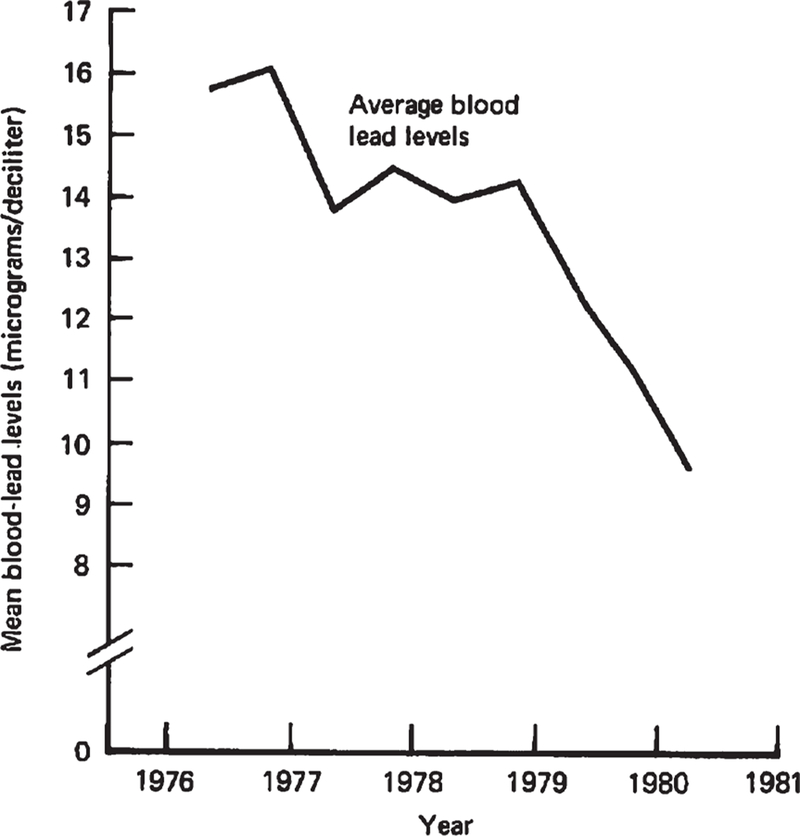
The average American’s blood-lead level in 1976 was 3 times greater than the current reference value for clinical attention (5 µg/dL). Surveillance of blood-lead levels across the U.S. began in 1976, with the second National Health and Nutrition Examination Survey (NHANES-II). At that time, according to NHANES-II estimates, the majority of Americans had blood-lead levels that are now deemed harmful; 85.0% of white children and 97.7% of black children aged 1 to 5 had blood-lead levels greater than 2 times the current reference value [14]. Reproduced from Annest et al. [148], US-Centers for Disease Control and Prevention.
The influence of historic childhood lead exposures on degenerative disease rates in the U.S. may not become apparent for another decade at least, as the children with the highest exposures enter the age at which degenerative disease endpoints begin to emerge, at and above age 65 years [150]. It is not clear what the magnitude of additional risk childhood lead exposure may confer on aging individuals, or indeed, if this additional risk may be offset by improvements in diet, education, cardiovascular health or physical activity that may otherwise be driving the current downward trends seen in degenerative disease rates. The decline in dementia rates witnessed over the past few decades is consistent, however, with there now being increasingly lower levels of lead in the environment. Both child and adult lead exposures have been dropping steadily in the U.S. since the late 1970s, following the removal of lead from gasoline and food cans, the general deindustrialization of the economy, and the gradual strengthening of air quality standards and pollution abatement technology. Indeed, the prevalence of American adults with elevated blood-lead levels (≥25 µg/dL) has declined nearly three-fold over the last twenty years (Fig. 4) [151]. Lower adult lead exposures could plausibly have contributed to recent declines in national dementia rates. They could also have contributed to a parallel trend seen, in at least one multi-decadal Swiss study, of lower rates of brain amyloid-β burden in elders autopsied across the years of 1972–2006 [152], a phenomenon likely unrelated to increases in cognitive reserve. (Declines in blood-lead levels in the Swiss population over the past three decades mirror those of the U.S. and the U.K.) [153]. Whether the aging of children highly exposed to lead will reverse these trends remains to be seen.
Fig. 4.
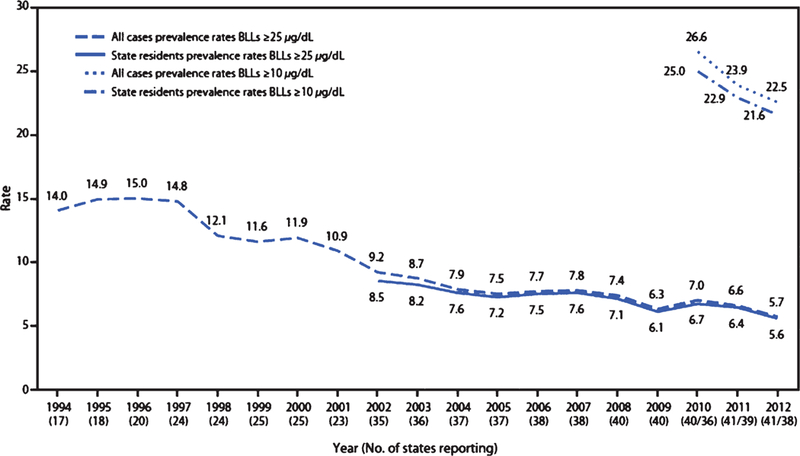
Adult blood-lead levels have continued to decline over the past two decades. This figure depicts the U.S. national prevalence rate (per 100,000 employed adults aged ≥16 years) of reported cases of elevated blood-lead levels (≥10 µg/dL and ≥25 µg/dL) by year from the State Adult Blood Epidemiology and Surveillance Programs, United States, 1994–2012. Reproduced from Alarcon et al. [151], US-Centers for Disease Control and Prevention.
Implications for social justice and community preparedness
While exposure to lead was once a ubiquitous experience across the U.S., the highest exposures were always concentrated among poor and minority groups living in large cities or near lead-emitting industries [11, 13, 154]. Low-income and minority communities are also, incidentally, those at the greatest risk for degenerative brain disease, a phenomenon currently attributed to differences in educational and occupational attainment (cognitive reserve), physical health, particularly diabetes and cardiovascular disease, and exposure to childhood adversity and stress [155–158]. Recent expert panel reviews have concluded that African-Americans are now roughly twice as likely to develop AD as non-Hispanic Whites, and Hispanics one and a half times as likely [157].
Should childhood lead exposures result in greater rates of dementia and adult brain disease, historic exposures will add to the already high disease burdens experienced in low income and minority communities. As these communities also experience the greatest obstacles to receiving degenerative disease diagnosis, treatment, and care [157, 159], the long-term burdens from childhood lead exposures may fall particularly hard on disadvantaged communities. This will, in turn, present additional barriers to upward social mobility, as family members with brain diseases like dementia carry significant emotional and financial costs. In 2016, nearly 16 million families provided over 18 billion hours in unpaid care for family members with dementia (roughly 22 hours a week on average) [157]. In national surveys, most unpaid care providers report high levels of emotional and physical stress, with half reporting having to cut back on spending or saving due to the cost of providing care [157].
In the future, public responses to community-level lead exposure events occurring in low-income or minority communities may have to consider expanded diagnostic, treatment, or caregiving opportunities for degenerative brain disease to avoid exaggerating existing inequalities in health and social outcomes decades later.
Implications for preventative medicine
For most Americans, lead exposure is a thing of the past. Yet millions with high exposures in childhood are now entering their fourth and fifth decade of life. Is it possible to intervene with these individuals now to lower disease risk in the coming years? No intervention studies have yet considered whether it is possible to improve neurological outcomes in adults exposed to lead in childhood, but targeting such individuals with concerted intervention could hold promise for improved population-level prevention of brain disease. A number of interventions exist, for example, that show encouraging results for improving cognitive and neurological outcomes in individuals known to be at elevated risk owing to other factors, such as APOE status and cardiovascular disease. These interventions warrant research attention for use in lead-exposed populations and are reviewed in greater detail in the following section on future research needs.
RESEARCH NEEDED TO FILL KEY KNOWLEDGE GAPS
The evidence reviewed in this report should be considered preliminary and merely suggestive for a number of reasons.
First, the extant literature suffers from a number of limitations common to toxicological studies. Primarily, despite a wealth of mechanistic information provided by decades of neurotoxicological studies of lead-exposed animals, the entwinement of lead exposure and socioeconomic status in most developed countries limits the full identification of lead’s impact on child and adult outcomes that are also ultimately entwined with socioeconomic status, including: intellectual ability, physical illness, and educational and occupational attainment. Nearly all observational lead-studies adjust statistically for possible confounding by socioeconomic status, but sampling bias, uninvestigated interaction effects, and residual confounding remain common threats to validity [160]. Additionally, both experimental and observational toxicological research suffers from an over-emphasis on significant findings [161], with negligible associations, particularly in studies examine low-level lead exposures, receiving far less attention and follow-up investigation [160].
Second, there are a number of research gaps that need to be filled before we can accurately characterize the potential neurodegenerative risks facing lead-exposed children. Notably, animal model evidence suggesting a link between early-life lead exposure and adult neurodegenerative disease requires confirmation from adult follow-up studies in lead-test child cohorts, such as those listed in Table 1. This is a critical step, as findings from research in animal models of neurodegenerative disease have generally failed to replicate at expected levels in studies in humans subjects, particularly when interventions have been examined [162, 163]. As it will be many years before lead-tested child cohorts are sufficiently aged for degenerative disease endpoints to emerge, however, waiting for full confirmation of animal model findings will be neither practical nor ethical. The window for possible pre-morbid intervention will close within the next two decades for the most highly exposed Americans –those born in the early 1970’s. Research questions that may be investigated now include those concerning the magnitude of disease risk, the nature of disease mechanisms and moderating factors, and the possibility of post-exposure intervention to limit disease development.
Research questions concerning disease risk
Does adult lead exposure increase risk for Alzheimer’s disease or other dementias?
Adult lead exposure has been linked to the risk of developing PD [51, 52, 164] and of experiencing accelerated cognitive decline, a risk factor for dementia [63–68]. No study appears to have yet examined AD or dementia risk directly. Longitudinal or cross-sectional studies in lead-tested elderly individuals that include dementia diagnoses and measurements of symptom severity could significantly extend the evidence base on the long-term risks facing lead-exposed individuals and provide increased confirmation that lead exposure may drive such outcomes. Bakulski et al. [31] discuss potential approaches to measuring lead-dementia associations in adults, including the possibility of simultaneous measurement of AD pathology and lead-load in tissue samples donated to AD Research Centers.
