

Abstract
Sotos syndrome is an overgrowth syndrome characterized by distinctive facial features and intellectual disability caused by haploinsufficiency of the NSD1 gene. Genotype-phenotype correlations have been observed, with major anomalies seen more frequently in patients with 5q35 deletions than those with point mutations in NSD1. Though endocrine features have rarely been described, transient hyperinsulinemic hypoglycemia (HI) of the neonatal period has been reported as an uncommon presentation of Sotos syndrome. Eight cases of 5q35 deletions and one patient with an intragenic NSD1 mutation with transient HI have been reported. Here, we describe seven individuals with HI caused by NSD1 gene mutations with three having persistent hyperinsulinemic hypoglycemia. These patients with persistent HI and Sotos syndrome caused by NSD1 mutations, further dispel the hypothesis that HI is due to the deletion of other genes in the deleted 5q35 region. These patients emphasize that NSD1 haploinsufficiency is sufficient to cause HI, and suggest that Sotos syndrome should be considered in patients presenting with neonatal HI. Lastly, these patients help extend the phenotypic spectrum of Sotos syndrome to include HI as a significant feature.
Keywords: Sotos syndrome, hyperinsulinism, hypoglycemia, endocrine, sacrococcygeal teratoma, NSD1, overgrowth syndrome
Introduction
Sotos syndrome (OMIM #117550) is a congenital disorder characterized by distinctive facial features (frontal bossing, broad and prominent forehead, sparse frontotemporal hair, prominent jaw, malar flushing, downslanting palpebral fissures), intellectual disability including early developmental delay and/or mild to severe intellectual impairment, and pre- and postnatal overgrowth with height and/or head circumference ≥2 standard deviations above the mean. All three cardinal features are present in >90% of individuals diagnosed with Sotos syndrome [Leventopoulos et al., 2009; Tatton-Brown et al., 2005]. Other common clinical features include cardiac defects, advanced bone age, cranial imaging abnormalities, maternal preeclampsia, renal anomalies, scoliosis, seizures, neonatal hypotonia, neonatal jaundice, and joint hyperlaxity [Leventopoulos et al., 2009; Tatton-Brown et al., 2005].
Sotos syndrome is caused by haploinsufficiency of the nuclear receptor binding SET domain 1 (NSD1) gene. NSD1, located at chromosome 5q35, encodes a histone methyltransferase that has been implicated in the regulation of opening and closing of chromatin [Aasland et al., 1995; Tatton-Brown et al., 2005]. Approximately 50% of Sotos syndrome in Japanese patients is caused by 5q35 deletions, whereas 90% of Sotos syndrome in non-Japanese patients is caused by intragenic mutations [Douglas et al., 2005; Kamimura et al., 2003]. Genotype-phenotype correlations have been observed, with major anomalies seen more frequently in patients with 5q35 deletions than those with point mutations in NSD1 [Douglas, et al., 2005; Kamimura et al., 2003]. It is presumed that additional complications are caused by the deletion of other genes in this region [Mastou et al., 2013].
Though hypoglycemia has rarely been described in Sotos syndrome, transient HI of the neonatal period has been reported as an uncommon presentation of Sotos syndrome [Cole and Hughes, 1990; Cole and Hughes, 1994; Hook and Reynolds, 1967; Tatton-Brown et al., 2005]. Matsou et al. described five patients with Sotos syndrome caused by 5q35 deletions found to have transient HI [Matsou et al., 2012]. More recently, Nakamura et al. reported an additional patient with a 5q35 deletion and transient neonatal HI [Nakamura et al., 2014]. Carrasco Sallas et al. first described a patient with an intragenic NSD1 mutation and transient HI [Carrasco Sallas, et al., 2015]. A majority of the cases reported to date of Sotos syndrome and HI have been the result of microdeletions of NSD1 (7/8) and all have been transient (8/8).
Currently, there are limited theories on why individuals with Sotos syndrome may have HI. Because a majority of the cases reported in the literature to date have been microdeletions, one theory is that there are additional genes in the 5q35 deleted region that could be causing HI [Matsou et al., 2012; Nakamura et al., 2015]. However, since there is one case in the literature with HI and an NSD1 pathogenic intragenic mutation, another theory is that a defect in NSD1 is sufficient to cause HI [Matsou et al., 2012; Nakamura et al., 2015].
Here, we describe seven patients with NSD1 gene mutations with HI (persistent in at least three) and atypical features of Sotos syndrome.
Materials and Methods
Patients were identified and evaluated clinically at either The Children’s Hospital of Philadelphia, Rady Children’s Hospital San Diego, Children’s National Health System, or Cincinnati Children’s Hospital Medical Center. Genetic testing was completed in all patients on a clinical basis and Sotos syndrome was molecularly confirmed by either exome sequencing, NSD1 sequencing, or NSD1 deletion/duplication analysis. Other genetic causes of HI were ruled out by exome sequencing and/or next generation sequencing for congenital HI genes (which included ABCC8, KCNJ11, GLUD1, GCK, SLC16A1, UCP2, HNF1A, HNF4A, and HADH unless otherwise specified). Clinical features and biochemical data were retrospectively collected from the electronic medical charts of the individual patients. Calculation of Z scores for most measurements were performed using CDC growth charts, taking into account gestational age [Kuczmarski et al., 2002]. Z scores for head circumference greater than 3 years of age were calculated using WHO growth charts [de Onis et al., 2007]. Literature was reviewed in order to gather frequency of HI in individuals with molecularly confirmed Sotos syndrome. This study protocol was reviewed and approved by the Institutional Review Boards at each institution and written informed consent was obtained from parents of all patients.
Results
Clinical Reports
Patient 1:
This female proband was born at 33 6/7 weeks gestation via cesarean section due to placental abruption to a then 33 year old G2P0->P1 mother. The pregnancy was complicated by hypertension with superimposed preeclampsia. Corrected birth weight was 1.98 kg (z = −2.6), birth length was 45 cm (z = −1.9), head circumference 29.5 cm (z = −3.9). Her initial plasma glucose concentration was 24 mg/dL and an intravenous dextrose bolus was administered.
