

Abstract
To determine the incidence and risk of Parkinson disease (PD) in patients with Sjögren syndrome (SS) according to a nationwide population-based database.
In total, 12,640 patients in the SS cohort and 50,560 in the non-SS cohort were enrolled from Taiwan's National Health Insurance Research Database from 2000 to 2010. We used the Cox multivariable proportional hazards model to determine the risk factors for PD in the SS cohort.
We observed an increased incidence of PD in patients with SS, with a crude hazard ratio (HR) of 1.40 and an adjusted HR (aHR) of 1.23. The cumulative incidence of PD was 1.95% higher in the SS cohort than in the non-SS cohort. The SS cohort had an elevated HR under medication use, namely cevimeline and pilocarpine (crude HR, 1.28), hydroxychloroquine (crude HR, 1.43; aHR, 1.46), and methylprednisolone (crude HR, 2.21; aHR, 1.49). Patients receiving other non-hydroxychloroquine immunosuppressant therapies had a lower risk (aHR, 0.86) of PD. Furthermore, patients with SS aged 20 to 49 years had a 1.93-fold higher risk of PD than did those without SS (aHR, 1.93). The risk of PD was higher (aHR, 2.20) in patients with SS without comorbidities than in those with comorbidities. The aHR of PD significantly increased when the follow-up period exceeded 9 years (aHR, 1.93).
We determined an increased risk of PD in patients with SS. Further investigation is warranted to determine the possible underlying mechanisms and the potential role of non-hydroxychloroquine immunosuppressants in ameliorating PD.
Keywords: longitudinal health insurance database, national health insurance research database, Parkinson disease, Sjögren syndrome
1. Introduction
Sjögren syndrome (SS) is a systemic chronic inflammatory and autoimmune disorder characterized by lymphocytic infiltrates in exocrine organs, typically the lacrimal and salivary glands, causing dryness of the eyes and mouth. A recent study confirmed that primary SS has a systemic expression, including glandular and extraglandular manifestations such as neurological involvement.[1–4] For diseases that affect both the peripheral and central nervous systems, various neurological manifestations may occur in those with SS.[5–7] Parkinson disease (PD) is a progressive neurodegenerative disease with the cardinal symptoms of tremor, bradykinesia, rigidity, and postural instability.[8] The pathophysiology of PD is dopamine-cell death in the pars compacta region of the substantia nigra[9] that results in major disruptions in the connections to the thalamus and motor cortex.
Some immune system-mediated mechanisms have been proposed to illustrate the possible pathogenic mechanisms through which autoantibodies cause dopaminergic cell death.[10,11] Benkler et al[12] reported that inflammatory autoantibodies, namely antineuronal-cells, anti-brain lysate, anti-dsDNA, may contribute to the clinical manifestations of PD. The ICAM4, Myotilin, Fibronectin 1 and Pentatricopeptide repeat domain 2 can also be detected in PD sera.[13] Sera from patient with SS who have autonomic neuropathy is also related to circulating anti-ganglionic acetylcholine receptor antibodies.[14] We designed this study to assess the risk of PD in patients with SS and to recognize the associated risk factors. We also evaluated the association between SS and the risk of PD and observed a higher risk of PD in patients with SS.
2. Methods
2.1. Data source
The National Health Insurance (NHI) program was established on March 1, 1995 to provide comprehensive medical care to the 23.54 million residents of Taiwan.[15] The NHI Research Database (NHIRD), maintained by the National Health Research Institutes (NHRI), contains claims records from the universal NHI program. The NHIRD has been described in detail in previous studies.[16,17] The NHRI encrypts the original identification information to protect patient privacy before releasing the NHIRD for research purposes. In this study, we used a subset of the NHIRD containing health care data, namely the Longitudinal Health Insurance Database for Catastrophic Illness Patients (LHID-CIP), and the Registry of Beneficiaries. The diagnoses and procedures recorded in the NHIRD are coded according to the International Classification of Diseases, Ninth Revision, Clinical Modification (ICD-9-CM). The Ethics Review Board of China Medical University and Hospital in Taiwan approved this study (CMUH-104-REC2–115-CR3).
