

Abstract
Background
Alcoholic hepatitis is a form of alcoholic liver disease characterised by steatosis, necroinflammation, fibrosis, and complications to the liver. Typically, alcoholic hepatitis presents in people between 40 and 50 years of age. Alcoholic hepatitis can be resolved if people abstain from drinking, but the risk of death will depend on the severity of the liver damage and abstinence from alcohol. Glucocorticosteroids have been studied extensively in randomised clinical trials to assess their benefits and harms. However, the results have been contradictory.
Objectives
To assess the benefits and harms of glucocorticosteroids in people with alcoholic hepatitis.
Search methods
We identified trials through electronic searches in Cochrane Hepato‐Biliary's (CHB) Controlled Trials Register, CENTRAL, MEDLINE, Embase, LILACS, and Science Citation Index Expanded. We looked for ongoing or unpublished trials in clinical trials registers and pharmaceutical company sources. We also scanned reference lists of the studies retrieved. The last search was 18 January 2019.
Selection criteria
Randomised clinical trials assessing glucocorticosteroids versus placebo or no intervention in people with alcoholic hepatitis, irrespective of year, language of publication, or format. We considered trials with adults diagnosed with alcoholic hepatitis, which could have been established through clinical or biochemical diagnostic criteria or both. We defined alcoholic hepatitis as mild (Maddrey's score less than 32) and severe (Maddrey's score 32 or more). We allowed cointerventions in the trial groups, provided they were similar.
Data collection and analysis
We followed Cochrane methodology, performing the meta‐analyses using Review Manager 5. We presented the results of dichotomous outcomes as risk ratios (RR) and of continuous outcomes as mean difference (MD), with 95% confidence intervals (CI). We used both the fixed‐effect and the random‐effects models for meta‐analyses. Whenever there were significant discrepancies in the results, we reported the more conservative point estimate of the two. We considered a P value of 0.01 or less, two‐tailed, as statistically significant if the required information size was reached for our three primary outcomes (all‐cause mortality, health‐related quality of life, and serious adverse events during treatment) and our post hoc decision to include analyses of mortality at more time points. We presented heterogeneity using the I² statistic. If trialists used intention‐to‐treat analysis to deal with missing data, we used these data in our primary analysis; otherwise, we used the available data. We assessed the bias risk of the trials using bias risk domains and the certainty of the evidence using GRADE.
Main results
Sixteen trials fulfilled our inclusion criteria. All trials but one were at overall high risk of bias. Fifteen trials (one of which was an abstract) provided data for analysis (927 participants received glucocorticosteroids and 934 participants received placebo or no intervention). Glucocorticosteroids were administered orally or parenterally for a median 28 days (range 3 days to 12 weeks). The participants were between 25 and 70 years old, had different stages of alcoholic liver disease, and 65% were men. Follow‐up, when reported, was up to the moment of discharge from the hospital, until they died (median of 63 days), or for at least one year. There was no evidence of effect of glucocorticosteroids on all‐cause mortality up to three months following randomisation (random‐effects RR 0.90, 95% CI 0.70 to 1.15; participants = 1861; trials = 15; very low‐certainty evidence) or on health‐related quality of life up to three months, measured with the European Quality of Life – 5 Dimensions – 3 Levels scale (MD –0.04 points, 95% CI –0.11 to 0.03; participants = 377; trial = 1; low‐certainty evidence). There was no evidence of effect on the occurrence of serious adverse events during treatment (random‐effects RR 1.05, 95% CI 0.85 to 1.29; participants = 1861; trials = 15; very low‐certainty evidence), liver‐related mortality up to three months following randomisation (random‐effects RR 0.89, 95% CI 0.69 to 1.14; participants = 1861; trials = 15; very low‐certainty evidence), number of participants with any complications up to three months following randomisation (random‐effects RR 1.04, 95% CI 0.86 to 1.27; participants = 1861; very low‐certainty evidence), and number of participants of non‐serious adverse events up to three months' follow‐up after end of treatment (random‐effects RR 1.99, 95% CI 0.72 to 5.48; participants = 160; trials = 4; very low‐certainty evidence). Based on the information that we collected from the published trial reports, only one of the trials seems not to be industry‐funded, and the remaining 15 trials did not report clearly whether they were partly or completely funded by the industry.
Authors' conclusions
We are very uncertain about the effect estimate of no difference between glucocorticosteroids and placebo or no intervention on all‐cause mortality and serious adverse events during treatment because the certainty of evidence was very low, and low for health‐related quality of life. Due to inadequate reporting, we cannot exclude increases in adverse events. As the CIs were wide, we cannot rule out significant benefits or harms of glucocorticosteroids. Therefore, we need placebo‐controlled randomised clinical trials, designed according to the SPIRIT guidelines and reported according to the CONSORT guidelines. Future trials ought to report depersonalised individual participant data, so that proper individual participant data meta‐analyses of the effects of glucocorticosteroids in subgroups can be conducted.
Plain language summary
Glucocorticosteroids for people with alcoholic hepatitis
Review question
To assess the benefits and harms of glucocorticosteroids administered at any route, dose, and duration versus placebo or no intervention in people with alcoholic hepatitis in terms of death, health‐related quality of life, and complications.
Background
Excessive alcoholic consumption may damage the liver, causing alcoholic hepatitis. The first stage of liver damage in alcoholic hepatitis is usually reversible if people abstain from drinking, but the risk of the disease developing further and getting more complications increases with resumed drinking. A heavy drinker is considered a person who consumes more than 30 g (for men) or more than 20 g (for women) of alcohol per day. Only 10 to 35 people out of 100 heavy drinkers with evidence of excessive fat in the liver would most probably develop alcoholic hepatitis. With time, alcoholic hepatitis will cause liver fibrosis (scarring of the liver) or liver cirrhosis with complications (bleeding, infections, liver cancer, etc.).
Glucocorticosteroids are considered to have anti‐inflammatory effects (relieving pain, swelling (oedema), fever). They are administered to people with alcoholic hepatitis in order to repair their liver injury. However, the benefits and harms of glucocorticosteroids are not well studied in randomised clinical trials (studies where people are randomly put into one of two or more treatment groups), and therefore, it is uncertain if they should be used in clinical practice for people with alcoholic liver disease.
Search date
The date of the last search was 18 January 2019.
Study characteristics
Sixteen randomised clinical trials compared glucocorticosteroids with placebo or no intervention in people with alcoholic hepatitis. Fifteen trials provided data for analysis (927 participants received glucocorticosteroids and 934 participants received placebo or no intervention). Glucocorticosteroids were administered orally or as an injection for a median of 28 days (range 3 days to 12 weeks). The trial participants were between 25 and 70 years old, 65% were men, and had different stages of alcoholic liver disease. Trial participants were followed up to the moment of discharge from the hospital, or until they died (a median of 63 days), or for at least one year. Not all trials reported the follow‐up of participants. The trials were conducted in France, India, the UK, and the USA. Two trials administered pentoxifylline (a medicine used for diseases of the blood vessels) to both glucocorticosteroids and placebo intervention groups.
Funding
Based on the information that we collected from the published trial reports, only one of the trials seems not to be industry‐funded, and the remaining 15 trials did not report clearly whether they were partly or completely funded by the industry.
Reliability of the evidence
The overall reliability of the evidence was low for health‐related quality of life and very low for death due to any cause up to three months following entry in the trial; serious side effects during treatment; liver‐related death up to three months following entry in the trial; number of participants with any complications up to three months following entry in the trial, and number of participants non‐serious side effects up to three months' follow‐up after the end of treatment. All trials but one were at overall high risk of bias, which means that there is possibility of drawing wrong conclusions, exaggerating benefits, or underestimating harms of glucocorticosteroids because of the way the trials were conducted and analysed.
Key results
We could not determine whether glucocorticosteroids had a positive or negative effect on people with alcoholic liver disease. Despite available data on outcomes which included mortality, health‐related quality of life, and serious complications, we were unable to draw firm conclusions mainly because available data were still insufficient to produce robust results, trials were small, and the included participants differed in severity of disease. Therefore, we have very little confidence in our conclusions.
Summary of findings
Summary of findings for the main comparison. Glucocorticosteroids for people with alcoholic hepatitis.
| Glucocorticosteroids for people with alcoholic hepatitis | ||||||
|
Patient or population: participants with alcoholic hepatitis at high risk of mortality and morbidity Settings: hospitals and clinics Intervention: glucocorticosteroids Comparison: placebo or no intervention | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo or no intervention | Glucocorticosteroids | |||||
| All‐cause mortality: up to 3 months' follow‐up after randomisation | 299 per 1000 | 278 per 1000 (210 to 344) |
RR 0.90 (0.70 to 1.15) |
1861 (15 RCTs) |
⊕⊝⊝⊝ Very lowa | We downgraded for inconsistency because of selection bias in the trials: trials either included or excluded people with gastrointestinal haemorrhage, active peptic ulcer disease, pancreatitis, renal failure, bacterial infections. The OIS was 7870 participants. |
|
Health‐related quality of life: up to 3 months (measured with European Quality of Life – 5 Dimensions – 3 Levels (EQ‐5D‐3L)b scale) |
The mean value was 0.592 | The mean value was 0.553 (0.502 to 0.604) |
MD –0.04 (–0.11 to 0.03) | 377 (1 RCT) |
⊕⊕⊝⊝ Lowc |
— |
| Serious adverse events during treatment | 362 per 1000 | 381 per 1000 (398 to 467) |
RR 1.05 (0.85 to 1.29) |
1861 (15 RCTs) |
⊕⊝⊝⊝ Very lowd | The OIS was 4197 participants. |
| Liver‐related mortality: up to 3 months' follow‐up after randomisation | 299 per 1000 | 267 per 1000 (207 to 341) |
RR 0.89 (0.69 to 1.14) |
1861 (15 RCTs) |
⊕⊝⊝⊝ Very lowe | The OIS was 7987 participants. |
| Participants with any complication: up to 3 months following randomisation | 444 per 1000 | 462 per 1000 (382 to 564) |
RR 1.04 (0.86 to 1.27) |
1861 (15 RCTs) |
⊕⊝⊝⊝ Very lowf | The OIS was 5980 participants. |
| Participants with non‐serious adverse events: up to 3 months' follow‐up after randomisation | 52 per 1000 | 104 per 1000 (38 to 285) |
RR 1.99 (0.72 to 5.48) |
160 (4 RCTs) |
⊕⊝⊝⊝ Very lowg | The OIS was 2698 participants. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; MD: mean difference; OIS: optimal information size; RCT: randomised controlled trial; RR: risk ratio. | ||||||
| GRADE Working Group: certainty of evidence grades High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded three levels: one level due to within‐study risk of bias (high overall risk of bias in all the trials but one (Thursz 2015); one level due to inconsistency of the data (there is wide variation in the effect estimates across studies; there is little overlap of confidence intervals associated with the effect estimates; presence of moderate heterogeneity: I² = 45%; heterogeneity could be explained with selection bias); one level due to imprecision (the OIS was not reached). bEQ‐5D‐5L: a self‐report, multiple‐choice questionnaire that provides a simple descriptive profile and a single index value for health status. The EQ‐5D‐5L essentially consists of two pages: the EQ‐5D descriptive system (on page 2) and the EQ visual analogue scale (EQ VAS) (on page 3). The descriptive system comprises the following five dimensions: mobility, self‐care, usual activities, pain/discomfort, and anxiety/depression. Each dimension has five levels: no problems; slight problems; moderate problems; severe problems; and extreme problems. The EQ VAS records the respondent's self‐rated health on a vertical, visual analogue scale. A summary index with a maximum score of 1 can be derived from these five dimensions by conversion with a table of scores. The maximum score of 1 indicates the best health state, by contrast with the scores of individual questions, where higher scores indicate more severe or frequent problems. Utility values for perfect health and death are 1 and 0, respectively. In addition, there is a visual analogue scale to indicate the general health status with 100 indicating the best health status. cDowngraded two levels: one level due to within‐study risk of bias (high overall risk of bias in the trial); one level due to imprecision of effect estimates (fewer than 400 participants). dDowngraded three levels: one level due to within‐study risk of bias (high overall risk of bias in all the trials but one); one level due to inconsistency of the data (there was wide variation in the effect estimates across studies; there was little overlap of confidence intervals associated with the effect estimates; presence of moderate heterogeneity: I² = 36%; heterogeneity could be explained with selection bias); one level due to imprecision (the OIS was not reached). eDowngraded three levels: one level due to within‐study risk of bias (high overall risk of bias in all the trials but one); one level due to inconsistency of the data (there is wide variation in the effect estimates across studies; there is little overlap of confidence intervals associated with the effect estimates; presence of moderate heterogeneity: I² = 46%; heterogeneity could be explained with selection bias); one level due to imprecision (the OIS was not reached). fDowngraded three levels: one level due to within‐study risk of bias (high overall risk of bias in all the trials but one); one level due to inconsistency of the data (there was wide variation in the effect estimates across studies; there was little overlap of confidence intervals associated with the effect estimates; presence of moderate heterogeneity: I² = 41%; heterogeneity could be explained with selection bias); one level due to imprecision (the OIS was not reached). gDowngraded four levels: one level due to within‐study risk of bias (high overall risk of bias in all the trials but one); one level due to inconsistency of the data (there is little overlap of confidence intervals associated with the effect estimates); one level due to publication bias (only four trials with a small number of participants reported on non‐serious adverse events); one level due to imprecision (the OIS was not reached).
Background
Description of the condition
The term 'alcoholic hepatitis' was used for the first time in a paper by Beckett and colleagues in 1961 (Beckett 1961), but clinical jaundice after excessive ethanol consumption was reported in the literature long before that, in 1912 (French 1912; Gerber 1973). Most probably, these reports represented people with alcoholic hepatitis (Mendenhall 1984; Jensen 1994).
Alcoholic hepatitis is a serious form of alcoholic liver disease (injury of the liver due to excessive alcohol consumption) (WHO 2010).
The first stage of liver damage in alcoholic hepatitis is usually reversible if people abstain from drinking, but the risk of progression to fibrosis and cirrhosis increases with resumed drinking (Ellis 2012). The accumulation of fat in the hepatocytes causes disruption of the mitochondrial beta‐oxidation of fatty acids, accumulation of lipotoxic metabolites, and release of reactive oxygen species (Lieber 1999; Wu 1999; Petrasek 2013). Lipotoxic metabolites and reactive oxygen species lead to cell death and liver inflammation (Wu 1999; Petrasek 2013; WHO 2013). Alcoholic hepatitis is a histological form of alcoholic liver disease, characterised by steatosis (the earliest stage of alcoholic liver damage) and necroinflammation (EASL 2018). Alcoholic hepatitis can be resolved if people abstain from drinking, but the risk of death will depend on the severity of the liver damage and drinking patterns. In 20% to 40% of persistent heavy drinkers (defined as alcohol consumption per day of more than 30 g in men (EASL 2018) and more than 20 g in women (EASL 2018), alcoholic hepatitis and other complications may develop (WHO 2013).
Severe alcoholic hepatitis may be characterised by clinically clear signs of jaundice, coagulopathy, liver decompensation with ascites, portal hypertension, variceal bleeding, hepatorenal syndrome, hepatic encephalopathy, systemic inflammatory response syndrome, or sepsis (Becker 1996; EASL 2018). Typically, alcoholic hepatitis presents in people aged between 40 and 50 years. Among the risk factors of developing severe alcoholic hepatitis are being female, Hispanic ethnicity, various types of alcohol, binge drinking, poor nutrition, obesity, etc. (WHO 2010). Several composite prognostic scores exist to distinguish people with poor prognosis from those who can become abstinent, instituting supportive care, until recovery is achieved. Some of these scores, designed to predict mortality, are Maddrey's discriminant function (Maddrey 1978), the model of end‐stage liver disease (MELD) score (Dunn 2005); the Glasgow alcoholic hepatitis score (Forrest 2005); and the age, bilirubin, international normalised ratio, creatinine (ABIC) score (Dominguez 2008).
The Maddrey Discriminant Function is the most often used score in severe alcoholic hepatitis to identify people in potential need of glucocorticosteroids (also known as glucocorticoids, corticosteroids, or steroids). The one‐month survival of people with alcoholic hepatitis and with Maddrey's score higher than 32 varied between 50% and 65% (Carithers 1989; Phillips 2006). The Lille Model (www.lillemodel.com) is the only validated model so far to assess glucocorticosteroid response and is highly predictive of death at six months (P < 0.001) in people with severe alcoholic hepatitis (Louvet 2007). A Lille Model score greater than 0.45, calculated after seven days of treatment with prednisolone, means failure to respond to treatment and predicts a six‐month mortality of about 75% (Lefkowitch 2005).
Description of the intervention
Glucocorticosteroids are used as anti‐inflammatory drugs. Glucocorticosteroid agents mimic the endogenously produced glucocorticoid (cortisol) (Rhen 2005). Glucocorticosteroids, primarily regulated by corticotropin, are considered to have anti‐inflammatory effects as well as metabolic and immunogenic effects (Rhen 2005). It is agreed that the anti‐inflammatory effects of glucocorticosteroids are mediated primarily through repression of gene transcription (Schäcke 2002).
How the intervention might work
Glucocorticosteroids administered to people with alcoholic hepatitis repair the liver injury by decreasing the liver polymorphonuclear neutrophil (PMN) (effector cells) infiltrates and the level of proinflammatory mediators such as tumour necrosis factor‐alpha (TNF‐alpha), intercellular adhesion molecule 1, and interleukin (IL)‐6 and IL‐8 in the liver tissue (Taïeb 2000; Spahr 2001). The benefits of corticosteroids ensue from short‐term vascular changes (Schäcke 2002). However, adverse events have still been poorly reported (Christensen 1995; Rambaldi 2008).
Why it is important to do this review
Several randomised clinical trials have studied the benefits and harms of corticosteroids for people with alcoholic hepatitis to determine the best route of administration, dose, and duration. However, results have been contradictory. Some systematic reviews (Christensen 1995; Rambaldi 2008), and meta‐analyses of randomised clinical trials (Reynolds 1989; Imperiale 1990; Daures 1991; Christensen 1999; Mathurin 2011; Louvet 2018), have been published. The review authors explained their various conclusions regarding patient‐orientated outcomes as being due to differences in glucocorticosteroid regimens, trial quality, participants' characteristics, and clinical spectrum of the disease. Reynolds 1989 concluded that corticosteroid treatment could help only the most severely ill people with severe alcoholic hepatitis characterised by high levels of serum bilirubin, prolonged prothrombin times, and development of hepatic encephalopathy. Imperiale 1990 concluded that glucocorticosteroids reduced short‐term mortality in people with severe alcoholic hepatitis, provided that they also had hepatic encephalopathy but did not have severe gastrointestinal bleeding. Daures 1991 concluded that further randomised clinical trials were needed to confirm the benefits and harms of glucocorticosteroids, especially in people with severe alcoholic hepatitis. Christensen 1995, Christensen 1999, and Rambaldi 2006 could not find sufficient proof supporting the routine use of glucocorticosteroids in people with alcoholic hepatitis, including those with hepatic encephalopathy. Rambaldi 2008 concluded that glucocorticosteroids did not improve overall survival in people with alcoholic hepatitis. Based on the Trial Sequential Analysis of the subgroup of people with Maddrey's score of at least 32 or spontaneous hepatic encephalopathy, the required information size of 2420 people for the outcome mortality was far from reached, with only 249 participants randomised in the six trials (Rambaldi 2008). Using the Lille Model, Mathurin 2011 concluded that glucocorticosteroids significantly improved 28‐day survival in people with severe alcoholic hepatitis. Mathurin 2011 based the meta‐analysis on individual patient data from five selected randomised clinical trials and was accordingly at risk of preferential selection. In 2018, Louvet and colleagues published four meta‐analyses in one publication in which they assessed the effects of corticosteroids versus placebo or control, corticosteroids versus pentoxifylline, corticosteroids plus pentoxifylline versus corticosteroids plus placebo or control, and pentoxifylline versus placebo in four meta‐analyses (Louvet 2018). The conclusions Louvet and colleagues made was that corticosteroids reduced the risk of death within 28 days of treatment, but not in the next six months. However, Louvet 2018 did not contain new references to randomised clinical trials of interest to our review. The present review is an update of our previously published Cochrane systematic review, assessing the benefits and harms of glucocorticosteroids in people with severe alcoholic hepatitis with or without complications.
Objectives
To assess the benefits and harms of glucocorticosteroids in people with alcoholic hepatitis.
Methods
Criteria for considering studies for this review
Types of studies
We included randomised clinical trials in which glucocorticosteroids were assessed in people with alcoholic hepatitis, irrespective of year or language of publication or format.
We found no reports of harm in the quasi‐randomised or observational studies retrieved with our searches for randomised clinical trials (Excluded studies).
Types of participants
We included adults with alcoholic hepatitis, diagnosed according to the diagnostic work‐up used in the individual randomised clinical trial. Alcoholic hepatitis could have been established through clinical or biochemical diagnostic criteria, or both.
We considered alcoholic hepatitis as mild if a randomised participant had a Maddrey's score less than 32 (Maddrey's score = 4.6 × (prothrombin time – control time)(s) + serum bilirubin (mg per dL)) (Maddrey 1978). Usually, people with mild alcoholic hepatitis do not have concomitant gastrointestinal bleeding.
We considered alcoholic hepatitis as severe at any stage of the alcoholic liver disease with the presence of spontaneous hepatic encephalopathy; or Maddrey's score of 32 or higher. We also examined whether there was a difference in terms of initiation of treatment with glucocorticosteroids in trials using the Maddrey's score where severe alcoholic hepatitis was defined as 32 or higher.
Included trial participants diagnosed with severe alcoholic hepatitis could also manifest with hepatic encephalopathy, gastrointestinal bleeding, cirrhosis (e.g. classified with Child‐Pugh score – Child‐Pugh type C (Pugh 1973)), ascites, hepatorenal syndrome, hyponatraemia, or spontaneous bacterial peritonitis.
For studies not reporting the Maddrey's score, we used the classifications for mild and severe alcoholic hepatitis as provided by the trialists.
Types of interventions
Glucocorticosteroids administered by any route, dose, and duration versus placebo or no intervention.
We allowed cointerventions in the trial groups, provided they were the same.
Types of outcome measures
Primary outcomes
All‐cause mortality: up to three months' follow‐up after randomisation (the primary time point for drawing our main conclusion); at the end of treatment (post hoc analysis); and one year following randomisation (post hoc analysis).
Health‐related quality of life as defined by the trial authors.
Serious adverse events during treatment. We used the International Conference on Harmonisation (ICH) Guidelines for Good Clinical Practice's definition of a serious adverse event (ICH‐GCP 1997), that is, any untoward medical occurrence that resulted in death, was life threatening, required hospitalisation or prolongation of existing hospitalisation, resulted in persistent or significant disability or incapacity, or was a congenital anomaly or birth defect. We considered all other adverse events as non‐serious (see Secondary outcomes).
Secondary outcomes
Liver‐related mortality up to three months' follow‐up after randomisation.