She was transferred for HI management on day of life (DOL) 10 with seizure-like activity. Subsequent electroencephalogram (EEG) showed no electrographic seizures. She was noted to have a sacral mass at birth, and a spinal MRI noted a Type III sacrococcygeal teratoma (Figure 1) that was surgically resected at 5 weeks of age. Due to persistent hypoglycemia, a diagnostic fasting test was performed. A critical sample was obtained on DOL 23 and showed plasma insulin of <2.0 μIU/mL with plasma glucose of 50 mg/dL and suppressed plasma betahydroxybutyrate at <0.3 mmol/L. Administration of 1 mg glucagon at the time of hypoglycemia resulted in an increase of the plasma glucose to 102 mg/dL within 40 minutes. The cortisol and growth hormone responses to hypoglycemia were normal at 13.7 µg/dL and 11.5 ng/mL, respectively. Total and free carnitine levels were normal. Diazoxide was initiated at 5 mg/kg/day on DOL 24, with subsequent improvement of glycemic control. She required increased doses of diazoxide at 2, 15, and 23 months of age due to recurrent hypoglycemia. Her most recent fasting study at 21 months of age showed a drop in her plasma glucose levels to 66 mg/dL at 12 hours (goal plasma glucose level >70 mg/dL) with plasma betahydroxybutyrate at 0.2 mmol/L. She is currently maintained on 10 mg/kg/day diazoxide at 30 months of age.
Figure 1: Sacrococcygeal teratoma of Patient 1.
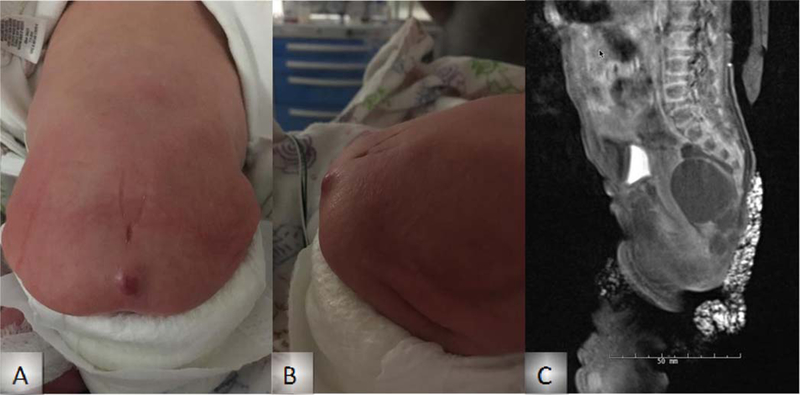
External views of lumbosacral region (A, B) and MRI (C).
She was seen by Clinical Genetics at 4 months of age and was noted to be scapholocephalic with bilateral posteriorly rotated ears, hypertelorism, downslanting palpebral fissures, anteverted nares with flat nasal bridge, short philtrum, downturned lip, and a thickened alveolar ridge (Figure 2A,B). She had poor oral feeding following her sacrococcygeal teratoma resection, and her growth parameters had plateaued with length at 0.01%ile (z=−3.72) and weight at <0.01%ile (z=−4.10). Sotos syndrome was considered, but because of her atypical presentation, exome sequencing was performed, which revealed a de novo NSD1 c.5332C>T (p.R1778*) pathogenic variant, consistent with Sotos syndrome. Congenital HI panel testing was negative.
Figure 2: Facial Images.
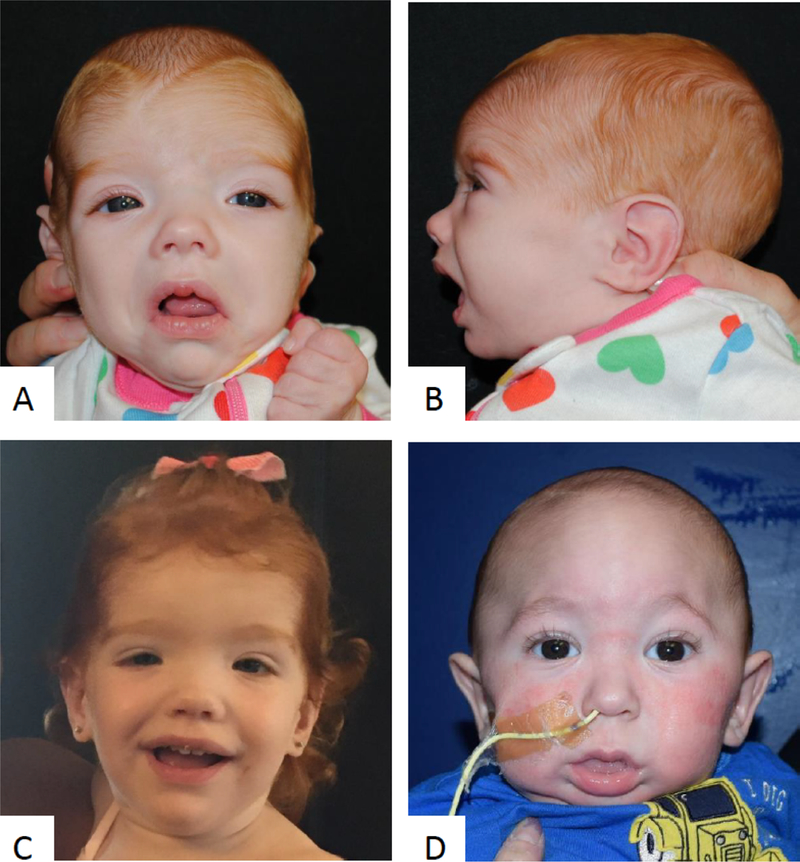
Patient 1 at 4 months old (A, B). Patient 1 at 18 months old (C). Patient 3 prior to craniosynostosis repair (D).