2.2. Patients
Patients newly diagnosed with SS (ICD-9-CM code 710.2) from 2000 to 2010 were identified from the LHID-CIP. The SS was diagnosed on the basis of ICD-9-CM codes, which were judged and determined by related specialists and physicians according to the standard clinical and laboratory criteria (ocular symptoms, oral symptoms, ocular signs, histopathology, salivary gland involvement, and autoantibodies, which met 4 of the 6 revised international classification criteria that were established by the American–European Consensus Group for diagnosing SS and 3 of the 4 objective criteria). The date of SS diagnosis was defined as the index date. Patients with a history of PD (ICD-9-CM 332) and aged <20 years were excluded. For the non-SS cohort, we randomly selected patients without a history of SS from the LHID-CIP, with exclusion criteria similar to those used for the SS cohort. Each patient with SS was frequency-matched with 4 patients without SS according to age (at 5-y intervals), sex, and the year of the index date.
2.3. Outcomes, comorbidities, and medications
The follow-up period began on the index date and continued until PD diagnosis; withdrawal from the NHI program; or December 31, 2011. Preexisting comorbidities included hypertension (ICD-9-CM codes 401–405), diabetes (ICD-9-CM code 250), hyperlipidemia (ICD-9-CM code 272), coronary artery disease (ICD-9-CM codes 410–414), head injury (ICD-9-CM codes 310.2, 800, 801, 803, 804, 850, 851, 853, and 854), depression (ICD-9-CM codes 296.2, 296.3, 296.82, 300.4, and 311), and stroke (ICD-9-CM codes 430–438). In addition, the use of non-hydroxychloroquine immunosuppressants (azathioprine, cyclosporine, mycophenolate, and tacrolimus), cholinergic agents (cevimeline and pilocarpine), hydroxychloroquine, and methylprednisolone was analyzed in the SS cohort.
2.4. Statistical analysis
We used the Chi-square test to compare the distributions of categorical variables and baseline comorbidities between the SS and non-SS cohorts. Differences in continuous variables between the cohorts were examined through the Student t test. The cumulative incidence of PD in the SS and non-SS cohorts was assessed through the Kaplan–Meier method; the log-rank test was used to compare the 2 cohorts. The incidence densities (per 1000 person-y) of PD were calculated for both cohorts.
Univariable and multivariable Cox proportional hazards regression models were used to calculate the hazard ratios (HRs) and 95% confidence intervals (CIs) of PD development in the SS cohort compared with the non-SS cohort, with stratification based on sex, age, comorbidities, and the follow-up period. The multivariable Cox models were simultaneously adjusted for age, sex, and the comorbidities of hypertension, diabetes, hyperlipidemia, coronary artery disease, head injury, depression, and stroke. Furthermore, we evaluated the effects of biological therapies and anti-SS drugs on the risk of PD in the SS cohort. All analyses were conducted using SAS Version 9.4 (SAS Institute Inc., Cary, NC), with the significance level set to 0.05 in 2-tailed tests.
3. Results
The SS and non-SS cohorts included 12,640 and 50,560 participants, respectively, with similar distributions of age and sex (Table 1). The majority of the participants were aged ≤49 years (39.4%; mean age, approximately 54 y), and 89.1% were women. Comorbidities of hyperlipidemia, coronary artery disease, head injury, and depression were more prevalent in the SS cohort than in the non-SS cohort (P < .05), but diabetes was more prevalent in the non-SS cohort. The mean follow-up period for PD in the SS and non-SS cohorts was 5.21 (standard deviation [SD], 3.15) and 5.18 (SD, 3.16) years, respectively. The Kaplan–Meier survival graph indicated that the cumulative incidence of PD was 1.95% higher in the SS cohort than in the non-SS cohort (P < .001; Fig. 1). The most common medications used for SS were cevimeline and pilocarpine (36.5%), hydroxychloroquine (27.1%) and non-hydroxychloroquine immunosuppressant therapies (17.5%).