Participants with any complication up to three months' follow‐up after randomisation (i.e. ascites, hepatorenal syndrome, spontaneous bacterial peritonitis, gastrointestinal bleeding, hepatic encephalopathy, non‐obstructive jaundice, systemic inflammatory response syndrome, sepsis, or hepatocellular carcinoma, or a combination of any of these).
Participants with non‐serious adverse events up to three months' follow‐up after randomisation.
Exploratory analysis
Participants with an increase of liver enzymes as defined by the trialists.
Participants with a decrease of prothrombin index as defined by the trialists.
Participants with a decrease of serum albumin as defined by the trialists.
Search methods for identification of studies
Electronic searches
We searched Cochrane Hepato‐Biliary's Controlled Trials Register (Gluud 2017; 18 January 2019), Cochrane Central Register of Controlled Trials (CENTRAL; 2019, Issue 1) in the Cochrane Library, MEDLINE Ovid (1946 to 18 January 2019), Embase Ovid (1974 to 18 January 2019), LILACS, and Science Citation Index Expanded (Web of Science; 1900 to 18 January 2019) (Royle 2003). We applied no language or document‐type restrictions. Appendix 1 shows the search strategies with the time spans of the searches.
Searching other resources
We searched online trials registries such as ClinicalTrials.gov (clinicaltrials.gov), European Medicines Agency (EMA; www.ema.europa.eu), World Health Organization (WHO) International Clinical Trial Registry Platform (www.who.int/ictrp), the Food and Drug Administration (FDA; www.fda.gov), eLibrary, and pharmaceutical company sources for ongoing or unpublished trials (last search 29 January 2019).
We handsearched the reference lists of articles from the computerised databases and relevant review articles.
Data collection and analysis
We followed the guidelines provided in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a), and the Cochrane Hepato‐Biliary Module (Gluud 2017). We performed the analyses using Review Manager 5 (Review Manager 2014) and Trial Sequential Analysis (Thorlund 2011; TSA 2011; Wetterslev 2017). We assessed the evidence according to Jakobsen and colleagues (Jakobsen 2014).
Selection of studies
We retrieved the full‐text publications that we considered as potentially eligible for inclusion after reading their titles and abstracts. Three review authors (CP, DV, GC) independently reviewed the full‐text publications for eligibility. The review authors assessed each publication to determine if trial participants and the interventions administered met the inclusion criteria. We included abstracts if there were sufficient data for analysis. We resolved disagreements by discussion or consulting any of the remaining review authors for arbitration.
Data extraction and management
Three review authors (CP, DV, GC) independently completed a data extraction form for all included trials, agreed on among the authors in advance. Authors extracted general information on the trial, such as publication title; place and year of publication; trial design; inclusion and exclusion criteria; preliminary sample size calculation reached or not; number of participants randomised in each trial and following treatment allocation; diagnostic work‐up; age (mean or median); sex or sex ratio; race; coinfection; type, dose, and route of administration of glucocorticosteroids and their possible link with adverse events; concurrent medications used; length of trial; and length of follow‐up. Three review authors (CP, DV, and GC) also extracted data on malnutrition whenever it was clearly defined by the trial authors.
The review authors resolved disagreements by discussion or asking the advice of the review arbitrator (CG).
Assessment of risk of bias in included studies
Three review authors (CP, DV, and GC) independently assessed the risk of bias of each included trial according to the recommendations in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011b), the Cochrane Hepato‐Biliary Module (Gluud 2017), and methodological studies (Schulz 1995; Moher 1998; Kjaergard 2001; Wood 2008; Savović 2012a; Savović 2012b; Lundh 2017). We used the following definitions in the assessment of risk of bias.
Allocation sequence generation
Low risk of bias: the study performed sequence generation using computer random number generation or a random number table. Drawing lots, tossing a coin, shuffling cards, and throwing dice were adequate if an independent person not otherwise involved in the study performed them.
Unclear risk of bias: the study authors did not specify the method of sequence generation.
High risk of bias: the sequence generation method was not random. We only included such studies for assessment of harms.
Allocation concealment
Low risk of bias: the participant allocations could not have been foreseen in advance of, or during, enrolment. A central and independent randomisation unit controlled allocation. The investigators were unaware of the allocation sequence (e.g. if the allocation sequence was hidden in sequentially numbered, opaque, and sealed envelopes).
Unclear risk of bias: the study authors did not describe the method used to conceal the allocation so the intervention allocations may have been foreseen before, or during, enrolment.
High risk of bias: it is likely that the investigators who assigned the participants knew the allocation sequence. We only included such studies for assessment of harms.
Blinding of participants and personnel
Low risk of bias: any of the following: no blinding or incomplete blinding, but the review authors judged that the outcome was not likely to have been influenced by lack of blinding; or blinding of participants and key study personnel ensured, and it was unlikely that the blinding could have been broken.
Unclear risk of bias: any of the following: insufficient information to permit judgement of 'low risk' or 'high risk;' or the trial did not address this outcome.
High risk of bias: any of the following: no blinding or incomplete blinding, and the outcome was likely to have been influenced by lack of blinding; or blinding of key study participants and personnel attempted, but likely that the blinding could have been broken, and the outcome was likely to be influenced by lack of blinding.
Blinded outcome assessment
Low risk of bias: any of the following: no blinding of outcome assessment, but the review authors judged that the outcome measurement was not likely to be influenced by lack of blinding; or blinding of outcome assessment ensured, and unlikely that the blinding could have been broken.
Unclear risk of bias: any of the following: insufficient information to permit judgement of 'low risk' or 'high risk;' or the trial did not address this outcome.
High risk of bias: any of the following: no blinding of outcome assessment, and the outcome measurement was likely to be influenced by lack of blinding; or blinding of outcome assessment, but likely that the blinding could have been broken, and the outcome measurement was likely to have been influenced by lack of blinding.
Incomplete outcome data
Low risk of bias: missing data were unlikely to make treatment effects depart from plausible values. The study used sufficient methods, such as multiple imputation, to handle missing data.
Unclear risk of bias: there was insufficient information to assess whether missing data in combination with the method used to handle missing data were likely to have induced bias on the results.
High risk of bias: the results were likely to be biased due to missing data.
Selective outcome reporting
Low risk of bias: the trial reported the following predefined outcomes: all‐cause mortality, serious adverse events, and liver‐related mortality. If the original trial protocol was available, the outcomes were those called for in that protocol. If the trial protocol was obtained from a trials registry (e.g. www.clinicaltrials.gov), the outcomes sought should have been those enumerated in the original protocol if the trial protocol was registered before or at the time that the trial was begun. If the trial protocol was registered after the trial was begun, those outcomes were not considered reliable.
Unclear risk of bias: not all predefined outcomes were reported fully, or it was unclear whether data on these outcomes were recorded or not.
High risk of bias: one or more predefined outcomes were not reported.
Other bias
Low risk of bias: the trial appeared free of other factors that could have put it at risk of bias.
Unclear risk of bias: the trial may or may not have been free of other factors that could have put it at risk of bias.
High risk of bias: there were other factors in the trial that could have put it at risk of bias.
Overall risk of bias
We judged each trial as having a low, unclear, or high risk of bias based on the definitions described above. We included a bias risk assessment combining all domains and judged the trials to be at low risk of bias if none of the trial domains was assessed at high or unclear risk of bias. Moreover, we considered trials with one or more domains with unclear or high risk of bias as trials at overall high risk of bias.
Measures of treatment effect
Dichotomous outcomes
We used risk ratio (RR) with 95% confidence interval (CI) and Trial Sequential Analysis‐adjusted CI for dichotomous outcomes.
Continuous outcomes
We used mean difference (MD) with 95% CI and Trial Sequential Analysis‐adjusted CI for health‐related quality of life. We planned to use the standardised mean difference (SMD) with 95% CI if trials used different measures for health‐related quality of life.
Unit of analysis issues
Trial participants as randomised per intervention group. In case of multiple treatment groups, we considered only the trial group to which glucocorticosteroids were administered versus the group that received placebo or no intervention. If a trial consisted of more than two groups (either parallel or factorial design), we compared the participants from all the glucocorticosteroid groups versus all participants from the placebo group(s). Had we been able to include a cross‐over trial from which we could extract data for analyses, we would have used the data from the first treatment period of the cross‐over trial.
Dealing with missing data
If dichotomous or continuous data were missing in a published report, whenever possible, we contacted the original investigators to request the missing data.
If trialists used intention‐to‐treat analysis to deal with missing data, we used these data in our primary analysis. Otherwise, we used the data that were available to us.
Dealing with missing data using sensitivity analysis
As some trials reported only per‐protocol analysis results, we included missing data by considering participants as treatment failures or treatment successes by imputing them according to the following two scenarios:
extreme‐case analysis favouring the experimental intervention ('best‐worse' case scenario): none of the participants who dropped out from the experimental group experienced the outcome, but all of the participants who dropped out from the control group experienced the outcome; including all randomised participants in the denominator;
extreme‐case analysis favouring the control ('worst‐best' case scenario): all participants who dropped out from the experimental group, but none from the control group experienced the outcome; including all randomised participants in the denominator.
For continuous outcomes (e.g. health‐related quality of life), we planned to perform a 'best‐worst' case scenario analysis assuming that all participants lost to follow‐up in the experimental group had an improved outcome (the group mean plus 1 standard deviation (SD)); and all those with missing outcomes in the control group had a worsened outcome (the group mean minus 1 SD) (Jakobsen 2014). We also planned to perform 'worst‐best' case scenario analysis assuming that all participants lost to follow‐up in the experimental group had a worsened outcome (the group mean minus 1 SD); and all those with missing outcomes in the control group had an improved outcome (the group mean plus 1 SD) (Jakobsen 2014).
We performed the two sensitivity scenario analyses only for our primary outcomes.
Assessment of heterogeneity
We addressed the presence of heterogeneity in both clinical and statistical ways.
To assess heterogeneity between the trials, we specifically examined the degree of heterogeneity observed in the results using the I² statistic (Higgins 2002). As thresholds for the interpretation of the I² statistic could be misleading, we used the following approximate guide for interpretation of heterogeneity provided in the Cochrane Handbook for Systematic Reviews of Interventions (Deeks 2011):
0% to 40%: might not be important;
30% to 60%: may represent moderate heterogeneity*;
50% to 90%: may represent substantial heterogeneity*;
75% to 100%: considerable heterogeneity*.
*The importance of the observed value of the I² statistic depends on the magnitude and direction of effects and the strength of evidence for heterogeneity (e.g. P value from the Chi² test, or a CI for I² statistic).
For the heterogeneity adjustment of the required information size in the Trial Sequential Analysis, we used diversity (D²) because the I² statistic used for this purpose underestimates the required information size (Wetterslev 2009).
Depending on the number of eligible trials, we planned to add covariates to a meta‐regression model to adjust for heterogeneity.
Assessment of reporting biases
We drew funnel plots to assess reporting biases from the individual trials by plotting the RR on a logarithmic scale against its standard error (Egger 1997; Sterne 2011).
For dichotomous outcomes, we tested asymmetry using the Harbord test in cases where Tau² was less than 0.1 (Harbord 2006), and we planned to use Rücker 2008 in cases where Tau² was more than 0.1. For continuous outcomes, we planned to use the regression asymmetry test (Egger 1997), and the adjusted rank correlation (Begg 1994).
Data synthesis
Meta‐analysis
We performed the meta‐analyses using Review Manager 5 (Review Manager 2014), and according to the recommendations of the Cochrane Handbook for Systematic Reviews of Interventions (Deeks 2011).
We presented the results of dichotomous outcomes of individual trials as RR with 95% CI and the results of the continuous outcomes as MD with 95% CI and Trial Sequential Analysis‐adjusted CI. We applied both the fixed‐effect model (DeMets 1987) and the random‐effects model (DerSimonian 1986) meta‐analyses. If there were statistically significant discrepancies in the results (e.g. one giving a significant intervention effect and the other no significant intervention effect), we reported the more conservative point estimate of the two (Jakobsen 2014). The more conservative point estimate is the estimate closest to zero effect. If the two point estimates were equal, we used the estimate with the widest CI as our main result of the two analyses. We considered a P value of 0.025 or less, two‐tailed, as statistically significant if the required information size was reached due to the three primary outcomes (Jakobsen 2014). Due to us expanding the number of analyses conducted, we post hoc made the alpha level even lower. We used the eight‐step procedure to assess if the thresholds for significance were crossed (Jakobsen 2014). We presented heterogeneity using the I² statistic (Higgins 2002). We presented the results of the individual trials and meta‐analyses in the form of forest plots.
Where data were only available from one trial (in our case continuous data on health‐related quality of life), we used Student's t‐test (Student 1908). We planned to use Fisher's exact test for dichotomous data in a single trial (Fisher 1922).
Trial Sequential Analysis
We applied Trial Sequential Analysis for both dichotomous and continuous outcomes (Thorlund 2011; TSA 2011; Wetterslev 2017), as cumulative meta‐analyses are at risk of producing random errors due to sparse data and repetitive testing of the accumulating data (Wetterslev 2008; Wetterslev 2017). To control random errors, we calculated the diversity‐adjusted required information size (DARIS) (i.e. the number of participants needed in a meta‐analysis to detect or reject a certain intervention effect) (Brok 2008; Wetterslev 2008; Brok 2009; Thorlund 2009; Wetterslev 2009; Thorlund 2010).
In our meta‐analysis, we based the DARIS for dichotomous outcomes on the event proportion in the control group; assumption of a plausible relative risk reduction of 20% of the risk observed in the included trials; a risk of type I error of 1% due to more than three outcomes, and as we decided to perform post hoc analyses on mortality at end of treatment and at one year following randomisation; a risk of type II error of 20%; and the diversity of the included trials in the meta‐analysis. For health‐related quality of life, we planned to estimate DARIS using a minimal relevant difference of 10% of the mean response observed in the control group; the SD; alpha of 1% (Jakobsen 2014); beta of 20%; and the diversity as estimated from the trials in the meta‐analysis (Wetterslev 2009). However, we did not conduct Trial Sequential Analysis because only one trial provided data on health‐related quality of life. We also calculated and reported the Trial Sequential Analysis‐adjusted CI (Thorlund 2011).
The underlying assumption of Trial Sequential Analysis is that testing for statistical significance may be performed each time a new trial is added to the meta‐analysis. We added the trials according to the year of publication, and, if more than one trial was published in a year, we added trials alphabetically according to the last name of the first author. On the basis of the DARIS, we constructed the trial sequential monitoring boundaries for benefit, harm, and futility (Wetterslev 2008; Thorlund 2011). These boundaries determine the statistical inference one may draw regarding the cumulative meta‐analysis that has not reached the DARIS; if the trial sequential monitoring boundary for benefit or harm is crossed before the DARIS is reached, firm evidence may be established and further trials may be superfluous. However, if the boundaries are not crossed, it is most probably necessary to continue doing trials in order to detect or reject a certain intervention effect. However, if the cumulative Z‐curve crosses the trial sequential monitoring boundaries for futility, no more trials may be needed.
A more detailed description of Trial Sequential Analysis can be found at www.ctu.dk/tsa/ (Thorlund 2011).
Subgroup analysis and investigation of heterogeneity
Whenever possible, we performed the following subgroup analyses for all‐cause mortality up to three months after randomisation.
Trials at low risk of bias compared to trials at high risk of bias.
Trials without for‐profit funding compared to trials at risk of for‐profit funding (Lundh 2017).
Trials with people with mild alcoholic hepatitis compared to trials with people with severe alcoholic hepatitis, following Maddrey's score lower than 32 or 32 or higher or presence of hepatic encephalopathy; or as provided by the trialists.
Trials with glucocorticosteroid dose 40 mg or less compared to trials with glucocorticosteroid dose more than 40 mg.
Trials with people with severe alcoholic hepatitis without cirrhosis compared to trials with people with severe alcoholic hepatitis with cirrhosis. If cirrhosis is classified by Child‐Pugh score, then we may be able to perform additional subgroup analyses in order to adjust for the clinical spectrum of the disease.
Trials with people with severe alcoholic hepatitis without hepatorenal syndrome compared to trials with people with severe alcoholic hepatitis with hepatorenal syndrome.
Trials with people with severe alcoholic hepatitis without ascites compared to trials with people with severe alcoholic hepatitis with ascites.
We did not perform any additional subgroup analyses to those planned in advance.
Sensitivity analysis
We planned to undertake additional sensitivity analyses to those specified under Dealing with missing data should we have considered it necessary (e.g. trials published as full‐paper articles, abstracts, and unpublished trials).
We compared our GRADE assessment of imprecision with that of Trial Sequential Analysis.
'Summary of findings' tables
We used GRADEpro GDT 2015 to create a 'Summary of findings' table for the following outcomes: all‐cause mortality: up to three months' follow‐up after randomisation; health‐related quality of life up to three months; serious adverse events during treatment; liver‐related mortality up to three months' follow‐up after randomisation; participants with any complication up to three months' follow‐up after randomisation; and number of participants with non‐serious adverse events up to three months' follow‐up after randomisation. The GRADE approach appraises the certainty of a body of evidence based on the extent to which one can be confident that an estimate of effect or association reflects the item being assessed. The certainty of a body of evidence considers within‐study risk of bias, indirectness of the evidence (population, intervention, control, outcomes), unexplained inconsistency (heterogeneity) of results (including problems with subgroup analyses); imprecision of results, and risk of publication bias (Balshem 2011; Guyatt 2011a; Guyatt 2011b; Guyatt 2011c; Guyatt 2011d; Guyatt 2011e; Guyatt 2011f; Guyatt 2011g; Guyatt 2011h; Guyatt 2013a; Guyatt 2013b; Guyatt 2013c; Mustafa 2013; Guyatt 2017).
We defined the levels of evidence as 'high,' 'moderate,' 'low,' or 'very low.' These grades are defined as follows.
High certainty: we are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different.
Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect.
Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect.
Results
Description of studies
See: Characteristics of included studies; and Characteristics of excluded studies tables.
Results of the search
We identified 1307 potentially relevant records through the electronic searches (Figure 1). Of these, 37 records that referred to 16 randomised clinical trials fulfilled our inclusion criteria. We found two trials published in abstract form (Mendenhall 1977; Richardet 1993), and 14 trials described as full‐text paper articles (Helman 1971; Porter 1971; Campra 1973; Blitzer 1977; Maddrey 1978; Shumaker 1978; Depew 1980; Theodossi 1982; Mendenhall 1984; Bories 1987; Carithers 1989; Ramond 1992; De 2014; Thursz 2015). Our searches retrieved some quasi‐randomised trials or observational studies that included administration of glucocorticosteroids to people with alcoholic hepatitis, but the studies did not report data of interest to our review. We identified no additional references by handsearching the reference lists of articles, retrieved through the computerised databases.
1.
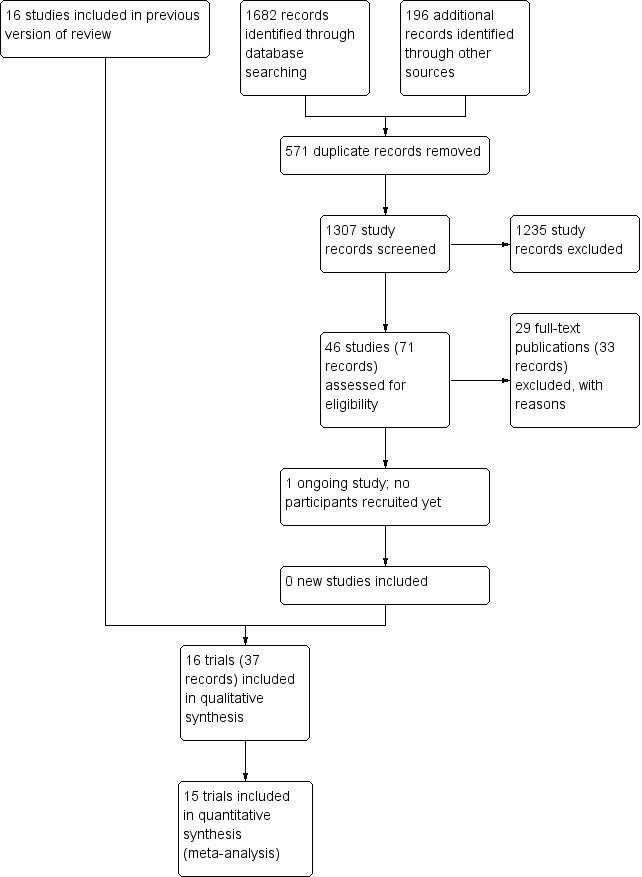
Study flow diagram.
We found one registered trial on clinicaltrials.gov comparing methylprednisolone versus placebo in severe alcoholic hepatitis (NCT03160651). However, the trial has not yet started recruitment of participants.
Included studies
Sixteen randomised clinical trials fulfilled our inclusion criteria (Helman 1971; Porter 1971; Campra 1973; Blitzer 1977; Mendenhall 1977; Maddrey 1978; Shumaker 1978; Depew 1980; Theodossi 1982; Mendenhall 1984; Bories 1987; Carithers 1989; Ramond 1992; Richardet 1993; De 2014; Thursz 2015). Two were three‐armed trials (Mendenhall 1977; Mendenhall 1984), one trial was a randomised trial with a two‐by‐two factorial design (Thursz 2015), one trial was a cross‐over trial (Richardet 1993), and the remaining were parallel, two‐group design trials. There were 1884 participants randomised in all trials. Some participants from Mendenhall 1977 (pilot trial or feasibility trial) continued participation in Mendenhall 1984. Three trials were conducted in France, one in India, two in the UK, and 10 in the USA (Characteristics of included studies table). All the trials reported the sex (65% of the participants were men) and age of the participants (range 25 years to 70 years). Four trials excluded women (Blitzer 1977; Mendenhall 1977; Mendenhall 1984; De 2014). Eleven trials reported to have included participants at different stages of alcoholic liver disease due to hepatitis, fibrosis, or cirrhosis (Helman 1971; Porter 1971; Campra 1973; Blitzer 1977; Maddrey 1978; Depew 1980; Theodossi 1982; Mendenhall 1984; Bories 1987; Ramond 1992; Thursz 2015). Most trials established diagnosis primarily through liver biopsy. One trial included only participants with liver cirrhosis in addition to alcoholic hepatitis (De 2014). The remaining trials did not provide information on the stage of disease. All the trials included participants with recent history of alcohol consumption, increase of serum bilirubin, liver enzymes, prolonged prothrombin time, and participants without previous treatment with glucocorticosteroids within the three months before the start of the trial. Ten trials performed liver biopsy whenever possible (Helman 1971; Porter 1971; Campra 1973; Blitzer 1977; Maddrey 1978; Shumaker 1978; Depew 1980; Bories 1987; Ramond 1992; Thursz 2015); however, it was an inclusion criterion in only one trial, performed at admission and after treatment (Helman 1971).