Are lead-exposed children at greater risk of heart or kidney disease in adulthood?
While adults exposed to lead develop hypertension and cardiovascular and kidney disease at greater rates [100–102, 107, 108], there have been no concerted evaluations of whether children exposed to lead are at elevated risk for these same diseases in adulthood. Recent long-term follow-ups in exposed young adults suggest that cardiovascular disease can emerge decades after lead exposure [105]. Whenever possible, follow-up studies in lead-tested children should evaluate risk for non-CNS physical disease outcomes, particularly cardiovascular disease and kidney disease. Harmful in their own right, such organ pathologies also increase the risk for degenerative brain disease.
What environmental or genetic factors influence individual variation in long-term outcomes following childhood lead exposure?
The most informative studies of lead toxicity include measures of susceptibility alongside measures of exposure and disease outcome [165].
On the genetic front, a number of polymorphisms have been identified that are believed to influence susceptibility to lead, including those that alter lead uptake, retention, and bioavailability (Table 2) [166]. Evidence of these genes’ role in lead toxi-codynamics comes from experimental studies using animal and cellular models to test hypotheses about molecular mechanisms [166] and, additionally, from large, genome-wide association studies, which have provided confirmation of a significant relationship between many of these genes and blood-lead levels [167, 168]. Thus far, only a few studies on lead-disease risk have included measures of genetic susceptibility. (Notable exceptions include studies of incident coronary heart disease [166], cognitive status [169, 170], and essential tremor [171] in older adults). Future studies on the long-term consequences of childhood lead exposure will be more informative for disease prevention if consideration can be given to also measuring genetic susceptibility. Many of the genes identified in lead toxicodynamics have also been implicated in degenerative disease risk, particularly APOE [172, 173], HMOX1 [174], and GST [175].
Table 2.
Genes with variants suspected to alter lead toxicodynamics
| Category of influence | Identified gene | Molecular activity coded for/possible lead interaction | Sample reference studies |
|---|---|---|---|
| May alter lead bioavailability | Hemochromatosis (HFE) | Cellular uptake of divalent metals. | [166, 212–216] |
| δ-aminolevulinic acid dehydratase (ALAD) | Heme synthesis/blood cell erythrocyte binding of lead. | [166, 168, 217–220] | |
| Vitamin D receptor (VDR) | Calcium homeostasis/lead retention in bone and blood. | [166, 221, 222] | |
| May alter lead toxic effect | Apolipoprotein E (APOE) | Lipid metabolism and antioxidative processes/magnification of lead cytotoxicity. | [223, 224] |
| Glutathione S-transferases (GSTs) | Detoxification catalyzation/reduction of lead-related oxidative stress. | [166, 225–227] | |
| Heme oxygenase-1 (HMOX1) | Heme degradation/alteration of lead-related inflammation and oxidative stress. | [166, 228] |
On the environmental front, psychosocial stressors and poverty have been proposed as modifiers of the long-term effects of early-life lead exposure [176]. In lead-exposed mice the presence of prenatal or postnatal stressors appears to amplify lead-related alterations in learning, impulsivity, HPA-axis activation, and dopaminergic and glutamatergic CNS dysregulation [177–180]. In humans, meanwhile, exposure to psychosocial stressors has been found to modify lead-associations with hypertension, cognitive impairment, and mental status in older adults and elderly men [181–183]. Lead impacts on child cognitive development have also been reported to be greater in children with lower socioeconomic status backgrounds [184–188]. Future studies linking childhood lead exposure to adult neurodegenerative disease should examine the role of socioeconomic status and recent or childhood psychosocial stressors as potential effect modifiers in addition to potential confounding variables [189].
Research questions concerning disease mechanisms
To what extent does childhood-lead stored in bone threaten adult health?
Lead mobilization from bone has been recorded in pregnant women, nursing mothers, peri and post-menopausal women, and in aged men and women with osteoporosis. Although there is no level of lead exposure considered “safe,” and mobilized lead has been found to reach significant levels for those highly exposed in childhood [96, 190, 191], the contribution of mobilized lead to adult disease has not been evaluated directly. To what extent does this physical mechanism explain the link between early-life lead exposure and later-life disease and dysfunction?
These questions are complicated by the possibility that childhood lead exposure may exert delayed harm through “silent” epigenetic dysregulation and through indirect effects on lifetime brain and cognitive reserve. Nevertheless, the primary question of mobilized lead’s influence on disease risk may be answered through experimental animal studies that compare the effects of early versus late-life lead exposure in the release rate of lead from bone and the magnitude of such release necessary for pathology to emerge. Differences in the mobilization of lead among similarly exposed study animals may allow for the teasing apart of disease risk resulting from delayed/silent effects versus acute effects from mobilized lead. Similar comparisons may be possible through follow-up studies in adult women entering menopause who were lead tested as children (Table 1).
To what extent do lead-related alterations of brain/cognitive reserve influence neurodegenerative disease risk following childhood exposures?
The potential of childhood lead exposure to significantly alter child levels of brain reserve (measured directly through head circumference and brain volume) and cognitive reserve (measured through proxies like intellectual ability and educational attainment) is well established. More evidence is needed, however, on the persistence of these alterations into midlife and old age. Structural and functional neuroimaging of lead-tested child cohorts (Table 1) at midlife could provide confirmation of the long-term persistence of neural and functional abnormalities and indicate differential risk of later susceptibility to degenerative disease. Reports from imaging studies involving members of the Cincinnati Lead Study suggest that brain volume losses and other structural abnormalities associated with childhood lead exposure persist to at least young adulthood [126].
No studies have yet directly considered the role of altered susceptibility to age-related brain disease (e.g., altered brain and cognitive reserve) as a potential mediator of the effects of early-life lead on late-life degenerative disease and cognitive decline. Neuroimaging of lead-tested child cohorts could provide such information when disease end-points emerge over the coming decades. In the nearer term, animal model studies could test this potential mechanism by incorporating measures of brain and cognitive reserve into studies linking lead exposure to disease pathology. Brain reserve can be measured directly, as macro or microstructural anatomical differences, while cognitive reserve may be measured through performance on learning or cognitive tasks adjusted for measures of brain pathology [127].
Do humans experience the same epigenetic changes following childhood lead exposure as those seen in animals exposed to lead early in life?
A wealth of studies in animal models have reported pathological alterations of epigenetic regulation in the brains of animals exposed to lead early in life [87]. In at least one study these alterations were found to remain dormant until midlife, after which they induced pathological AD-like protein accumulation [88]. If these animal findings hold in humans, children exposed to lead may experience pro-degenerative epigenetic alterations in adulthood and old age. This theory requires testing. To date only one follow-up study in a cohort of lead-tested children has examined epigenetic markers [83]. This small (n = 39) exploratory study reported a number of gene expression differences among individuals with differential lead exposure in childhood, many of which have been implicated in AD onset and pathology. These findings warrant replication in a larger sample. Based on findings from animal studies, we would predict lead-exposed children would show significant gene expression changes by midlife in genes related to neurodevelopment in early-life and implicated in degenerative disease in late-life. Kovacs et al. [5] provide a helpful review of the neurodevelopmental pathways thus far implicated in degenerative disease which may show dysregulation following early insult.
Research questions concerning post-exposure interventions
Are there interventions for lead-exposed children that can delay or limit long-term neurodegenerative consequences? As animal model studies continue to illuminate the pathways linking early-life lead exposure to adult neurodegenerative disease, it may be possible to evaluate whether, specifically, midlife or later interventions can improve outcomes in lead exposed subjects. Such evidence would inform studies on prevention and reversibility in lead-exposed children now entering midlife and old age and could inform studies on degenerative disease prevention more generally.
Several early-life interventions show promise for potentially limiting harm in lead-exposed children. Vitamin and mineral supplementation has been known for some time to limit child susceptibility to lead [192]. More recently, flavonol-rich dark cocoa supplementation was found to improve cognition and neuroinflammation markers in air pollution-exposed children [193], suggesting that adding chocolate to the diets of lead-exposed children could potentially provide some measure of neuroprotection –a speculation that requires testing. And, in rats, enriched cage environments have been reported to limit or reverse some of the developmental injuries from early lead exposure [194, 195], suggesting that children exposed to lead may see ameliorative benefits from enhanced educational opportunities or enriched home environments.
For those Americans entering midlife and old age now, a primary question is whether intervention may improve outcomes decades after lead exposure has ceased. Analysis of known modifiable risk factors for dementia by the Lancet Commission on Dementia Prevention, Intervention, and Care may shed some light on this question. The Commission estimated last year that one third of all dementia cases could be prevented if known adult risk factors, including physical inactivity, midlife hypertension, diabetes, depression, and obesity, were eliminated [196]. A recent U.S.-National Academies of Sciences, Engineering and Medicine (NASEM) committee commissioned by the U.S.-National Institute on Aging identified three specific interventions against cognitive decline and dementia that are currently supported by “encouraging although inconclusive” evidence (Table 3) and eight promising interventions for which there was not enough evidence to determine impact [197].
Table 3.
The US-National Academies of Sciences, Engineering and Medicine (NASEM) committee conclusions on interventions against cognitive decline and dementia [197]
| Level of evidentiary support | Specific Intervention |
|---|---|
| Encouraging although inconclusive evidence | Cognitive training, Blood pressure management for people with hypertension, and Increased physical activity. |
| Not enough evidence to determine impact | Diabetes treatment, Depression treatment, Dietary interventions, Lipid-lowering treatment/statins, Sleep quality interventions, Social engagement interventions, and Vitamin B12 plus folic acid supplementation. |
A full review of possible interventions against degenerative brain disease is outside the scope of this report. However, the best evidence now suggests that multimodal intervention, including a combination of physical, nutritional, social, and cognitive interventions, may prove the most effective for altering long-term risks [196]. It should be possible to test many of these interventions in experimental animal studies to see if they may reverse some of the long-term consequences of early lead exposure. While many of these interventions are not easily adaptable for use in animal studies, those that are, including increased physical activity, sleep quality adjustments, and some dietary interventions, could, in theory, counteract or limit long-term harm following early lead exposure. Rodents allowed to run show enhanced learning ability, for example, with accompanying hippocampal neurogenesis [198, 199]. Early physical activity in mice has, additionally, been linked to life-long neurogenic benefits [200].