Table 1.
Characteristics of patients with and without Sjögren syndrome.
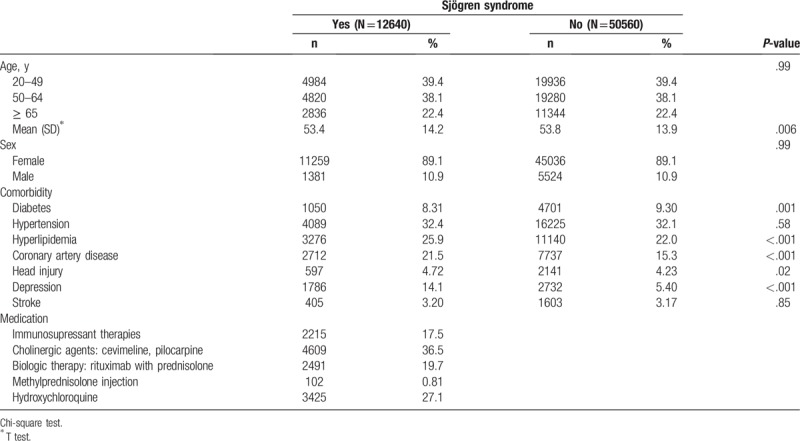
Figure 1.
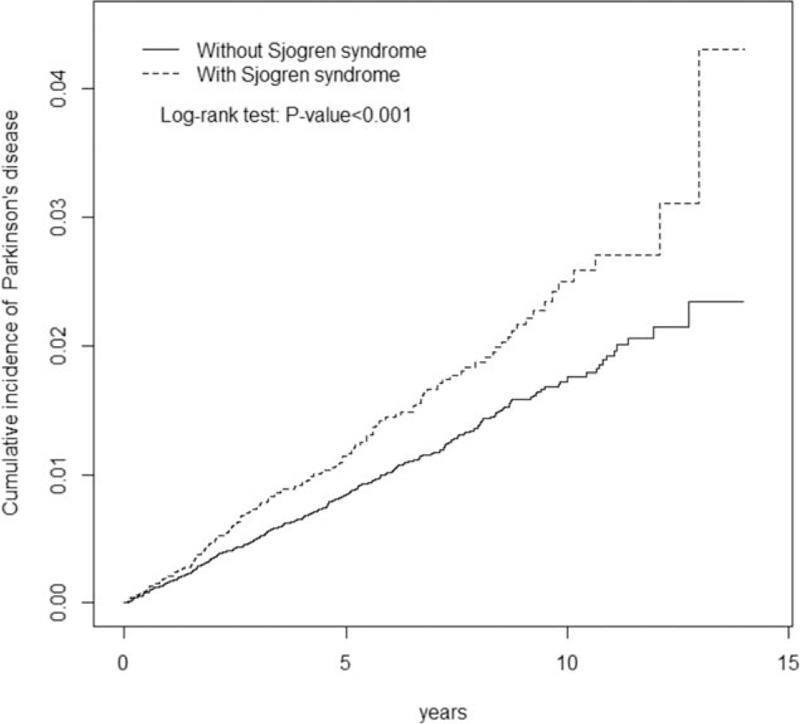
Cumulative incidence of Parkinson disease compared between patients with and without Sjögren syndrome.