Ten trials reported the period of trial enrolment (range of one year to five years; median of three years) (Campra 1973; Blitzer 1977; Mendenhall 1977; Maddrey 1978; Depew 1980; Mendenhall 1984; Bories 1987; Carithers 1989; De 2014; Thursz 2015). The earliest trial began participant recruitment in 1966 (Campra 1973), and the most recently published trial began recruitment in 2011 and completed it in 2014 (Thursz 2015).
Three trials followed participants up to one year (Mendenhall 1984; De 2014; Thursz 2015). The remaining trials followed their participants to the moment of discharge from the hospital or until death occurred, with a median duration of follow‐up of 63 days (range 28 to 120).
We could extract data for analysis from all 16 trials but one (Richardet 1993). We contacted Richardet and colleagues in 2006, but received no reply. In the remaining 15 trials, 182 participants had mild alcoholic hepatitis and 1679 had severe alcoholic hepatitis. The analyses of the 15 trials accounted for 927 participants randomised to glucocorticosteroids, and 934 participants randomised to placebo or no intervention.
Based on the information that we collected from the published trial reports, three of the trials were not industry‐funded (Porter 1971; Ramond 1992; Thursz 2015), and the remaining 13 trials did not report clearly if they were partly or completely funded by the industry.
Experimental interventions
Glucocorticosteroids (prednisolone or 6‐methylprednisolone in equivalent dose of prednisolone) were administered orally or parenterally at different dose regimens and different durations. Twelve trials assessed oral glucocorticosteroids using prednisolone 40 mg or greater (Helman 1971; Mendenhall 1977; Maddrey 1978; Shumaker 1978; Depew 1980; Mendenhall 1984; Bories 1987; Carithers 1989; Ramond 1992; Richardet 1993; De 2014; Thursz 2015). Three trials also allowed parenteral administration to participants who were not able to swallow (Shumaker 1978; Carithers 1989; Ramond 1992). Two trials assessed oral glucocorticosteroids using prednisolone less than 40 mg (Campra 1973; Blitzer 1977), and in one trial the initial therapy was parenteral and then it was administered orally (Porter 1971). One trial used only parenteral (intravenous) glucocorticosteroids (Theodossi 1982).
The median duration of glucocorticosteroid administration was 28 days with a range of three days (Theodossi 1982) to 11 weeks (De 2014): one week (Richardet 1993), three weeks (Mendenhall 1977), four weeks (Ramond 1992; Thursz 2015), 26 days (Blitzer 1977), one month (Maddrey 1978; Mendenhall 1984; Bories 1987), five weeks (Shumaker 1978; Carithers 1989), six weeks (Helman 1971; Campra 1973; Depew 1980), 45 days (Porter 1971). Ten trials tapered the dose of prednisolone until it was stopped (Helman 1971; Porter 1971; Campra 1973; Blitzer 1977; Mendenhall 1977; Shumaker 1978; Depew 1980; Mendenhall 1984; Carithers 1989; De 2014).
Control interventions
Twelve trials used placebos that were identical in appearance to the glucocorticosteroid intervention (Helman 1971; Porter 1971; Blitzer 1977; Mendenhall 1977; Maddrey 1978; Shumaker 1978; Depew 1980; Mendenhall 1984; Carithers 1989; Ramond 1992; De 2014; Thursz 2015), and four trials used no intervention (Campra 1973; Theodossi 1982; Bories 1987; Richardet 1993).
Cointerventions
Two trials administered pentoxifylline to both glucocorticosteroids and placebo intervention groups (De 2014; Thursz 2015). There seemed to be no interaction between the intervention effects of pentoxifylline and glucocorticosteroids (De 2014; Thursz 2015).
Outcomes
The Characteristics of included studies tables details the outcomes reported in the individual trials. Five trials reported on outcomes with a follow‐up period up to three months after randomisation (Helman 1971; Mendenhall 1977; Bories 1987; De 2014; Thursz 2015). Twelve trials reported on outcomes at the end of treatment or at the moment of discharge from the hospital (Helman 1971; Porter 1971; Campra 1973; Blitzer 1977; Maddrey 1978; Shumaker 1978; Depew 1980; Theodossi 1982; Bories 1987; Carithers 1989; Ramond 1992; Richardet 1993). Three trials exceeded the 12‐month follow‐up period (Mendenhall 1984; De 2014; Thursz 2015).
Only one trial reported health‐related quality of life, using the European Quality of Life – 5 dimensions (EQ‐5D) score registered to Eudra CT 2009‐013897‐42 and ISRCTN 88782125 and it was reported in all the groups at three months' follow‐up after randomisation, and at one year (Thursz 2015; see Notes in Characteristics of included studies table).
None of the trials provided usable data for meta‐analyses of our exploratory outcomes.
For further details on trial characteristics, see Characteristics of included studies table.
Excluded studies
We excluded 29 publications from the final assessment with the reasons for their exclusion provided in the Characteristics of excluded studies table.
Among the excluded studies were two trials that used a nutritional intervention in the control group (Lesesne 1978; Cabré 2000). Although nutritional intervention as an overall intervention does not seem to influence all‐cause mortality or serious adverse events (Feinberg 2017), including the Cabré 2000 and Lesesne 1978 trials in our review would not have affected our results noticeably because these trials were small and had very few events.
Risk of bias in included studies
Allocation
Random sequence generation
We assessed the random sequence generation as low risk of bias in eight trials (Porter 1971; Campra 1973; Blitzer 1977; Maddrey 1978; Carithers 1989; Ramond 1992; De 2014; Thursz 2015), and as unclear in the remaining trials (Helman 1971; Mendenhall 1977; Shumaker 1978; Depew 1980; Theodossi 1982; Mendenhall 1984; Bories 1987; Richardet 1993).
Allocation concealment
We assessed allocation concealment as low risk of bias in ten trials (Helman 1971; Porter 1971; Campra 1973; Blitzer 1977; Shumaker 1978; Theodossi 1982; Mendenhall 1984; Carithers 1989; Ramond 1992; Thursz 2015), and as unclear in the remaining trials (Mendenhall 1977; Maddrey 1978; Depew 1980; Bories 1987; Richardet 1993; De 2014).
Blinding
Three trials were at high risk of performance bias as they were open‐label trials, without blinding of participants or investigators (Campra 1973; Theodossi 1982; Bories 1987), and one trial used placebo, but there was no description of it and we judged the risk of bias as unclear (Richardet 1993). Twelve trials were blinded, using identical placebo, and hence, at low risk of bias (Helman 1971; Porter 1971; Blitzer 1977; Mendenhall 1977; Maddrey 1978; Shumaker 1978; Depew 1980; Mendenhall 1984b: Carithers 1989; Ramond 1992; De 2014; Thursz 2015).
We assessed four trials at low risk of detection bias (Porter 1971; Shumaker 1978; De 2014; Thursz 2015), one trial at high risk of bias (Carithers 1989), and the remaining 11 trials as unclear risk of detection bias (Helman 1971; Campra 1973; Blitzer 1977; Mendenhall 1977; Maddrey 1978; Depew 1980; Theodossi 1982; Mendenhall 1984; Bories 1987; Ramond 1992; Richardet 1993).
Incomplete outcome data
We classed four trials at high risk of attrition bias because they did not account for participants with missing outcomes (Porter 1971; Blitzer 1977; Theodossi 1982; Thursz 2015 (the latter regarding one‐year follow‐up results)). Twelve trials were assessed as having low risk of attrition bias (Helman 1971; Campra 1973; Mendenhall 1977; Maddrey 1978; Shumaker 1978; Depew 1980; Mendenhall 1984; Bories 1987; Carithers 1989; Ramond 1992; De 2014; Thursz 2015 (the latter regarding follow‐up to end of treatment and up to three‐month follow‐up)). We judged one trial at unclear risk of bias (Richardet 1993).
Selective reporting
There were three trials at high risk of bias (Helman 1971; Mendenhall 1977; Mendenhall 1984), and one trial at unclear risk of bias (Richardet 1993). We found a registered protocol for only one trial (Thursz 2015). The remaining 11 trials reported all‐cause mortality, serious adverse events, and liver‐related mortality. Thus, 12 trials were at low risk of selective reporting bias.
Other potential sources of bias
We identified no other biases in 15 of the included trials. One trial was published as an abstract; we assessed this domain at unclear risk of other potential source of bias (Richardet 1993).
Overall risk of bias
We judged all trials but one (Thursz 2015) at high risk of bias. Figure 2 and Figure 3 show our assessment of risk of bias of the published trial reports (Characteristics of included studies table).
2.
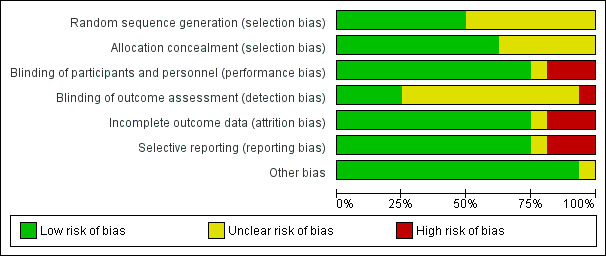
Risk of bias graph: review authors' judgements about each risk of bias domain presented as percentages across all included studies.
3.
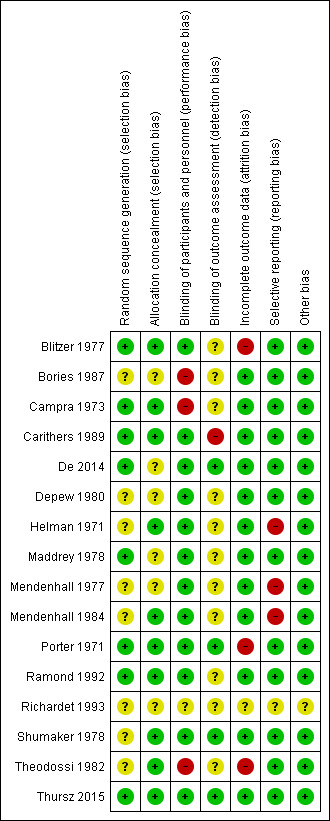
Risk of bias summary: review authors' judgements about each risk of bias domain for each included study.
Effects of interventions
See: Table 1
Primary outcomes
All‐cause mortality
Up to three months following randomisation
In total, 258/927 (27.8%) participants in the glucocorticosteroid group died versus 279/934 (29.9%) participants in the control group. There was no evidence of effect of glucocorticosteroids on all‐cause mortality (random‐effects RR 0.90, 95% CI 0.70 to 1.15; participants = 1861; trials = 15; I² = 45% (moderate heterogeneity; Analysis 1.1). We rated the certainty of the evidence as low (Table 1).
1.1. Analysis.
Comparison 1 Glucocorticosteroids versus placebo/no intervention, Outcome 1 All‐cause mortality.
We observed a similar result with the Trial Sequential Analysis showing that the cumulative Z‐curve did not cross the trial sequential monitoring boundaries for benefit or harm, nor enter the trial sequential monitoring area for futility in order to include an intervention effect of 20% relative risk reduction (Figure 4). The Trial Sequential analysis‐adjusted CI was 0.36 to 2.32.
4.
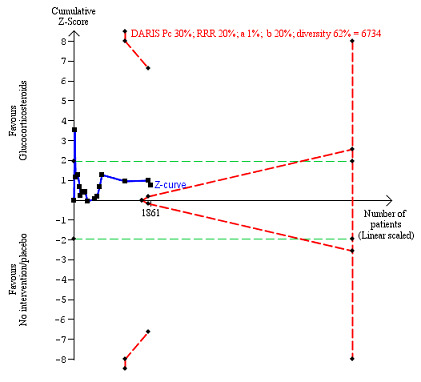
All‐cause mortality up to three months after randomisation. Fifteen trials provided data. The diversity‐adjusted required information size (DARIS) was calculated based on all‐cause mortality of 30% in the control group; relative risk reduction (RRR) in the glucocorticosteroid group of 20%; type I error of 1%; and type II error of 20% (80% power). Trial diversity was 62%. The required information size was 6734 participants. The cumulative Z‐curve (blue line) did not cross the trial sequential monitoring boundaries for benefit or harm (red inward sloping lines) and did not enter the trial sequential monitoring area for futility (inner‐wedge with red outward sloping lines). The green dotted lines show the conventional boundaries of the naive alpha of 5% equal to Z‐scores of +1.96 and –1.96.
Visual inspection of the funnel plot suggested publication bias or small‐trial bias on all‐cause mortality, but when using the Harbord 2006 test, we found no evidence of bias (P = 0.31) (Figure 5).
5.
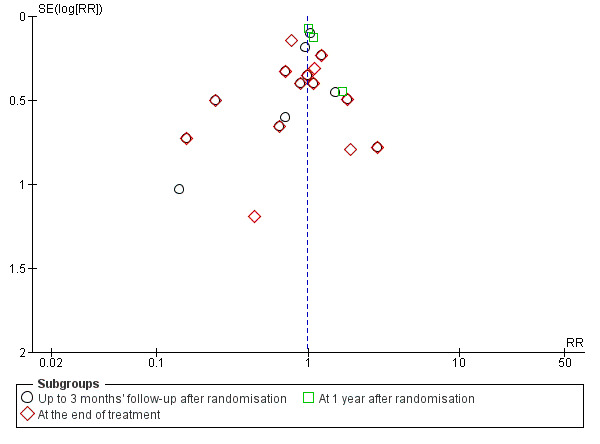
Funnel plot of comparison 1. Glucocorticosteroids versus no intervention/placebo, outcome 1.1 all‐cause mortality.
'Best‐worst' case scenario analysis
The 'best‐worst' case scenario analysis on mortality up to three months after randomisation produced two different results. While there was no evidence of effect of glucocorticosteroids with the random‐effects model (RR 0.82, 95% CI 0.64 to 1.05; I² = 47%), there was evidence of beneficial effect with the fixed‐effect model (RR 0.74, 95% CI 0.65 to 0.84; participants = 1861; trials = 15; I² = 47%; Analysis 3.1). Heterogeneity in both analyses was moderate.
3.1. Analysis.
Comparison 3 Sensitivity analysis: all‐cause mortality, Outcome 1 Best‐worst scenario all‐cause mortality up to 3 months' follow‐up after randomisation.
'Worst‐best' case scenario analysis
The 'worst‐best' case scenario analysis on mortality up to three months after randomisation produced two different results. While there was no evidence of effect of glucocorticosteroids with the random‐effects model (RR 0.97, 95% CI 0.73 to 1.29; I² = 62%), there was evidence of a harmful effect with the fixed‐effect model (RR 1.21, 95% CI 1.06 to 1.37; I² = 62%; Analysis 3.2).
3.2. Analysis.
Comparison 3 Sensitivity analysis: all‐cause mortality, Outcome 2 Worst‐best scenario analysis: all‐cause mortality up to 3 months' follow‐up after randomisation.
Our Trial Sequential Analysis assessment of imprecision coincided with assessment of imprecision with GRADE for all‐cause mortality: three‐months following randomisation.
At the end of treatment (post hoc analysis)
Treatment lasted for a median of 28 days (range 3 days to 12 weeks). In total, 162/907 (17%) participants in the glucocorticosteroid group died versus 202/917 (22%) participants in the control group. There was no evidence of effect of glucocorticosteroids on all‐cause mortality (random‐effects RR 0.87, 95% CI 0.66 to 1.15; participants = 1824; trials = 14; I² = 42%; moderate heterogeneity; Analysis 1.1.1). We rated the certainty of the evidence as low (not presented in Table 1).
We observed a similar result with the Trial Sequential Analysis showing that the cumulative Z‐curve did not cross the trial sequential monitoring boundaries for benefit or harm, and did not enter the trial sequential monitoring area for futility in order to exclude an intervention effect of 20% RRR (Figure 6). The Trial Sequential Analysis‐adjusted CI was CI 0.29 to 2.68.
6.
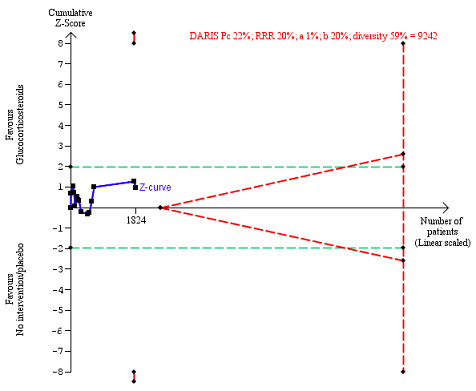
All‐cause mortality at the end of treatment (median 28 days (range 3 days to 12 weeks) (post hoc analysis). Fourteen trials provided data. The diversity‐adjusted required information size (DARIS) was calculated based on all‐cause mortality of 22% in the control group; relative risk reduction (RRR) in the glucocorticosteroid group of 20%; type I error of 1%; and type II error of 20% (80% power). Trial diversity was 59%. The required information size was 9242 participants. The cumulative Z‐curve (blue line) did not cross the trial sequential monitoring boundaries for benefit or harm (red inward sloping lines) and did not enter the trial sequential monitoring area for futility (inner‐wedge with red outward sloping lines). The green dotted lines show the conventional boundaries of the naive alpha of 5% equal to Z‐scores of +1.96 and –1.96.
Visual inspection of the funnel plot suggested publication bias or small‐trial bias on all‐cause mortality at the end of treatment, but when using the Harbord 2006 test, we found no evidence of bias (P = 0.84) (Figure 5).
A sensitivity analysis of full‐text articles (RR 0.85, 95% CI 0.64 to 1.11; participants = 1795; studies = 13; I² = 41%) and abstract (RR 2.83, 95% CI 0.61 to 13.06; participants = 29; studies = 1; I² = 0%) did not affect the overall result of mortality at the end of treatment (analysis not shown).
One year following randomisation (post hoc analysis)
Three of the included trials provided data on all‐cause mortality one year following randomisation (Mendenhall 1984; De 2014; Thursz 2015). In total, 274/668 (41%) participants in the glucocorticosteroid group died versus 265/664 (40%) participants in the control group. There was no evidence of effect of glucocorticosteroids on all‐cause mortality (random‐effects RR 1.03, 95% CI 0.91 to 1.17; participants = 1343; trials = 3; I² = 0%; no heterogeneity among the trials; Analysis 1.1.3). We rated the certainty of the evidence as moderate (not presented in Table 1).
We observed a similar result with the Trial Sequential Analysis showing that the cumulative Z‐curve entered the area of futility, which excludes an intervention effect of 20% RRR (Figure 7). The Trial Sequential analysis‐adjusted CI was CI 0.85 to 1.25.
7.
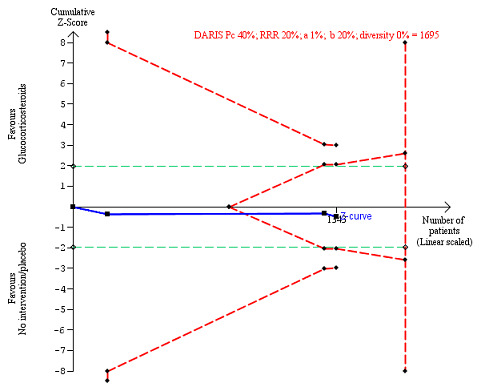
All‐cause mortality up to 1 year (post hoc analysis). Three trials provided data. The diversity‐adjusted required information size (DARIS) was calculated based on mortality in the control group of 40%; relative risk reduction (RRR) of 20% in the glucocorticosteroid group; type I error of 1%; and type II error of 20% (80% power). Trial diversity was 0%. The required information size was 1695 participants. The cumulative Z‐curve (blue line) did not cross the trial sequential monitoring boundaries for benefit or harm (red inward sloping lines). The cumulative Z‐curve crossed the inner‐wedge futility line (red outward sloping lines). The green dotted lines show the conventional boundaries of the naive alpha of 5% equal to Z‐scores of +1.96 and –1.96.
Subgroup analysis and investigation of heterogeneity: all‐cause mortality up to three months after randomisation
Trials at low risk of bias compared to trials at high risk of bias
Thursz 2015 was the only trial at low risk of bias. There was no significant difference (P = 0.32) between the subgroups of trials including one trial at low risk of bias (RR 1.03, 95% CI 0.84 to 1.26; participants = 1103; studies = 1; I² = 0%; Analysis 2.1.1; P = ) and the remaining 14 trials at high risk of bias (RR 0.86, 95% CI 0.63 to 1.17; participants = 758; studies = 14; I² = 48%; Analysis 2.1.2).
2.1. Analysis.
Comparison 2 Subgroup analysis: all‐cause mortality up to 3 months' follow‐up after randomisation, Outcome 1 Bias risk.
Trials without for‐profit funding compared to trials at risk of for‐profit funding
Thursz 2015 was the only trial which seemed not to have received industry funding. There was no significant difference (P = 0.32) between the subgroups of trials including one trial at low risk of bias ((RR 1.03, 95% CI 0.84 to 1.26; participants = 1103; Analysis 2.1.1) and the remaining 14 trials at high risk of bias ((RR 0.86, 95% CI 0.63 to 1.17; participants = 758; studies = 14; I² = 48%; Analysis 2.1.2).
Trials with people with mild alcoholic hepatitis compared to trials with severe alcoholic hepatitis, following Maddrey's score lower than 32 or 32 or higher or presence of hepatic encephalopathy; or as provided by the trialists
There was no significant difference (P = 0.75) between the subgroups (mild alcoholic hepatitis: RR 1.02, 95% CI 0.58 to 1.80; participants = 182; trials = 4; I² = 0%; Analysis 2.3.1) and severe alcoholic hepatitis (RR 0.92, 95% CI 0.73 to 1.16; participants = 1679; trials = 14; I² = 37%; Analysis 2.3.2).
2.3. Analysis.
Comparison 2 Subgroup analysis: all‐cause mortality up to 3 months' follow‐up after randomisation, Outcome 3 Severity of alcoholic hepatitis.
Trials with glucocorticosteroid dose 40 mg or less compared to trials with glucocorticosteroid dose more than 40 mg
There was no significant difference (P = 0.22) between the subgroups of the trials with glucocorticosteroid dose less than or equal to 40 mg (RR 0.75, 95% CI 0.50 to 1.14; participants = 1547; trials = 10; I² = 58%; Analysis 2.4.1) and trials with glucocorticosteroid dose more than 40 mg (RR 1.02, 95% CI 0.79 to 1.30; participants = 314; trials = 5; I² = 0%; Analysis 2.4.2).
2.4. Analysis.
Comparison 2 Subgroup analysis: all‐cause mortality up to 3 months' follow‐up after randomisation, Outcome 4 Glucocorticosteroid (prednisolone) dose.
Trials with people with severe alcoholic hepatitis without cirrhosis compared to trials with people with severe alcoholic hepatitis with cirrhosis
There was no significant difference (P = 0.83) between the subgroups of the trials with severe alcoholic hepatitis without cirrhosis (RR 0.79, 95% CI 0.18 to 3.48; participants = 123; trials = 3; I² = 77%; Analysis 2.5.1) and trials with people with severe alcoholic hepatitis with cirrhosis (RR 0.92, 95% CI 0.74 to 1.16; participants = 1738; studies = 12; I² = 35%; Analysis 2.5.2).