Finally, calcium supplementation has been shown to reduce bone loss in postmenopausal women [201] and bone-lead remobilization in pregnant and nursing mothers [202]. This suggests that such supplementation could be effective against bone-lead remobilization in aging adults. It should be possible to test these and other interventions in animal models and adult follow-ups in lead tested child cohorts.
CONCLUSIONS
Lead is a profound neurotoxin that disrupts child brain development in myriad ways. Follow-up studies in lead-exposed children suggest that early lead-related deficits in brain health and cognitive function persist and may even enhance by adulthood. In adults exposed to lead, toxic injuries to the brain appear to be pro-degenerative, leading to faster rates of cognitive decline, the presence of biomarkers of AD, and a greater odds of developing PD. Studies in animal models, meanwhile, implicate early lead exposure in a host of AD-like pathologic changes detectible in old age, changes which appear to be driven, in part, by epigenetic modifications that trigger or exacerbate pathological protein accumulation and undermine typical compensatory responses. Under-studied but plausible biological mechanisms also link childhood lead exposure to adult degenerative disease through potentially-mediating cardiovascular and kidney pathologies and through the potential remobilization during menopause and osteoporosis of lead stored in bone. Lead-related deficits in brain (structural) and cognitive (functional) reserve also suggest greater susceptibility to normal age-related cognitive loses for aging lead-exposed children.
Taken together the accumulated evidence suggests that America’s most highly-lead exposed children, who were born in the 1960s, 1970s, and 1980s, may be at greater risk of neurodegenerative disease as they age. Such risk could act to retard or even reverse the improvements in dementia rates seen in developed countries over the past few decades—improvements that could have reflected, in part, the results of lowered lead exposures in aging adults. These risks will also be borne most acutely by minority and socially disadvantaged populations, who experienced higher lead burdens in childhood and now see the greatest obstacles to receiving dementia diagnosis and care.
Better characterization of the magnitude and nature of the risks facing adults exposed to lead as children will inform efforts to intervene with at-risk individuals before disease end points emerge. Yet the window for intervention grows smaller every day, and interventions that show promise already exist. Now is the time to focus increased attention on this issue. While we seek to safeguard today’s children from the harms of lead exposure, let us not forget yesterday’s children, whose needs may grow in the coming years.
ACKNOWLEDGMENTS
This research received support from the U.S.-National Institute on Aging grant R01 AG032282 and the U.S.-National Institute of Environmental Health Sciences grant F31 ES029358.
The author thanks Dr. Terrie Moffitt, Dr. Avshalom Caspi, Dr. Edward Levin, Dr. Gregory Samanez-Larkin, and Dr. Amber Beckley for their review and guidance on this manuscript. He also thanks Dr. Jean Harry, Dr. David Bellinger, Dr. Marc Weisskopf, and Dr. Nasser Zawia for their helpful correspondence.
Footnotes
The author’s disclosure is available online https://www.j-alz.com/manuscript-disclosures/18-0267r1).
REFERENCES
- [1].Jagust W (2016) Early life sets the stage for aging. Proc Natl Acad SciUSA 113, 9148–9150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Borenstein AR, Copenhaver CI, Mortimer JA (2006) Early-life risk factors for Alzheimer disease. Alzheimer Dis Assoc Disord 20, 63–72. [DOI] [PubMed] [Google Scholar]
- [3].Mehler MF, Gokhan S (2000) Mechanisms underlying neural cell death in neurodegenerative diseases: Alterations of a developmentally-mediated cellular rheostat. Trends Neurosci 23, 599–605. [DOI] [PubMed] [Google Scholar]
- [4].Heindel JJ, Vandenberg LN (2015) Developmental origins of health and disease: A paradigm for understanding disease rtiology and prevention. Curr Opin Pediatr 27, 248–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kovacs GG, Adle-Biassette H, Milenkovic I, Cipriani S, van Scheppingen J, Aronica E (2014) Linking pathways in the developing and aging brain with neurodegeneration. Neuroscience 269, 152–172. [DOI] [PubMed] [Google Scholar]
- [6].Walhovd KB, Krogsrud SK, Amlien IK, Bartsch H, Bjørnerud A, Due-Tønnessen P, Grydeland H, Hagler DJ, Håberg AK, Kremen WS, Ferschmann L, Nyberg L, Panizzon MS, Rohani DA, Skranes J, Storsve AB, Sølsnes AE, Tamnes CK, Thompson WK, Reuter C, Dale AM, Fjell AM (2016) Neurodevelopmental origins of lifespan changes in brain and cognition. Proc Natl Acad SciUSA 113, 9357–9362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Rahimi J, Kovacs GG (2014) Prevalence of mixed pathologies in the aging brain. Alzheimers Res Ther 6, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Brookmeyer R, Johnson E, Ziegler-Graham K, Arrighi HM (2007) Forecasting the global burden of Alzheimer’s disease. Alzheimers Dement 3, 186–191. [DOI] [PubMed] [Google Scholar]
- [9].Nriagu JO (1990) The rise and fall of leaded gasoline. Sci Total Environ 92, 13–28. [Google Scholar]
- [10].Annest JL, Mahaffey KR, Cox DH, Roberts J (1982) Blood lead levels for persons 6 months-74 years of age: United States, 1976–80. Adv Data, 1–23. [PubMed]
- [11].Moody H, Darden JT, Pigozzi BW (2016) The racial gap in childhood blood lead levels related to socioeconomic position of residence in metropolitan Detroit. Sociol Race Ethn 2, 200–218. [Google Scholar]
- [12].Brody DJ, Pirkle JL, Kramer RA, Flegal KM, Matte TD, Gunter EW, Paschal DC (1994) Blood lead levels in the US population: Phase 1 of the Third National Health and Nutrition Examination Survey (NHANES III, 1988 to 1991). JAMA 272, 277–283. [DOI] [PubMed] [Google Scholar]
- [13].Pirkle JL, Brody DJ, Gunter EW, Kramer RA, Paschal DC, Flegal KM, Matte TD (1994) The decline in blood lead levels in the United States: The National Health and Nutrition Examination Surveys (NHANES). JAMA 272, 284–291. [PubMed] [Google Scholar]
- [14].Bellinger DC (2008) Very low lead exposures and children’s neurodevelopment. Curr Opin Pediatr 20, 172–177. [DOI] [PubMed] [Google Scholar]
- [15].Bellinger DC (2017) Childhood lead exposure and adult outcomes. JAMA 317, 1219–1220. [DOI] [PubMed] [Google Scholar]
- [16].Robbins N, Zhang Z-F, Sun J, Ketterer ME, Lalumandier JA, Shulze RA (2010) Childhood lead exposure and uptake in teeth in the Cleveland area during the era of leaded gasoline. Sci Total Environ 408, 4118–4127. [DOI] [PubMed] [Google Scholar]
- [17].Seeley WW, Crawford RK, Zhou J, Miller BL, Greicius MD (2009) Neurodegenerative diseases target large-scale human brain networks. Neuron 62, 42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ahmed RM, Devenney EM, Irish M, Ittner A, Naismith S, Ittner LM, Rohrer JD, Halliday GM, Eisen A, Hodges JR, Kiernan MC (2016) Neuronal network disintegration: Common pathways linking neurodegenerative diseases. J Neurol Neurosurg Psychiatry 87, 1234–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].James BD, Wilson RS, Boyle PA, Trojanowski JQ, Bennett DA, Schneider JA (2016) TDP-43 stage, mixed pathologies, and clinical Alzheimer’s-type dementia. Brain 139, 2983–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Schneider JA, Arvanitakis Z, Leurgans SE, Bennett DA (2009) The neuropathology of probable Alzheimer disease and mild cognitive impairment. Ann Neurol 66, 200–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kamel F, Umbach DM, Hu H, Munsat TL, Shefner JM, Taylor JA, Sandler DP (2005) Lead exposure as a risk factor for amyotrophic lateral sclerosis. Neurodegener Dis 2, 195–201. [DOI] [PubMed] [Google Scholar]
- [22].Fang F, Kwee LC, Allen KD, Umbach DM, Ye W, Watson M, Keller J, Oddone EZ, Sandler DP, Schmidt S, Kamel F (2010) Association between blood lead and the risk of amyotrophic lateral sclerosis. Am J Epidemiol 171, 1126–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Li Y, Li Y, Li X, Zhang S, Zhao J, Zhu X, Tian G (2017) Head injury as a risk factor for dementia and Alzheimer’s disease: A systematic review and meta-analysis of 32 observational studies. PLoS One 12, e0169650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Stern MB (1991) Head trauma as a risk factor for Parkinson’s disease. Mov Disord 6, 95–97. [DOI] [PubMed] [Google Scholar]
- [25].Gavett BE, Stern RA, Cantu RC, Nowinski CJ, McKee AC (2010) Mild traumatic brain injury: A risk factor for neurodegeneration. Alzheimers Res Ther 2, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Chen C-Y, Hung H-J, Chang K-H, Hsu CY, Muo C-H, Tsai C-H, Wu T-N (2017) Long-term exposure to air pollution and the incidence of Parkinson’s disease: A nested case-control study. PLoS One 12, e0182834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Power MC, Adar SD, Yanosky JD, Weuve J (2016) Exposure to air pollution as a potential contributor to cognitive function, cognitive decline, brain imaging, and dementia: A systematic review of epidemiologic research. Neurotoxicology 56, 235–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Jung C-R, Lin Y-T, Hwang B-F (2015) Ozone, particulate matter, and newly diagnosed Alzheimer’s disease: A population-based cohort study in Taiwan. J Alzheimers Dis 44, 573–584. [DOI] [PubMed] [Google Scholar]
- [29].Salama M, Arias-Carrión O (2011) Natural toxins implicated in the development of Parkinson’s disease. Ther Adv Neurol Disord 4, 361–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wirdefeldt K, Adami H, Cole P, Trichopoulos D, Mandel J (2011) Epidemiology and etiology of Parkinson’s disease: A review of the evidence. Eur J Epidemiol 26, S1–58. [DOI] [PubMed] [Google Scholar]