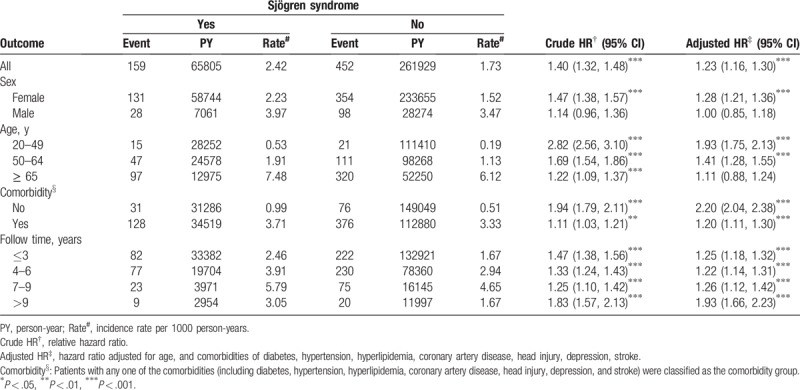
The overall incidence of PD was 1.40-fold higher in the SS cohort than in the non-SS cohort (2.42 vs 1.73 per 1000 person-y; 95% CI, 1.32–1.48). After adjustment for age, sex, and comorbidities, participants with SS had a higher risk of PD than did those without SS (adjusted HR [aHR], 1.23; 95% CI, 1.16–1.30; Table 2]. The sex-specific risk of PD was significantly higher in women (aHR, 1.28; 95% CI, 1.21–1.36), and the incidence of PD decrease with age. The risk of PD was 1.93- and 1.14-fold higher in participants aged 20 to 49 (aHR, 1.93; 95% CI, 1.75–2.13) and 50 to 64 (aHR = 1.41; 95% CI = 1.28–1.55) years, respectively, than in those without SS. Furthermore, the risk of PD was significantly higher in participants with SS than in those without SS after stratification without any comorbidities (aHR, 2.20; 95% CI, 2.04–2.38) and with at least one comorbidity (aHR, 1.20; 95% CI, 1.11–1.30). The aHR of PD significantly increased with the follow-up period of more than 9 years (aHR, 1.93; 95% CI, 1.66–2.23). The SS cohort had a higher risk of PD than did the non-SS cohort throughout the follow-up period.
Table 2.
Incidence and hazard ratio of Parkinson disease in patients with and without Sjögren syndrome.
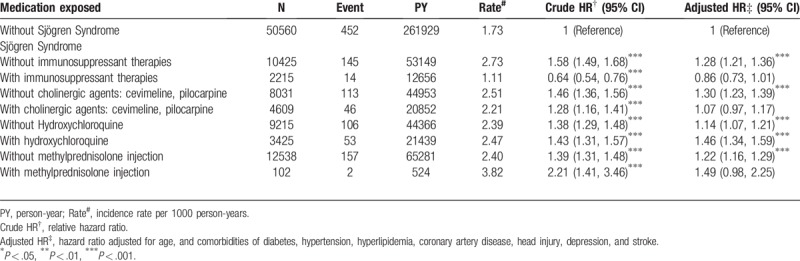
Compared with the participants without SS, the participants with SS to whom hydroxychloroquine was administered had a significantly higher risk of PD (aHR, 1.46; 95% CI, 1.34–1.59). Moreover, the risk of PD was higher in participants with SS to whom cevimeline and pilocarpine (aHR, 1.30; 95% CI, 1.23–1.39), hydroxychloroquine (aHR, 1.14; 95% CI, 1.07–1.21), and methylprednisolone (aHR, 1.22; 95% CI, 1.16–1.29) were not administered. Participants under other non-hydroxychloroquine immunosuppressant therapies had a relatively lower risk of PD (aHR, 0.86; 95% CI, 0.73–1.01; Table 3).
Table 3.
Incidence and adjusted hazard ratio of Parkinson disease stratified by medication in patients with Sjögren syndrome.
4. Discussion
As per our review of relevant literature, no large-scale cohort study has investigated the association between SS and PD. This study was the 1st extensive analysis of a nationwide population database of participants with SS and PD. Our findings revealed that SS increased the risk of PD. The aHR of PD significantly increased for the participants aged 20 to 49 years and gradually decreased for those aged >49 years. The crude HR of 1.40 (95% CI, 1.38–1.48), with an aHR of 1.23 (95% CI, 1.16–1.30), for PD development was higher in the participants with SS. The participants with SS also had a higher risk of PD during the long-term follow-up period, and the participants without comorbidities had a notably elevated HR for PD (aHR, 2.20).