2.5. Analysis.
Comparison 2 Subgroup analysis: all‐cause mortality up to 3 months' follow‐up after randomisation, Outcome 5 Alcoholic hepatitis without or with cirrhosis.
As only two trials classified cirrhosis by Child‐Pugh score (Bories 1987; De 2014), and we did not know what classification system the remaining trials had used, we could not perform a subgroup analysis in order to adjust for the clinical spectrum of the disease.
Trials with people with severe alcoholic hepatitis without hepatorenal syndrome compared to trials with people with severe alcoholic hepatitis with hepatorenal syndrome
There was no significant difference (P = 0.64) between the subgroups of the trials with people with severe alcoholic hepatitis without hepatorenal syndrome (RR 1.00, 95% CI 0.85 to 1.17; participants = 1382; studies = 8; I² = 0%; Analysis 2.6.1) compared to trials with people with severe alcoholic hepatitis with hepatorenal syndrome (RR 0.56, 95% CI 0.05 to 6.49; participants = 129; studies = 2; I² = 88%; Analysis 2.6.2). Five trials did not clearly describe the presence of hepatorenal syndrome (Blitzer 1977; Mendenhall 1977; Mendenhall 1984; Bories 1987; Ramond 1992).
2.6. Analysis.
Comparison 2 Subgroup analysis: all‐cause mortality up to 3 months' follow‐up after randomisation, Outcome 6 Alcoholic hepatitis without or with hepatorenal syndrome.
Trials with people with severe alcoholic hepatitis without ascites compared to trials with people with severe alcoholic hepatitis with ascites
As we did not have data on trials with participants not having ascites, we could analyse only the subgroup of trials including participants with ascites (RR 0.82, 95% CI 0.60 to 1.12; participants = 729; trials = 13; I² = 48%; Analysis 2.7.1). In addition, two trials did not clearly describe the presence of ascites (Mendenhall 1977; Thursz 2015).
2.7. Analysis.
Comparison 2 Subgroup analysis: all‐cause mortality up to 3 months' follow‐up after randomisation, Outcome 7 Alcoholic hepatitis without or with ascites.
Health‐related quality of life
Up to three months
Only one trial reported quality of life at a follow‐up period of up to three months, using responses to the European Quality of Life – 5 Dimensions – 3 Levels (EQ‐5D‐3L) (Thursz 2015). We applied the Student's t‐test for the glucocorticosteroids versus the placebo group. We observed no difference between the two groups (MD –0.04 points, 95% CI –0.11 to 0.03; Analysis 1.2). We rated the certainty of the evidence as low (Table 1). We did not perform Trial Sequential Analysis.
1.2. Analysis.
Comparison 1 Glucocorticosteroids versus placebo/no intervention, Outcome 2 Health‐related quality of life.
Up to one year
Only one trial reported quality of life at a follow‐up period of up to one year, using responses to the EQ‐5D‐3L (Thursz 2015). We applied the Student's t‐test for the glucocorticosteroids versus the placebo group. We observed no difference between the two groups (MD 0.00 points; 95% CI –0.11 to 0.10; Analysis 1.2). We rated the certainty of the evidence as low (not presented in Table 1). We did not perform Trial Sequential Analysis.
As the data for health‐related quality of life came from one and the same trial, we could not perform sensitivity analyses.
Serious adverse events during treatment
Fifteen trials reported number of participants with serious adverse events during treatment. In total, 361/927 (38%) participants in the glucocorticosteroid group had serious adverse events during treatment versus 338/934 (36%) participants in the control group. There was no evidence of effect of glucocorticosteroids on the occurrence of serious adverse events (random‐effects RR 1.05, 95% CI 0.85 to 1.29; participants = 1861; trials = 15; I² = 36%; moderate heterogeneity; Analysis 1.3). We rated the certainty of the evidence as very low (Table 1).
1.3. Analysis.
Comparison 1 Glucocorticosteroids versus placebo/no intervention, Outcome 3 Serious adverse events during treatment.
We observed a similar result with the Trial Sequential Analysis showing that the cumulative Z‐curve entered the area of futility which excludes an intervention effect of 20% RRR (Figure 8). The Trial Sequential analysis‐adjusted CI was 0.60 to 1.82.
8.
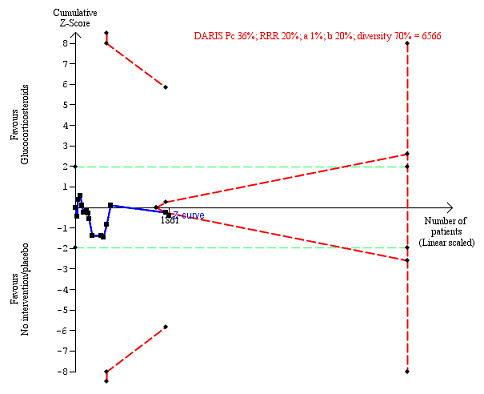
Serious adverse events during treatment. There are 15 trials providing data. The diversity‐adjusted required information size (DARIS) was calculated based on an incidence rate of serious adverse events in the control group of 36%; relative risk reduction (RRR) of 20% in the glucocorticosteroid group; type I error of 1%; and type II error of 20% (80% power). Trial diversity was 70%. The required information size was 6566 participants. The cumulative Z‐curve (blue line) did not cross the trial sequential monitoring boundaries for benefit or harm (red inward sloping lines), but it entered the trial sequential monitoring area for futility (inner‐wedge futility line red outward sloping lines) indicating that sufficient information was provided. The green dotted lines show the conventional boundaries of the naive alpha of 5% equal to Z‐scores of +1.96 and –1.96.
Table 2 shows the number of participants with the most often occurring serious adverse events in 14 included trials; mortality was not included. Table 3 presents the most often occurring serious adverse events in Thursz 2015 because this trial did not specify the individual number of participants with a serious adverse event.
1. Number of participants with most often occurring serious adverse events during treatment.
| Trial | Gastrointestinal haemorrhage | Hepatorenal syndrome (with or without hepatic failure) | Septicaemia | Hepatocellular carcinoma | ||||
| Prednisolone | Control | Prednisolone | Control | Prednisolone | Control | Prednisolone | Control | |
| Helman 1971 | — | — | — | 3 | — | — | — | — |
| Porter 1971 | 4 | 2 | — | — | — | — | — | — |
| Campra 1973 | 3 | 5 | — | 4 | — | — | — | — |
| Blitzer 1977 | 3 | 2 | — | 2 | 2 fungal | — | — | 1 |
| Mendenhall 1977 | Not reported | — | — | — | — | — | — | — |
| Maddrey 1978 | 1 | 1 | 3 | 6 | — | — | — | — |
| Shumaker 1978 | 3 | 3 | — | — | 2 | — | — | |
| Depew 1980 | 2 | 1 | — | — | 2 | 1 | — | — |
| Theodossi 1982 | 11 | 6 | — | — | 7 | 6 | — | — |
| Bories 1987 | 3 | 3 | — | 2 | — | — | — | — |
| Carithers 1989 | 2 | 4 | — | — | 1 | — | — | — |
| Mendenhall 1984 | — | — | — | — | — | — | — | 2 |
| Ramond 1992 | 1 | 2 | — | — | 1 | 1 | — | — |
| De 2014 | 2 | 3 | 3 | — | 3 | 1 | — | — |
Richardet 1993 is missing from the table as no data were provided for quantitative analysis.
For Thursz 2015, see Table 3.
2. Most often occurring serious adverse events in Thursz trial: number of events.
| Type of adverse event | Prednisolone group | Control group |
| Gastrointestinal haemorrhage plus variceal bleeding |
40 | 28 |
| Infections | 74 | 43 |
| – lung | 38 | 17 |
| – sepsis | 14 | 14 |
We constructed a funnel plot for publication bias, and using the Harbord 2006 test, we found no evidence of reporting bias (P = 0.63).
'Best‐worst' case scenario analysis
There was no evidence of effect of glucocorticosteroids on serious adverse events during treatment, with neither of the models (random‐effects model: RR 1.00, 95% CI 0.83 to 1.21; participants = 1861; studies = 15; I² = 28%; not important heterogeneity; fixed‐effect model: RR 0.99, 95% CI 0.89 to 1.11; participants = 1861; I² = 28%; not important heterogeneity; Analysis 4.1).
4.1. Analysis.
Comparison 4 Sensitivity analysis: serious adverse events, Outcome 1 Best‐worse scenario of serious adverse events during treatment.
'Worst‐best' case scenario analysis
While there was evidence of a harmful effect of glucocorticosteroids with the fixed‐effect model (RR 1.18, 95% CI 1.05 to 1.31; participants = 1861; I² = 38%), there was no evidence of effect of glucocorticosteroids with the random‐effects model (RR 1.11, 95% CI 0.91 to 1.36; I² = 38%; Analysis 4.2).
4.2. Analysis.
Comparison 4 Sensitivity analysis: serious adverse events, Outcome 2 Worst‐best scenario of serious adverse events during treatment.
Our Trial Sequential Analysis assessment of imprecision coincided with assessment of imprecision with GRADE for serious adverse events during treatment.
A sensitivity analysis of full‐text articles (RR 1.03, 95% CI 0.84 to 1.27; participants = 1832; studies = 14; I² = 36%) and abstract (RR 2.83, 95% CI 0.61 to 13.06; participants = 29; studies = 1; I² = 0%) did not affect the serious adverse events during treatment (analysis not shown).
Secondary outcomes
Liver‐related mortality up to three months' follow‐up after randomisation
In total, 257/927 (27.7%) participants in the glucocorticosteroid group died versus 279/934 (29.9%) participants in the control group. There was no evidence of effect of glucocorticosteroids on liver‐related mortality (random‐effects RR 0.89, 95% CI 0.69 to 1.14; participants = 1861; trials = 15; I² = 46%; moderate heterogeneity; Analysis 1.4). We rated the certainty of the evidence as very low (Table 1).
1.4. Analysis.
Comparison 1 Glucocorticosteroids versus placebo/no intervention, Outcome 4 Liver‐related mortality: up to 3 months' follow‐up after randomisation.
We observed a similar result with the Trial Sequential Analysis showing that the cumulative Z‐curve did not cross the trial sequential monitoring boundaries for benefit or harm, nor enter the trial sequential monitoring area for futility in order to include an intervention effect of 20% RRR (Figure 9). The Trial Sequential analysis‐adjusted CI was 0.32 to 2.45.
9.
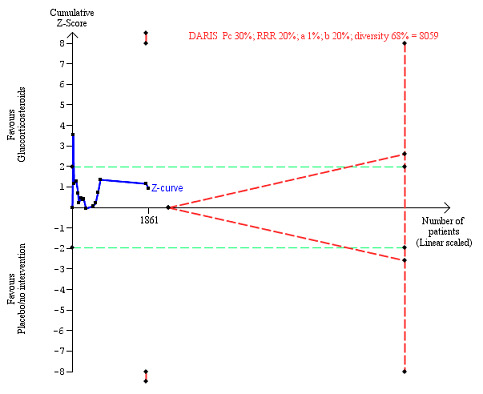
Liver‐related mortality up to three months after randomisation. Fifteen trials provided data. The diversity‐adjusted required information size (DARIS) was calculated based on liver‐related mortality of 30% in the control group; relative risk reduction (RRR) in the glucocorticosteroid group of 20%; type I error of 1%; and type II error of 20% (80% power). Trial diversity was 68%. The required information size was 8059 participants. The cumulative Z‐curve (blue line) did not cross the trial sequential monitoring boundaries for benefit or harm (red inward sloping lines) and did not enter the trial sequential monitoring area for futility (inner‐wedge with red outward sloping lines). The green dotted lines show the conventional boundaries of the naive alpha of 5% equal to Z‐scores of +1.96 and –1.96.
Our Trial Sequential Analysis assessment of imprecision coincided with assessment of imprecision with GRADE for liver‐related mortality up to three months following randomisation.
Participants with any complication up to three months' follow‐up after randomisation
In total, 440/927 (47%) participants in the glucocorticosteroid group had one or more complications versus 414/934 (44%) participants in the control group. There was no evidence of effect of glucocorticosteroids on the number of participants with any complications (random‐effects RR 1.04, 95% CI 0.86 to 1.27; participants = 1861; I² = 42%; moderate heterogeneity; Analysis 1.5). We rated the certainty of the evidence as very low, mainly due to within‐study bias, inconsistency, and imprecision (Table 1).
1.5. Analysis.
Comparison 1 Glucocorticosteroids versus placebo/no intervention, Outcome 5 Participants with any complication up to 3 months' follow‐up.
We observed a similar result with the Trial Sequential Analysis showing that the cumulative Z‐curve did not cross the trial sequential monitoring boundaries for benefit or harm but it crossed the trial sequential monitoring area for futility in order to include an intervention effect of 20% RRR (Figure 10). The Trial Sequential analysis‐adjusted CI was 0.67 to 1.63.
10.
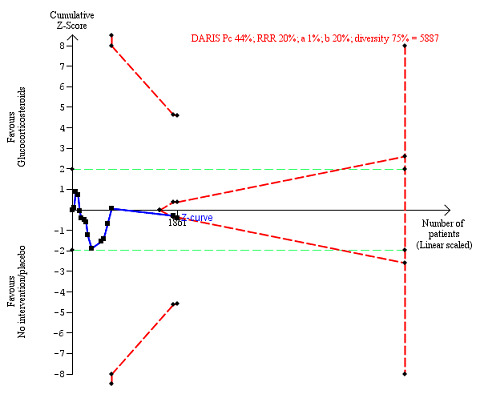
Any complications up to three months' follow‐up after randomisation. Fifteen trials provided data. The diversity‐adjusted required information size (DARIS) was calculated based on any complications of 44% in the control group; relative risk reduction (RRR) in the glucocorticosteroid group of 20%; type I error of 1%; and type II error of 20% (80% power). Trial diversity was 75%. The required information size was 5887 participants. The cumulative Z‐curve (blue line) did not cross the trial sequential monitoring boundaries for benefit or harm (red inward sloping lines). The cumulative Z‐curve crossed the inner‐wedge futility line (red outward sloping lines). The green dotted lines show the conventional boundaries of the naive alpha of 5% equal to Z‐scores of +1.96 and –1.96.
Our Trial Sequential Analysis assessment of imprecision coincided with assessment of imprecision with GRADE for number of participants with any complication up to three months' follow‐up after randomisation.
Participants with non‐serious adverse events up to three months' follow‐up after randomisation
Only four trials reported non‐serious adverse events such as Cushingoid symptoms, vertigo, and fungal lesions. There was no evidence of effect of glucocorticosteroids on number of participants with non‐serious adverse events (random‐effects RR 1.99, 95% CI 0.72 to 5.48; participants = 160; trials = 4; I² = 0%; no heterogeneity; Analysis 1.6). We rated the certainty of the evidence as very low to low (Table 1).
1.6. Analysis.
Comparison 1 Glucocorticosteroids versus placebo/no intervention, Outcome 6 Participants with non‐serious adverse events up to 3 months' follow‐up after randomisation.
We observed a similar result with the Trial Sequential Analysis showing that the cumulative Z‐curve did not cross the trial sequential monitoring boundaries for benefit or harm, nor did it enter the trial sequential monitoring area for futility in order to include an intervention effect of 50% RRR (Figure 11). The Trial Sequential Analysis‐adjusted CI was 0.01 to 249.60.
11.
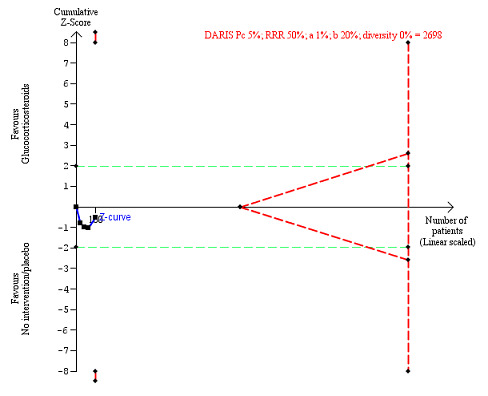
Non‐serious adverse events up to three months after randomisation. Four trials provided data. The diversity‐adjusted required information size (DARIS) was calculated based on non‐serious adverse events of 5% in the control group; relative risk reduction (RRR) in the glucocorticosteroid group of 50%; type I error of 1%; and type II error of 20% (80% power). Trial diversity was 0%. The required information size was 2698 participants. The cumulative Z‐curve (blue line) did not cross the trial sequential monitoring boundaries for benefit or harm (red inward sloping lines) and did not enter the trial sequential monitoring area for futility (inner‐wedge with red outward sloping lines). The green dotted lines show the conventional boundaries of the naive alpha of 5% equal to Z‐scores of +1.96 and –1.96.
Our Trial Sequential Analysis assessment of imprecision coincided with assessment of imprecision with GRADE for number of people with non‐serious adverse events up to three months following randomisation.
Exploratory outcomes at the end of treatment
No trial reported on number of participants with change of level of liver enzymes, prothrombin index, or serum albumin at the end of treatment. This is why we could not perform the planned exploratory analyses. Instead, post hoc, we decided to present in a tabular way the extracted information on level of liver enzymes reported in the trials by Campra 1973; Maddrey 1978; Theodossi 1982; and Carithers 1989 (Appendix 2); prothrombin index or international normalised ratio reported in the trials by Campra 1973; Maddrey 1978; Theodossi 1982; Carithers 1989; Ramond 1992 (Appendix 3); and level of serum albumin (and bilirubin post hoc) reported in the trials by Campra 1973; Maddrey 1978; Depew 1980; Theodossi 1982; Carithers 1989; and Ramond 1992 (Appendix 4; Appendix 5).
'Summary of findings' table
We presented the key results on the outcomes all‐cause mortality, health‐related quality of life, serious adverse events, liver‐related mortality, all complications, and non‐serious adverse events in Table 1. We assessed the evidence as being very low for all listed outcomes but health‐related quality of life for which the evidence was low. We downgraded the evidence because of within‐study risk of bias, inconsistency of the data, imprecision, and publication bias. We presented the results obtained at predefined primary time points.
Discussion
Summary of main results
We included 16 randomised clinical trials comparing glucocorticosteroids versus placebo or no intervention in people with alcoholic hepatitis. Fifteen trials provided data for analyses. Our meta‐analyses showed no beneficial or detrimental effects of glucocorticosteroids on any of our outcomes. In general, serious and non‐serious adverse events as well as complications were poorly reported or the information was unclear, and hence, these analyses may be subject to outcome reporting bias (Ioannidis 2009). Trial Sequential Analyses showed similar results. Based on methodological concerns, we classified the certainty of the evidence as low for health‐related quality of life, and very low for all the remaining primary and secondary outcomes. We assessed the certainty of evidence for all‐cause mortality at one‐year follow‐up (post hoc analysis) as moderate. As only one trial was at low risk of bias, it is more likely that that the trials at high risk of bias were overestimating benefits and overlooking harms.
Overall completeness and applicability of evidence
The trial participants varied according to severity of alcoholic hepatitis and the trials were published between 1971 and 2015. However, only 1861 participants were included. During this time period, glucocorticosteroid interventions varied regarding dose and duration. The small number of trials and trial participants, with the exception of Thursz 2015, the poor trial design and reporting, all make the results of our review inconclusive. The high risk of bias of almost all trials undermined the precision of our meta‐analyses results.
We were unable to assess if ethnicity had any influence on our results, as data were either lacking or insufficient. The same applied for the nutritional status of the participants, as only one trial reported on it (Mendenhall 1984). Mathurin and coworkers proposed that people with alcoholic hepatitis with Maddrey's score of at least 32 should likely benefit from glucocorticosteroids (Mathurin 2011). However, we found no significant effect of glucocorticosteroids in this subgroup of trial participants.
This review is applicable to people with alcoholic hepatitis at different stages of the disease. Our meta‐analyses and Trial Sequential Analyses seem to provide no evidence of benefit of glucocorticosteroids on all‐cause mortality at one‐year follow‐up after randomisation. It is also unlikely that glucocorticosteroids may have a beneficial effect on mortality at the end of treatment and three months following randomisation; however, due to mainly imprecision (the CI crossed the clinical decision threshold between recommending and not recommending treatment and the required number of participants was far from reached), we could not exclude the possibility of a short‐term beneficial or harmful effect. We could not say if glucocorticosteroids may have influenced infection and gastrointestinal bleeding as we had no data for meta‐analysis. However, Thursz and colleagues' analysis showed an increase in the number of these complications in treated participants (Thursz 2015). The only worst‐best sensitivity analysis for all‐cause mortality and serious adverse events showed a tendency of harmful effect of glucocorticosteroids compared to the best‐worse sensitivity analysis showing no difference in effect.
Quality of the evidence
The certainty of the evidence reflects only the quality of the included trials, and this is why we could not be certain in our conclusions. We judged the overall certainty of evidence as low for health‐related quality of life to very low for all outcomes except for all‐cause mortality at one year after randomisation, for which the certainty of the evidence was moderate (not presented in Table 1). The randomisation procedures were insufficiently reported in 15 of the trials. In addition to downgrading the trials for within‐study risk of bias, we also downgraded the trials for imprecision of effect estimates due to the number of participants included in the trials (all but one of the 14 trials had fewer than 400 participants), and for inconsistency of the results (there was wide variation in the effect estimates across the trials; there was little overlap of CIs associated with the effect estimates; and we assessed heterogeneity of the data as moderate with I² statistics of 36% to 46%, which could be explained with selection bias). We found no statistical evidence of publication bias or small‐study bias.
In spite of the certainty of the evidence being very low or low, we are reasonably confident in our recommendations regarding implications for practice and for research. This ensues from our analysis results and is based on the knowledge that trials at high risk of bias overestimate benefits and underestimate harms. Therefore, we found no supporting evidence for using glucocorticosteroids in clinical practice. There is definitely a need for more transparent reporting of individual participant data (NTAWG 2015; Garattini 2016).
Potential biases in the review process
The strengths of our review are that we have conducted our review following the recommendations of Cochrane Hepato‐Biliary and the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011c; Gluud 2017). We included only randomised clinical trials in our review. This creates a bias towards focusing on benefits as short‐term randomised trials often overlook harms. We attempted to minimise possible selection biases by using a comprehensive search strategy. We combined searches in electronic databases with extensive manual searches. In addition, we also searched conference proceedings and abstract books, irrespective of language. We think it is unlikely that we have missed any published trials, but we cannot exclude the possibility that we have missed unpublished trials. Visual inspection of the funnel plots suggests publication bias or small‐study bias on all‐cause mortality at the end of treatment and following three months after randomisation in contrast to the statistical result with Harbord 2006 test (Figure 5). We wrote to pharmaceutical companies and regulatory authorities. We made extensive attempts to avoid risk of system and random errors. We assessed the evidence with GRADE approach.