- [31].Bakulski KM, Rozek LS, Dolinoy DC, Paulson HL, Hu H (2012) Alzheimer’s disease and environmental exposure to lead: The epidemiologic evidence and potential role of epigenetics. Curr Alzheimer Res 9, 563–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Gunnarsson L-G, Bodin L (2017) Parkinson’s disease and occupational exposures: A systematic literature review and meta-analyses. Scand J Work Environ Health 43, 197–209. [DOI] [PubMed] [Google Scholar]
- [33].Machado A, Herrera AJ, de Pablos RM, Espinosa-Oliva AM, Sarmiento M, Ayala A, Venero JL, Santiago M, Villara´n RF, Delgado-Cortés MJ, Argüelles S, Cano J (2014) Chronic stress as a risk factor for Alzheimer’s disease. Rev Neurosci 25, 785–804. [DOI] [PubMed] [Google Scholar]
- [34].Bressler JP, Goldstein GW (1991) Mechanisms of lead neurotoxicity. Biochem Pharmacol 41, 479–484. [DOI] [PubMed] [Google Scholar]
- [35].Mason LH, Harp JP, Han DY (2014) Pb neurotoxicity: Neuropsychological effects of lead toxicity. BioMed Res Int 2014, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lidsky TI, Schneider JS (2003) Lead neurotoxicity in children: Basic mechanisms and clinical correlates. Brain 126, 5–19. [DOI] [PubMed] [Google Scholar]
- [37].Leggett RW (1993) An age-specific kinetic model of lead metabolism in humans. Environ Health Perspect 101, 598–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lefauconnier JM, Bernard G, Mellerio F, Sebille A, Cesarini E (1983) Lead distribution in the nervous system of 8-month-old rats intoxicated since birth by lead. Experientia 39, 1030–1031. [DOI] [PubMed] [Google Scholar]
- [39].Grandjean P (1978) Regional distribution of lead in human brains. Toxicol Lett 2, 65–69. [Google Scholar]
- [40].Gilbert SG, Rice DC (1987) Low-level lifetime lead exposure produces behavioral toxicity (spatial discrimination reversal) in adult monkeys. Toxicol Appl Pharmacol 91, 484–490. [DOI] [PubMed] [Google Scholar]
- [41].Rice DC, Gilbert SG (1990) Sensitive periods for lead-induced behavioral impairment (nonspatial discrimination reversal) in monkeys. Toxicol Appl Pharmacol 102, 101–109. [DOI] [PubMed] [Google Scholar]
- [42].Wu J, Basha MR, Brock B, Cox DP, Cardozo-Pelaez F, McPherson CA, Harry J, Rice DC, Maloney B, Chen D, Lahiri DK, Zawia NH (2008) Alzheimer’s disease (AD) like pathology in aged monkeys following infantile exposure to environmental metal lead (Pb): Evidence for a developmental origin and environmental link for AD. J Neurosci 28, 3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Bihaqi SW, Zawia NH (2013) Enhanced taupathy and AD-like pathology in aged primate brains decades after infantile exposure to Lead (Pb). Neurotoxicology 39, 95–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Bihaqi SW, Bahmani A, Subaiea GM, Zawia NH (2014) Infantile exposure to lead and late-age cognitive decline: Relevance to AD. Alzheimers Dement 10, 187–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Bihaqi SW, Bahmani A, Adem A, Zawia NH (2014) Infantile postnatal exposure to lead (Pb) enhances tau expression in the cerebral cortex of aged mice: Relevance to AD. Neurotoxicology 44, 114–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Behl M, Zhang Y, Shi Y, Cheng J, Du Y, Zheng W (2010) Lead-induced accumulation of beta-amyloid in the choroid plexus: Role of low density lipoprotein receptor protein-1 and protein kinase C. Neurotoxicology 31, 524–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Behl M, Zhang Y, Monnot AD, Jiang W, Zheng W (2009) Increased beta-amyloid levels in the choroid plexus following lead exposure and the involvement of low-density lipoprotein receptor protein-1. Toxicol Appl Pharmacol 240, 245–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Bolin CM, Basha R, Cox D, Zawia NH, Maloney B, Lahiri DK, Cardozo-Pelaez F (2006) Exposure to lead and the developmental origin of oxidative DNA damage in the aging brain. FASEB J 20, 788–790. [DOI] [PubMed] [Google Scholar]
- [49].Dosunmu R, Alashwal H, Zawia NH (2012) Genome-wide expression and methylation profiling in the aged rodent brain due to early-life Pb exposure and its relevance to aging. Mech Ageing Dev 133, 435–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Gu H, Robison G, Hong L, Barrea R, Wei X, Farlow MR, Pushkar YN, Du Y, Zheng W (2012) Increased β-amyloid deposition in Tg-SWDI transgenic mouse brain following in vivo lead exposure. Toxicol Lett 213, 211–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Coon S, Stark A, Peterson E, Gloi A, Kortsha G, Pounds J, Chettle D, Gorell J (2006) Whole body lifetime occupational lead exposure and risk of Parkinson’s disease. Environ Health Perspect 114, 1872–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Weisskopf MG, Weuve J, Nie H, Saint-Hilaire M-H, Sudarsky L, Simon DK, Hersh B, Schwartz J, Wright RO, Hu H (2010) Association of cumulative lead exposure with Parkinson’s disease. Environ Health Perspect 118, 1609–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Gorell JM, Johnson CC, Rybicki BA, Peterson EL, Kortsha GX, Brown GG, Richardson RJ (1997) Occupational exposures to metals as risk factors for Parkinson’s disease. Neurology 48, 650–658. [DOI] [PubMed] [Google Scholar]
- [54].Seidler A, Hellenbrand W, Robra BP, Vieregge P, Nischan P, Joerg J, Oertel WH, Ulm G, Schneider E (1996) Possible environmental, occupational, and other etiologic factors for Parkinson’s disease: A case-control study in Germany. Neurology 46, 1275–1284. [DOI] [PubMed] [Google Scholar]
- [55].Firestone JA, Lundin JI, Powers KM, Smith-Weller T, Franklin GM, Swanson PD, Longstreth WT, Checkoway H (2010) Occupational factors and risk of Parkinson’s disease: A population-based case-control study. Am J Ind Med 53, 217–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Palacios N, Fitzgerald K, Roberts AL, Hart JE, Weisskopf MG, Schwarzschild MA, Ascherio A, Laden F (2014) A prospective analysis of airborne metal exposures and risk of Parkinson disease in the nurses’ health study cohort. Environ Health Perspect 122, 933–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Willis AW, Evanoff BA, Lian M, Galarza A, Wegrzyn A, Schootman M, Racette BA (2010) Metal emissions and urban incident Parkinson disease: A community health study of Medicare beneficiaries by using geographic information systems. Am J Epidemiol 172, 1357–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Khalil N, Morrow LA, Needleman H, Talbott EO, Wilson JW, Cauley JA (2009) Association of cumulative lead and neurocognitive function in an occupational cohort. Neuropsychology 23, 10–19. [DOI] [PubMed] [Google Scholar]
- [59].Weuve J, Korrick SA, Weisskopf MG, Weisskopf MA, Ryan LM, Schwartz J, Nie H, Grodstein F, Hu H (2009) Cumulative exposure to lead in relation to cognitive function in older women. Environ Health Perspect 117, 574–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Shih RA, Glass TA, Bandeen-Roche K, Carlson MC, Bolla KI, Todd AC, Schwartz BS (2006) Environmental lead exposure and cognitive function in community-dwelling older adults. Neurology 67, 1556–1562. [DOI] [PubMed] [Google Scholar]
- [61].Grashow R, Spiro A, Taylor KM, Newton K, Shrairman R, Landau A, Sparrow D, Hu H, Weisskopf M (2013) Cumulative lead exposure in community-dwelling adults and fine motor function: Comparing standard and novel tasks in the VA Normative Aging Study. Neurotoxicology 35, 154–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Weisskopf MG, Proctor SP, Wright RO, Schwartz J, Spiro A, Sparrow D, Nie H, Hu H (2007) Cumulative lead exposure and cognitive performance among elderly men. Epidemiology 18, 59–66. [DOI] [PubMed] [Google Scholar]
- [63].Bandeen-Roche K, Glass TA, Bolla KI, Todd AC, Schwartz BS (2009) The longitudinal association of cumulative lead dose with cognitive function in community-dwelling older adults. Epidemiology 20, 831–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Shih RA, Hu H, Weisskopf MG, Schwartz BS (2007) Cumulative lead dose and cognitive function in adults: A review of studies that measured both blood lead and bone lead. Environ Health Perspect 115, 483–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Weisskopf MG, Wright RO, Schwartz J, Spiro A, Sparrow D, Aro A, Hu H (2004) Cumulative lead exposure and prospective change in cognition among elderly men: The VA Normative Aging Study. Am J Epidemiol 160, 1184–1193. [DOI] [PubMed] [Google Scholar]
- [66].Power MC, Korrick S, Tchetgen Tchetgen EJ, Nie LH, Grodstein F, Hu H, Weuve J, Schwartz J, Weisskopf MG (2014) Lead exposure and rate of change in cognitive function in older women. Environ Res 129, 69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Schwartz BS, Lee B-K, Bandeen-Roche K, Stewart W, Bolla K, Links J, Weaver V, Todd A (2005) Occupational lead exposure and longitudinal decline in neurobehavioral test scores. Epidemiology 16, 106–113. [DOI] [PubMed] [Google Scholar]
- [68].Schwartz BS, Stewart WF, Bolla KI, Simon PD, Bandeen-Roche K, Gordon PB, Links JM, Todd AC (2000) Past adult lead exposure is associated with longitudinal decline in cognitive function. Neurology 55, 1144–1150. [DOI] [PubMed] [Google Scholar]
- [69].Wilson RS, Leurgans SE, Boyle PA, Bennett DA (2011) Cognitive decline in prodromal Alzheimer disease and mild cognitive impairment. Arch Neurol 68, 351–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Amieva H, Jacqmin-Gadda H, Orgogozo J-M, Le Carret N, Helmer C, Letenneur L, Barberger-Gateau P, Fabrigoule C, Dartigues J-F (2005) The 9 year cognitive decline before dementia of the Alzheimer type: A prospective population-based study. Brain 128, 1093–1101. [DOI] [PubMed] [Google Scholar]
- [71].Elias MF, Beiser A, Wolf PA, Au R, White RF, D’Agostino RB (2000) The preclinical phase of alzheimer disease: A 22-year prospective study of the Framingham Cohort. Arch Neurol 57, 808–813. [DOI] [PubMed] [Google Scholar]