Patient 2:
Patient 2 was female and born at 33 5/7 weeks via cesarean section due to presence of sacrococcygeal teratoma to a then 37 year old G3P2->P3 mother. The pregnancy was complicated by a fetal cystic sacrococcygeal teratoma. Corrected birth weight was 2.50 kg (z=−1.7), birth length was 53.5 cm (z=0.2), and head circumference was 36.0 cm (z=−0.7). She had tumor debulking on DOL 1 and remained in the NICU due to complications of prematurity. Hypoglycemia was not reported in the newborn period.
She was evaluated by Clinical Genetics at 2 months of age due to a concern for hypotonia. Physical examination at this time was notable for dolichocephaly, flat nasal bridge, high arched palate, slightly downslanting palpebral fissures, and an open mouth. Genetic testing was sent, including a chromosome SNP microarray, Prader-Willi methylation testing, and NSD1 sequencing and deletion/duplication analysis. The chromosome SNP microarray and Prader-Willi testing were negative and NSD1 sequencing revealed a c.5036C>G (p.S1679*) pathogenic variant, consistent with a diagnosis of Sotos syndrome.
Endocrinology was consulted at 1 week of age due to hypoglycemia (plasma glucose 52 mg/dL) that was first noted post-operatively after resection of her sacrococcygeal teratoma. It was noted that she had received systemic steroids between DOL3 and DOL26 for hypotension, and she passed her follow-up adrenocorticotropic hormone (ACTH) stimulation test. Thyroid function tests were normal at 2 months of age with TSH at 3.16 uIU/mL, thyroxine at 6.7 µg/dL, free thyroxine index 1.8 µg/dL, and T3 uptake 27.1%. She had an MRI of her pituitary gland that was normal.
At 5 months of age she was admitted for feeding intolerance, and subsequent laboratory studies showed hypoglycemia with a plasma glucose concentration of 45 mg/dL. Plasma insulin was <2.0 μIU/mL with plasma glucose of 44 mg/dL and suppressed plasma betahydroxybutyrate (<0.3 mmol/L). Administration of 1 mg glucagon at the time of hypoglycemia resulted in an increase of the plasma glucose to 76 mg/dL within 40 minutes. On the critical sample, the cortisol and growth hormone concentrations were low at 1.1 µg/dL, and 4.17 ng/mL, respectively. Total and free carnitine levels were normal. Diazoxide was initiated at 5 mg/kg/day at 5 months of life, with subsequent improvement of glycemic control. She has been lost to follow-up.
Patient 3:
Patient 3 was a male born at 37 4/7 weeks via spontaneous vaginal delivery to a surrogate mother due to maternal Mayer-Rokitansky-Küster-Hauser syndrome using maternal eggs and paternal sperm. The pregnancy was complicated by gestational diabetes and scant bleeding early in the pregnancy. APGAR scores were 8 at 1 minute and 9 at 5 minutes. Birth weight was 3.50 kg (z=−0.1), birth length was 51 cm (z=1.1), and head circumference was 35.6 cm (z=−0.1). He was initially admitted to the NICU due to a concern for transient tachypnea and was found to be hypoglycemic. He also received 2 days of phototherapy for jaundice.
He was transferred to a tertiary care center on DOL 7 due to truncal hypotonia, poor feeding, lethargy, and craniosynostosis. Clinical Genetics was consulted on DOL 7 where physical examination was remarkable for prominent metopic and sagittal ridge (Figure 2D), prominent crus, slightly flattened pinnas, enlarged scrotum, and hypotonia. Genetic testing, including a chromosome SNP microarray and a craniosynostosis gene panel, was normal.
On DOL 9 persistent hypoglycemia was noted with plasma glucose concentration in the range of 54–81 mg/dL. A diagnostic fasting test on DOL 15 showed plasma insulin of <2.0 μIU/mL with plasma glucose of 46 mg/dL. Administration of 1 mg glucagon at the time of hypoglycemia resulted in an increase of the plasma glucose to 78 mg/dL within 40 minutes. On the critical sample, the cortisol and growth hormone were at 1.7 µg/dL, and 8.58 ng/mL, respectively, but subsequent corticotropin-releasing hormone stimulation testing ruled out hypopituitarism. Total and free carnitine levels were normal. Diazoxide was initiated at 5 mg/kg/day on DOL 18 but was subsequently discontinued on DOL 27 due to respiratory distress with fluid overload. His plasma glucose concentration remained >70 mg/dL on his home feeding regimen. Prior to discharge at 2 months of age, he underwent a second fasting study and was able to fast for 18 hours without plasma glucose dropping below 50 mg/dL. After 18 hours, he had a plasma glucose level of 57 mg/dl, insulin levels of <2 µIU/L, and plasma betahydroxybutyrate of 0.4 mmol/L. The results were consistent with persistent HI with inadequate ketone response to hypoglycemia. At 12 months of age, his parents continue to monitor plasma glucose levels, and he has not had any plasma glucose levels <70 mg/dL.
Exome sequencing was initiated on DOL 36 and revealed a de novo heterozygous NSD1 c.3004_3005del (p.Lys1002fs) pathogenic variant, consistent with a diagnosis of Sotos syndrome. He also had a negative congenital HI panel.
Patient 4:
Patient 4 was the male product of a dizygotic twin gestation to a then 36 year old G2P0->P2 mother. The pregnancy was conceived via assisted reproductive technology and complicated by intrauterine growth restriction. He was born at born at 30 weeks gestation via cesarean section for breech presentation and a concern for poor growth. APGAR scores were 6 at 1 minute and 8 at 5 minutes. Corrected birth weight was 0.890 kg (z=−3.3), birth length was 34.9 cm (z=−5.4), and birth head circumference was 25 cm (z=−3.5). Patient 4 was hypoglycemic at birth with plasma glucose concentration between 20–39 mg/dL and was placed on intravenous dextrose. Other complications of prematurity in the neonatal period included respiratory distress requiring CPAP and intubation, difficulty with feeding, hyperbilirubinemia, anemia and thrombocytopenia requiring transfusions, patent ductus arteriosus (PDA) closed with ligation, a small patent foramen ovale (PFO) vs atrial septal defect (ASD), umbilical hernia, and bilateral hydroceles.