Limitations of our review were the small number of trials and the small total number of participants. Having in mind that hepatitis C viral disease was discovered as late as 1989, we might have run the risk that the included trials initiated before 1989 did not include participants with only alcoholic hepatitis (Houghton 2009). Furthermore, our results were hampered by the quality of the included trials as well as imprecision and severe inconsistency. Even though all trials provided data on mortality, data on other serious adverse events and complications were rarely reported, which calls into question the reliability of the two latter analyses. Only one trial reported quality of life. Moreover, by including primarily randomised clinical trials we have focused on potential beneficial effects and overlooked the many known harms connected with the administration of glucocorticosteroids. Again, these flaws in our review make us suspect that benefits are overestimated and harms are underestimated.
When conducting our Trial Sequential Analyses, we used plausible parameters to calculate our required information sizes. However, we only used 80% power (beta = 20%). Had we used 90% power (beta = 10%) or less, which is relevant in meta‐analyses where one does not want to discharge a potentially relevant intervention, then we would have obtained larger required information sizes and wider Trial Sequential Analyses‐adjusted CIs (Garattini 2016; Castellini 2017). Accordingly, the imprecision may be worse than signalled by our analyses.
Agreements and disagreements with other studies or reviews
The meta‐analysis by Christensen 1995 found no effect of glucocorticosteroids versus placebo on mortality. The review included data from 13 trials with 659 participants randomised. Rambaldi 2008 updated the meta‐analysis by Christensen 1995, adding two more trials with 62 participants randomised. Hence, Rambaldi 2008 concluded that depending on the estimation of the information size, their review lacked another 1000 to 2000 participants randomised to glucocorticosteroids versus placebo in order to be able to either demonstrate or reject a clinically relevant 20% mortality reduction.
Our review published in 2017 (Pavlov 2017) included two new trials (De 2014; Thursz 2015), compared with the previous review version published in a paper journal (Rambaldi 2008). The review by Pavlov 2017 excluded two of the trials from the Rambaldi 2008 as they assessed glucocorticosteroids versus nutrition (Lesesne 1978; Cabré 2000). In addition, two trial reports turned out to be the same trial (Shumaker 1978; Galambos 1984), and thus, they are counted as one trial (Pavlov 2017). We did not identify any new trials for the update of this current review.
Our systematic review of pair‐wise comparison randomised clinical trials is in agreement with the meta‐analysis by Buzzetti 2017. In this network meta‐analysis, the authors found no significant effects of glucocorticosteroids on mortality at maximal follow‐up and up to 90 days of follow‐up.
Our review now includes 1861 participants. The Thursz 2015 trial included 1103 participants and found "a reduction in the 28‐day mortality in the prednisolone‐treated group on logistic regression model analysis, but there was not clear evidence of benefit, sustained beyond this point." Mathurin 2011 performed "analysis of individual data from five randomised clinical trials which showed that corticosteroids significantly improved 28‐day survival in patients with severe alcoholic hepatitis." In our present aggregate meta‐analysis, based on the certainty of evidence, we could not determine whether there was an effect or not of glucocorticosteroids on mortality at 'end of treatment,' which is quite close to 28 days. The review by Louvet 2018 (see Why it is important to do this review section), assessed the effects of corticosteroids versus placebo or control, corticosteroids versus pentoxifylline, corticosteroids plus pentoxifylline versus corticosteroids plus placebo or control, and pentoxifylline versus placebo in four meta‐analyses. However, the number of participants with severe alcoholic hepatitis providing individual participants' data from the six included in the meta‐analysis trials, comparing corticosteroids versus placebo or control, was too small to draw a firm conclusion on the beneficial or harmful effects of glucocorticosteroids. In addition, the control intervention of two of the included six studies was nutrition or antioxidants, which did not compare well with the placebo group of participants. The conclusions Louvet and colleagues made was that "corticosteroids used to reduce risk of death within 28 days of treatment, but not in the following six months. This loss of efficacy over time indicates a need for new therapeutic strategies to improve medium‐term outcomes." Louvet and colleagues did not assess the risk of bias and the quality of the included trials which adds further to the unreliability of their conclusions.
Clinical guidelines recommend prescribing glucocorticosteroids as follows: AASLD 2010 reads: "Patients with severe disease (Maddrey's Discriminant Function (MDF) score of ≥ 32, with or without hepatic encephalopathy) and lacking contraindications to steroid use should be considered for a four‐week course of prednisolone (40 mg/day for 28 days, typically followed by discontinuation or a 2‐week taper) (Class I, level A)" and EASL 2012 reads: "First‐line therapy in patients with severe alcoholic hepatitis includes corticosteroids or, in case of ongoing sepsis, pentoxifylline (Recommendation B1)." In the absence of active infection, EASL 2018 suggests the use of corticosteroids (prednisolone 40 mg/day or methylprednisolone 32 mg/day) for people with severe alcoholic hepatitis to reduce short‐term mortality (Grade A1). However, in our present meta‐analysis, we could not rule out a beneficial or harmful effect of glucocorticosteroids in people with severe alcoholic hepatitis.
Authors' conclusions
Implications for practice.
We are very uncertain about the effect estimate of no difference between glucocorticosteroids and placebo or no intervention on all‐cause mortality and serious adverse events during treatment because the risk of bias was high and the certainty of the evidence was very low. Our confidence in the effect of glucocorticosteroids on health‐related quality of life is low. Due to inadequate reporting, we cannot exclude increases in adverse events. As the confidence intervals, except for one‐year all‐cause mortality, were wide, we cannot rule out significant benefits or harms of glucocorticosteroids.
Implications for research.
As there could be some people with alcoholic hepatitis who could benefit from glucocorticosteroids, it could be of use for researchers to study further the effects of glucocorticosteroids in randomised clinical trials on short‐term all‐cause mortality. Additional evidence evaluating the effect on health‐related quality of life is also needed. Future trials ought to be designed according to the SPIRIT guidelines (www.spirit‐statement.org/) and reported according to the CONSORT guidelines (www.consort‐statement.org). Future trials ought to report individual participant data, so that proper individual participant data meta‐analyses of the effects of glucocorticosteroids in subgroups can be conducted (NTAWG 2015; Garattini 2016).
What's new
| Date | Event | Description |
|---|---|---|
| 19 February 2019 | New search has been performed | We have revised the whole review so that it reflects current Cochrane methodology. We have excluded evaluation of imprecision with Trial Sequential Analysis from the GRADE assessment. In the previously published review, assessment of imprecision with Trial Sequential Analysis was presented in the 'Summary of findings' table. |
| 28 January 2019 | New citation required but conclusions have not changed | No new randomised clinical trials identified for the review update. Discrepancies occurred in GRADE assessment and in conclusions in certainty of evidence for the outcomes: serious adverse events (from low to very low); liver‐related mortality up to three months following randomisation (from low to very low); and any complication, up to three months following randomisation (from low to very low). |
| 28 January 2019 | New search has been performed | Search for new trials performed 18 January 2019 |
Notes
Cochrane Reviews can be expected to have a high percentage of overlap in the methods section because of standardised methods. In addition, overlap may be observed across two of our protocols as they share at least four common authors.
Acknowledgements
Cochrane Review Group funding acknowledgement: the Danish State is the largest single funder of Cochrane Hepato‐Biliary through its investment in the Copenhagen Trial Unit, Centre for Clinical Intervention Research, Copenhagen, Denmark. Disclaimer: the views and opinions expressed in this review are those of the authors and do not necessarily reflect those of the Danish State or the Copenhagen Trial Unit.
Peer reviewers: Kurinchi S Gurusamy, UK; Michael Ronan Lucey, USA. Contact editor: Dario Conte, Italy. Sign‐off editor: Agostino Colli, Italy.
Appendices
Appendix 1. Search strategies
| Database | Search performed | Search strategy |
| Cochrane Hepato‐Biliary Controlled Trials Register | January 2019 | (glucocortico* or steroid* or dexamethasone or prednis* or hydrocortisone or corticosteroid* or cortiso* or budesonide* or beclomethasone*) AND (alcohol* and (liver or hepati*)) |
| Cochrane Central Register of Controlled Trials (CENTRAL) in the Cochrane Library | Issue 1, 2019 | #1 MeSH descriptor: [Adrenal Cortex Hormones] explode all trees #2 (glucocortico* or steroid* or dexamethasone or prednis* or hydrocortisone or corticosteroid* or cortiso* or budesonide* or beclomethasone*) #3 #1 or #2 #4 MeSH descriptor: [Hepatitis, Alcoholic] explode all trees #5 (alcohol* and (liver or hepati*)) #6 #4 or #5 #7 #3 and #6 |
| MEDLINE Ovid | 1946 to January 2019 | 1. exp Adrenal Cortex Hormones/ 2. (glucocortico* or steroid* or dexamethasone or prednis* or hydrocortisone or corticosteroid* or cortiso* or budesonide* or beclomethasone*).mp. [mp=title, abstract, original title, name of substance word, subject heading word, keyword heading word, protocol supplementary concept word, rare disease supplementary concept word, unique identifier] 3. 1 or 2 4. exp Hepatitis, Alcoholic/ 5. (alcohol* and (liver or hepati*)).mp. [mp=title, abstract, original title, name of substance word, subject heading word, keyword heading word, protocol supplementary concept word, rare disease supplementary concept word, unique identifier] 6. 4 or 5 7. 3 and 6 8. (random* or blind* or placebo* or meta‐analys*).mp. [mp=title, abstract, original title, name of substance word, subject heading word, keyword heading word, protocol supplementary concept word, rare disease supplementary concept word, unique identifier] 9. 7 and 8 |
| Embase Ovid | 1974 to January 2019 | 1. exp corticosteroid/ 2. (glucocortico* or steroid* or dexamethasone or prednis* or hydrocortisone or corticosteroid* or cortiso* or budesonide* or beclomethasone*).mp. [mp=title, abstract, heading word, drug trade name, original title, device manufacturer, drug manufacturer, device trade name, keyword] 3. 1 or 2 4. exp alcohol liver disease/ 5. (alcohol* and (liver or hepati*)).mp. [mp=title, abstract, heading word, drug trade name, original title, device manufacturer, drug manufacturer, device trade name, keyword] 6. 4 or 5 7. 3 and 6 8. (random* or blind* or placebo* or meta‐analys*).mp. [mp=title, abstract, heading word, drug trade name, original title, device manufacturer, drug manufacturer, device trade name, keyword] 9. 7 and 8 |
| Science Citation Index Expanded (Web of Science) | 1900 to January 2019 | #5 217 #4 AND #3 #4 1,347,943 TS=(random* or blind* or placebo* or meta‐analys*) #3 1,060 #2 AND #1 #2 36,574 TS=(alcohol* and (liver or hepati*)) #1 425,242 TS=(glucocortico* or steroid* or dexamethasone or prednis* or hydrocortisone or corticosteroid* or cortiso* or budesonide* or beclomethasone*) |
| eLibrary | 1999 to January 2019 | (glucocortico* or steroid* or dexamethasone or prednis* or hydrocortisone or corticosteroid* or budesonide*) AND (alcohol* and (liver or hepati*)) |
Appendix 2. Level of liver enzymes (at the end of treatment)
| Study | Glucocorticosteroids | Placebo/no intervention | ||||
| Mean | SD | n | Mean | SD | n | |
| Campra 1973 | 62.5 | 3.72 | 22 | 79.2 | 5.83 | 28 |
| Maddrey 1978 | 34.5 | 4 | 23 | 41.7 | 4.1 | 27 |
| Theodossi 1982 | 164.0 | — | 28 | 118 | — | 32 |
| Carithers 1989 | 74.9 | 4.19 | 36 | 119.9 | 7.79 | 31 |
| —: data not reported; n: number of participants; SD: standard deviation. | ||||||
Appendix 3. Prothrombin index (seconds)
| Study | Glucocorticosteroids | Placebo/no intervention | ||||
| Mean | SD | n | Mean | SD | n | |
| Percentage of normal | ||||||
| Campra 1973 | 77.5 | 2.5 | 36 | 83.1 | 2 | 31 |
| Ramond 1992 | 48 | 0.5 | 32 | 45 | 0.5 | 29 |
| At the end of treatment | ||||||
| Maddrey 1978 | 14 | 0.5 | 23 | 15.5 | 0.9 | 27 |
| Theodossi 1982 | 13 | — | 28 | 10 | — | 32 |
| Carithers 1989 | 15.5 | 1.25 | 36 | 16.25 | 1 | 31 |
| —: data not reported; n: number of participants; SD: standard deviation. | ||||||
Appendix 4. Level of serum albumin (g/L)
| Study | Glucocorticosteroids | Placebo/no intervention | ||||
| Mean | SD | n | Mean | SD | n | |
| Campra 1973 | 33.3 | 1.48 | 22 | 30.08 | 0.66 | 28 |
| Maddrey 1978 | 33 | 2 | 23 | 30 | 2 | 27 |
| Depew 1980 | 32 | 0.17 | 15 | 25.8 | 0.13 | 13 |
| Carithers 1989 | 27 | 0.35 | 36 | 29 | 0.4 | 31 |
| Ramond 1992 | 33 | 0.17 | 32 | 30 | 0.37 | 29 |
| n: number of participants; SD: standard deviation. | ||||||
Appendix 5. Level of bilirubin (μmol/L) (at the end of treatment)
| Study | Glucocorticosteroids | Placebo/no intervention | ||||
| Mean | SD | n | Mean | SD | n | |
| Campra 1973 | 43.2 | 2.9 | 22 | 63.95 | 6.84 | 28 |
| Maddrey 1978 | 64.98 | 4.97 | 24 | 66.69 | 3.69 | 31 |
| Depew 1980 | 68.4 | 6.5 | 15 | 136.8 | 11.79 | 13 |
| Carithers 1989 | 125 | 8.12 | 36 | 190 | 17.9 | 31 |
| Ramond 1992 | 100 | 7.1 | 32 | 150 | 18.8 | 29 |
| n: number of participants; SD: standard deviation | ||||||
Appendix 6. Age (years)
| Study | Glucocorticosteroids | Placebo/no intervention | ||||
| Mean | SD | n | Mean | SD | n | |
| Porter 1971 | 44.6 | 4.4 | 11 | 49.5 | 8.9 | 9 |
| Campra 1973 | 43.1 | 11.1 | 22 | 42.7 | 8.1 | 28 |
| Blitzer 1977 | 47.2 | — | 17 | 48.4 | — | 16 |
| Maddrey 1978 | 40 | 8.5 | 25 | 42.3 | 11.1 | 32 |
| Shumaker 1978 | 45.5 | — | 15 | 44.5 | — | 13 |
| Depew 1980 | 49.8 | 2.1 | 15 | 48.2 | 2.3 | 13 |
| Mendenhall 1984 | 51.5 | 8.2 | 90 | 50.4 | 9.2 | 88 |
| Bories 1987 | 41 | — | 24 | 49 | — | 21 |
| Carithers 1989 | 43.1 | 2 | 36 | 44.4 | 1.7 | 31 |
| Ramond 1992 | 48.1 | 1.3 | 33 | 48.2 | 1.6 | 32 |
| De 2014 | 42.7 | 0.4 | 31 | 41.3 | 7.8 | 31 |
| Thursz 2015 | 48.6 | 9.8 | 277 | 47.9 | 9.2 | 276 |
| Thursz 2015 | 49.3 | 10.6 | 274 | 48.8 | 10.3 | 276 |
| —: data not reported; n: number of participants; SD: standard deviation. | ||||||
Data and analyses
Comparison 1. Glucocorticosteroids versus placebo/no intervention.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 All‐cause mortality | 15 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Up to 3 months' follow‐up after randomisation | 15 | 1861 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.70, 1.15] |
| 1.2 At the end of treatment | 14 | 1824 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.66, 1.15] |
| 1.3 At 1 year after randomisation | 3 | 1343 | Risk Ratio (M‐H, Random, 95% CI) | 1.03 [0.91, 1.17] |
| 2 Health‐related quality of life | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 2.1 Up to 3 months | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Up to 1 year | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Serious adverse events during treatment | 15 | 1861 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.85, 1.29] |
| 4 Liver‐related mortality: up to 3 months' follow‐up after randomisation | 15 | 1861 | Risk Ratio (M‐H, Random, 95% CI) | 0.89 [0.69, 1.14] |
| 5 Participants with any complication up to 3 months' follow‐up | 15 | 1861 | Risk Ratio (M‐H, Random, 95% CI) | 1.04 [0.86, 1.27] |
| 6 Participants with non‐serious adverse events up to 3 months' follow‐up after randomisation | 4 | 160 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.99 [0.72, 5.48] |
Comparison 2. Subgroup analysis: all‐cause mortality up to 3 months' follow‐up after randomisation.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Bias risk | 15 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Trials at low risk | 1 | 1103 | Risk Ratio (M‐H, Random, 95% CI) | 1.03 [0.84, 1.26] |
| 1.2 Trials at high risk | 14 | 758 | Risk Ratio (M‐H, Random, 95% CI) | 0.86 [0.63, 1.17] |
| 2 Trials without for‐profit funding compared to trials at risk of for‐profit funding | 15 | 1861 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.70, 1.15] |
| 2.1 Trials without for‐profit funding | 1 | 1103 | Risk Ratio (M‐H, Random, 95% CI) | 1.03 [0.84, 1.26] |
| 2.2 Trials at risk of for‐profit funding | 14 | 758 | Risk Ratio (M‐H, Random, 95% CI) | 0.86 [0.63, 1.17] |
| 3 Severity of alcoholic hepatitis | 15 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 Mild alcoholic hepatitis | 4 | 182 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.58, 1.80] |
| 3.2 Severe alcoholic hepatitis | 14 | 1679 | Risk Ratio (M‐H, Random, 95% CI) | 0.92 [0.73, 1.16] |
| 4 Glucocorticosteroid (prednisolone) dose | 15 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 ≤ 40 mg | 10 | 1547 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.50, 1.14] |
| 4.2 > 40 mg | 5 | 314 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.79, 1.30] |
| 5 Alcoholic hepatitis without or with cirrhosis | 15 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 Without cirrhosis | 3 | 123 | Risk Ratio (M‐H, Random, 95% CI) | 0.79 [0.18, 3.48] |
| 5.2 With cirrhosis | 12 | 1738 | Risk Ratio (M‐H, Random, 95% CI) | 0.92 [0.74, 1.16] |
| 6 Alcoholic hepatitis without or with hepatorenal syndrome | 10 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 With hepatorenal syndrome | 8 | 1382 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.85, 1.17] |
| 6.2 Without hepatorenal syndrome | 2 | 129 | Risk Ratio (M‐H, Random, 95% CI) | 0.56 [0.05, 6.49] |
| 7 Alcoholic hepatitis without or with ascites | 14 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 With ascites | 13 | 729 | Risk Ratio (M‐H, Random, 95% CI) | 0.82 [0.60, 1.12] |
| 7.2 Unclear if they had ascites | 1 | 29 | Risk Ratio (M‐H, Random, 95% CI) | 2.83 [0.61, 13.06] |
2.2. Analysis.
Comparison 2 Subgroup analysis: all‐cause mortality up to 3 months' follow‐up after randomisation, Outcome 2 Trials without for‐profit funding compared to trials at risk of for‐profit funding.
Comparison 3. Sensitivity analysis: all‐cause mortality.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Best‐worst scenario all‐cause mortality up to 3 months' follow‐up after randomisation | 15 | 1861 | Risk Ratio (M‐H, Random, 95% CI) | 0.82 [0.64, 1.05] |
| 2 Worst‐best scenario analysis: all‐cause mortality up to 3 months' follow‐up after randomisation | 15 | 1861 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.21 [1.06, 1.37] |
Comparison 4. Sensitivity analysis: serious adverse events.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Best‐worse scenario of serious adverse events during treatment | 15 | 1861 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.83, 1.21] |
| 2 Worst‐best scenario of serious adverse events during treatment | 15 | 1861 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.18 [1.05, 1.31] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Blitzer 1977.
| Methods | Prospective, double‐blind randomised trial Country: USA Dates: 1971–1973 Intention‐to‐treat analysis: no Sample size calculation: not reported |
|
| Participants |
Demographic characteristics Age (mean): prednisolone group: 47.2 years; control group: 48.4 years Sex: 100% men Inclusion criteria and degree of severity People with alcoholic hepatitis meeting the following criteria after ≥ 5 days in hospital: recent history of heavy alcohol consumption (> 1 pint whiskey per day or alcoholic equivalent); hepatomegaly based on physical examination (palpable > 5 cm below the costal margin) or liver scan or both; total serum bilirubin > 5 mg/100 mL; and ≥ 2 abnormalities of AST > 100 Reitman‐Frankel units/mL, serum albumin concentration < 3 g/100 mL, or prothrombin time > 2 s greater than control value. Liver biopsies: performed whenever possible, but were not required for admission to the study. 14 biopsies proved alcoholic hepatitis. Neither positive PPD skin tests nor active tuberculosis excluded people from randomisation. No positive PPD skin tests, and 1 active tuberculosis continued to receive isoniazid and para‐aminosalicylic acid throughout the study. If serious life‐threatening infection present, patients' entry into study was postponed until it was eradicated. People with history of peptic ulcer, active peptic ulcer disease, or gastrointestinal bleeding were included. Severity of disease: not clearly described; however, participants probably had moderate‐to‐severe alcoholic hepatitis, since they presented people with alcoholic hepatitis who met the described criteria. Exclusion criteria People treated with adrenocorticosteroid in the 6 months prior to admission or who showed evidence of psychotic behaviour precluding their co‐operation Randomisation procedure Random, sealed‐envelope technique Number of participants randomised: 33 Prednisolone group: n = 17 Control group: n = 16 |
|
| Interventions |
Experimental group: oral prednisolone 10 mg 4 times a day for 14 days, 5 mg 4 times a day for 4 days, 2.5 mg 4 times a day for 4 days, and 2.5 mg twice a day for 4 days Control group: placebo tablets; same schedule as prednisolone group Additional interventions to the trial groups: participants encouraged to eat standard hospital 2600‐calorie diet and were offered supplements when caloric intake seemed inadequate. Low‐protein, low‐sodium, and other special diets used as clinical situation dictated. Duration of treatment: 26 days Follow‐up after randomisation: 9 weeks |
|
| Outcomes | Mortality Liver biochemistry Liver histology Adverse events |
|
| Notes | Quote: "There were no significant differences between them [participants] with respect to mean age, sex, race, duration of hospitalization prior to entry into the study, frequency of histologically proved cirrhosis, or to the histologic severity of the alcoholic hepatitis." Letter sent to authors in March 2000. No answer received. No further attempts were made as the trial was conducted between 1971 and 1973. 1 participant received placebo treatment during trial. At the end of the therapy, due to lack of improvement, the ward physician requested the code be broken. The participant received a 7‐day course of prednisolone. He died 17 days later; his death was included in the mortality data of the control group on an intention‐to‐treat basis. On the 26th day of treatment, 3 participants in control group and 1 in glucocorticosteroid group received the alternative medication on a double‐blind basis. Quote: "Both prednisolone and placebo tablets were kindly supplied by the Upjohn Co., Kalamazoo, Michigan." |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were assigned by random, sealed‐envelope technique to receive either placebo or steroid." |
| Allocation concealment (selection bias) | Low risk | Quote: "… sealed‐envelope technique to receive either placebo or steroid." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Only the pharmacist was aware of the type of therapy which any individual patient was receiving." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote: "5 participants, who had each received less than 5 days of therapy, were subsequently excluded from analysis. Of these, three had left the hospital against medical advice or withdrew from the study, and in two participants experimental therapy had been stopped following gastrointestinal haemorrhage. One bled after 4 days of therapy from a gastric varix and the other from an unknown site after three days of treatment. On breaking the code at the end of the investigation, it was learned that all five participants had been in the steroid group … Furthermore, the addition of two deaths among the five excluded participants …" 3/17 (9%) people in prednisolone group and 0/16 (0%) people in control group dropped out. |
| Selective reporting (reporting bias) | Low risk | No protocol available. However, all‐cause mortality, serious adverse events, and liver‐related mortality were reported. |
| Other bias | Low risk | None suspected |
Bories 1987.