- [72].Stewart WF, Schwartz BS, Davatzikos C, Shen D, Liu D, Wu X, Todd AC, Shi W, Bassett S, Youssem D (2006) Past adult lead exposure is linked to neurodegeneration measured by brain MRI. Neurology 66, 1476–1484. [DOI] [PubMed] [Google Scholar]
- [73].Weisskopf MG, Hu H, Sparrow D, Lenkinski RE, Wright RO (2007) Proton magnetic resonance spectroscopic evidence of glial effects of cumulative lead exposure in the adult human hippocampus. Environ Health Perspect 115, 519–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Hsieh T-J, Chen Y-C, Li C-W, Liu G-C, Chiu Y-W, Chuang H-Y (2009) A proton magnetic resonance spectroscopy study of the chronic lead effect on the basal ganglion and frontal and occipital lobes in middle-age adults. Environ Health Perspect 117, 941–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Jiang Y-M, Long L-L, Zhu X-Y, Zheng H, Fu X, Ou S-Y, Wei D-L, Zhou H-L, Zheng W (2008) Evidence for altered hippocampal volume and brain metabolites in workers occupationally exposed to lead: A study by magnetic resonance imaging and 1H magnetic resonance spectroscopy. Toxicol Lett 181, 118–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Londono AC, Castellanos FX, Arbelaez A, Ruiz A, Aguirre-Acevedo DC, Richardson AM, Easteal S, Lid-bury BA, Arcos-Burgos M, Lopera F (2014) An 1H-MRS framework predicts the onset of Alzheimer’s disease symptoms in PSEN1 mutation carriers. Alzheimers Dement 10, 552–561. [DOI] [PubMed] [Google Scholar]
- [77].Mueller SG, Schuff N, Yaffe K, Madison C, Miller B, Weiner MW (2010) Hippocampal atrophy patterns in mild cognitive impairment and Alzheimer’s disease. Hum Brain Mapp 31, 1339–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Henneman WJP, Sluimer JD, Barnes J, van der Flier WM, Sluimer IC, Fox NC, Scheltens P, Vrenken H, Barkhof F (2009) Hippocampal atrophy rates in Alzheimer disease. Neurology 72, 999–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Haraguchi T, Ishizu H, Takehisa Y, Kawai K, Yokota O, Terada S, Tsuchiya K, Ikeda K, Morita K, Horike T, Kira S, Kuroda S (2001) Lead content of brain tissue in diffuse neurofibrillary tangles with calcification (DNTC): The possibility of lead neurotoxicity. Neuroreport 12, 3887–3890. [DOI] [PubMed] [Google Scholar]
- [80].Niklowitz WJ, Mandybur TI (1975) Neurofibrillary changes following childhood lead encephalopathy. J Neuropathol Exp Neurol 34, 445–455. [DOI] [PubMed] [Google Scholar]
- [81].Niklowitz WJ (1975) Neurofibrillary changes after acute experimental lead poisoning. Neurology 25, 927–934. [DOI] [PubMed] [Google Scholar]
- [82].White RF, Diamond R, Proctor S, Morey C, Hu H (1993) Residual cognitive deficits 50 years after lead poisoning during childhood. Br J Ind Med 50, 613–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Mazumdar M, Xia W, Hofmann O, Gregas M, Sui SH, Hide W, Yang T, Needleman HL, Bellinger DC (2012) Prenatal lead levels, plasma amyloid β levels, and gene expression in young adulthood. Environ Health Perspect 120, 702–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Reuben A, Caspi A, Belsky DW, Broadbent J, Harrington H, Sugden K, Houts RM, Ramrakha S, Poulton R, Moffitt TE (2017) Association of childhood blood lead levels with cognitive function and socioeconomic status at age 38 years and with IQ change and socioeconomic mobility between childhood and adulthood. JAMA 317, 1244–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Eid A, Zawia N (2016) Consequences of lead exposure, and it’s emerging role as an epigenetic modifier in the aging brain. Neurotoxicology 56, 254–261. [DOI] [PubMed] [Google Scholar]
- [86].Rahman A, Brew BJ, Guillemin GJ (2011) Lead dysregulates serine/threonine protein phosphatases in human neurons. Neurochem Res 36, 195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Bihaqi SW, Zawia NH (2012) Alzheimer’s disease biomarkers and epigenetic intermediates following exposure to Pb in vitro. Curr Alzheimer Res 9, 555–562. [DOI] [PubMed] [Google Scholar]
- [88].Basha MR, Wei W, Bakheet SA, Benitez N, Siddiqi HK, Ge Y-W, Lahiri DK, Zawia NH (2005) The fetal basis of amyloidogenesis: Exposure to lead and latent overexpression of amyloid precursor protein and beta-amyloid in the aging brain. J Neurosci 25, 823–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Huang H, Bihaqi SW, Cui L, Zawia NH (2011) In vitro Pb exposure disturbs the balance between Aβ production and elimination: The role of AβPP and neprilysin. Neurotoxicology 32, 300–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Eid A, Bihaqi SW, Renehan WE, Zawia NH (2016) Developmental lead exposure and lifespan alterations in epigenetic regulators and their correspondence to biomarkers of Alzheimer’s disease. Alzheimers Dement (Amst) 2, 123–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Giedraitis V, Sundelüf J, Irizarry MC, Gårevik N, Hyman BT, Wahlund L-O, Ingelsson M, Lannfelt L (2007) The normal equilibrium between CSF and plasma amyloid beta levels is disrupted in Alzheimer’s disease. Neurosci Lett 427, 127–131. [DOI] [PubMed] [Google Scholar]
- [92].Janelidze S, Stomrud E, Palmqvist S, Zetterberg H, Westen D van, Jeromin A, Song L, Hanlon D, Hehir CAT, Baker D, Blennow K, Hansson O (2016) Plasma β-amyloid in Alzheimer’s disease and vascular disease. Sci Rep 6, srep26801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Rabinowitz MB (1991) Toxicokinetics of bone lead. Environ Health Perspect 91, 33–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].ATSDR (Agency for Toxic Substances and Disease Registry) (2007) Toxicological profile for lead, U.S. Centers for Disease Control, Atlanta, GA. [Google Scholar]
- [95].Hu H, Rabinowitz M, Smith D (1998) Bone lead as a biological marker in epidemiologic studies of chronic toxicity: Conceptual paradigms. Environ Health Perspect 106, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Silbergeld EK, Schwartz J, Mahaffey K (1988) Lead and osteoporosis: Mobilization of lead from bone in postmenopausal women. Environ Res 47, 79–94. [DOI] [PubMed] [Google Scholar]
- [97].Gulson B, Mizon K, Korsch M, Taylor A (2016) Revisiting mobilisation of skeletal lead during pregnancy based on monthly sampling and cord/maternal blood lead relationships confirm placental transfer of lead. Arch Toxicol 90, 805–816. [DOI] [PubMed] [Google Scholar]
- [98].Smith DR, Osterloh JD, Flegal AR (1996) Use of endogenous, stable lead isotopes to determine release of lead from the skeleton. Environ Health Perspect 104, 60–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Vaziri ND (2008) Mechanisms of lead-induced hypertension and cardiovascular disease. Am J Physiol Heart Circ Physiol 295, H454–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Navas-Acien A, Guallar E, Silbergeld EK, Rothenberg SJ (2007) Lead exposure and cardiovascular disease—A systematic review. Environ Health Perspect 115, 472–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Weisskopf M, Sparrow D, Hu H, Power MC (2015) Biased exposure–health effect estimates from selection in cohort studies: Are environmental studies at particular risk? Environ Health Perspect 123, 1113–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Vaziri ND, Sica DA (2004) Lead-induced hypertension: Role of oxidative stress. Curr Hypertens Rep 6, 314–320. [DOI] [PubMed] [Google Scholar]
- [103].Kennelly SP, Lawlor BA, Kenny RA (2009) Blood pressure and dementia – A comprehensive review. Ther Adv Neurol Disord 2, 241–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Gump BB, Stewart P, Reihman J, Lonky E, Darvill T, Matthews KA, Parsons PJ (2005) Prenatal and early child-hood blood lead levels and cardiovascular functioning in 9½ year old children. Neurotoxicol Teratol 27, 655–665. [DOI] [PubMed] [Google Scholar]
- [105].Lanphear BP, Rauch S, Auinger P, Allen RW, Hornung RW (2018) Low-level lead exposure and mortality in US adults: A population-based cohort study. Lancet Public Health 3, e177–e184. [DOI] [PubMed] [Google Scholar]
- [106].Ekong EB, Jaar BG, Weaver VM (2006) Lead-related nephrotoxicity: A review of the epidemiologic evidence. Kidney Int 70, 2074–2084. [DOI] [PubMed] [Google Scholar]
- [107].Kurella M, Chertow GM, Fried LF, Cummings SR, Harris T, Simonsick E, Satterfield S, Ayonayon H, Yaffe K (2005) Chronic kidney disease and cognitive impairment in the elderly: The Health, Aging, and Body Composition Study. J Am Soc Nephrol 16, 2127–2133. [DOI] [PubMed] [Google Scholar]
- [108].Bugnicourt J-M, Godefroy O, Chillon J-M, Choukroun G, Massy ZA (2013) Cognitive disorders and dementia in CKD: The neglected kidney-brain axis. J Am Soc Nephrol 24, 353–363. [DOI] [PubMed] [Google Scholar]
- [109].Landrigan PJ, Sonawane B, Butler RN, Trasande L, Callan R, Droller D (2005) Early environmental origins of neurodegenerative disease in later life. Environ Health Perspect 113, 1230–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Jagust W (2013) Vulnerable neural systems and the borderland of brain aging and neurodegeneration. Neuron 77, 219–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Langston JW, Forno LS, Tetrud J, Reeves AG, Kaplan JA, Karluk D (1999) Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine exposure. Ann Neurol 46, 598–605. [DOI] [PubMed] [Google Scholar]
- [112].Stern Y (2012) Cognitive reserve in ageing and Alzheimer’s disease. Lancet Neurol 11, 1006–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Schofield PW, Logroscino G, Andrews HF, Albert S, Stern Y (1997) An association between head circumference and Alzheimer’s disease in a population-based study of aging and dementia. Neurology 49, 30–37. [DOI] [PubMed] [Google Scholar]