He was transferred to a tertiary care center at 4 months of age for management of hypoglycemia and respiratory distress. During his stay at his birth hospital, he passed an ACTH stimulation test. At the time of admission, his hypoglycemia was being managed with continuous nasogastric tube feeds. A diagnostic fast showed plasma insulin of <2.0 μIU/mL with plasma glucose of 39 mg/dL. Administration of 1 mg glucagon at the time of hypoglycemia resulted in an increase of plasma glucose to 71 mg/dL within 40 minutes. On the critical sample, the cortisol and growth hormone were 10.1 μg/dL and 6.41 ng/mL, respectively. Total and free carnitine levels were normal. Diazoxide was initiated but was subsequently discontinued due to development of severe pulmonary hypertension. His hypoglycemia was managed with continuous nasoduodenal tube feeds, secondary to reflux. He was discharged home at 6 months of age on continuous gastric tube feeds.
He was readmitted at 14 months of age, and diazoxide was re-initiated at a dose of 8 mg/kg/day, which he tolerated and was able to fast for 8 hours while maintaining plasma glucose >70 mg/dL. He was discharged home on overnight continuous gastrostomy tube feeds. He required increased doses of diazoxide at 16 months of age due to recurrent hypoglycemia. At 4 years of age, diazoxide was discontinued and resolution of the HI was demonstrated by a repeat fasting study, during which he maintained his plasma glucose concentration >70 mg/dL for 15 hours (>50 mg/dL for 17 hours), generated appropriate ketones, and did not have a glycemic response to glucagon.
Genetic testing for HI genes (ABCC8, GCK, GLUD1, and KCNJ11) and chromosome SNP microarray were both normal. Exome sequencing was sent due to his history of IUGR, global developmental delays, dysmorphic features, HI, congenital kyphosis, epilepsy, tethered cord s/p repair, and prominent supratentorial ventricles with slight thinning of the corpus callosum and brainstem. Exome sequencing results revealed a de novo NSD1 c.6050G>A (p.R2017Q) pathogenic variant, consistent with a diagnosis of Sotos syndrome.
Patient 5:
Patient 5 was male and the product of a twin gestation born at 35 1/7 weeks to a then 35 year old mother via cesarean section due to HELLP (hemolysis, elevated liver enzymes, low platelet count) syndrome. The pregnancy was conceived by assisted reproductive technology, specifically intracytoplasmic sperm injection. The pregnancy was complicated by subchorionic bleeding at 4 months, and pruritic urticarial rash at 33 weeks, and HELLP syndrome. APGAR scores were 8 at 1 minute and 9 at 5 minutes. Corrected birth weight was 2.720 kg (z=−1.4), length was 46 cm (z=−1.5), and head circumference was 33 cm (z=−1.3). Of note, his twin sister’s corrected birth weight was 2.280 kg (z=−2.1), length was 43 cm (z=−3.0), and head circumference was 32 cm (z=−1.8).
Patient 5 was admitted to the NICU after birth due to poor feeding, hypotonia, and hypoxia. He had hypoglycemia during the first hour after birth, but no further hypoglycemia for the following 5 weeks while on continuous nasogastric feedings, which were initiated due to feeding difficulty, emesis with bolus feedings, and dysphagia with silent aspiration.
At five weeks of life, he had a fundoplication with gastrostomy. He began to have recurrent episodes of severe hypoglycemia with plasma glucose concentrations <40 mg/dL. A fasting study was not performed as he was having hypoglycemia prior to feeds. Critical samples were obtained with the first showing plasma insulin of 2 μIU/mL at a plasma glucose of 34 mg/dL, and the second sample showing plasma insulin of 1 μU/mL with plasma glucose of 32 mg/dL and negative urine ketones. Plasma betahydroxybutyrate was not able to be assessed due to difficulties with blood collection. Glucagon (33 μg/kg) administered 30 minutes after plasma glucose was 35 mg/dL, resulted in an increase of plasma glucose to 114 mg/dL 42 minutes later. On the critical sample, the cortisol and growth hormone were 6.7 μg/dL and 11.8 ng/mL respectively. Total and free carnitine levels were normal. Diazoxide was initiated at 10 mg/kg/day at five weeks of life, with subsequent improvement of glycemic control.
Patient 5 was seen by Clinical Genetics during NICU admission. Physical examination showed central hypotonia, a relatively large head circumference, otherwise normal growth parameters, and bi-temporal narrowing. Studies revealed a normal brain on MRI, a hemodynamically insignificant ventral septal defect (VSD), and a normal chromosome SNP microarray. He had a laparoscopic gastrostomy and fundoplication, and a muscle biopsy showed type II fiber predominance and mitochondrial cristae abnormalities including lamellar bodies. Mitochondrial DNA sequencing revealed variants of uncertain significance in COX2 and ATP6, mitochondrial DNA deletions studies were negative, and quantitative testing of mitochondrial DNA showed mild to moderate depletion.
Patient 5 was readmitted 3 days following discharge at 7 weeks due to respiratory distress, decreased responsiveness, and pallor at his follow-up Endocrinology appointment requiring bag-mask ventilation and oxygen in the clinic and eventually intubation when transferred to the emergency department and intensive care unit. His point of care glucose in the clinic was repeatedly high (over 400 mg/dL). Diazoxide was temporarily held but then restarted when the patient developed recurrent hypoglycemia. He remained hospitalized for four weeks until his respiratory status improved. By the time of discharge at 11 weeks of life he was stably euglycemic on 7 mg/kg/day of diazoxide. Diazoxide was gradually weaned and discontinued by 4 months of age, but he then had recurrent hypoglycemia at age 6 months to 7 months, requiring the addition of powdered carbohydrate to his feedings to maintain euglycemia.