The treatment of dry mouth caused by salivary gland hypofunction aims to alleviate symptoms. Participants with SS who received cevimeline and pilocarpine, which act as sialogogues agents, did not increase the HR for PD in our study (aHR, 1.07). However, there was an increasing of the HR for those who did not receive the treatment (aHR, 1.30). In addition, the administration of hydroxychloroquine yielded similar results. The participants with SS who were treated with or without hydroxychloroquine had a higher risk of PD (aHR, 1.46 and 1.14, respectively). Intravenous pulse steroid therapy with methylprednisolone is sometimes used in participants with SS because of its antiinflammatory effect. We observed that participants with SS who did not receive methylprednisolone treatment had a higher risk (aHR, 1.22). Using other immunosuppressant therapies (azathioprine, cyclosporine, mycophenolate, and tacrolimus) yielded a relatively lower risk of PD (aHR, 0.86) than did use other treatments.
Dopamine depletion from the basal ganglia results in major disruptions in the connections to the thalamus and motor cortex, facilitating the development of PD. Previous studies have proposed that inflammation plays a crucial role in the pathogenesis of PD and its neurodegeneration probably involves either programmed cell death (apoptosis) or necrosis.[18–20] Patients with SS have been hypothesized to have an increased risk of PD, and the etiology of PD may be associated with inflammatory autoantibodies,[10,11,21,22] which also play a crucial role in systemic diseases, such as diabetes,[23] cardiovascular diseases,[24] and Alzheimer disease.[25]
The SS is a chronic autoimmune disease that is characterized by the presence of certain autoantibodies, namely anti-Ro/SSA and anti-La/SSB,[26] which, according to some criteria, are required for SS diagnosis. Patients may also have high antinuclear antibodies, rheumatoid factor, and anti-acetylcholine receptors.[27–29] At present, neither a cure for SS nor a specific treatment is known to permanently restore gland secretion. We observed that the risk of PD was directly proportional to the follow-up period of participants with SS.
Another concern is whether immunosuppressive agents are beneficial in preventing PD in patients with autoimmune diseases. A nationwide population-based study that investigated systemic lupus erythematosus (SLE) and PD reported a contrasting association, concluding that the regular use of immunosuppressants could have potential benefit of preventing neurodegeneration and, thus, the development of PD.[30] Rugbjerg et al suggested that, for patients with rheumatoid arthritis (RA), administering immunosuppressant therapy reduced the risk of PD by 30%.[31] In our study, the participants with SS who received non-hydroxychloroquine immunosuppressant therapies had a relatively lower risk of PD, whereas those who used other treatments had a higher risk of PD. Recent studies also demonstrated that immunotherapies have benefit for neuropathies.[32–34] We reasonably supposed that non-hydroxychloroquine immunosuppressive agents play a key role in reducing the risk of PD. Nevertheless, participants with SS still had a significantly higher risk for a long follow-up period of more than 9 years (aHR, 1.93). Further research is required to determine the association between connective tissue diseases and PD and between non-hydroxychloroquine immunosuppressive agents and PD.
Our study had some limitations. First, the NHIRD does not include comprehensive information on clinical, laboratory, and imaging examinations as well as autoantibodies that can be used as diagnostic biomarkers (anti-Ro/SSA and anti-La/SSB). We could not obtain information on a family history of PD, smoking habits, alcohol consumption, occupation, and lifestyle, all of which may be risk factors for PD. Furthermore, we could not analyze the further possible pathogenesis of PD in the participants with SS. Uncertainty regarding whether outpatients with SS comply with instructions for medication use was the 2nd limitation. Third, this study was a retrospective cohort study; this type of study typically has low statistical quality because of possible biases associated with adjustments. Fourth, the participants in our study were predominantly Asian, and other ethnic populations were not well represented in our study. Fifth, we usually administer cholinergic agents for SS patients with symptoms of dry mouth and immunosuppressive agents for those who have extraglandular manifestation; however, data on the timing, dosage, and duration of antiinflammatory or immunosuppressive agents were insufficient. In our NHI program, we could not distinguish whether the SS was primary or secondary to another autoimmune disease.
