

Supplemental Digital Content is available in the text
Keywords: dravet syndrome, SCN1B, SCN2B, variant
Abstract
Previous research identified SCN1B variants in some cases of Dravet syndrome (DS). We investigated whether SCN1B and SCN2B variants are commonly happened in DS patients without SCN1A variants. A total of 22 DS patients without SCN1A variants and 100 healthy controls were enrolled in this genetic study. DNA from DS patients was sequenced by Sanger method in whole exons of SCN1B and SCN2B genes. We identified two exon variants (c.351C>T, p.G117G and c.467C>T, p.T156M), which were present both in 1000 egenomes database and in healthy controls with a frequency of 0.54% and 4%, 0.06% and 0%, respectively. Additionally, eight intron or 3 prime UTR variants showing benign clinical significance have also been identified. Our results suggest that variants of SCN1B and SCN2B may not be common causes of DS according to our data. Further large sample-size cohort studies are needed to confirm our conclusion.
1. Introduction
Dravet syndrome (DS) is a kind of archetypal genetic infantile epileptic encephalopathy with genetic etiology in most patients.[1–3] DS is characterized by an onset, in the first year of an infant's life, of febrile or afebrile clonic or tonic–clonic seizures.[1] DS patients are antiepileptic drug resistant and later ataxia, psychomotor development, metal decline are observed in DS patients.[4,5]
DS has variants in SCN1A gene in 70%–80% of the patients[1,2,6] and has rare variants in GABRG2, GABRB3, GABRA1, PCDH19, HCN1, and STXBP1 genes.[4,7] More recently, SCN1B variants were also identified in DS patients without SCN1A variants.[8–10] However, these variants of SCN1B are found in individual patients, but not in cohort patients. A study found no variant of SCN1B in a cohort of 54 DS patients in whom SCN1A exons variant had been excluded.[5]
Sodium channels are composed of one α subunit and two β submits.[11,12]SCN1B encodes the β1 subunit of voltage-gated sodium channel 1, which contributes to regulate sodium channel gating and control the channel Nav 1.1 expression and localization.[12]SCN2B encodes the β2 subunit and results showed that SCN2B-null neurons have reduced voltage-gated sodium channel expression in cell surface, which implies the modulation function of SNC2B in neurons.[13]
In this study, we tested the hypothesis that SCN1B and SCN2B variants confer susceptibility to DS with SCN1A-variant free patients.
2. Methods
2.1. Subjects
Patients enrolled in this study were recruited from the Xiangya hospital and the Second Xiangya Hospital of Central South University according to the international epilepsy classification in previous studies.[1,2] The whole blood was collected from the subjects, and DNA was extracted by the phenol-chloroform method.[14–16] The diagnostic criteria for DS were (1) the onset is before 1 year of age, (2) the seizure type is clonic and often unilateral instead of generalized tonic or tonic–clonic, (3) the seizure episodes are more prolonged and frequent, even when treated, and (4) the body temperature is not very high.[17–19] First, patients were screened by SCN1A exons variant according to previous studies.[20,21] Exclusion criteria were patients who did not have their blood drawn for sequencing and who were not diagnosed with DS. Then patients without SCN1A exons variant were enrolled in this study to investigate the variants of SCN1B and SCN2B. Healthy controls without any illness were randomly selected from Medical Examination Center of the Xiangya Hospital. A standardized questionnaire was administered to collect clinical details such as symptoms, seizure types and frequency, and AEDs used. Patient characteristics were collected from clinical records, including gender, age, age at onset, familial or sporadic, electroencephalogram, magnetic resonance imaging, computed tomography, duration of seizure, frequency of seizure, and current medication. The Ethics Committee of Institute of Clinical Pharmacology of Central South University approved the study. The Chinese Clinical Trial Register approved the clinical study admission (Registration Number: ChiCTR-TCH-0000813).