At 6 months of age, a repeat MRI of the brain showed diffuse thinning of the corpus callosum, moderate-to-severe delays in myelination, and ventriculomegaly. At 10 months of age, exome sequencing results revealed a de novo NSD1 c.3548_3549delCT (p.S1183X) pathogenic variant, consistent with a diagnosis of Sotos syndrome.
Patient 6:
Patient 6 was a female born at 27 weeks gestation to a then 25 year old mother via cesarean section due to fetal distress. The pregnancy was a monochorionic-monoamniotic twin gestation and was complicated by maternal hyperthyroidism for which she received beta blockers during pregnancy. Corrected birth weight was 0.930 kg (z=−4.0), length was 36 cm (z=−7.7), and head circumference was 25 cm (z=−8.7). Patient 6 and her twin stayed in the NICU due to prematurity. Patient 6’s twin died at DOL 40 due to cardiac arrest in the setting of intestinal perforation and sepsis. Patient 6 remained in the NICU for 5 months due to bronchopulmonary dysplasia. She required intubation for 2 months and had a Nissen fundoplication, hiatal hernia repair, and gastrostomy tube placement at 5 months of age.
Genetic workup was initiated due to her developmental delays at 2 years old with a normal karyotype and chromosome SNP microarray. At three years of age, exome sequencing identified a novel de novo pathogenic missense variant (c.5182 G>C, p.A1728P) in of NSD1.
Growth parameters at the age of 4 years included a weight 10.8 kg (z=−2.4), height 91.5 cm (z=−2.5), head circumference 52.3 cm (z=2.1). On examination, she was noted to have relative macrocephaly, triangular face with frontal bossing, pointed chin, downslanting palpebral fissures, and a depressed nasal bridge. She had single palmar creases with clinodactyly of the fifth fingers bilaterally. She was also noted to have mild generalized hypotonia and joint hypermobility.
At 4 years, the patient was referred to Gastroenterology due to persistent feeding intolerance and abdominal distension. She was found to have hypoglycemia with plasma glucose of 36 mg/dL after >24 hours of fasting for endoscopy. Plasma glucose normalized after glucose infusion. Subsequent diagnostic fast showed plasma glucose of 34 mg/dL and insulin of 1.6 μIU/mL. She continued to have episodic hypoglycemia. She was started on 10 mg/kg/day diazoxide with good response. She remained on diazoxide for 5 days after which she was transitioned to nearly continuous enteral feeds with 4 hour windows with stable plasma glucose. She was discharged home on this regimen. Of note, review of her medical labs indicated multiple asymptomatic hypoglycemic episodes prior to 4 years of age.
Previous exome sequencing data were reanalyzed and no additional variants related to the patient’s phenotype were identified, specifically in genes associated with congenital HI.
Patient 7:
Patient 7 was born at 38.5 weeks of gestation to a 29-year-old mother with a pregnancy notable for maternal depression and anxiety requiring for which Zoloft was prescribed. There was a subchorionic hemorrhage in the first trimester and sparse prenatal care. He was born vaginally with a weight of 3.36 kg (z=−0.3), length of 53.3 cm (z=1.3), and a head circumference of 33 cm (z=−1.4). He was limp and apneic and needed some resuscitative efforts (high-flow nasal cannula oxygen). Apgar scores were 5, 7, and 8 at 1, 5, and 10 minutes. Initial blood glucose was 18 mg/dL, and he was stabilized with glucose infusion and enteral feedings.
On DOL 3, he had jerky movements suspected to be seizures, and was transferred to a tertiary care facility for video EEG, which noted to excessive dyssynchrony, asymmetry and discontinuity of background, but no seizures. MRI showed normal brain parenchyma and hemorrhagic subdural collections interpreted as related to birthing. Despite 22 cal/oz enteral feedings, plasma glucose was <50 mg/dL and intravenous 20% dextrose infusions were used to counteract hypoglycemia. A diagnostic fast when plasma glucose was 26 mg/dL showed an insulin concentration of 1.4 μIU/mL and suppressed betahydroxybutyrate, consistent with HI. He also had an undetectable cortisol level during the hypoglycemic event and an ACTH stimulation test (1 μg) showed a blunted response (peak cortisol 7.4 ug/dL), leading to initiation of oral hydrocortisone. He had no other evidence of hypopituitarism. He was started on diazoxide 10 mg/kg/day and 24 cal/oz feedings with glucose infusions weaned over the following 3 days. He was discharged at 14 days of age, but he was then readmitted at 5.5 months of age due to respiratory distress. Plasma glucose levels during this admission were between 72 mg/dL and 107 mg/dL. At this time, he was maintained on 3.7 mg/kg/day of diazoxide. Endocrinology was consulted and recommended discontinuing the diazoxide and that pre-prandial glucose be tested several times per day. He is currently 7.5 months old and has not been seen for follow-up since the admission at 5.5 months.
While in the intensive care nursery, the infant was enrolled in a rapid whole genome sequencing research protocol, and results showed a de novo c.5431C>T (p.Arg1811Ter) pathogenic variant in NSD1. The variant was confirmed with Sanger sequencing. He saw a clinical geneticist at 4 months of age, at which time his weight was 7.17 kg (51%, z= +0.03), length was 67.3 cm (91%, z= +1.36) and head circumference was 40.5 cm (12%, z= −1.16). He exhibited apparent hypertelorism, downslanting palpebral fissures, periocular fullness with infraorbital creases, exotropia and pointed chin. He did not have a tall or broad forehead.