5. Conclusion
In conclusion, our study was the 1st to determine an increased risk of PD in patients with SS and that non-hydroxychloroquine immunosuppressant therapy may reduce this risk. Further research is warranted to determine the possible underlying mechanisms and the potential role of non-hydroxychloroquine immunosuppressants in ameliorating PD.
Author contributions
Conceptualization: Uei-Han Ju, Feng-Cheng Liu, Chia-Hung Kao, Tse-Yen Yang.
Data curation: Feng-Cheng Liu, Cheng-Li Lin, Chia-Hung Kao.
Formal analysis: Cheng-Li Lin.
Funding acquisition: Chia-Hung Kao.
Investigation: Uei-Han Ju, Chin-Sheng Lin, Wen-Yen Huang, Te-Yu Lin, Chih-Hao Shen, Yu-Ching Chou, Kuen-Tze Lin, Chao-Hsien Chen.
Methodology: Cheng-Li Lin.
Project administration: Chin-Sheng Lin, Wen-Yen Huang, Te-Yu Lin, Chih-Hao Shen, Kuen-Tze Lin.
Resources: Te-Yu Lin, Yu-Ching Chou, Chao-Hsien Chen.
Software: Cheng-Li Lin.
Supervision: Uei-Han Ju, Feng-Cheng Liu, Yu-Ching Chou, Chia-Hung Kao, Chao-Hsien Chen, Tse-Yen Yang.
Validation: Uei-Han Ju, Feng-Cheng Liu, Te-Yu Lin, Cheng-Li Lin, Chia-Hung Kao, Chao-Hsien Chen.
Visualization: Chia-Hung Kao.
Writing – original draft: Uei-Han Ju, Feng-Cheng Liu, Chin-Sheng Lin, Wen-Yen Huang, Yu-Ching Chou, Cheng-Li Lin, Kuen-Tze Lin, Chia-Hung Kao, Chao-Hsien Chen, Tse-Yen Yang.
Writing – review & editing: Uei-Han Ju, Feng-Cheng Liu, Chin-Sheng Lin, Wen-Yen Huang, Yu-Ching Chou, Cheng-Li Lin, Kuen-Tze Lin, Chia-Hung Kao, Chao-Hsien Chen, Tse-Yen Yang.
Tse-Yen Yang orcid: 0000-0002-3165-132X.
Footnotes
Abbreviations: aHR = adjusted hazard ratio, CI = confidence interval, ICD-9-CM = International Codes of Disease Ninth Edition Clinical Modification, LHID2000 = Longitudinal Health Insurance Database, NHI = National Health Insurance, NHIRD = National Health Insurance Research Database, PD = Parkinson disease, SS = Sjögren syndrome.
T-YY and C-HC contributed equally to this work.
Financial support and conflict of interest disclosure: Miss C-L L have received her salary from the Management Office for Health Data, China Medical University Hospital, Taichung, Taiwan. The other authors have no received the other financial support. No additional external funding received for this study. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. This work was supported by grants from the Ministry of Health and Welfare, Taiwan (MOHW108-TDU-B-212–133004), China Medical University Hospital, Academia Sinica Stroke Biosignature Project (BM10701010021), MOST Clinical Trial Consortium for Stroke (MOST 107–2321-B-039–004), Tseng-Lien Lin Foundation, Taichung, Taiwan, and Katsuzo and Kiyo Aoshima Memorial Funds, Japan. The above list of funding was supported by the Management Office for Health Data, China Medical University Hospital, Taichung. The funders had no role in study design, data collection, and analysis, decision to publish, or preparation of the manuscript.
The authors have no conflicts of interest to disclose.