2.2. Variant analysis
DNA was extracted from the whole blood of the subjects according to previous standard procedures.[15] The whole coding exons of SCN1B, SCN2B, and SCN1A were screened for variants by PCR amplification and Sanger sequencing according to previous research (Table 1).[22,23] ABI 3700 Genetic Analyzer analyzed PCR products. PubMed database (https://www.ncbi.nlm.nih.gov/pubmed/) was used to evaluate single nuclear acid variant information including the position, nucleotide changes, function changes, clinical significance, and 1000G frequency. SIFT database (http://provean.jcvi.org/index.php) and PolyPhen-2 database (http://genetics.bwh.harvard.edu/pph2/index.shtml) were used to predict the variant function.
Table 1.
Primer sequences of SCN1B and SCN2B genes.

3. Results
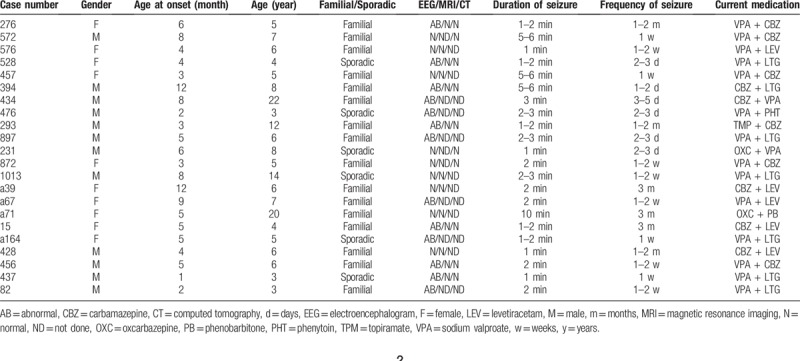
Of the total 48 DS patients, 26 patients had SCN1A variants (Table S1). Twenty-two patients with DS without SCN1A exon variant were enrolled in further screening the exon variants of SCN1B and SCN2B. One hundred healthy controls also had their exon variants of SCN1B and SCN2B analyzed. The characteristics of DS patients were shown in Table 2. The 22 DS patients include 12 men and 10 women in the age range of 7.5 ± 5.1 months. The age of onset was 5.4 ± 2.99 months. The medications include valproic acid, topiramate, carbamazepine, oxcarbazepine, phenobarbital, and levetiracetam.
Table 2.
Characteristics of Dravet syndrome patients.
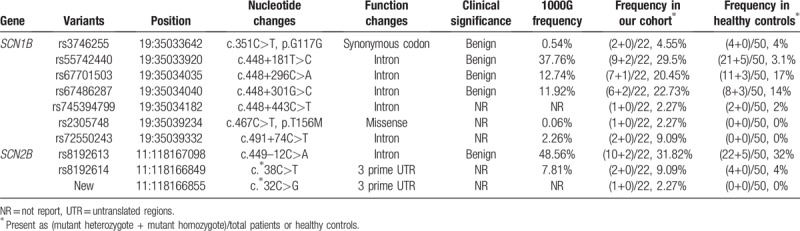
In general, no pathogenic SCN1B and SCN2B variants were detected in our study cohort. We found one exon variant (c.351 C>T, p.G117G), which is a synonymous codon with benign clinical significance and is present in 1000 genomes database with a frequency of 0.54% (Fig. 1). And this mutant heterozygote was also found in four healthy controls (4%). We also found one missense variant changing the coding amino acid from threonine to methionine (c.467C>T, p.T156M) in one DS patient and the frequency of this variant is 0.06% in 1000 genomes database and 0% in healthy control. SIFT and PolyPhen-2 database to predict the function of missense variant and results showed damaging and neutral, respectively. We found no exon variant of SCN2B. Moreover, we also found six intron variants and two 3 prime untranslated regions (UTR) variants in patients and healthy controls (Fig. 2 and Table 3). Interestingly, we found one new variant in 3 prime UTR region of SCN2B, which was not in the dbSNP and 1000G database. This 3 prime UTR variant (c. ∗32C>T) was found in one DS patient and was not present in healthy controls.