Clinical Data
Table 1 summarizes the genetic data. The clinical features, laboratory data, and treatments used to treat hypoglycemia are presented in Table 2 and Table 3. Four patients were male and three patients were female. The NSD1 pathogenic variants were identified by exome or genome sequencing in 6/7 of the patients and by NSD1 sequencing in 1/7 of the patients. Maternal complications of pregnancy were present in all patients. Five of the seven patients were born prematurely with two being small for gestational age. Some of the patients had clear or suspected neurological features with 4/7 having hypotonia, two of which had a sacrococcygeal teratoma. Albeit minor or related to prematurity, five patients had cardiovascular anomalies and two patients had minor genitourinary anomalies. In most cases, hypoglycemia was noted shortly after birth but a diagnosis of hyperinsulinemia was not always found after birth. All patients were treated with diazoxide with some response.
Table 1:
NSD1 Pathogenic variants
| Patient | Gene | Coding DNA | Variant | Zygosity | Inheritance | Classification |
|---|---|---|---|---|---|---|
| 1 | NSD1 | c.5332C>T | p.R1778* | Heterozygous | de novo | Pathogenic Variant |
| 2 | NSD1 | c.5036 C>G | p.S1679* | Heterozygous | de novo | Pathogenic Variant |
| 3 | NSD1 | c. 3004_3005del | p.K1002fs | Heterozygous | de novo | Pathogenic Variant |
| 4 | NSD1 | c.6050G>A | p.R2017Q | Heterozygous | de novo | Pathogenic Variant |
| 5 | NSD1 | c.3548_3549delCT | p.S1183* | Heterozygous | de novo | Pathogenic Variant |
| 6 | NSD1 | c.5182G>C | p.A1782P | Heterozygous | de novo | Pathogenic Variant |
| 7 | NSD1 | c.5431C>T | p.R1811* | Heterozygous | de novo | Pathogenic Variant |
Table 2:
Clinical Features of Patients with Sotos Syndrome and HI
| Patient 1 | Patient 2 | Patient 3 | Patient 4ss | Patient 5 | Patient 6 | Patient 7 | Totals | |
|---|---|---|---|---|---|---|---|---|
| Gender | Female | Female | Male | Male | Male | Female | Male | |
| Clinical Features | ||||||||
| Central nervous system | ||||||||
| Hypotonia | + | − | + | − | + | + | − | 4/7 |
| Other | Tethered cord; seizures; prominent supratentorial ventricles; diffuse thinning of corpus callosum | Diffuse thinning of corpus callosum; moderate to severe delays in myelination; ventriculomegaly | Periventricular leukomalacia | Abnormal EEG (background dyssyncrhony) | ||||
| Cardiology | ||||||||
| Cardiac defect | - | PDA; small PFO vs ASD | LV enlargement | PDA; small PFO vs ASD | VSD | PDA; ASD | - | 5/7 |
| Genitourinary | ||||||||
| - | Pelvicaliectasis | - | Hydroceles (bilaterally) | - | - | - | 2/7 | |
| Other clinical features | ||||||||
| Sacrococcygeal teratoma; scoliosis | Sacrococcygeal teratoma; obstructive sleep apnea | Metopic craniosynostosis | Congenital kyphosis; umbilical hernia | - | Bronchopulmonary dysplasia; recurrent aspiration pneumonia; hiatal hernia | - | ||
| Atypical features | ||||||||
| Failure to thrive; Sacrococcygeal teratoma | Sacrococcygeal teratoma | Metopic craniosynostosis | Failure to thrive | |||||
EEG- electroencephalogram; PDA- patent ductus arteriosis; PFO- patent foramen ovale; ASD- atrial septal defect; VES- ventricular septal defect
Table 3:
Hyperinsulinemic Hypoglycemia Data in Patients with Sotos and HI
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | Patient 6 | Patient 7 | |
|---|---|---|---|---|---|---|---|
| Hypoglycemia Data | |||||||
| Onset of hypoglycemia after birth | Shortly after birth | 5 months | Shortly after birth | Birth | 1 month | Possibly birth | Birth |
| Representative data of insulin [uIU/mL]/ glucose [mg/dL] (age) | <2.0/48 (day 23) | <2.0/69 (5 months) | <2.0/49 (day 17) | <2/29 (day 112) | <2/34 (5 weeks) | 1.6/34 (4 years) | 1.4/14 (day 5) |
| Irritability | Yes | No | Unknown | Yes | No | No | No |
| Seizures | Yes | No | Unknown | No | No | No | |
| Initial treatment for hypoglycemia | IV | – | IV | IV | IV | IV | IV |
| Diazoxide Data | |||||||
| IVG duration (days) | 30 | – | 27 | Unknown | – | Unknown | 7 |
| Maximum IV glucose rate (mg/kg/min) | 9 | – | 5 | Unknown | – | Unknown | Unknown |
| Diazoxide treatment (maximum dose, duration) | Yes (10 mg/kg/day, ongoing at 30 months) | Yes (5 mg/kg/day, ongoingat 5 months) | Yes (5 mg/kg/day, 9 days) | Yes (10mg/kg/day, 4 days; 8mg/kg/day, 32 months) | Yes (5 mg/kg/day, until 4 months) | Yes (10 mg/kg/day, 5 days at 4 years of age) | Yes (10 mg/kg/day) |
| Duration of feeding tube | Ongoing at 14 months | Ongoing at 5 months | None | Ongoing at 3 years 10 months | Removed at 3 years | Ongoing at 4 years | None |
| Duration of HI | Ongoing at 25 months | Ongoing at 5 months | 2 months | 4 years | 7 months | 4 years | Ongoing at 4 months |
Discussion
To our knowledge, these seven patients are among the first to be described with HI and Sotos syndrome caused by point mutations in NSD1. Although hypoglycemia has been described as a minor feature in Sotos syndrome, there are a few publications regarding genotype-phenotype correlations. Recently, data have been published on 8 individuals with HI and Sotos syndrome, with 7/8 patients having a 5q35 microdeletion and 8/8 patients having transient neonatal HI, with 3/8 requiring diazoxide treatment [Matsou et al., 2013; Carrasco Salas et al., 2015; Nakamura et al., 2015]. In our cohort, at least 3/7 patients (Patient 1, Patient 4, and Patient 6) had persistent HI, a clinical feature not previously reported in the Sotos syndrome literature, with HI being present beyond 1 year of life. Additionally, 2/7 patients are currently less than 1 year of age, so determination of persistent versus transient HI cannot yet be determined. Most of these patients did not present with classic features of Sotos syndrome, and most obtained a molecular diagnosis via exome sequencing. These patients demonstrate a broader spectrum of clinical features in Sotos syndrome, especially in the context of HI.