References
- [1].Retamozo S, Brito-Zerón P, Gheitasi H, et al. Systemic therapy of Sjögren syndrome. Connective Tissue Dis 2016;1:383–98. [Google Scholar]
- [2].Ramos-Casals M, Tzioufas A, Font J. Primary Sjögren's syndrome: new clinical and therapeutic concepts. Ann Rheumatic Dis 2005;64:347–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Lopez-Miguel A, Teson M, Martin-Montanez V, et al. Clinical and molecular inflammatory response in Sjogren syndrome-associated dry eye patients under desiccating stress. Am J Ophthalmol 2016;161:133–41. e131-132. [DOI] [PubMed] [Google Scholar]
- [4].Ngo DY, Thomson WM, Nolan A, et al. The lived experience of Sjogren's syndrome. BMC Oral Health 2016;16:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Mori K, Iijima M, Koike H, et al. The wide spectrum of clinical manifestations in Sjogren's syndrome-associated neuropathy. Brain 2005;128(Pt 11):2518–34. [DOI] [PubMed] [Google Scholar]
- [6].Delalande S, de Seze J, Fauchais AL, et al. Neurologic manifestations in primary Sjogren syndrome: a study of 82 patients. Medicine (Baltimore) 2004;83:280–91. [DOI] [PubMed] [Google Scholar]
- [7].Bougea A, Anagnostou E, Konstantinos G, et al. A systematic review of peripheral and central nervous system involvement of rheumatoid arthritis, systemic lupus erythematosus, primary Sjogren's syndrome, and associated immunological profiles. Int J Chronic Dis 2015;2015:910352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Morris KAHR. Parkinson's disease: chameleons and mimics. Pract Neurol 2015;15:14–25. [DOI] [PubMed] [Google Scholar]
- [9].Obeso JA, Rodriguez-Oroz MC, Benitez-Temino B, et al. Functional organization of the basal ganglia: therapeutic implications for Parkinson's disease. Movement Disord 2008;23Suppl 3:S548–559. [DOI] [PubMed] [Google Scholar]
- [10].Ralf C, Kunas AM, Kesselring Jürg, Villiger PM. Antidopaminergic antibodies in a patient with a complex autoimmune disorder and rapidly progressing Parkinson's disease. J Allergy Clin Immunol 1995;96:688–90. [DOI] [PubMed] [Google Scholar]
- [11].Monahan AJ, Warren M, Carvey PM. Neuroinflammation and peripheral immune infiltration in Parkinson's disease: an autoimmune hypothesis. Cell Transplant 2008;17:363–72. [PubMed] [Google Scholar]
- [12].Benkler M, Agmon-Levin N, Hassin-Baer S. Immunology, autoimmunity, and autoantibodies in Parkinson's disease. Clin Rev Allergy Immunol 2012;42:164–71. [DOI] [PubMed] [Google Scholar]
- [13].Han M, Nagele E, DeMarshall C, et al. Diagnosis of Parkinson's disease based on disease-specific autoantibody profiles in human sera. PLoS One 2012;7:e32383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Mukaino A, Nakane S, Higuchi O, et al. Insights from the ganglionic acetylcholine receptor autoantibodies in patients with Sjogren's syndrome. Modern Rheumatol 2016;1–40. [DOI] [PubMed] [Google Scholar]
- [15].National Health Insurance Administration, Ministry of Health and Welfare, Taiwan. Universal Health Coverage in Taiwan. Available at: https://www.nhi.gov.tw/DL.aspx?sitessn=293&u=LzAwMS9VcGxvYWQvMjkzL3JlbGZpbGUvMC8xNjEwNC8yMDE4MDYwOF91bnZlcnNhbGhlYWx0aGNvdmVyYWdlLnBkZg%3d%3d&n=MjAxODA2MDhfVW52ZXJzYWxIZWFsdGhDb3ZlcmFnZS5wZGY%3d Accessed March 19, 2019. [Google Scholar]
- [16].Chuang YW, Yu MC, Lin CL, et al. Risk of peripheral arterial occlusive disease in patients with rheumatoid arthritis. A nationwide population-based cohort study. Thromb Haemost 2016;115:439–45. [DOI] [PubMed] [Google Scholar]