- [114].Sumowski JF, Rocca MA, Leavitt VM, Riccitelli G, Comi G, DeLuca J, Filippi M (2013) Brain reserve and cognitive reserve in multiple sclerosis: What you’ve got and how you use it. Neurology 80, 2186–2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Sumowski JF, Rocca MA, Leavitt VM, Dackovic J, Mesaros S, Drulovic J, DeLuca J, Filippi M (2014) Brain reserve and cognitive reserve protect against cognitive decline over 4.5 years in MS. Neurology 82, 1776–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Espinosa PS, Kryscio RJ, Mendiondo MS, Schmitt FA, Wekstein DR, Markesbery WR, Smith CD (2006) Alzheimer’s disease and head circumference. J Alzheimers Dis 9, 77–80. [DOI] [PubMed] [Google Scholar]
- [117].Whitwell JL (2010) The protective role of brain size in Alzheimer disease. Expert Rev Neurother 10, 1799–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Schofield PW, Mosesson RE, Stern Y, Mayeux R (1995) The age at onset of Alzheimer’s disease and an intracranial area measurement: A relationship. Arch Neurol 52, 95–98. [DOI] [PubMed] [Google Scholar]
- [119].Graves AB, Mortimer JA, Larson EB, Wenzlow A, Bowen JD, McCormick WC (1996) Head circumference as a measure of cognitive reserve. Association with severity of impairment in Alzheimer’s disease. Br J Psychiatry 169, 86–92. [DOI] [PubMed] [Google Scholar]
- [120].Poletti M, Emre M, Bonuccelli U (2011) Mild cognitive impairment and cognitive reserve in Parkinson’s disease. Parkinsonism Relat Disord 17, 579–586. [DOI] [PubMed] [Google Scholar]
- [121].Stern Y (2006) Cognitive reserve and Alzheimer disease. Alzheimer Dis Assoc Disord 20, S69–74. [DOI] [PubMed] [Google Scholar]
- [122].Ballew C, Khan LK, Kaufmann R, Mokdad A, Miller DT, Gunter EW (1999) Blood lead concentration and children’s anthropometric dimensions in the Third National Health and Nutrition Examination Survey (NHANES III), 1988–1994. J Pediatr 134, 623–630. [DOI] [PubMed] [Google Scholar]
- [123].Rothenberg SJ, Schnaas L, Perroni E, Hernández RM, Martínez S, Hernández C (1999) Pre- and postnatal lead effect on head circumference: A case for critical periods. Neurotoxicol Teratol 21, 1–11. [DOI] [PubMed] [Google Scholar]
- [124].Schell LM, Denham M, Stark AD, Parsons PJ, Schulte EE (2009) Growth of infants’ length, weight, head and arm circumferences in relation to low levels of blood lead measured serially. Am J Hum Biol 21, 180–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Kafourou A, Touloumi G, Makropoulos V, Loutradi A, Papanagiotou A, Hatzakis A (1997) Effects of lead on the somatic growth of children. Arch Environ Health 52, 377–383. [DOI] [PubMed] [Google Scholar]
- [126].Cecil KM, Brubaker CJ, Adler CM, Dietrich KN, Altaye M, Egelhoff JC, Wessel S, Elangovan I, Hornung R, Jarvis K, Lanphear BP (2008) Decreased brain volume in adults with childhood lead exposure. PLoS Med 5, e112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Barulli D, Stern Y (2013) Efficiency, capacity, compensation, maintenance, plasticity: Emerging concepts in cognitive reserve. Trends Cogn Sci 17, 502–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Myung W, Lee C, Park JH, Woo S-Y, Kim S, Kim S, Chung JW, Kang HS, Lim S-W, Choi J, Na DL, Kim SY, Lee J-H, Han S-H, Choi SH, Kim SY, Carroll BJ, Kim DK (2017) Occupational attainment as risk factor for progression from mild cognitive impairment to Alzheimer’s disease: A CREDOS study. J Alzheimers Dis 55, 283–292. [DOI] [PubMed] [Google Scholar]
- [129].Stern Y, Albert S, Tang M-X, Tsai W-Y (1999) Rate of memory decline in AD is related to education and occupation: Cognitive reserve? Neurology 53, 1942–1942. [DOI] [PubMed] [Google Scholar]
- [130].Sharp ES, Gatz M (2011) The relationship between education and dementia: An updated systematic review. Alzheimer Dis Assoc Disord 25, 289–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Siedlecki KL, Stern Y, Reuben A, Sacco RL, Elkind MSV, Wright CB (2009) Construct validity of cognitive reserve in a multi-ethnic cohort: The Northern Manhattan Study. J Int Neuropsychol Soc 15, 558–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Bleecker ML, Ford DP, Celio MA, Vaughan CG, Lindgren KN (2007) Impact of cognitive reserve on the relationship of lead exposure and neurobehavioral performance. Neurology 69, 470–476. [DOI] [PubMed] [Google Scholar]
- [133].Needleman HL, Riess JA, Tobin MJ, Biesecker GE, Green-house JB (1996) Bone lead levels and delinquent behavior. JAMA 275, 363–369. [PubMed] [Google Scholar]
- [134].Mendelsohn AL, Dreyer BP, Fierman AH, Rosen CM, Legano LA, Kruger HA, Lim SW, Courtlandt CD (1998) Low-level lead exposure and behavior in early childhood. Pediatrics 101, E10. [DOI] [PubMed] [Google Scholar]
- [135].Mazumdar M, Bellinger DC, Gregas M, Abanilla K, Bacic J, Needleman HL (2011) Low-level environmental lead exposure in childhood and adult intellectual function: A follow-up study. Environ Health 10, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Needleman HL, Schell A, Bellinger D, Leviton A, Allred EN (1990) The long-term effects of exposure to low doses of lead in childhood. N Engl J Med 322, 83–88. [DOI] [PubMed] [Google Scholar]
- [137].Brubaker CJ, Schmithorst VJ, Haynes EN, Dietrich KN, Egelhoff JC, Lindquist DM, Lanphear BP, Cecil KM (2009) Altered myelination and axonal integrity in adults with childhood lead exposure: A diffusion tensor imaging study. Neurotoxicology 30, 867–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Potter GG, Helms MJ, Plassman BL (2008) Associations of job demands and intelligence with cognitive performance among men in late life. Neurology 70, 1803–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Ott A, Slooter A, Hofman A, van Harskamp F, Witteman J, Van Broeckhoven C, van Duijn C, Breteler M (1998) Smoking and risk of dementia and Alzheimer’s disease in a population-based cohort study: The Rotterdam Study. Lancet 351, 1840–1843. [DOI] [PubMed] [Google Scholar]
- [140].Richards M, Jarvis MJ, Thompson N, Wadsworth MEJ (2003) Cigarette smoking and cognitive decline in midlife: Evidence from a prospective birth cohort study. Am J Public Health 93, 994–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Nyberg J, Åberg MAI, Schiöler L, Nilsson M, Wallin A, Torén K, Kuhn HG (2014) Cardiovascular and cognitive fitness at age 18 and risk of early-onset dementia. Brain 137, 1514–1523. [DOI] [PubMed] [Google Scholar]
- [142].Fishbein DH, Todd AC, Ricketts EP, Semba RD (2008) Relationship between lead exposure, cognitive function, and drug addiction: Pilot study and research agenda. Environ Res 108, 315–319. [DOI] [PubMed] [Google Scholar]
- [143].Valles R, Cardon AL, Heard HM, Bratton GR, Nation JR (2003) Morphine conditioned place preference is attenuated by perinatal lead exposure. Pharmacol Biochem Behav 75, 295–300. [DOI] [PubMed] [Google Scholar]
- [144].Rocha A, Valles R, Cardon AL, Bratton GR, Nation JR (2004) Self-administration of heroin in rats: Effects of low-level lead exposure during gestation and lactation. Psychopharmacology (Berl) 174, 203–210. [DOI] [PubMed] [Google Scholar]
- [145].Satizabal CL, Beiser AS, Chouraki V, Chêne G, Dufouil C, Seshadri S (2016) Incidence of dementia over three decades in the framingham heart study. N Engl J Med 374, 523–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Matthews FE, Arthur A, Barnes LE, Bond J, Jagger C, Robinson L, Brayne C, Medical Research Council Cognitive Function and Ageing Collaboration (2013) A two-decade comparison of prevalence of dementia in individuals aged 65 years and older from three geographical areas of England: Results of the Cognitive Function and Ageing Study I and II. Lancet 382, 1405–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Langa KM, Larson EB, Crimmins EM, Faul JD, Levine DA, Kabeto MU, Weir DR (2017) A comparison of the prevalence of dementia in the United States in 2000 and 2012. JAMA Intern Med 177, 51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Annest J (1983) Trends in the blood lead levels of the US population: The Second National Health and Nutrition Examination Survey (NHANES II) 1976–1980. In: Lead versus Health: Sources and Effects of Low Level Lead Exposure, Rutter M, Jones RR, eds. John Wiley and Sons, pp. 33–58. [Google Scholar]
- [149].Needleman HL (2000) The removal of lead from gasoline: Historical and personal reflections. Environ Res 84, 20–35. [DOI] [PubMed] [Google Scholar]
- [150].Filley CM, Kelly J, Heaton RK (1986) Neuropsychologic features of early-and late-onset Alzheimer’s disease. Arch Neurol 43, 574–576. [DOI] [PubMed] [Google Scholar]
- [151].Alarcon WA (2015) Elevated blood lead levels among employed adults — United States, 1994–2012. Morb Mortal Wkly Rep 62, 52–75. [DOI] [PubMed] [Google Scholar]
- [152].Kövari E, Herrmann FR, Bouras C, Gold G (2014) Amyloid deposition is decreasing in aging brains An autopsy study of 1,599 older people. Neurology 82, 326–331. [DOI] [PubMed] [Google Scholar]
- [153].Wietlisbach V, Rickenbach M, Berode M, Guillemin M (1995) Time trend and determinants of blood lead levels in a Swiss population over a transition period (1984–1993) from leaded to unleaded gasoline use. Environ Res 68, 82–90. [DOI] [PubMed] [Google Scholar]
- [154].Mielke HW (1999) Lead in the inner cities. Am Sci 87, 62–73. [Google Scholar]
- [155].Zhang Z, Hayward MD, Yu Y-L (2016) Life course pathways to racial disparities in cognitive impairment among older Americans. J Health Soc Behav 57, 184–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Gurland BJ, Wilder DE, Lantigua R, Stern Y, Chen J, Killeffer EH, Mayeux R (1999) Rates of dementia in three ethnoracial groups. Int J Geriatr Psychiatry 14, 481–493. [PubMed] [Google Scholar]
- [157].Alzheimer’s Association (2017) 2017 Alzheimer’s Disease facts and figures. Alzheimers Dement 13, 325–373. [Google Scholar]
- [158].Chin AL, Negash S, Hamilton R (2011) Diversity and disparity in dementia: The impact of ethnoracial differences in Alzheimer disease. Alzheimer Dis Assoc Disord 25, 187–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Clark PC, Kutner NG, Goldstein FC, Peterson-Hazen S, Garner V, Zhang R, Bowles T (2005) Impediments to timely diagnosis of Alzheimer’s disease in African Americans. J Am Geriatr Soc 53, 2012–2017. [DOI] [PubMed] [Google Scholar]