| Methods | Randomised controlled trial Country: France Dates: 1979–1982 Intention‐to‐treat analysis: yes Sample size calculation: not reported. |
|
| Participants |
Demographic characteristics Age (mean): prednisolone group: 41 years (range 26 to 68 years); control group: 49 years (range 30 to 70 years) Sex: prednisolone group: 16 men and 8 women; control group: 11 men and 10 women Inclusion criteria and degree of severity Not stated clearly, but mean level of bilirubin ≥ 147 (SD 30.78) mmol/L Alcohol consumption: men: 155 (SD 46) g/day; women: 140 (SD 32) g/day Exclusion criteria 48 excluded due to infections (n = 45), diabetes (n = 2), and tuberculosis (n = 1) Randomisation procedure Random number table Number of participants randomised: 45 Prednisolone group: n = 24 Control group: n = 21 |
|
| Interventions |
Experimental group: oral prednisolone 40 mg/day Control group: no intervention Additional interventions to the trial groups: 1500 calories and protein 50 g/day. Encephalopathy treated with lactulose and neomycin. In case of infection, participants received antibiotics. Duration of treatment: 1 month Duration of follow‐up: 3 months after randomisation |
|
| Outcomes | Mortality Liver histology Adverse events |
|
| Notes | Letter sent to authors in March 2000. No answer received | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "By random number table." |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Not blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts |
| Selective reporting (reporting bias) | Low risk | No protocol available. However, all‐cause mortality, serious adverse events, and liver‐related mortality were reported. |
| Other bias | Low risk | None suspected |
Campra 1973.
| Methods | Prospective randomised control trial Country: USA Date: 1971 Intention‐to‐treat analysis: no Sample size calculation: not reported |
|
| Participants |
Demographic characteristics Age (mean): prednisolone group 43.1 (SD 11.1) years; control group 42.7 (SD 8.1) years Sex: prednisolone group: 40 (8%) men; control group: 35 (9%) men Inclusion criteria and degree of severity Clinical diagnosis of severe acute alcoholic liver disease, absence of contraindication to corticosteroids therapy, no history of liver disease. Liver biopsy not required for inclusion since some participants had prothrombin time < 50% of normal value. Severity of disease: no clear definition Exclusion criteria People with other known illness or illnesses Randomisation procedure Previously prepared sealed envelopes Number of participants randomised: 50 participants entered trial, but 5 subsequently withdrawn when additional data favoured another diagnosis. 45 analysed (see 'Risk of bias' table). Prednisolone group: n = 20 Control group: n = 25 |
|
| Interventions |
Experimental group: oral prednisone 0.5 mg/kg bodyweight daily for 3 weeks; then 0.25 mg/kg bodyweight daily for 3 weeks Control group: no intervention Additional interventions to the trial groups: vitamin supplements, folic acids; high calorie, high protein diet if tolerated. In people with encephalopathy, protein intake was reduced to 20 g or 40 g and neomycin 500 mg 4 times daily was given. In case of bleeding, vomiting, and extreme anorexia, people received 5% or 10% dextrose solutions. Duration of treatment: 6 weeks Duration of follow‐up: hospital stay after randomisation: prednisolone group: 42–92 days, mean 47 days; control group: 43–95 days, mean 48 days |
|
| Outcomes | Mortality Liver biochemistry Liver histology Adverse events |
|
| Notes | Letter sent to authors in March 2000. AG Redeker answered in January 2001 (see the 'Risk of bias' table) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "… using previously prepared sealed envelopes, patients were randomly allocated to one of the two treatment groups." |
| Allocation concealment (selection bias) | Low risk | Information obtained through personal communication with the authors in 2001 read: "they [envelopes] were never in the possession of the investigators, but were kept by the department secretary who opened them upon request." However, the publication reads: "using previously prepared sealed envelopes, patients were randomly allocated to one of the two treatment groups." |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "the trial was not double blind." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "… all statistical analyses and interpretation were done under supervision of Dr. John Weiner of the Department of Biostatics, University of Southern California School of Medicine." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "50 patients entered the trial, but five were subsequently withdrawn when additional data favoured another diagnosis. In one case (group 2), jaundice proved to be caused by hepatitis B … the patient died … 2 of these patients were in group 2, one patient in group 1; all survived. The fifth patient was removed from the trial when peptic ulcer was diagnosed after 15 days of prednisolone therapy." Total: prednisolone group: n = 22; control group: n = 28 |
| Selective reporting (reporting bias) | Low risk | No protocol available. However, all‐cause mortality, serious adverse events, and liver‐related mortality were reported. |
| Other bias | Low risk | Not suspected |
Carithers 1989.
| Methods | Randomised, multicentre, double‐blind, placebo‐controlled, clinical trial Country: USA Dates: 1979–1984 Intention‐to‐treat analysis: yes Sample size calculation: reported (calculated that 62 patients should be entered to have 95% chance of detecting a difference in survival between the 2 groups). |
|
| Participants |
Demographic characteristics Age (mean): methylprednisolone group: 43.1 (SD 2.0) years; control group: 44.4 (SD 1.7) years Sex: methylprednisolone group: 20 (57%) men; control group: 21 (68%) men Inclusion criteria and degree of severity History of long‐standing alcoholism and clinical features of alcoholic hepatitis evaluated by 1 principal investigator within 3 days of admission; clinical evidence of spontaneous hepatic encephalopathy (assessed using standard clinical criteria and present when correctable causes of encephalopathy had been excluded) or a discriminant function value > 32 or both; negative hepatitis B surface antigen within the first 3 days of hospitalisation; and no history of previous viral hepatitis Exclusion criteria Gastrointestinal haemorrhage requiring transfusions; diabetes requiring insulin administration; active infection requiring treatment; clinical and laboratory evidence of acute pancreatitis; history of recent head trauma; known prior heroin addiction; or pre‐existing chronic renal disease with a serum creatinine > 175 mmol/L Randomisation procedure Random code sequence generated and kept by an independent source (see 'Risk of bias' table) Number of participants randomised: 67 Prednisolone group: n = 36 (2 refused, 1 was excluded from analysis); 33 analysed Control group: n = 31 |
|
| Interventions |
Experimental group: methylprednisolone 32 mg/day (equivalent to 40 mg prednisolone). Single dose of 8 tablets of 4 mg each morning for 28 days. In participants unable to take oral medications, intravenous infusions of study drug administered daily (methylprednisolone sodium succinate (SoluMedrol) or identical placebo. After 4 weeks, 4 tablets administered daily for 1 week followed by 2 tablets daily for 1 week; then therapy discontinued. Control group: placebo; identical tablets Additional treatment: participants offered 3000‐caIorie diet. Protein (1–1.5 g/kg bodyweight) provided when no evidence of hepatic encephalopathy. Protein restricted to ≤ 20 g/day and lactulose therapy instituted in participants with signs of hepatic encephalopathy. Ascites managed with sodium restriction or by addition of spironolactone in participants who did not respond with diuresis within 5 days. Fluid intake restricted in participants with hyponatraemia. B‐compIex multivitamins and folic acid 1 mg given daily. Participants who developed tremulousness or delirium tremens received diazepam or oxazepam. Duration of treatment: 5 weeks; 28 days at 32 mg/day then 16 mg/day for 7 days Duration of follow‐up after randomisation: at discharge |
|
| Outcomes | Mortality Liver biochemistry Adverse events |
|
| Notes | Letter sent to authors in March 2000. No answer received | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "… Random sequences for drug or placebo were submitted to the Upjohn Company (Kalamazoo, Michigan), which provided methylprednisolone (Medrol) in 4‐mg tablets and identical placebo tablets as well as intravenous preparations of methylprednisolone sodium succinate (SoluMedrol) and placebo. A random code was prepared for each of the four participating institutions such that within each group of 10 patients, 5 would receive methylprednisolone and 5 placebo. The random code sequence was kept by an independent source." |
| Allocation concealment (selection bias) | Low risk | Quote: "the random code sequence was kept by an independent source." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "neither the principal investigators nor their associates were aware of which regimen patients received throughout the trial." |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: "Data obtained at initial evaluation and follow‐up were recorded on standardized data collection forms that were submitted to the statistical coordinating center at the end of each study … A study overview committee, chaired by Dr. Hyman Zimmerman, reviewed the ongoing results of the study on a yearly basis from reports generated by the statistical coordinating center, which had access to the randomisation codes." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 3% Quote: "One patient who received methylprednisolone was belatedly discovered to have had neither encephalopathy nor elevation of discriminant function sufficient to meet the entry criteria and was excluded from analysis. Of the remaining 66 patients, 64 remained in the hospital for the duration of the study. Two methylprednisolone recipients refused to continue in the study. The first patient signed out of the hospital after 10 days on the trial and was alive at the end of the study. The second patient was discharged at his insistence after 15 days on the trial and was given the study drug to take at home, but he never returned for follow‐up. His status at the end of the study was unknown. He was the only patient lost to follow‐up." Prednisolone group: 2/36; control group: 0/31 |
| Selective reporting (reporting bias) | Low risk | No protocol available. However, all‐cause mortality, serious adverse events, and liver‐related mortality were reported. |
| Other bias | Low risk | Not suspected |
De 2014.
| Methods | Randomised controlled clinical trial Country: Medical College and Hospital, Kolkata, India Dates: January 2010 to August 2012 Study protocol approved by the institutional ethical committee. |
|
| Participants |
Demographic characteristics Age (mean): pentoxifylline + prednisolone group: 42.73 (SD 0.43) years; pentoxifylline group: 41.33 (SD 7.81) years Sex: 100% men Inclusion criteria and degree of severity History of chronic alcohol intake > 50 g/day with clinical and biochemical features of severe alcoholic hepatitis (MDF score ≥ 32 and AST:ALT > 2:1 with absolute value of AST < 500 IU/L and ALT < 200 IU/L MELD score, GAHS, and Child‐Pugh score calculated for all included participants. Exclusion criteria Other potential aetiology of liver injury (acute or chronic viral hepatitis, autoimmune liver disease, Wilson's disease) even in the background of chronic alcohol intake, positive for HIV antibodies or history of abstinence from alcohol in the last month, infection, sepsis or spontaneous bacterial peritonitis, acute pancreatitis, gastrointestinal bleeding, hepatorenal syndrome or any other severe associated disease such as uncontrolled diabetes mellitus, systemic hypertension, heart failure, pulmonary disease or malignancy at the time of inclusion or in the previous 3 months. Randomisation procedure Computer‐generated randomisation table Number of participants randomised: 62 Pentoxifylline + prednisolone group: n = 31 (1 voluntary dropped out) Pentoxifylline group: n = 31 (1 vertigo and withdrew) |
|
| Interventions |
Experimental group: prednisolone (Wysolone, Wreath, Mumbai, India) 40 mg once daily for 4 weeks + pentoxifylline (Trental tablets, Sanofi Aventis, Mumbai, India) tablets 400 mg 3 times daily for first 4 weeks Control group: placebo tablet for 4 weeks + pentoxifylline 400 mg 3 times daily orally first 4 weeks Duration of treatment: 11 weeks (12 weeks in group 1 (the control) and 11 weeks in group 2 (the experimental)) Quote: "After the initial 4 weeks, the study was opened and the patients allocated to different groups were revealed. Patients in Group 1 (PTX [pentoxifylline]) who tolerated the drug well, continued to receive the medication at the same dose for the next 8 weeks and then stopped. After 4 weeks of initial therapy, the dose of prednisolone in Group 2 was tapered by 5 mg/week over a period of 7 weeks and then stopped and received PTX like Group 1 patients." (Thus, we could use only 3 months.) Duration of follow‐up: 12 months |
|
| Outcomes | Mortality Adverse events Morbidity |
|
| Notes | Quote: "One patient in Group 1 developed severe vertigo within 7 days after starting PTX, and one patient in Group 2 withdrew voluntarily from the study and hence they were excluded. A total of 60 patients, 30 in each group, were considered for the final analysis." Letter sent to SK Mandal 12 December 2016. No reply received Quote: "Prednisolone tablet (Wysolone, Wreath, Mumbai, India) and PTX (trental tablets, Sanofi Aventis, Mumbai, India" |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "The recruited patients were then divided into 2 groups by a computer generated randomization table." |
| Allocation concealment (selection bias) | Unclear risk | Unclear |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The investigator, who allocated the patients to the groups, administered the drugs and collected the clinical and laboratory data, as well as statisticians were all blinded regarding the nature of the pharmacotherapy." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "as well as statisticians were all blinded regarding the nature of the pharmacotherapy." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 1.6% Quote: "One patient in Group 1 developed severe vertigo within 7 days after starting PTX [pentoxifylline], and one patient in Group 2 withdrew voluntarily from the study and hence they were excluded." |
| Selective reporting (reporting bias) | Low risk | No protocol available. However, all‐cause mortality, serious adverse events, and liver‐related mortality were reported. |
| Other bias | Low risk | Not suspected |
Depew 1980.
| Methods | Randomised, double‐blind, controlled clinical trial Country: USA Dates: 1977–1979 Study approved by the Human Experimentation Committee of the John Wesley County Hospital. Intention‐to‐treat analysis: yes Sample size calculation: reported |
|
| Participants |
Demographic characteristics Age (mean): prednisolone group: 49.8 (SD 2.1) years; control group: 48.2 (SD 2.3) years Sex: prednisolone group: 10 (67%) men; control group: 6 (43%) men Hepatic encephalopathy: prednisolone group: 100%; control group: 100% Ascites: prednisolone group: 87%; control group: 92% Inclusion criteria and degree of severity Alcohol abusers from lower socioeconomic strata with a clinical diagnosis of severe alcoholic hepatitis manifested by hepatomegaly, leukocytosis, and a serum bilirubin > 5 mg/dL, spontaneous hepatic encephalopathy occurring in absence of gastrointestinal haemorrhage, sedation, diuretic usage, or major electrolyte disturbances. Exclusion criteria Quote: "Severe diabetes, active tuberculosis, and serious bacterial infection prevented participation in the trial" Liver biopsy was not required. Randomisation procedure Unclear as not described (see 'Risk of bias' table) Number of participants randomised: 28 Prednisolone group: n = 15 Control group: n = 13 |
|
| Interventions |
Prednisolone group: prednisolone 40 mg daily orally Control group: placebo Additional treatment: supportive measures were attention to fluid and electrolyte balance, multiple vitamin supplementation, and parenteral glucose administration when food intake was poor. Encephalopathy treated with catharsis, protein restriction, and oral neomycin Duration of treatment: 28 days followed by tapered withdrawal over the ensuing 14 days Duration of follow‐up: 6 weeks |
|
| Outcomes | Mortality Liver biochemistry Liver histology Adverse events |
|
| Notes | Letter sent to authors in March 2000. No answer received. No further attempts were made as the trial was conducted between 1977–1979. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described. Quote: "All patients fulfilling the criteria who gave informed consent were randomised into two treatment protocols" and "… to avoid introducing bias based on the presence of the hepatorenal syndrome, the randomisation procedure was stratified to distinguish those with a serum creatinine greater than 2.5 mg/dL." |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Neither the principal investigators nor the physicians attending the patients were aware of the identity of the coded drugs. Provision was made for breaking the code if serious complications developed which could be related to steroid therapy." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No withdrawals and dropouts |
| Selective reporting (reporting bias) | Low risk | No protocol available. However, all‐cause mortality, serious adverse events, and liver‐related mortality were reported. |
| Other bias | Low risk | None suspected |
Helman 1971.
| Methods | Randomised controlled trial Country: USA Intention‐to‐treat analysis: yes Sample size calculation: not reported |
|
| Participants |
Demographic characteristics Age (mean): 47.8 years (range 30–67 years) Sex: 12 men and 25 women Inclusion criteria and degree of severity Diagnosis of alcoholic hepatitis confirmed by percutaneous needle biopsy. 70% of participants had anaemia on admission attributed to folate deficiency, blood loss, alcoholism, and haemolysis Exclusion criteria Biopsy could not be obtained within the first week of hospitalisation, gastrointestinal bleeding requiring transfusion, or if PPD was positive. Authors reported that participants were classified into three groups, according to clinical severity of their disease. Quote: "Group I were severely ill – manifesting precoma or coma, group 2 were moderately ill, but no evidence of encephalopathy, group 3 were asymptomatic ambulatory patients." Randomisation procedure Random, double‐blind technique. Determined by hospital pharmacist, without informing physicians, nurses, or patients until completion of the study Number of participants randomised: 37, divided in 3 groups according to severity of disease Prednisolone group: n = 20 Control group: n = 17 |
|
| Interventions |
Experimental group: prednisolone 40 mg daily Control group: daily lactose placebo Additional intervention: bed rest, high‐protein (100 g) and high‐calorie diet (3000 kcal) when tolerated and vitamin supplementation including folic acid. Sodium restriction instituted and all participants with ascites and oedema were treated with diuretics. Duration of treatment: 6 weeks: 4 weeks and 2‐week period tapered Duration of follow‐up after randomisation: 4 months |
|
| Outcomes | Mortality Liver biochemistry Liver histology Adverse events |
|
| Notes | Quote: "tablets 40 mg of prednisolone daily or lactose placebo (provided by Upjohn Co, Kalamazoo, Mich)." Letter sent to authors in March 2000. No answer received |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described. Quote: "patients were selected by a random, double‐blind technique …" |
| Allocation concealment (selection bias) | Low risk | Quote: "Drug treatment was randomly determined by the hospital pharmacist, without informing physicians, nurses, or patients until completion of the study." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "… The treatment code was broken during the study in only one case because of a medical emergency." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No withdrawals or dropouts |
| Selective reporting (reporting bias) | High risk | No protocol available. However, all‐cause mortality and liver‐related mortality were reported. |
| Other bias | Low risk | |
Maddrey 1978.
| Methods | Randomised, double‐blind clinical trial with parallel‐group design (3 groups) Country: USA Intention‐to‐treat analysis: no Sample size calculation: not reported |
|
| Participants |
Demographic characteristics Age (mean): prednisolone group: 40 (SD 8.5) years; control group: 42.3 (SD 11.1) years Sex: prednisolone group: 12 (50%) men; control group: 23 (74%) men Inclusion criteria and degree of severity History of long‐standing and recent alcoholism referred to Liver Service (The Johns Hopkins Hospital). Percutaneous liver biopsy performed unless precluded by coagulation abnormalities. Alcoholic hepatitis defined histologically as an inflammatory hepatic disease with cell swelling and hydropic change, cell necrosis, and polymorphonuclear leukocytic infiltration. Exclusion criteria Active gastrointestinal haemorrhage, pancreatitis, history of peptic ulcer, active infection, presence of hepatitis B infection, or history of previous viral hepatitis. MDF. People had wedged hepatic venous pressure determination. Randomisation procedure Random drug sequences Number of participants randomised: 57 Participants randomised into 3 groups based on apparent severity of disease. Group A (moderately ill), serum bilirubin > 3 mg/dL; hepatomegaly; and clotting factors adequate to allow liver biopsy Group B (more severely ill), hyperbilirubinaemia and hepatomegaly as in A with additional presence of ascites or hepatic encephalopathy (or both), but coagulation studies adequate for liver biopsy Group C (severely ill), hyperbilirubinaemia and hepatomegaly as in A and B, with or without ascites or hepatic encephalopathy (or both), but coagulation abnormalities precluded liver biopsy. Prednisolone group: n = 25 Control group: n = 32 |
|
| Interventions |
Experimental group: prednisolone 40 mg/day orally; 8 × 5‐mg tablets every morning Control group: identical placebo tablets Additional interventions to the trial groups: offered 3000 calorie diet. Protein 1–1.5 g/kg provided for people with no evidence of hepatic encephalopathy. In people with encephalopathy, protein restriction to ≤ 20 g/day and lactulose therapy. Ascites managed with sodium restriction alone or with the addition of spironolactone in people who did not respond with diuresis in 5 days. All participants initially received thiamine 100 mg intramuscularly. B‐complex multivitamins and folic acid given daily Duration of treatment: 28–32 days Follow‐up: until discharge |
|
| Outcomes | Early mortality Complications of therapy Liver function and haematological tests Wedged hepatic venous pressure Factors associated with a fatal outcome Discriminant function analysis |
|
| Notes | Study supported by Research Grant AA00201 from the National Institute of Alcohol Abuse and Alcoholism of the National Institutes of Health, and by Grant RR‐35 from the Clinical Research Centers Program, United States Public Health Service.