Since these patients all have point mutations in NSD1 as the cause of Sotos syndrome, they conclusively disprove the hypothesis that HI in Sotos syndrome is due to the deletion of additional genes in the 5q35 deleted region.
These data implicate a role for NSD1 in glucose homeostasis. NSD1 is a histone methyltransferase that has been implicated in the regulation of chromatin and gene expression. Furthermore, NSD1 is expressed in human pancreatic beta cells in both bulk islet cell analyses and single-cell RNA-sequencing data [Ackermann et al., 2016; Wang et al., 2016]. In addition, chromatin immunoprecipiation for NSD1-mediated histone methylation on chromatin marks in normal human islets found enrichment of the activating H3K36m3 mark and low levels of the repressive H4K20me1 mark at the insulin locus [Wilson et al. 2009]. This heavily supports the hypothesis that NSD1 regulates islet insulin expression. Matsou et al. did indeed speculate that NSD1 may be associated with beta cell-specific transcription factors that suppress the expression of insulin, and, that NSD1 haploinsufficiency may cause excessive expression of insulin [Matsou et al., 2013]; however, they were not able to exclude the possibility that another gene in the 5q35 region was the cause of HI. These patients, along with an additional patient described by Carrasco Salas et al. (2016), support the hypothesis that haploinsufficiency of NSD1 results in dysregulated insulin expression.
Interestingly, in 6/7 of our patients the diagnosis of Sotos syndrome was made by exome sequencing, since they did not convincingly fit a diagnosis of Sotos syndrome. We speculate that this is due to the atypical presentation of Sotos syndrome likely seen in patients who have HI as a clinical manifestation. When Patient 1 was seen by Clinical Genetics, her growth had plateaued. Although some facial features of Patient 1 were suggestive of Sotos syndrome, given the severe failure to thrive and the history of sacrococcygeal teratoma, exome sequencing was sent. Patient 3 was initially consulted for hypotonia, a known feature of Sotos syndrome, and craniosynostosis, a reported, though, atypical feature of Sotos syndrome. Patient 4 presented with failure to thrive at 1 month of age, a clinical feature not typical for Sotos syndrome. Lastly, it is important to note that since exome sequencing was completed in 6/7 patients, this would have identified any single gene causes of HI in those patients.
All individuals with Sotos syndrome with HI reported in the literature have been described as having transient HI. However, at least three of our patients have persistent HI, with Patient 1 being treated with diazoxide at 30 months of age and Patients 4 and 6 at 4 years of age. Importantly, all of the patients who underwent diazoxide treatment were responsive, suggesting that the potassium channel subunits encoded by KCNJ11 and ABCC8 are functional in these patients and that NSD1 does significantly alter expression of the potassium channel in the beta cell. Although all of the patients in this series have evidence of perinatal stress, which is known to be associated with transient HI, it would not be a cause of persistent HI. Similarly, Patients 5 and 6 had a fundoplication, which is known to be associated with post-prandial hyperinsulinemic hypoglycemia. We do not have sufficient clinical data for Patient 5 to definitively exclude fundoplication as a cause of his hypoglycemia. However, Patient 6 had fasting hyperinsulinemic hypoglycemic, which is not associated with fundoplication and is likely due to Sotos syndrome. Altogether, the frequency of persistent HI in our cohort suggests that the HI is likely due to NSD1 changes.
Lastly, overgrowth syndromes have often been associated with tumor risk. Although the developmental function of NSD1 is largely unknown, it can be speculated that because haploinsufficiency of NSD1 induces overgrowth, NSD1 likely acts as a corepressor of genes that promote growth [Sotos JF, 2014]. Benign and malignant tumors have been described previously in patients with Sotos syndrome. Malignant tumors present at various sites and at different ages, with the overall risk estimated to be ~3%, however given the wide range of site and age, no specific tumor screening guidelines are currently recommended. There have been no genotype-phenotype correlations on tumors and Sotos syndrome [Villani et al., 2017]. Benign tumors have been reported, though infrequently, with only two cases of teratomas being reported to date, neither of which were associated with HI [Leonard NJ, 2000]. It currently remains unclear, whether, if any connection exists between sacrococcygeal teratomas and HI in our patients.
In conclusion, the data reported herein support the hypothesis that HI is a clinical feature that should raise the index of suspicion for Sotos syndrome and that point mutations in NSD1 are sufficient to cause HI. Additionally, this report widens the spectrum of clinical features observed in Sotos syndrome, and in the context of HI, Sotos syndrome should be considered even if overgrowth is lacking. It also emphasizes the utility of further investigation to determine exactly how mutations in NSD1 play a role in insulin secretion.
Acknowledgements
The authors thank Benjamin Saben, MD for his assistance with data collection and the patients and their families for participating in this work. JMK acknowledges support from NIH K08 CA193915, St. Baldrick’s Foundation, and Alex’s Lemonade Stand Foundation.
Footnotes
Financial Disclosure: The authors have no financial relationships relevant to this article to disclose.
Conflict of Interest: The authors have no conflicts of interest to disclose.