- [17].Wang CC, Chang CT, Lin CL, et al. Spinal cord injury is associated with an increased risk of atrial fibrillation: a population-based cohort study. Heart Rhythm 2016;13:416–23. [DOI] [PubMed] [Google Scholar]
- [18].Savitt JM, Dawson VL, Dawson TM. Diagnosis and treatment of Parkinson disease: molecules to medicine. J Clin Invest 2006;116:1744–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lang AE. The progression of Parkinson disease: a hypothesis. Neurology 2007;68:948–52. [DOI] [PubMed] [Google Scholar]
- [20].Atkin G, Paulson H. Ubiquitin pathways in neurodegenerative disease. Front Mol Neurosci 2014;7:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Orr CF, Rowe DB, Halliday GM. An inflammatory review of Parkinson's disease. Progress Neurobiol 2002;68:325–40. [DOI] [PubMed] [Google Scholar]
- [22].Chen H, O’Reilly EJ, Schwarzschild MA, et al. Peripheral inflammatory biomarkers and risk of Parkinson's disease. Am J Epidemiol 2008;167:90–5. [DOI] [PubMed] [Google Scholar]
- [23].Notkins AL, Lernmark A. Autoimmune type 1 diabetes: resolved and unresolved issues. J Clin Invest 2001;108:1247–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Liang KP, Kremers HM, Crowson CS, et al. Autoantibodies and the risk of cardiovascular events. J Rheumatol 2009;36:2462–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Oscar L, Lopez BSR, Jacob Huff F, et al. Serum autoantibodies in patients with Alzheimer's disease and vascular dementia and in nondemented control subjects. Stroke 1992;23:1078–83. [DOI] [PubMed] [Google Scholar]
- [26].Santiagoa ML, Seisdedos MR, Garcia Salinas RN, et al. Usefulness of antibodies and minor salivary gland biopsy in the study of Sicca syndrome in daily clinical practice. Reumatol Clín 2015;11:156–60. [DOI] [PubMed] [Google Scholar]
- [27].Robinson CP, Brayer J, Yamachika S, et al. Transfer of human serum IgG to nonobese diabetic Igmu null mice reveals a role for autoantibodies in the loss of secretory function of exocrine tissues in Sjogren's syndrome. Proc Natl Acad Sci U S A 1998;95:7538–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Dawson LTA, Smith P, Gordon T. Antimuscarinic antibodies in Sjögren's syndrome: where are we, and where are we going? Arthritis Rheum 2005;52:2984–95. [DOI] [PubMed] [Google Scholar]
- [29].Bacman S, Perez Leiros C, Sterin-Borda L, et al. Autoantibodies against lacrimal gland M3 muscarinic acetylcholine receptors in patients with primary Sjogren's syndrome. Invest Ophthalmol Vis Sci 1998;39:151–6. [PubMed] [Google Scholar]
- [30].Liu FC, Huang WY, Lin TY, et al. Inverse association of Parkinson disease with systemic lupus erythematosus: a nationwide population-based study. Medicine (Baltimore) 2015;94:e2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Rugbjerg K, Friis S, Ritz B, et al. Autoimmune disease and risk for Parkinson disease: a population-based case-control study. Neurology 2009;73:1462–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kampylafka EI, Alexopoulos H, Dalakas MC, et al. Immunotherapies for neurological manifestations in the context of systemic autoimmunity. Neurotherapeutics 2016;13:163–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Oaklander AL. Immunotherapy prospects for painful small-fiber sensory neuropathies and ganglionopathies. Neurotherapeutics 2016;13:108–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Louvet C, Maqdasy S, Tekath M, et al. Infundibuloneurohypophysitis associated with Sjogren syndrome successfully treated with mycophenolate mofetil: a case report. Medicine (Baltimore) 2016;95:e3132. [DOI] [PMC free article] [PubMed] [Google Scholar]