Prednisolone and placebo tablets provided by the Division of Steroid Research, The Upjohn Company, Kalamazoo, Mich. However, no further details were provided. Letter sent to authors in March 2000. No answer received. No further attempts made. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomised for treatment within three groups based on apparent severity of disease. Random drug sequences were arranged within each group." |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Prednisolone (5 mg) or identical placebo tablets were given in a single dose of 8 pills each morning for 28 to 32 days. (Prednisolone (5 mg) and identical placebo tablets were provided by the Division of Steroid Research, The Upjohn Company, Kalamazoo, Mich.). The investigators were not aware of which regimen the patient was receiving until the completion of the study." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 3.5% dropped out or were withdrawn. Quote: "Two additional patients were removed from the study after randomisation. One patient who was randomised to the placebo group bled from oesophageal varices before receiving the study drug. He subsequently stopped bleeding and survived. Another patient had an episode of upper gastrointestinal haemorrhage presumably from oesophageal varices after receiving prednisolone for 9 days and the drug was stopped." Prednisolone group: 1/25; control group: 1/32 |
| Selective reporting (reporting bias) | Low risk | No protocol available. However, all‐cause mortality, serious adverse events, and liver‐related mortality were reported. |
| Other bias | Low risk | None suspected |
Mendenhall 1977.
| Methods | Prospective, randomised clinical trial (3 intervention groups) Country: USA Intention‐to‐treat analysis: not mentioned Sample size calculation: not reported |
|
| Participants |
Demographic characteristics Age: not reported Sex: not reported; most probably men as they came from V.A. (Veteran Affairs) Medical Centers Inclusion criteria and degree of severity History of daily ethanol ingestion > 100 g/day for ≥ 1 year; hepatomegaly (> 12 cm) and significant jaundice (bilirubin > 5 mg %). Liver biopsy obtained in about 70% of participants to confirm diagnosis Exclusion criteria Not described Randomisation procedure Not described, but mentioned that "regimens were chosen randomly and blinded so that neither physician nor patient was aware of the treatment modality." Number of participants randomised: 46 Prednisolone group: n = 12 (all severe alcoholic hepatitis) Control group: n = 17 (all severe alcoholic hepatitis) Oxandrolone group: n = 17 |
|
| Interventions |
Experimental group: prednisolone 60 mg/day × 5, then decreased over a 16‐day period Control group: placebo Experimental group 2: oxandrolone (not included in the review) Additional treatment: supportive care Duration of treatment: 21 days Duration of follow‐up: to discharge |
|
| Outcomes | Mortality Liver biochemistry |
|
| Notes | Letter sent to study authors in 2006. No answer received. Only published as an abstract |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "… regimens were chosen randomly." |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "blinded so that neither physician nor patient was aware of the treatment modality." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "one additional mortality withdrew from the study on the 8 day" (not mentioned from what group out of 50 participants 17 participants treated with oxandrolone |
| Selective reporting (reporting bias) | High risk | No protocol available. However, all‐cause mortality was reported. |
| Other bias | Low risk | None suspected |
Mendenhall 1984.
| Methods | Co‐operative, multicentre, randomised clinical trial (3 intervention groups) Country: USA Dates: 1980–1983 Intention‐to‐treat analysis: yes Sample size calculation: reported |
|
| Participants |
Demographic characteristics Age (mean): prednisolone group: 51.5 (SD 8.2) years; control group 50.4 (SD 9.2) years Sex: 100% men Inclusion criteria and degree of severity Men hospitalised at 6 Veterans Administration Medical Centers in whom diagnosis of moderate or severe alcoholic hepatitis was based on conventional clinical and laboratory changes of this disease. Histological confirmation not required, so severely ill people not excluded. Severity classified by degree of jaundice (bilirubin) and coagulopathy (prothrombin time) Exclusion criteria Conditions that contradicted corticosteroid therapy: severe infections, active peptic ulcer disease, or insulin‐dependent diabetes mellitus, or if they had taken corticosteroids within the preceding 3 months; positive test for hepatitis B surface antigen; clinical or historical evidence of recent parenteral drug abuse, intractable congestive heart failure, neoplasms that commonly metastasise to the liver, or non‐alcoholic liver diseases Randomisation procedure Assignment made by Coordinating Center (Hines, Ill) was balanced within each hospital, and according to disease severity Number of participants randomised: 178 (prednisolone and placebo) + 85 (n = oxandrolone) Prednisolone group: n = 90 (moderate 46, severe 44) Control group: n = 88 (moderate 45, severe 43) Oxandrolone group: 85 |
|
| Interventions | 132 participants with moderate disease and 131 with severe disease were randomly assigned to 1 of 3 treatments: prednisolone, oxandrolone, or placebo Experimental group: prednisolone Dose: 60 mg/day for 4 days; 40 mg/day for 4 days; 30 mg/day for 4 days; 20 mg/day for 4 days; 10 mg/day for 7 days; 5 mg/day for 7 final days Control group: placebo Experimental group 2: oxandrolone (not included in the review) Duration of treatment: 30 days When possible, participants were evaluated monthly at outpatient clinics. If alcoholic hepatitis recurred and required rehospitalisation, the person was reassigned to the same therapy for 30 days with his permission. Duration of follow‐up after randomisation: 1 year (350 days for prednisolone group) |
|
| Outcomes | Mortality Liver complications Liver biochemistry Adverse events |
|
| Notes | Matching placebos prepared by Upjohn Company and G.D. Searle and Company. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Treatment assignment was made by the Coordinating Center (Hines, Ill.). The random assignment of treatments was balanced within each hospital, as well as according to disease severity." |
| Allocation concealment (selection bias) | Low risk | Quote: "Treatment assignment were made by the Coordinating Center (Hines, Ill)." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Medication was packed into unit dose kits at the Veterans Administration Center Pharmacy(Albuquerque, N.M.). The patient, physician and the local hospital pharmacy had no knowledge of the specific medication in use." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 4.5% Quote: "Ten patients withdrew from the study before completing treatment (5 given placebo, 3 prednisolone). However, these patients were included in the outcome assessment." Prednisolone group: 3/90; control group: 5/88 Quote: "324 days … 37 patients were lost to follow‐up: 13 given placebo, 11 prednisolone." |
| Selective reporting (reporting bias) | High risk | No protocol available. However, all‐cause mortality and liver‐related mortality were reported. |
| Other bias | Low risk | None suspected |
Porter 1971.
| Methods | Prospective, double‐blind, controlled pilot trial Country: USA Intention‐to‐treat analysis: no Sample size calculation: not reported |
|
| Participants |
Demographic characteristics Age (mean): prednisolone group: 44.6 (SD 4.4) years; control group: 49.5 (SD 8.9) years; overall range 27–61 years) Sex: prednisolone group: 13 (64%) men; control group: 7 (67%) men Inclusion criteria and degree of severity History of recent heavy alcohol ingestion, serum bilirubin concentration ≥ 5 mg/100 mL, clinical and laboratory deterioration over the first 5 hospital days, striking lack of improvement in the patient's clinical and biochemical status over first 5 hospital days, or rapid marked deterioration in < 24 hours For admission to the study all three absolute criteria were required. In addition, ≥ 2 major criteria or 1 major and ≥ 4 minor criteria had to be met. Major criteria: liver biopsy showing alcoholic hepatitis; hepatic encephalopathy (including asterixis); persistent or progressive azotaemia not explained by another process; and total bilirubin levels > 20 mg/100 mL. Minor criteria: fever not obviously secondary to another process; white blood count > 12,000 not obviously secondary to another process; anorexia or nausea or vomiting; palpable hepatomegaly; palpable splenomegaly; oesophageal varices; spider angiomas, oedema or ascites; palmar erythema; and prothrombin time prolonged ≥ 3 seconds over control. The Australia antigen was absent from the serum of all 16 participants in whom it was sought. Before the trial, a percutaneous needle biopsy of the liver was performed if the prothrombin time was not prolonged >4 seconds over control and there was no clinical bleeding tendency. Exclusion criteria Active gastrointestinal bleeding, pancreatitis, radiological evidence of peptic‐ulcer disease, active or questionably active pulmonary tuberculosis, and potentially life‐threatening bacterial infection Randomisation procedure Number drawn from a pool Number of participants randomised: 23 (20 analysed). 23 accepted to participate, but 3 died within 36 hours of start of therapy, and were excluded from analysis before code was broken, and did not receive adequate medication. Final series consisted of 20 participants Prednisolone group: n = 11 Control group: n = 9 |
|
| Interventions |
Prednisolone group: 6‐methyl‐prednisolone 50 mg (equivalent to prednisolone 50 mg, or hydrocortisone 200 mg) in 3 divided doses, parenterally for the first 10 days. If clinical improvement occurred over this interval and if nausea and vomiting were absent the drug was administered orally, and the dose gradually tapered (decreased every second day by 4 mg for the 11th to the 18th days, by 2 mg for the 19th to 30th days and every third day by 2 mg for the 31st to 45th days). If there was no clinical improvement within 10 days, the initial parenteral dose of 40 mg daily was continued until improvement or death occurred. Control group: placebo (lactose) Additional treatment: early in study only participants with a positive intermediate strength PPD test or a suspicious chest x‐ray were given isoniazid; however, later in study, all participants received isoniazid. Received general supportive care required in hepatic decompensation. Special attention given to fluid and electrolyte balance, prompt treatment of hepatic encephalopathy, and repeated evaluation for infection. Most participants had daily estimation of the caloric and protein intake by a hospital dietitian. People unable to take oral nutrition received glucose ≥ 400 calories/day parenterally Duration of treatment and of follow‐up: 45 days after randomisation |
|
| Outcomes | Mortality Liver biochemistry Liver histology Adverse events |
|
| Notes | Country: USA Letter sent to study authors in March 2000. No answer received. Quote: "Twenty‐three patients were accepted for studying. However, three died within 36 hours of the start of the therapy Quote: and were excluded from analysis before the code was broken because they did not receive adequate medication." Supported in part by a gastroenterology‐research training grant (AM‐05099) from the National Institute of Arthritis and Metabolic Diseases (a portion of this work was conducted within the Clinical Research Center of the University of Washington, with support by a grant MO1 FR‐37 from the National Institutes of Health. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "the case was randomised into one of the two treatment groups. Both the steroid (6‐methyl prednisolone, or Medrol) and the placebo (lactose) were packaged and coded by number in both parenteral and oral forms (prepared and supplied through the courtesy of the Upjohn Co, Kalamazoo, Mich) and randomisation was achieved by a number drawn from a pool. Neither patients nor physicians knew which form of treatment was used until the study had been completed, when the code was broken." |
| Allocation concealment (selection bias) | Low risk | Quote: "the case was randomised into one of the two treatment groups. Both the steroid (6‐methyl prednisolone, or Medrol) and the placebo (lactose) were packaged and coded by number in both parenteral and oral forms (prepared and supplied through the courtesy of the Upjohn Co, Kalamazoo, Mich) …" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Neither patients nor physicians knew which form of treatment was used until the study had been completed, when the code was broken." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "At the conclusion of the study, all needle biopsy and post‐mortem liver specimens were coded and read in blind review by the same observer." |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 13% Quote: "Twenty three patients were accepted for study, three died within 36 hours of the start of therapy and were excluded from analysis before the code was broken because they didn't receive adequate medication. The final series thus consisted of 20 patients." |
| Selective reporting (reporting bias) | Low risk | No protocol available. However, all‐cause mortality, serious adverse events, and liver‐related mortality were reported. |
| Other bias | Low risk | None suspected |
Ramond 1992.
| Methods | Randomised, double‐blind trial Country: France Dates: March 1987 to June 1990 Intention‐to‐treat analysis: yes Study approved by hospital ethics committees |
|
| Participants |
Demographic characteristics 124 people with alcohol dependence were admitted to 2 centres. Age (mean): prednisolone group: 48.1 (SD 1.3) years; control group: 48.2 (SD 1.6) years Sex: prednisolone group: 10 men; control group: 9 men Randomisation procedure Computer‐generated random code Inclusion criteria and degree of severity Biopsy‐confirmed alcoholic hepatitis (characterised by hyaline necrosis and infiltration of polymorphonuclear leukocytes) and spontaneous hepatic encephalopathy or a MDF > 32 (or both) 8 people died before inclusion in the trial. Exclusion criteria Gastrointestinal bleeding or bacterial infection excluded unless they could be effectively treated within 48 hours; presence of hepatitis B surface antigen; presence of HIV antibodies; refusal of liver biopsy; non‐alcoholic hepatitis at histology Number of participants randomised: 65 (4 excluded) (see 'Notes') Prednisolone group: n = 33 (32 analysed) Control group: n = 32 (29 analysed) |
|
| Interventions |
Prednisolone group: prednisolone (Solupred) 40 mg (prednisolone 40 mg equivalent methylprednisolone 32 mg) given in single dose of 2 tablets each morning for 28 days. If participant was unable to take oral medication, received intravenous infusions of prednisolone (Hydrocortancyl) Control group: single dose of 2 tablets Additional treatment: provided with 3000 kcal containing 1 g protein/kg. Participants with hepatic encephalopathy received lactulose therapy. Ascites managed with sodium restriction or by adding spironolactone to the treatment regimen. Received B complex multivitamins, folic acid, and antacids daily Duration of treatment: 28 days Duration of follow‐up: 8 weeks |
|
| Outcomes | Mortality Liver biochemistry Adverse events |
|
| Notes | Letter sent to study authors in March 2000. No answer received. Trial stopped at the first interim analysis after inclusion of 61 out of the planned 130 participants. Authors used an alpha error < 0.025. This is too high a value to prevent early stopping at a random high. Quote: "Drug therapy was interrupted by the attending physician if there was severe bacterial infection or gastrointestinal bleeding, or if a corticosteroid‐related complication was suspected … in patients with such complications the remaining tablets of the study drug were replaced with placebo tablets provided by the pharmacist (the only person who knew which regimen the patient had received first). The principal investigator and their associates were not aware of randomisation procedure or of the medication that the patients were receiving throughout the trial." Quote: "65 patients were randomly assigned, but 4 were excluded – one patient assigned to receive prednisolone was found to have anguilluliasis and her treatment was stopped one day after her inclusion in the study. Three patients assigned to placebo were found not to have satisfied the inclusion criteria. These 4 patients were alive at the end of treatment." Prednisolone tablets and placebo were provided by Laboratoire Houdé (Paris). |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "a random code was prepared by computer for each participating centre … There was a different code prepared for men and women in each center, so that within each group of six patients (male and female), three patients received prednisolone and three received placebo." |
| Allocation concealment (selection bias) | Low risk | Quote: "a random code was prepared by computer for each participating centre. Random sequences of drug or placebo were prepared by the pharmacist at each hospital." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "a random code was prepared by computer for each participating centre. Random sequences of drug or placebo were prepared by the pharmacist at each hospital." Quote: "prednisolone (Solupred) in 20 mg tablets and identical placebo were provided by the pharmacists at each hospital." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 1.5% 1 woman left the hospital and then "she was re hospitalised 56 days after enrolment and left again the following day. She was the only patient lost to follow up." Prednisolone group: 1/33; control group: 0/32 |
| Selective reporting (reporting bias) | Low risk | No protocol available. However, all‐cause mortality, serious adverse events, and liver‐related mortality were reported. |
| Other bias | Low risk | Not suspected |
Richardet 1993.
| Methods | Randomised clinical trial with a cross‐over design Country: France Intention‐to‐treat analysis: not mentioned Sample size calculation: not reported |
|
| Participants |
Demographic characteristics No information Inclusion criteria and degree of severity Non‐infected people with histologically confirmed alcoholic hepatitis. All participants had severe hepatic failure (prothrombin time < 50%, or bilirubin > 5.6 mg/dL, or encephalopathy) Randomisation procedure Not mentioned Number of participants randomised: 23 Glucocorticosteroid group: n = 12 Control group: n = 11 |
|
| Interventions |
Prednisolone group: prednisolone 40 mg daily Control group: placebo 40 mg daily Duration of treatment: Prednisolone group: 1 week of treatment followed by 1 week of no treatment Control group: 1 week of no treatment followed by 1 week of treatment After that, both groups received glucocorticosteroids for 3 weeks. Duration of follow‐up: at discharge from hospital (3 months) |
|
| Outcomes | Mortality Liver biochemistry |
|
| Notes | Letter sent to study authors in 2006. No answer received. Only published as abstract |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No information |
| Selective reporting (reporting bias) | Unclear risk | No information |
| Other bias | Unclear risk | No information |
Shumaker 1978.
| Methods | Prospective, double‐blind, randomised clinical trial Country: USA Intention‐to‐treat analysis: not mentioned, but presumably used Sample size calculation: not reported. |
|
| Participants |
Demographic characteristics Age (mean): prednisolone group/control group: 45,5/44,5 Sex: prednisolone group: 25% men; control group: 44% men Inclusion criteria and degree of severity History of recent heavy alcoholic ingestion, serum bilirubin > 5 mg %, hospitalisation for ≥ 5 days without improvement in liver tests; or rapid deterioration of the clinical condition during a 24‐hour period while under observation. In addition, minimum of 2 major criteria or 1 major and 2 minor criteria to be included. Major criteria: liver biopsy showing alcoholic hepatitis (with or without Mallory bodies), hepatic encephalopathy, azotaemia unexplained by another process (blood urea nitrogen > 20 mg % or creatinine > 1.5 mg %), hyperbilirubinaemia (> 20 mg %) and prothrombin time prolonged > 4 seconds over control; and unresponsive to parenteral administration of vitamin K. Minor criteria included fever not obviously secondary to another process, white blood count > 12,000, hepatomegaly (span > 14 cm), splenomegaly, or liver stigmas (spider telangiectasias, palmar erythema, ascites, oedema, etc.) Positive hepatitis B antigen did not exclude them from the study if a percutaneous liver biopsy confirmed alcoholic hepatitis. Exclusion criteria AST > 500 IU/L; active gastrointestinal bleeding; pancreatitis; x‐ray evidence of peptic ulcer disease; active or questionably active tuberculosis; active infection; or severe psychiatric disorder Randomisation procedure Predetermined code provided by the drug manufacturer Number of participants randomised: 27 Prednisolone group: n = 12 Control group: n = 15 |
|
| Interventions |
Prednisolone group: 6‐methylprednisolone 80 mg (equivalent to prednisolone 100 mg) for 4–7 days; medication was then tapered on a flexible schedule with cessation of therapy planned for 4 weeks unless death or complications Control group: placebo Additional interventions to the trial groups: both groups received comparable supportive care required in hepatic decompensation. All participants with positive tuberculin tests were treated with isoniazid 300 mg daily and pyridoxine 50 mg daily. Duration of treatment: 5 weeks; participants were placed on treatment for 4–7 days. Then the medication was tapered on flexible schedule with cessation of therapy planned for 4 weeks unless death or complication intervened. Duration of follow‐up: to hospital discharge |
|
| Outcomes | Mortality Liver histology Adverse events |
|
| Notes | Letter sent to study authors in March 2000. No answer received Quote: "The patient was then randomised into a predetermined code provided by the drug manufacturer. (Upjohn Co., Kalamazoo, MI, prepared and supplied the medication and placebo." |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "The patient was then randomised into a predetermined code provided by the drug manufacturer. Immediately prior to randomisation, patients were stratified into two categories based on the presence or abstinence of criteria permitting liver biopsy" the purpose of this procedure was to provide comparable case material for both steroid and placebo control groups.in the absence of other contradictions, patients with prothrombin times less than four seconds prolonged were placed in the "Biopsy feasible" group (n = 10) whether or not they agreed to a biopsy. All other patients constituted the "Biopsy‐ Disallowed" group (n = 17)." |
| Allocation concealment (selection bias) | Low risk | Quote: "The patient was then randomised into a predetermined code provided by the drug manufacturer." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind Quote: "80 mg of 6‐methylprednisolone or equivalent number of placebo tablets (or parenteral therapy of the same dosage intravenously if gastrointestinal function precluded oral intake." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Clinical evaluation was carried out by junior staff physicians blinded to treatment status of the patients." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 3.7 % Quote: "a steroid treated patient voluntarily withdrew from the study after eight days but was retained for statistical purposes." Prednisolone group: 1/12; control group: 0/15 |
| Selective reporting (reporting bias) | Low risk | No protocol available. However, all‐cause mortality, serious adverse events, and liver‐related mortality were reported. |
| Other bias | Low risk | None suspected |
Theodossi 1982.
| Methods | Randomised controlled trial Country: UK Intention‐to‐treat analysis: no Sample size calculation: not reported |
|
| Participants |
Demographic characteristics Age: not mentioned Sex: prednisolone group: 19 men/8 women; control group: 12 men/16 women Inclusion criteria and degree of severity Patients had to satisfy the following criteria: history of alcohol intake ≥ 80 g/day for ≥ 5 years; serum bilirubin > 80 pmol/L (normal up to 20 µmol/L); serum AST level ≥ 2 × upper limit of normal (normal up to 40 IU/L); prothrombin time prolonged by ≥ 9 seconds. Gastrointestinal bleeding, renal failure, and sepsis did not invalidate entry. Exclusion criteria Hepatoma and other diseases such as recent myocardial infarction, accompanying cerebrovascular accident including evidence of subdural haematoma, and active tuberculosis Randomisation procedure Random sealed envelope Number of participants: 60 (55 analysed). Referred from other hospitals because of the severity of their illness. 5 excluded from the analyses because of doubts in initial diagnosis Prednisolone group: n = 28 (analysed n = 27) Control group: n = 32 (analysed n = 28) |
|
| Interventions |
Prednisolone group: intravenous methylprednisolone 1 g daily (equivalent to 1.25 g prednisolone) for 3 days Control group: no intervention Additional treatment: participants who were too ill to take the standard hospital diet received a ≥ 2000 calories as intravenous 20% glucose. Encephalopathy treated with protein restriction (maximum of 20 g/day), lactulose (15–30 mL twice daily), and daily magnesium sulphate enemas Duration of treatment: 3 days Duration of follow‐up: little difference between groups in mean length of stay in hospital (prednisolone group: 24.2 days; control group: 28.1 days) |
|
| Outcomes | Mortality Liver biochemistry Adverse events |
|
| Notes | Letter sent to study authors in March 2000. No answer received Quote: "Of the 60 patients who satisfied the entry criteria, one in the treatment group and four in the control group were excluded from the final analysis because subsequent findings in four cases cast doubt on the initial diagnosis, and one patient was later found to have been given corticosteroids at the referring hospital. Thus there were 27 patients in the treatment and 28 in the control group." |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients … referred from other hospitals because of the severity of their illness. Patients were allocated by random sealed envelope technique to a control or treatment group …" |
| Allocation concealment (selection bias) | Low risk | Quote: "Patients were allocated by random sealed envelope technique to a control or treatment group, …" |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Not blinded Quote: "Patients were allocated by random sealed envelope technique to a control or treatment group, the latter receiving intravenous methylprednisolone 1 g daily for three days." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 8% Quote: "Of the 60 patients who satisfied the entry criteria, one in the treatment group and four in the control group were excluded from the final analysis because subsequent findings in four cases cast doubt on the initial diagnosis, and one patient was later found to have been given corticosteroids at the referring hospital. Thus, there were 27 patients in the treatment and 28 in the control group." Prednisolone group: 1/28; control group: 4/32 |
| Selective reporting (reporting bias) | Low risk | No protocol available. However, all‐cause mortality, serious adverse events, and liver‐related mortality were reported. |
| Other bias | Low risk | None suspected |
Thursz 2015.