References
- Aasland R, Gibson TJ, Stewart AF. 1995. The PHD finger: implications for chromatin-mediated transcriptional regulation. Trends Biochem Sci 20:56–59. [DOI] [PubMed] [Google Scholar]
- Ackermann AM, Wang Z, Schug J, Naji A, Kaestner KH. 2016. Integration of ATAC-seq and RNA-seq identifies human alpha cell and beta cell signature genes. Molecular Metabolism 5(3): 233–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasco Salas P, Palma Milla C, Lezana Rosales JM, Benito C, Franco Freire S, Lopez Siles J. 2016. Hyperinsulinemic hypoglycemia in a patient with an intragenic NSD1 mutation. Am J Med Genet Part A 170A:544–546. [DOI] [PubMed] [Google Scholar]
- Cole TR, Hughes HE. 1990. Sotos syndrome. J Med Genet 27:571–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole TR, Hughes HE. 1994. Sotos syndrome: A study of the diagnostic criteria and natural history. J Med Genet 31:20–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Onis M, Onyango A, Borghi E, Siyam A, Pinol A, et al. 2007. WHO Child Growth standards: Head circumference-for age, arm circumference-for-age, tricepts skinfold-for -age and subscapular skinfold-for-age: Methods and development. World Health Organization 307: 44–49. [Google Scholar]
- Douglas J, Tatton-Brown K, Coleman K, Guerrero S, Berg J, Cole TR, Fitzpatrick D, Gillerot Y, Hughes HE, Pilz D, Raymond FL, Temple IK, Irrthum A, Schouten JP, Rahman N. 2005. Partial NSD1 deletions cause 5% of Sotos syndrome and are readily identifiable by multiplex ligation dependent probe amplification. J Med Genet 42:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hook EB, Reynolds JW. 1967. Cerebral gigantism: Endocrinological and clinical observations of six patients including a congenital giant, concordant monozygotic twins, and a child who acheived adult gigantic size. J Pediatr 70:900–914. [DOI] [PubMed] [Google Scholar]
- Kamimura J, Endo Y, Kurotaki N, Kinoshita A, Miyake N, Shimokawa O, Harada N, Visser R, Ohashi H, Miyakawa K, Gerritsen J, Innes AM, Lagace L, Frydman M, Okamoto N, Puttinger R, Raskin S, Resic B, Culic V, Yoshiura K, Ohta T, Kishino T, Ishikawa M,Niikawa N, Matsumoto N 2003. Identification of eight novel NSD1 mutations in Sotos syndrome. J Med Genet 40:126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuczmarski RJ, Ogden CL, Guo SS, et al. 2002. 2000 CDC growth charts for the United States: Methods and development. National Center for Health Statistics. Vital Health Stat 11(246). [PubMed] [Google Scholar]
- Leonard NJ. 2000. Sacrococcygeal Teratoma in Two Cases of Sotos Syndrome. Am J Med Genet 95:182–184. [DOI] [PubMed] [Google Scholar]
- Leventopoulos G, Kitsiou-Tzeli S, Kritikos K, Psoni S, Mavrou A, Kanavakis E, Fryssira H. 2009. A clinical study of Sotos syndrome patients with review of the literature. Pediatr Neurol 40:357–364. [DOI] [PubMed] [Google Scholar]
- Matsuo T, Ihara K, Ochiai M, Kinjo T, Yoshikawa Y, Kojima-Ishii K, Noda M, Mizumoto H, Misaki M, Minagawa K, Tominaga K, Hara T. 2013. Hyperinsulinemic hypoglycemia of infancy in Sotos syndrome. Am J Med Genet Part A 161A:34–37. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Takagi M, Yoshihashi H, Miura M, Narumi S, Hasegawa T, Miyake Y, Hasegawa Y. 2015. A case with neonatal hyperinsulinemic hypoglycemia: It is a characteristic complication of sotos syndrome. Am J Med Genet Part A 167A:1171–1174. [DOI] [PubMed] [Google Scholar]
- Toda N, Ihara K, Kojima-Ishii K, Ochiai M, Ohkubo K, Kawamoto Y, Kohno Y, Kumasaka S, Kawase A, Ueno Y, Futatani T, Miyazawa T, Nagaoki Y, Nakata S, Misaki M, Arai H, Kawai M, Sato M, Yada Y, Takahashi N, Komatsu A, Maki K, Watabe S, Sumida Y, Kuwashima M, Mizumoto H, Sato K, Hara T. 2017. Hyperinsulinemic hypoglycemia in Beckwith–Wiedemann, Sotos, and Kabuki syndromes: A nationwide survey in Japan. Am J Med Genet Part A 173A:360–367. [DOI] [PubMed] [Google Scholar]
- Tatton-Brown K, Douglas J, Coleman K, Baujat G, Cole TR, Das S, Horn D, Hughes HE, Temple IK, Faravelli F, Waggoner D, Turkmen S, Cormier-Daire V, Irrthum A, Rahman N. 2005. Genotype–phenotype associations in Sotos syndrome: Analysis of 266 individuals with NSD1 aberrations. Am J Hum Genet 77:193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotos JF. 2014. Sotos syndrome 1 and 2. Ped Endocrin Rev 12:2–16. [PubMed] [Google Scholar]
- Villani A, Greer MLC, Kalish JM, Nakagawara A, Nathanson KL, Pajtler KW, Pfister SM, Walsh MF, Wasserman JD, Zelley K, and Kratz CP. 2017. Recommendations for Cancer Surveillance in Individuals with RASopathies and Other Rare Genetic Conditions with Increased Cancer Risk. Clin Cancer Res 23(12): 83–90. [DOI] [PubMed] [Google Scholar]
- Wang YJ, Avrahami D, Golson M, Kaestner KH. 2016. Single-Cell Transcriptomics of the Human Endocrine Pancreas. Diabetes 65(10): 3028–3038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson LM, Wong SH, Yu N, Geras-Raaka E, Raaka BM, Gershengorn MC. 2009. Insulin but not glucagon gene is silenced in human pancreas-derived mesenchymal stem cells. Stem Cells 27(11): 2703–2711. [DOI] [PMC free article] [PubMed] [Google Scholar]