Figure 1.
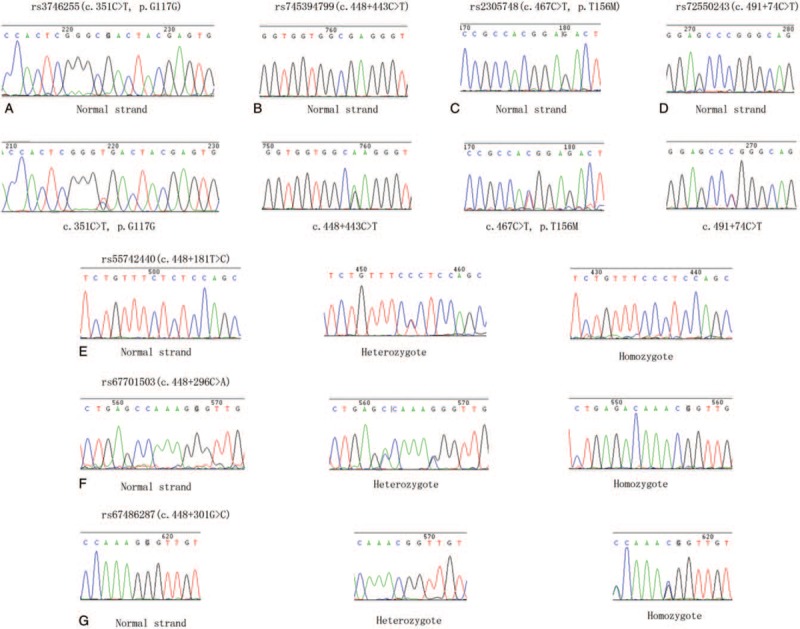
The identified SCN1B variants in Dravet syndrome. (a) rs3746255(c.351C>T, p.G117G); (b) rs745394799(c.448+443C>T); (c) rs2305748(c.467C>T, p.T156M); (d) rs72550243(c.491+74C>T); (e) rs55742440(c.448+181T>C); (f) rs67701503(c.448+296C>A); (g) rs67486287(c.448+301G>C).
Figure 2.
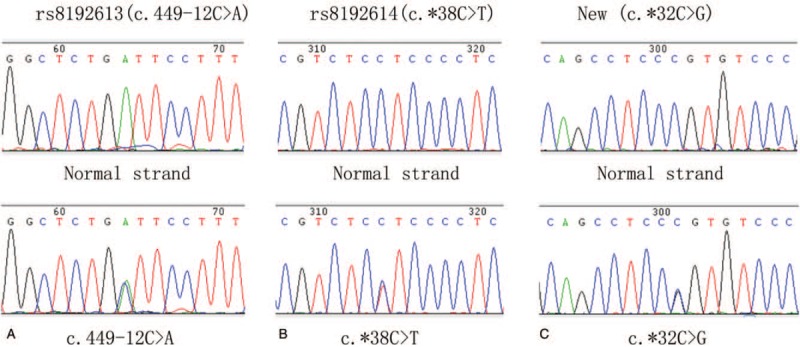
The identified SCN2B variants in dravet syndrome. (a) rs8192613(c.449-12C>A); (b) rs8192614(c.∗38C>T); (c) New(c.∗32C>G).
Table 3.
Characteristics of SCN1B and SCN2B mutations identified in our cohort.
4. Discussion
DS has variants in SCN1A gene in 70%–80% of the patients.[1,2,6] We screened 48 DS patients and found 26 DS patients (54.2%) had SCN1A variants (Table S1), which were also reported in previous studies and database.[30–32] Studies reported causative variants of SCN1B p.R125C in two infants with DS[8,10] and SCN1B p.lle106Phe in one of 67 DS patients.[9] In order to find the pathogenesis of DS except SCN1A variants, we carried out this study to investigate the variants of SCN1B and SCN2B variants in DS patients.