- [160].Wilson IH, Wilson SB (2016) Confounding and causation in the epidemiology of lead. Int J Environ Health Res 26, 467–482. [DOI] [PubMed] [Google Scholar]
- [161].Wandall B, Hansson SO, Rudén C (2007) Bias in toxicology. Arch Toxicol 81, 605–617. [DOI] [PubMed] [Google Scholar]
- [162].Cuadrado-Tejedor M, García-Osta A (2014) Current animal models of Alzheimer’s disease: Challenges in translational research. Front Neurol 5, 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Franco R, Cedazo-Minguez A (2014) Successful therapies for Alzheimer’s disease: Why so many in animal models and none in humans? Front Pharmacol 5, 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Ratner MH, Farb DH, Ozer J, Feldman RG, Durso R (2014) Younger age at onset of sporadic Parkinson’s disease among subjects occupationally exposed to metals and pesticides. Interdiscip Toxicol 7, 123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Todd AC, Wetmur JG, Moline JM, Godbold JH, Levin SM, Landrigan PJ (1996) Unraveling the chronic toxicity of lead: An essential priority for environmental health. Environ Health Perspect 104, 141–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Ding N, Wang X, Weisskopf MG, Sparrow D, Schwartz J, Hu H, Park SK (2016) Lead-related genetic loci, cumulative lead exposure and incident coronary heart disease: The Normative Aging Study. PLoS One 11, e0161472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Warrington NM, Zhu G, Dy V, Heath AC, Madden PAF, Hemani G, Kemp JP, Mcmahon G, St Pourcain B, Timpson NJ, Taylor CM, Golding J, Lawlor DA, Steer C, Montgomery GW, Martin NG, Davey Smith G, Evans DM, Whitfield JB (2015) Genome-wide association study of blood lead shows multiple associations near ALAD. Hum Mol Genet 24, 3871–3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Ng E, Lind PM, Lindgren C, Ingelsson E, Mahajan A, Morris A, Lind L (2015) Genome-wide association study of toxic metals and trace elements reveals novel associations. Hum Mol Genet 24, 4739–4745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Weuve J, Kelsey KT, Schwartz J, Bellinger D, Wright RO, Rajan P, Spiro A, Sparrow D, Aro A, Hu H (2006) Delta-aminolevulinic acid dehydratase polymorphism and the relation between low level lead exposure and the Mini-Mental Status Examination in older men: The Normative Aging Study. Occup Environ Med 63, 746–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Rajan P, Kelsey KT, Schwartz JD, Bellinger DC, Weuve J, Spiro A, Sparrow D, Smith TJ, Nie H, Weisskopf MG, Hu H, Wright RO (2008) Interaction of the δ-aminolevulinic acid dehydratase (ALAD) polymorphism and lead burden on cognitive function: The VA Normative Aging Study. J Occup Environ Med 50, 1053–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Louis ED, Applegate L, Graziano JH, Parides M, Slavkovich V, Bhat HK (2005) Interaction between blood lead concentration and δ-amino-levulinic acid dehydratase gene polymorphisms increases the odds of essential tremor. Mov Disord 20, 1170–1177. [DOI] [PubMed] [Google Scholar]
- [172].Saunders AM, Strittmatter WJ, Schmechel D, George-Hyslop PHS, Pericak-Vance MA, Joo SH, Rosi BL, Gusella JF, Crapper-MacLachlan DR, Alberts MJ, Hulette C, Crain B, Goldgaber D, Roses AD (1993) Association of apolipoprotein E allele ‹4 with late-onset familial and sporadic Alzheimer’s disease. Neurology 43, 1467–1467. [DOI] [PubMed] [Google Scholar]
- [173].Coon KD, Myers AJ, Craig DW, Webster JA, Pearson JV, Lince DH, Zismann VL, Beach TG, Leung D, Bryden L, Halperin RF, Marlowe L, Kaleem M, Walker DG, Ravid R, Heward CB, Rogers J, Papassotiropoulos A, Reiman EM, Hardy J, Stephan DA (2007) A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer’s disease. J Clin Psychiatry 68, 613–618. [DOI] [PubMed] [Google Scholar]
- [174].Schipper HM, Song W (2015) A heme oxygenase-1 transducer model of degenerative and developmental brain disorders. Int J Mol Sci 16, 5400–5419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Sultana R, Butterfield DA (2004) Oxidatively modified GST and MRP1 in Alzheimer’s disease brain: Implications for accumulation of reactive lipid peroxidation products. Neurochem Res 29, 2215–2220. [DOI] [PubMed] [Google Scholar]
- [176].Bellinger DC (2000) Effect modification in epidemiologic studies of low-level neurotoxicant exposures and health outcomes. Neurotoxicol Teratol 22, 133–140. [DOI] [PubMed] [Google Scholar]
- [177].Virgolini MB, Rossi-George A, Lisek R, Weston DD, Thiruchelvam M, Cory-Slechta DA (2008) CNS effects of developmental Pb exposure are enhanced by combined maternal and offspring stress. Neurotoxicology 29, 812–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Amos-Kroohs RM, Graham DL, Grace CE, Braun AA, Schaefer TL, Skelton MR, Vorhees CV, Williams MT (2016) Developmental stress and lead (Pb): Effects of maternal separation and/or Pb on corticosterone, monoamines, and blood Pb in rats. Neurotoxicology 54, 22–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Virgolini MB, Chen K, Weston DD, Bauter MR, Cory-Slechta DA (2005) Interactions of chronic lead exposure and intermittent stress: Consequences for brain catecholamine systems and associated behaviors and HPA axis function. Toxicol Sci 87, 469–482. [DOI] [PubMed] [Google Scholar]
- [180].Graham DL, Grace CE, Braun AA, Schaefer TL, Skelton MR, Tang PH, Vorhees CV, Williams MT (2011) Effects of developmental stress and lead (Pb) on corticosterone after chronic and acute stress, brain monoamines, and blood Pb Levels in rats. Int J Dev Neurosci 29, 45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Peters JL, Kubzansky L, McNeely E, Schwartz J, Spiro A, Sparrow D, Wright RO, Nie H, Hu H (2007) Stress as a potential modifier of the impact of lead levels on blood pressure: The normative aging study. Environ Health Perspect 115, 1154–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Peters JL, Weisskopf MG, Spiro A, Schwartz J, Sparrow D, Nie H, Hu H, Wright RO, Wright RJ (2010) Interaction of stress, lead burden, and age on cognition in older men: The VA Normative Aging Study. Environ Health Perspect 118, 505–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Glass TA, Bandeen-Roche K, McAtee M, Bolla K, Todd AC, Schwartz BS (2009) Neighborhood psychosocial hazards and the association of cumulative lead dose with cognitive function in older adults. Am J Epidemiol 169, 683–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Tong S, McMichael AJ, Baghurst PA (2000) Interactions between environmental lead exposure and sociodemographic factors on cognitive development. Arch Environ Health 55, 330–335. [DOI] [PubMed] [Google Scholar]
- [185].Dietrich KN, Succop PA, Berger OG, Hammond PB, Bornschein RL (1991) Lead exposure and the cognitive development of urban preschool children: The Cincinnati Lead Study cohort at age 4 years. Neurotoxicol Teratol 13, 203–211. [DOI] [PubMed] [Google Scholar]
- [186].Bellinger D, Leviton A, Waternaux C, Needleman H, Rabinowitz M (1988) Low-level lead exposure, social class, and infant development. Neurotoxicol Teratol 10, 497–503. [DOI] [PubMed] [Google Scholar]
- [187].Winneke G, Kraemer U (1984) Neuropsychological effects of lead in children: Interactions with social background variables. Neuropsychobiology 11, 195–202. [DOI] [PubMed] [Google Scholar]
- [188].Weiss B, Bellinger DC (2006) Social ecology of children’s vulnerability to environmental pollutants. Environ Health Perspect 114, 1479–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Bellinger DC (2004) Assessing environmental neurotoxicant exposures and child neurobehavior: Confounded by confounding? Epidemiology 15, 383–384. [DOI] [PubMed] [Google Scholar]
- [190].Riess ML, Halm JK (2007) Lead poisoning in an adult: Lead mobilization by pregnancy? J Gen Intern Med 22, 1212–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Thompson GN, Robertson EF, Fitzgerald S (1985) Lead mobilization during pregnancy. Med J Aust 143, 131. [DOI] [PubMed] [Google Scholar]
- [192].Mahaffey KR (1990) Environmental lead toxicity: Nutrition as a component of intervention. Environ Health Perspect 89, 75–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Calderón-Garcidueñas L, San Juan Chávez V, Vacaseydel-Aceves NB, Calderón-Sánchez R, Macías-Escobedo E, Frías C, Giacometto M, Velasquez L, Félix-Villarreal R, Martin JD, Draheim C, Engle RW (2016) Chocolate, air pollution and children’s neuroprotection: What cognition tools should be at hand to evaluate interventions? Front Pharmacol 7, 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Schneider JS, Lee MH, Anderson DW, Zuck L, Lidsky TI (2001) Enriched environment during development is protective against lead-induced neurotoxicity. Brain Res 896, 48–55. [DOI] [PubMed] [Google Scholar]
- [195].Guilarte TR, Toscano CD, McGlothan JL, Weaver SA (2003) Environmental enrichment reverses cognitive and molecular deficits induced by developmental lead exposure. Ann Neurol 53, 50–56. [DOI] [PubMed] [Google Scholar]
- [196].The Lancet Neurology (2017) Pointing the way to primary prevention of dementia. Lancet Neurol 16, 677. [DOI] [PubMed] [Google Scholar]
- [197].The National Academies of Sciences, Engineering, and Medicine (2017) Preventing cognitive decline and dementia: A way forward, The National Academies Press, Washington, DC. [PubMed] [Google Scholar]
- [198].van Praag H, Shubert T, Zhao C, Gage FH (2005) Exercise enhances learning and hippocampal neurogenesis in aged mice. J Neurosci 25, 8680–8685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].van Praag H, Christie BR, Sejnowski TJ, Gage FH (1999) Running enhances neurogenesis, learning, and long-term potentiation in mice. Proc Natl Acad SciUSA 96, 13427–13431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Shevtsova O, Tan Y-F, Merkley CM, Winocur G, Wojtowicz JM (2017) Early-age running enhances activity of adult-born dentate granule neurons following learning in rats. eNeuro 4, ENEURO.0237–17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Shea B, Wells G, Cranney A, Zytaruk N, Robinson V, Griffith L, Ortiz Z, Peterson J, Adachi J, Tugwell P, Guyatt G (2002) Meta-analysis of calcium supplementation for the prevention of postmenopausal osteoporosis. Endocr Rev 23, 552–559. [DOI] [PubMed] [Google Scholar]