In our study, we found one exon variant of SCN1B (c.351 C>T, p.G117G), which is a synonymous codon with benign clinical significance and is present in 1000 genomes database with a frequency of 0.54%. We also found one missense variant of SCN1B (c.467C>T, p.T156M) presented in 1000 genomes database with a frequency of 0.06%. We used SIFT and PolyPhen-2 database to predict the function of missense variant and results showed damaging and neutral, respectively. Other intron variants of SCN1B and SCN2B found in our cohort were not related to the pathogenesis of DS.
Voltage-gated sodium channels are composed of a large α-subunit and two smaller β-subunits (β1 and β2).[22] Functional studies found the SCN1B gene which encodes that the β1-subunit regulates dorsal root ganglion nociceptor excitability in vivo.[24]SCN1B variants (p.C121W, p.I70 E74del, p.R85C, p.R85H, and p.R125L) were also reported in genetic epilepsy with febrile seizure plus and absence epilepsy patients.[10,25–27] Our results found that SCN1B variants are not a common cause of DS, which is consistent with previous studies.[5]
SCN2B gene encodes β2-subunit of voltage-gated sodium channel and the gene consists of 4 exons and is highly conserved in the coding region.[22]SCN2B variant might be associated with the risk of Brugada syndrome but not with the idiopathic generalized epilepsy in previous research.[15,22,28,29] We first screened the exon region of SCN2B in DS patients without SCN1A variant. There was no research about SCN2B variant in DS patients. Our research found no exon variant of SCN2B in DS patients.
SCN1A gene variants were found in 70%–80% of the DS patients, and SCN1B and SCN2B encoding β1/β2 subunits regulate sodium channel gating and control the channel Nav 1.1 expression and localization. Therefore, SCN1B and SCN2B variants may influence the channel Nav 1.1 function, inducing the seizure of DS patients. Lots of SCN1A variants were identified in DS patients at previous study and in our study. However, we found no combined variant of SCN1A, SCN1B and SCN2B, and SCN1B and SCN2B variant was rarely identified in DS patients. The roles of SCN1B and SCN2B variants contributed to the etiology of DS patients were needed in large sample-size cohort studies and mechanism function studies.
Our study suggested that SCN1B and SCN2B variants are uncommon in DS patients according to our data. Further large sample-size cohort studies are needed to confirm our conclusion.
Acknowledgments
We thank the supported grants of the National Natural Science Foundation of China (Nos. 81503166, 81603208, and 81671299), Hunan Provincial Natural Science Foundation of China (2018JJ3718), Omics-based precision medicine of epilepsy being entrusted by Key Research Project of the Ministry of Science and Technology of China (No. 2016YFC0904400), and the Science and Technology Department Funds of Hunan Province Key Project (2016JC2057).
Author contributions
Conceptualization: Jiao-E Gong, Qiang Qu, Li-Min Yang, Jian Qu.
Data collection: Jiao-E Gong, Hong-Yu Long, Shao-Hua Lu, Qiang Qu, Li-Min Yang.
Formal analysis: Hong-Mei Liao, Xiang-Min Li, Li-Min Yang, Jian Qu.
Funding acquisition: Qiang Qu, Jian Qu.
Investigation: Jiao-E Gong, Xiang-Min Li, Li-Li Long, Luo Zhou.
Methodology: Hong-Mei Liao, Xiang-Min Li, Li-Li Long, Luo Zhou, Shao-Hua Lu.
Resources: Hong-Yu Long.
Software: Hong-Yu Long.
Supervision: Hong-Mei Liao.
Writing – original draft: Hong-Yu Long, Li-Li Long, Luo Zhou, Shao-Hua Lu, Qiang Qu.
Writing – review & editing: Li-Li Long, Luo Zhou, Shao-Hua Lu, Qiang Qu, Li-Min Yang, Wen-Ping Gu, Bo Xiao, Jian Qu.
Supplementary Material
Footnotes
Abbreviations: CBZ = carbamazepine, CT = computed tomography, DS = Dravet syndrome, EEG = electroencephalogram, LEV = levetiracetam, MRI = magnetic resonance imaging, OXC = oxcarbazepine, PB = phenobarbitone, PHT = phenytoin, TPM = topiramate, UTR = 3 prime untranslated regions, VPA = sodium valproate.
Conflict of interests: The authors have no funding and conflicts of interest to disclose.
Supplemental Digital Content is available for this article.
References
- [1].Dravet C. The core Dravet syndrome phenotype. Epilepsia 2011;52Suppl 2:3–9. [DOI] [PubMed] [Google Scholar]
- [2].Harkin LA, McMahon JM, Iona X, et al. The spectrum of SCN1A-related infantile epileptic encephalopathies. Brain 2007;130(Pt 3):843–52. [DOI] [PubMed] [Google Scholar]
- [3].Zou R, Wang S, Zhu L, et al. Calgary score and modified Calgary score in the differential diagnosis between neurally mediated syncope and epilepsy in children. Neurol Sci 2017;38:143–9. [DOI] [PubMed] [Google Scholar]
- [4].Steel D, Symonds JD, Zuberi SM, et al. Dravet syndrome and its mimics: beyond SCN1A. Epilepsia 2017;58:1807–16. [DOI] [PubMed] [Google Scholar]
- [5].Kim YO, Dibbens L, Marini C, et al. Do variants in SCN1B cause Dravet syndrome? Epilepsy Res 2013;103:97–100. [DOI] [PubMed] [Google Scholar]
- [6].Xiang J, Jiang Y. Regulation of Cu-Zn superoxide dismutase on SCN2A in SH-SY5Y cells as a potential therapy for temporal lobe epilepsy. Mol Med Rep 2014;9:16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Le SV, Le PHT, Le TKV, et al. A variant in GABRB3 associated with Dravet syndrome. Am J Med Genet A 2017;173:2126–31. [DOI] [PubMed] [Google Scholar]
- [8].Mukherjee D, Mukherjee S, Niyogi P, et al. Dravet syndrome with SCN1B gene variant: a rare entity. Neurol India 2017;65:801–3. [DOI] [PubMed] [Google Scholar]
- [9].Ogiwara I, Nakayama T, Yamagata T, et al. A homozygous variant of voltage-gated sodium channel beta(I) gene SCN1B in a patient with Dravet syndrome. Epilepsia 2012;53:e200–3. [DOI] [PubMed] [Google Scholar]
- [10].Patino GA, Claes LR, Lopez-Santiago LF, et al. A functional null variant of SCN1B in a patient with Dravet syndrome. J Neurosci 2009;29:10764–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Brackenbury WJ, Djamgoz MB, Isom LL. An emerging role for voltage-gated Na+ channels in cellular migration: regulation of central nervous system development and potentiation of invasive cancers. Neuroscientist 2008;14:571–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Chen C, Westenbroek RE, Xu X, et al. Mice lacking sodium channel beta1 subunits display defects in neuronal excitability, sodium channel expression, and nodal architecture. J Neurosci 2004;24:4030–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Auerbach DS, Jones J, Clawson BC, et al. Altered cardiac electrophysiology and SUDEP in a model of Dravet syndrome. PLoS ONE 2013;8:e77843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chen J, Li B, Yang Y, et al. Variants of the TGFBR2 gene in Chinese patients with Marfan-related syndrome. Clin Invest Med 2010;33:E14–21. [DOI] [PubMed] [Google Scholar]
- [15].Qu J, Lu SH, Lu ZL, et al. Pharmacogenetic and case-control study on potassium channel related gene variants and genetic generalized epilepsy. Medicine (Baltimore) 2017;96:e7321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Tang FY, Xie XW, Ling GH, et al. Endothelial nitric oxide synthase and nicotinamide adenosine dinucleotide phosphate oxidase p22phox gene (C242T) polymorphisms and systemic lupus erythematosus in a Chinese Population. Lupus 2010;19:192–6. [DOI] [PubMed] [Google Scholar]