- [202].Ettinger AS, Hu H, Mauricio-Hernandez-Avila (2007) Dietary calcium supplementation to lower blood lead levels in pregnancy and lactation. J Nutr Biochem 18, 172–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Cecil KM, Dietrich KN, Altaye M, Egelhoff JC, Lindquist DM, Brubaker CJ, Lanphear BP (2011) Proton magnetic resonance spectroscopy in adults with childhood lead exposure. Environ Health Perspect 119, 403–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Searle AK, Baghurst PA, van Hooff M, Sawyer MG, Sim MR, Galletly C, Clark LS, McFarlane AC (2014) Tracing the long-term legacy of childhood lead exposure: A review of three decades of the Port Pirie Cohort study. Neurotoxicology 43, 46–56. [DOI] [PubMed] [Google Scholar]
- [205].Stokes L, Letz R, Gerr F, Kolczak M, McNeill FE, Chettle DR, Kaye WE (1998) Neurotoxicity in young adults 20 years after childhood exposure to lead: The Bunker Hill experience. Occup Environ Med 55, 507–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Ernhart CB, Morrow-Tlucak M, Wolf AW, Super D, Drotar D (1989) Low level lead exposure in the prenatal and early preschool periods: Intelligence prior to school entry. Neurotoxicol Teratol 11, 161–170. [DOI] [PubMed] [Google Scholar]
- [207].Canfield RL, Henderson CR, Cory-Slechta DA, Cox C, Jusko TA, Lanphear BP (2003) Intellectual impairment in children with blood lead concentrations below 10 µg per deciliter. N Engl J Med 348, 1517–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Jusko TA, Henderson CR, Lanphear BP, Cory-Slechta DA, Parsons PJ, Canfield RL (2008) Blood lead concentrations < 10 µg/dL and child intelligence at 6 years of age. Environ Health Perspect 116, 243–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Schnaas L, Rothenberg SJ, Perroni E, Martínez S, Hernández C, Hernández RM (2000) Temporal pattern in the effect of postnatal blood lead level on intellectual development of young children. Neurotoxicol Teratol 22, 805–810. [DOI] [PubMed] [Google Scholar]
- [210].Schnaas L, Rothenberg SJ, Flores M-F, Martinez S, Hernandez C, Osorio E, Velasco SR, Perroni E (2006) Reduced intellectual development in children with prenatal lead exposure. Environ Health Perspect 114, 791–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Wasserman GA, Liu X, Lolacono NJ, Factor-Litvak P, Kline JK, Popovac D, Morina N, Musabegovic A, Vrenezi N, Capuni-Paracka S, Lekic V, Preteni-Redjepi E, Hadzialjevic S, Slavkovich V, Graziano JH (1997) Lead exposure and intelligence in 7-year-old children: The Yugoslavia Prospective Study. Environ Health Perspect 105, 956–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Kayaalti Z, Kaya-Akyüzlü D, Söylemez E, Söylemezoğlu (2015) Maternal hemochromatosis gene H63D single-nucleotide polymorphism and lead levels of placental tissue, maternal and umbilical cord blood. Environ Res 140, 456–461. [DOI] [PubMed] [Google Scholar]
- [213].Fan G, Du G, Li H, Lin F, Sun Z, Yang W, Feng C, Zhu G, Li Y, Chen Y, Jiao H, Zhou F (2014) The effect of the hemochromatosis (HFE) genotype on lead load and iron metabolism among lead smelter workers. PLoS One 9, e101537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Szymańska-Chabowska A, Łaczmański Ł, Jędrychowska I, Chabowski M, Gać P, Janus A, Goslawska K, Smyk B, Solska U, Mazur G, Poręba R (2015) The relationship between selected VDR, HFE and ALAD gene polymorphisms and several basic toxicological parameters among persons occupationally exposed to lead. Toxicology 334, 12–21. [DOI] [PubMed] [Google Scholar]
- [215].Nigg JT, Elmore AL, Natarajan N, Friderici KH, Nikolas MA (2016) Variation in an iron metabolism gene moderates the association between blood lead levels and attention-deficit/hyperactivity disorder in children. Psychol Sci 27, 257–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Wright RO, Silverman EK, Schwartz J, Tsaih S-W, Senter J, Sparrow D, Weiss ST, Aro A, Hu H (2004) Association between hemochromatosis genotype and lead exposure among elderly men: The normative aging study. Environ Health Perspect 112, 746–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Miyaki K, Lwin H, Masaki K, Song Y, Takahashi Y, Muramatsu M, Nakayama T (2009) Association between a polymorphism of Aminolevulinate dehydrogenase (ALAD) gene and blood lead levels in Japanese subjects. Int J Environ Res Public Health 6, 999–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Zhao Y, Wang L, Shen H-B, Wang Z-X, Wei Q-Y, Chen F (2007) Association between delta-aminolevulinic acid dehydratase (ALAD) polymorphism and blood lead levels: A meta-regression analysis. J Toxicol Environ Health A 70, 1986–1994. [DOI] [PubMed] [Google Scholar]
- [219].Kelada SN, Shelton E, Kaufmann RB, Khoury MJ (2001) Delta-aminolevulinic acid dehydratase genotype and lead toxicity: A HuGE review. Am J Epidemiol 154, 1–13. [DOI] [PubMed] [Google Scholar]
- [220].Scinicariello F, Murray HE, Moffett DB, Abadin HG, Sexton MJ, Fowler BA (2007) Lead and delta-aminolevulinic acid dehydratase polymorphism: Where does it lead? A meta-analysis. Environ Health Perspect 115, 35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Rezende VB, Barbosa F, Montenegro MF, Sandrim VC, Gerlach RF, Tanus-Santos JE (2008) Haplotypes of vitamin D receptor modulate the circulating levels of lead in exposed subjects. Arch Toxicol 82, 29–36. [DOI] [PubMed] [Google Scholar]
- [222].Jhun MA, Hu H, Schwartz J, Weisskopf MG, Nie LH, Sparrow D, Vokonas PS, Park SK (2015) Effect modification by vitamin D receptor genetic polymorphisms in the association between cumulative lead exposure and pulse pressure: A longitudinal study. Environ Health 14, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Mannila MN, Mahdessian H, Franco-Cereceda A, Eggertsen G, Faire U de, Syvänen A-C, Eriksson P, Hamsten A, Hooft FM van ‘t (2013) Identification of a functional apolipoprotein E promoter polymorphism regulating plasma apolipoprotein E concentration. Arterioscler Thromb Vasc Biol 33, 1063–1069. [DOI] [PubMed] [Google Scholar]
- [224].Theppeang K, Glass TA, Bandeen-Roche K, Todd AC, Rohde CA, Links JM, Schwartz BS (2008) Associations of bone mineral density and lead levels in blood, tibia, and patella in urban-dwelling women. Environ Health Perspect 116, 784–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Coral-Vázquez RM, Romero Arauz JF, Canizales-Quinteros S, Coronel A Valencia Villalvazo EY, Hernández Rivera J, Ramírez Regalado B, Rojano Mejía D, Canto P (2013) Analysis of polymorphisms and haplotypes in genes associated with vascular tone, hypertension and oxidative stress in Mexican-Mestizo women with severe preeclampsia. Clin Biochem 46, 627–632. [DOI] [PubMed] [Google Scholar]
- [226].Eum K-D, Seals RM, Taylor KM, Grespin M, Umbach DM, Hu H, Sandler DP, Kamel F, Weisskopf MG (2015) Modification of the association between lead exposure and amyotrophic lateral sclerosis by iron and oxidative stress related gene polymorphisms. Amyotroph Lateral Scler Front Degener 16, 72–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Sirivarasai J, Wananukul W, Kaojarern S, Chanprasertyothin S, Thongmung N, Ratanachaiwong W, Sura T, Sritara P (2013) Association between inflammatory marker, environmental lead exposure, and glutathione S-transferase gene. BioMed Res Int 2013, 474963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Park SK, Hu H, Wright RO, Schwartz J, Cheng Y, Sparrow D, Vokonas PS, Weisskopf MG (2009) Iron metabolism genes, low-level lead exposure, and QT interval. Environ Health Perspect 117, 80–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [229].Antony PMA, Diederich NJ, Krüger R, Balling R (2013) The hallmarks of Parkinson’s disease. FEBS J 280, 5981–5993. [DOI] [PubMed] [Google Scholar]
- [230].O’Flaherty EJ (1995) Physiologically based models for bone-seeking elements. V. Lead absorption and disposition in childhood. Toxicol Appl Pharmacol 131, 297–308. [DOI] [PubMed] [Google Scholar]
- [231].Hunter DJ, Sambrook PN (2000) Bone loss: Epidemiology of bone loss. Arthritis Res 2, 441–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [232].Riggs BL, Wahner HW, Seeman E, Offord KP, Dunn WL, Mazess RB, Johnson KA, Melton LJ (1982) Changes in bone mineral density of the proximal femur and spine with aging. J Clin Invest 70, 716–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [233].Chalmers J, Ho KC (1970) Geographical variations in senile osteoporosis: The association with physical activity. J Bone Joint Surg Br 52-B, 667–675. [PubMed]
- [234].Tsaih SW, Korrick S, Schwartz J, Lee ML, Amarasiri-wardena C, Aro A, Sparrow D, Hu H (2001) Influence of bone resorption on the mobilization of lead from bone among middle-aged and elderly men: The Normative Aging Study. Environ Health Perspect 109, 995–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].Gertz BJ, Shao P, Hanson DA, Quan H, Harris ST, Genant HK, Chesnut CH, Eyre DR (1994) Monitoring bone resorption in early postmenopausal women by an immunoassay for cross-linked collagen peptides in urine. J Bone Miner Res 9, 135–142. [DOI] [PubMed] [Google Scholar]
- [236].Silbergeld EK (1991) Lead in bone: Implications for toxicology during pregnancy and lactation. Environ Health Perspect 91, 63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [237].Gulson BL, Mizon KJ, Palmer JM, Korsch MJ, Taylor AJ, Mahaffey KR (2004) Blood lead changes during pregnancy and postpartum with calcium supplementation. Environ Health Perspect 112, 1499–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]