- [17].Dravet C. Dravet syndrome history. Dev Med Child Neurol 2011;53Suppl 2:1–6. [DOI] [PubMed] [Google Scholar]
- [18].Scheffer IE, Berkovic S, Capovilla G, et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017;58:512–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Fisher RS, Cross JH, French JA, et al. Operational classification of seizure types by the International League Against Epilepsy: Position Paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017;58:522–30. [DOI] [PubMed] [Google Scholar]
- [20].Binini N, Sancini G, Villa C, et al. Identification of two variants in cis in the SCN1A gene in a family showing genetic epilepsy with febrile seizures plus (GEFS+) and idiopathic generalized epilepsy (IGE). Brain Res 2017;1677:26–32. [DOI] [PubMed] [Google Scholar]
- [21].Gontika MP, Konialis C, Pangalos C, et al. Novel SCN1A and GABRA1 gene variants with diverse phenotypic features and the question on the existence of a broader spectrum of Dravet syndrome. Child Neurol Open 2017;4: 2329048X17706794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Haug K, Sander T, Hallmann K, et al. The voltage-gated sodium channel beta2-subunit gene and idiopathic generalized epilepsy. Neuroreport 2000;11:2687–9. [DOI] [PubMed] [Google Scholar]
- [23].Claes L, Del-Favero J, Ceulemans B, et al. De novo variants in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet 2001;68:1327–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lopez-Santiago LF, Brackenbury WJ, Chen C, et al. Na+ channel Scn1b gene regulates dorsal root ganglion nociceptor excitability in vivo. J Biol Chem 2011;286:22913–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Audenaert D, Claes L, Ceulemans B, et al. A deletion in SCN1B is associated with febrile seizures and early-onset absence epilepsy. Neurology 2003;61:854–6. [DOI] [PubMed] [Google Scholar]
- [26].Fendri-Kriaa N, Kammoun F, Salem IH, et al. New variant c.374C>T and a putative disease-associated haplotype within SCN1B gene in Tunisian families with febrile seizures. Eur J Neurol 2011;18:695–702. [DOI] [PubMed] [Google Scholar]
- [27].Scheffer IE, Harkin LA, Grinton BE, et al. Temporal lobe epilepsy and GEFS+ phenotypes associated with SCN1B variants. Brain 2007;130(Pt 1):100–9. [DOI] [PubMed] [Google Scholar]
- [28].Riuro H, Beltran-Alvarez P, Tarradas A, et al. A missense variant in the sodium channel beta2 subunit reveals SCN2B as a new candidate gene for Brugada syndrome. Hum Mutat 2013;34:961–6. [DOI] [PubMed] [Google Scholar]
- [29].Dulsat G, Palomeras S, Cortada E, et al. Trafficking and localisation to the plasma membrane of Nav 1.5 promoted by the beta2 subunit is defective due to a beta2 variant associated with Brugada syndrome. Biol Cell 2017;109:273–91. [DOI] [PubMed] [Google Scholar]
- [30].Do TT, Vu DM, Huynh TT, et al. SCN1A gene variant and adaptive functioning in 18 Vietnamese children with Dravet syndrome. J Clin Neurol 2017;13:62–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ishii A, Watkins JC, Chen D, et al. Clinical implications of SCN1A missense and truncation variants in a large Japanese cohort with Dravet syndrome. Epilepsia 2017;58:282–90. [DOI] [PubMed] [Google Scholar]
- [32].Meng H, Xu HQ, Yu L, et al. The SCN1A variant database: updating information and analysis of the relationships among genotype, functional alteration, and phenotype. Hum Mutat 2015;36:573–80. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.