| Methods | Multicentre, randomised trial with a 2 × 2 factorial design (09/MRE09/59). Country: UK (65 hospitals) Dates: January 2011 to February 2014 Intention‐to‐treat analysis: yes Sample size calculation: reported |
|
| Participants |
Demographic characteristics Age (mean): prednisolone plus placebo (n = 274) 49.3 ± 10.6); prednisolone plus pentoxifylline (n = 277) 48.6 ± 9.8; control group: placebo plus pentoxifylline (276) 47.9 ± 10.2; placebo plus placebo (276) 48.8 ± 10.3 Sex: glucocorticosteroid groups: 359 (65.6%) men; control groups: 326 (59.8%) men Hepatic encephalopathy: glucocorticosteroid groups: 152 (28%); control groups: 143 (26%) Inclusion criteria and degree of severity People abusing alcohol with a clinical diagnosis of severe alcoholic hepatitis manifested by hepatomegaly, leukocytosis, serum bilirubin > 5 mg/dL, spontaneous hepatic encephalopathy; aged ≥ 18 years; clinical diagnosis of alcoholic hepatitis; mean alcohol consumption > 80 g/day for men and > 60 g/day for women; serum bilirubin > 80 μmol/L (4.7 mg/dL); discriminant function ≥ 32 Exclusion criteria Jaundice for > 3 months; cessation of alcohol consumption for > 2 months before randomisation; presence of other causes of liver disease; serum AST > 500 IU/L or serum ALT > 300 IU/L; previous entry into the study within the preceding 6 months Randomisation procedure Web‐based computer system Number of participants randomised: 1103; data from 1053 were available for the primary end‐point analysis |
|
| Interventions | Participants randomised to 1 of 4 groups: pentoxifylline‐matched placebo + prednisolone‐matched placebo; pentoxifylline‐matched placebo + prednisolone; pentoxifylline + prednisolone‐matched placebo; or pentoxifylline + prednisolone Experimental groups: group 2 received prednisolone 40 mg daily + pentoxifylline‐matched placebo (n = 277); group 4 received prednisolone 40 mg daily + pentoxifylline 400 mg 3 times daily (n = 274) Control groups: group 1 received pentoxifylline‐matched placebo + prednisolone‐matched placebo (n = 276); group 3 received pentoxifylline 400 mg 3 times daily + prednisolone matched placebo (n = 276) Additional interventions to the trial groups: standard supportive care and nutritional support. Clinician made decision regarding other treatments, such as terlipressin for people with developing hepatorenal failure, acid suppression for prophylaxis against gastrointestinal haemorrhage, antibiotics, and vitamin supplementation. People with renal failure (defined as creatinine level > 500 μmol/L (> 5.7 mg/dL) or requirement for renal‐replacement therapy), active gastrointestinal bleeding, or untreated sepsis, and people requiring inotropic support with adrenaline or noradrenaline, were excluded unless the condition stabilised within the first 7 days after admission to hospital. Duration of treatment: 28 days Duration of follow‐up: 1 year |
|
| Outcomes | Mortality Adverse events Quality of life (using the EQ‐5D score registered to Eudra CT 2009‐013897‐42 and ISRCTN 88782125) |
|
| Notes | European Quality of Life – 5 Dimension – 5 Level Scale (EQ‐5D‐5L): self‐report, multiple choice questionnaire that provides a simple descriptive profile and a single index value for health status. Essentially consists of 2 pages: the EQ‐5D descriptive system (page 2) and the EQ VAS (page 3). The descriptive system comprises: mobility, self‐care, usual activities, pain/discomfort, and anxiety/depression. Each dimension has 5 levels: no problems, slight problems, moderate problems, severe problems, and extreme problems. The EQ VAS records the respondent's self‐rated health on a vertical, VAS. The EQ‐5D‐5L takes a few minutes to complete. A summary index with a maximum score of 1 can be derived from these 5 dimensions by conversion with a table of scores. The maximum score of 1 indicates the best health state, by contrast with the scores of individual questions, where higher scores indicate more severe or frequent problems. In addition, there is a VAS to indicate the general health status with 100 indicating the best health status. Study approved by the Multicenter Research Ethics Committee (reference number 09/MRE09/59), and clinical trial authorisation received from the Medicines and Healthcare Products Regulatory Agency (funded by the National Institute for Health Research Health Technology Assessment program; STOPAH EudraCT number, 2009‐013897‐42, and Current Controlled Trials number, ISRCTN88782125. Trial was conducted and reported according to the protocol, the Medicines for Human Use (Clinical Trials) Regulations 2004, as amended in 2006, the European Union Clinical Trials Directive (Directive 2001/20/EC) guidelines, the principles of the International Conference on Harmonisation Good Clinical Practice under the oversight of University Hospital Southampton NHS Foundation Trust, and the provisions of the Declaration of Helsinki. Letter sent to M Thursz 12 October 2016. No reply received yet |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "… A web‐based computer system (Tenalea, Forms‐Vision) was used to enrol eligible patients and randomly assign them to study groups. The randomization schedule was created with the use of Stata software, version 11 (StataCorp). Randomization was performed with a block size of four, with stratification according to geographic area and risk category. The high‐risk category consisted of patients who had an occurrence of gastrointestinal bleeding, renal impairment, or sepsis before randomisation. All other patients were assigned to the intermediate‐risk category." |
| Allocation concealment (selection bias) | Low risk | Quote: "The randomization schedule was created with the use of Stata software, version 11 (StataCorp). Randomization was performed with a block size of four, with stratification according to geographic area and risk category. Treatment allocation was blinded to site staff and the patient by providing each patient with a unique four‐digit patient pack number." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind Quote: "The treatment arm was also concealed to investigators and researchers. Only the study statisticians were unblinded and this was for analysis purposes only." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "An independent data monitoring and ethics committee, whose members were aware of the group assignments, was convened to review the conduct of the trial and to analyze primary end‐point data, using prespecified stopping guidelines, after the recruitment of 200, 400, and 800 patients, to avoid continued recruitment in the event that a definitive result had been achieved. Data collected by site investigative teams were submitted to the clinical trials unit and analysed by study statisticians. The first author wrote the first draft of the manuscript, with substantial contributions from the coauthors. All the authors vouch for the accuracy and completeness of the data and analyses." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Outcome data at the specific time points were reported. Quote: "At the time the trial was stopped, 33 patients who underwent randomization during the last 90 days of the trial could not be included in the 90‐day or 12‐month analyses. In addition, there were 159 patients who underwent randomization within 90 days to 12 months before the end of trial who could not be included in 12‐month analyses. The four groups were well matched with regard to their baseline characteristics, including laboratory values (See Table 1 in the published article). At 28 days, 16% of the patients had died, 1% had been lost to follow‐up, and 2% had withdrawn from the study. At 90 days, 29% of the patients (285 of 968 patients) had died, 5% had been lost to follow‐up, 3% had withdrawn, and 4% had not completed follow‐up owing to cessation of the study. At 1 year, 56% of the patients (421 of 747 patients) had died or undergone liver transplantation (the latter were 3 patients), 8% had been lost to follow‐up, 4% had withdrawn, and 20% had not completed follow‐up owing to cessation of the study due to limitations on funding." Quote: "Owing to limitations on funding, the trial was stopped after all enrolled patients had completed at least 28 days of follow‐up." Quote: "This project was funded by the National Institute for Health Research (NIHR) Health Technology Assessment programme. The NIHR Clinical Research Network provided research nurse support and the Imperial College Biomedical Research Centre also provided funding." |
| Selective reporting (reporting bias) | Low risk | Protocol was available, and data on all protocol outcomes such as all‐cause mortality, serious adverse events, liver‐related mortality, and quality of life were reported. |
| Other bias | Low risk | None suspected |
ALT: alanine aminotransferase; AST: aspartate aminotransferase; EQ‐5D: European Quality of Life‐5 dimensions; GAHS: Glasgow alcoholic hepatitis score; MDF: Maddrey's Discriminant Function; MELD: model for end‐stage liver disease; n: number of participants; PPD: purified protein derivative; SD: standard deviation; VAS: visual analogue scale.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Alvarez 2004 | Observational study (patient series). 13 participants with severe alcoholic hepatitis treated with systemic glucocorticosteroids and total enteral nutrition. |
| Cabré 2000 | Randomised trial of glucocorticosteroids versus nutrition in people with alcoholic hepatitis. Participants received oral or intravenous prednisolone or enteral nutrition (2000 kcal/day of a chemically defined polymeric enteral diet enriched in branched‐chain amino acids). |
| Christensen 1981 | Quasi‐randomised clinical study |
| Copenhagen 1969 | Meta‐analysis |
| Daures 1991 | Meta‐analysis |
| Dhanda 2016 | Prospective single‐centre cohort of people with severe alcoholic hepatitis treated with steroids; incidence and significance of infection. |
| Galambos 1984 | Reported in article through private contacts as part of Shumaker 1978. |
| Gill 1984 | Trial randomised 10 people with severe alcoholic hepatitis to prednisolone, testosterone, and amino acid supplement versus no intervention. |
| Goldis 2000 | Observational study (patient series); the authors used a control group from the same centre. |
| Hozo 1996 | Trial randomised people with alcoholic liver cirrhosis to glucocorticosteroids versus placebo. |
| Imperiale 1990 | Meta‐analysis |
| Lee 2016 | Review |
| Lesesne 1978 | Randomised trial of glucocorticosteroids versus nutrition in people with alcoholic hepatitis. Participants received glucocorticosteroids plus permission to eat as they wanted or a maximum of 600 kcal/day as intravenous glucose, while the control group received caloric supplements of at least 1600 kcal/day. |
| Mal 1991 | Abstract about influence of corticosteroids on the level of serum tumour necrosis factor concentrations |
| Mathurin 2018 | Trial randomised people with alcoholic hepatitis to receive selonsertib 18 mg versus placebo infliximab versus placebo. All participants received prednisone 40 mg orally. |
| Mendenhall 1993 | Chapter on alcoholic hepatitis in a book |
| Moreno 2014 | Multicentral study with 2 groups of comparison of intensive enteral nutrition with complete nutrition. Both groups received prednisolone. |
| Morris 2005 | Observational study (patient series) |
| Naganuma 2014 | Trial of granulocytapheresis and leukocytapheresis for the treatment of severe alcoholic hepatitis |
| Naveau 2004 | Trial randomised people with alcoholic hepatitis to receive infliximab versus placebo. All participants received prednisone. |
| Phillips 2001 | Trial randomised participants to antioxidants versus glucocorticosteroids. |
| Poynard 1991 | Meta‐analysis on alcoholic hepatitis |
| Reynolds 1989 | Narrative review on alcoholic hepatitis |
| Schlichting 1976 | Quasi‐randomised clinical study |
| Singal 2018 | Review |
| Spahr 2002 | Trial randomised people with alcoholic hepatitis to receive infliximab versus placebo. All participants received prednisone. |
| Stewart 2002 | Trial stratified participants by gender and glucocorticosteroid use, and then randomised participants to receive antioxidants versus placebo. |
| Szabo 2018 | Trial randomised people with alcoholic hepatitis to receive prednisolone 32 mg orally daily for 28 days versus a combination of anakinra + pentoxifylline + zinc orally |
| Tygstrup 1979 | Meta‐analysis |
Characteristics of ongoing studies [ordered by study ID]
NCT03160651.
| Trial name or title | Corticosteroids in severe alcoholic hepatitis patients with early spontaneous improvement |
| Methods | Interventional (clinical trial). Double‐blind randomised trial: Investigator, participants, and care providers will be masked. Only statisticians and pharmacist will not be masked. |
| Participants | Participants with alcoholic hepatitis: aged ≥ 18 years of either sex Inclusion criteria: clinical syndrome of alcoholic hepatitis; recent jaundice or in recent aggravation (< 3 months), serum bilirubin > 5 mg/dL, history of excess alcohol abuse (> 40 g/day); alcoholic hepatitis confirmed by liver biopsy (histological criteria of alcoholic hepatitis defined according to EASL clinical practice guidelines: steatosis, hepatocyte ballooning, and an inflammatory infiltrate with PMNs); spontaneous liver function improvement, defined by a decrease in Maddrey Discriminant Function and serum bilirubin > 10% between admission and day 7 after admission < 2 weeks since admission to hospital; Maddrey Discriminant Function ≥ 32; people must voluntarily sign and date an informed consent form, approved by an Institutional Review Board/Independent Ethics Committee prior to the initiation of any screening or study‐specific procedures; be able to understand and adhere to the study visit schedule and all other protocol requirements; people with significant hepatic encephalopathy will not be excluded. In this case, the person should be accompanied by a legal representative who will decide participation in the clinical study and sign informed consent form. Exclusion criteria: other causes of liver disease including viral hepatitis (positive hepatitis B surface antigen, HCV RNA positive), autoimmune hepatitis, biliary obstruction; other disease compromising 90‐day survival; positive HIV serology; uncontrolled infection. All participants will be screened for infection involving chest radiography, urinalysis, PMNs count in ascites (if ascites present). All other sign or clinical suspicion of infection with or without antibiotherapy will be recorded as an infection. Positive culture and initiation of antibiotics with clinical or radiological signs of infection, as well as clinical suspicion, will be recorded as infection. People with evidence of sepsis will be treated for a minimum of 2 days with appropriate antibiotics. Once the local principal investigator considers that the sepsis is under control, the person may be rescreened and randomised. Uncontrolled gastrointestinal bleeding judged as controlled for ≥ 5 days; serum creatinine > 2.5 mg/dL, under renal replacement therapy or under terlipressin (or other vasoactive drugs); pentoxyphilline therapy; pregnant or lactating women |
| Interventions | Parallel assignment of methylprednisolone or placebo Active comparator: methylprednisolone: methylprednisolone 32 mg/day for 28 days Placebo comparator: matching placebo for 28 days |
| Outcomes |
Primary outcomes Mortality at 90 days Secondary outcome Mortality at 28 days Incidence of infections during the study period (90 days) |
| Starting date | Estimated study start date: June 2017 |
| Contact information | Contact: Christophe Moreno, MD, PhD +32 2 5553714christophe.moreno@erasme.ulb.ac.be Contact: Françoise Smits, Nurse +32 2 5554478francoise.smits@erasme.ulb.ac.be Sponsors and collaborators: Erasme University Hospital Principal investigator: Christophe Moreno, MD, PhD; Erasme University Hospital |
| Notes | Estimated primary completion date: June 2020 Estimated study completion date: January 2021 |
EASL: European Association for the Study of the Liver; HCV: hepatitis C virus; MDF: Maddrey's Discriminant Function; PMN: polymorphonuclear neutrophil.
Differences between protocol and review
Review author team changed.
We removed the word 'alcohol' from the outcome "Alcohol liver‐related mortality up to three months' follow‐up after end of treatment" as it was superfluous.
-
Outcomes
All‐cause mortality is now better defined. Duration of treatment varied across the trials and also mortality data for up to three‐months' follow‐up. This is why we have modified all‐cause mortality to all‐cause mortality at the end of treatment, up to three months' follow‐up after randomisation, and one year following randomisation. Thus, our primary time point has become "all‐cause mortality up to three months' follow‐up after randomisation."
Trials also reported data on liver‐related mortality, any complication, and non‐serious adverse events up to three months' follow‐up after randomisation. Thus, three months' follow‐up after randomisation has also become our primary time point for the latter outcomes. However, serious adverse events were reported mostly during the treatment period.
Regarding exploratory outcomes, we created tables, as we did not have sufficient data for analysis.
Originally we wrote in the protocol that "We will consider trials published before or after 1989 carefully, as the Maddrey's score was modified in 1989 in order to stratify severe alcoholic hepatitis and define the group of people to be treated." However, it made also sense to use the definitions of the trialists for mild and severe alcoholic hepatitis and we wrote: "For studies not reporting the Maddrey's score, we used the classifications for mild and severe alcoholic hepatitis as provided by the trialists."
As we did not have trials at low risk of bias, we calculated the diversity‐adjusted required information size (DARIS) for our Trial Sequential Analysis using data from all included trials.
We calculated and reported the Trial Sequential Analysis‐adjusted CI as a supplement to the naive 95% CI.
We changed the risk of type I error from 2.5% (as originally planned based due to the three primary outcomes) into type I error of 1%, as we performed Trial Sequential Analysis on all primary and secondary outcomes, including post‐hoc time points.
Differences between previously published review version and this version
'Quality of evidence' was modified into 'certainty of evidence.'
As per current Cochrane recommendations, we were advised to keep in separate assessments of imprecision with Trial Sequential Analysis and GRADE in the 'Summary of findings' table. Therefore, we assessed imprecision with Trial Sequential Analysis and GRADE as sensitivity analysis.
Serious adverse events during treatment; liver‐related mortality up to three months following randomisation; and number of participants with any complications up to three months following randomisation in the GRADE 'Summary of findings' table.
As per current Cochrane recommendations, we were advised not to include 'for‐profit bias' risk domain in the overall bias risk assessment tool. Therefore, we removed the domain. We planned to perform a subgroup analysis with trials without for‐profit funding compared to trials at risk of for‐profit funding instead. However, only one trial seemed not be industry funded and the remaining trials did not clearly report on industry funding.
We performed a subgroup analysis on risk of bias (only one trial (Thursz 2015) fell into the group of trials at low risk of bias).
Contributions of authors
CP, DV, and GC: drafted the review. DN, ET, and CG: revised the review. CP and DV: are the guarantors of the review. All authors approved the review.
Sources of support
Internal sources
The Cochrane Hepato‐Biliary Group Editorial Team Office, Denmark.
External sources
No sources of support supplied
Declarations of interest
CP: none. DV: none. GC: none. ET: none. DN: none. CG: none.
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Blitzer 1977 {published data only}
- Blitzer BL, Mutchnick MG, Joshi PH, Phillips MM, Fessel JM, Conn HO. Adrenocorticosteroid therapy in alcoholic hepatitis. A prospective, double‐blind randomized study. American Journal of Digestive Diseases 1977;22(6):477‐84. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Blitzer BL, Mutchnick MG, Joshi PH, Phillips MM, Fessel JM, Conn HO. Adrenocorticosteroid therapy in alcoholic hepatitis. A prospective, double‐blind randomized study. Gastroenterology 1973;64:880. [DOI] [PubMed] [Google Scholar]
Bories 1987 {published data only}
- Bories P, Guedj JY, Mirouze D, Yousfi A, Michel H. Prednisolone treatment of alcoholic hepatitis: forty‐five patients [Traitement de l'hepatite alcoolique aigue par la prednisolone. Quarante‐cinq malades]. La Presse Medicale 1987;16:769‐72. [MEDLINE: ] [PubMed] [Google Scholar]
Campra 1973 {published data only}
- Campra JL, Hamlin EM Jr, Kirshbaum RJ, Olivier M, Redeker AG, Reynolds TB. Prednisone therapy of acute alcoholic hepatitis. Report of a controlled trial. Annals of Internal Medicine 1973;79(5):625‐31. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Carithers 1989 {published data only}
- Erratum: methylprednisolone therapy in patients with severe alcoholic hepatitis: randomized multicenter trial (American Journal of Gastroenterology, Vol. 85, No. 4 (473)). American Journal of Gastroenterology 1990; Vol. 85, issue 6:776. [CRS: 6800131000045704; EMBASE: 1990185566]
- Carithers RJ, Herlong HF, Diehl AM, Shaw EW, Combes B, Fallon HJ, et al. Corticosteroid therapy of alcoholic hepatitis: how many studies will it take?. Hepatology (Baltimore, Md.) 1990;12(3):619‐20. [CENTRAL: 844269; CRS: 6800100000024896; CN‐00844269] [DOI] [PubMed] [Google Scholar]
- Carithers RL Jr, Herlong HF, Diehl AM, Shaw EW, Combes B, Fallon HJ, et al. Methylprednisolone therapy in patients with severe alcoholic hepatitis. A randomized multicenter trial. Annals of Internal Medicine 1989;110:685‐90. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Maddrey WC, Carithers R Jr, Herlong HF, Combes B, Diehl AM, Shaw E, et al. Prednisolone therapy in patients with severe alcoholic hepatitis: results of a multicenter trial. Hepatology (Baltimore, Md.) 1986;6(5):1202. [Google Scholar]
De 2014 {published data only}
- De BK, Mandal SK, Sau D, Mani S, Chatterjee S, Mondal SS, et al. Pentoxifylline plus prednisolone versus pentoxifylline only for severe alcoholic hepatitis: a randomised controlled clinical trial. Annals of Medical and Health Sciences Research 2014;4(5):810‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Depew 1980 {published data only}
- Depew W, Boyer T, Omata M, Redeker A, Reynolds T. Double‐blind controlled trial of prednisolone therapy in patients with severe acute alcoholic hepatitis and spontaneous encephalopathy. Gastroenterology 1980;78(3):524‐9. [MEDLINE: ] [PubMed] [Google Scholar]
- Depew WT, Boyer TD, Omata M, Redeker AG, Reynolds TB. Double‐blind controlled trial of corticosteroid‐therapy in severe alcoholic hepatitis with encephalopathy. Clinical Research 1978;26(2):A150. [CRS: 6800131000045795] [Google Scholar]
Helman 1971 {published data only}
- Helman RA, Temko MH, Nye SW, Fallon HJ. Alcoholic hepatitis. Natural history and evaluation of prednisolone therapy. Annals of Internal Medicine 1971;74(3):311‐21. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Maddrey 1978 {published data only}
- Maddrey WC, Boitnott JK, Bedine MS. Corticosteroid treatment of alcoholic liver disease: a controlled trial. Gastroenterology 1977;72(5 II):A‐148. [CRS: 6800131000045726; EMBASE: 0978117751] [Google Scholar]
- Maddrey WC, Boitnott JK, Bedine MS, Weber FL Jr, Mezey E, White RI Jr. Corticosteroid therapy of alcoholic hepatitis. Gastroenterology 1978;75(2):193‐9. [MEDLINE: ] [PubMed] [Google Scholar]
Mendenhall 1977 {published data only}
- Mendenhall CL, Goldberg S. Risk factors and therapy in alcoholic hepatitis (AH). Gastroenterology 1977;72(5):1100. [Google Scholar]
Mendenhall 1984 {published data only}
- Mendenhall C, The Cooperative Study on Alcoholic Hepatitis. Survival after steroid treatment (T) for alcoholic hepatitis (AH). Hepatology (Baltimore, Md.) 1983;3(5):850. [CENTRAL: 221691; CRS: 6800100000005197; CN‐00221691] [Google Scholar]
- Mendenhall CL, Anderson S, Garcia‐Pont P, Goldberg S, Kiernan T, Seeff LB, et al. Short‐term and long‐term survival in patients with alcoholic hepatitis treated with oxandrolone and prednisolone. New England Journal of Medicine 1984;311(23):1464‐70. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Mendenhall CL, Roselle GA, Gartside P, Moritz T. Relationship of protein calorie malnutrition to alcoholic liver disease: a reexamination of data from two Veterans Administration Cooperative Studies. Alcoholism, Clinical and Experimental Research 1995;19(3):635‐41. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Porter 1971 {published data only}
- Porter HP, Simon FR, Pope CE II, Volwiler W, Fenster LF. Corticosteroid therapy in severe alcoholic hepatitis. A double‐blind drug trial. New England Journal of Medicine 1971;284(24):1350‐5. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Ramond 1992 {published data only}
- Mathurin P, Duchatelle V, Ramond MJ, Degott C, Bedossa P, Erlinger S, et al. Long term survival and prognostic factors in patients with severe biopsy‐proved alcoholic hepatitis (AH) treated by prednisolone: randomized trial, new cohort and simulation. Hepatology (Baltimore, Md.) 1994;20(4):319A. [Google Scholar]
- Mathurin P, Duchatelle V, Ramond MJ, Degott C, Bedossa P, Erlinger S, et al. Survival and prognostic factors in patients with severe alcoholic hepatitis treated with prednisolone. Gastroenterology 1996;110(6):1847‐53. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Ramond M‐J, Poynard T, Rueff B, Carey WD. Steroids in alcoholic hepatitis: another salvo of data. American Journal of Gastroenterology 1992;87(9):1219‐20. [CRS: 6800131000045543; EMBASE: 1992277170; CN‐00196579] [PubMed] [Google Scholar]
- Ramond MJ, Poynard T, Rueff B, Horwitz RJ. Prednisolone for severe alcoholic hepatitis. Annals of Internal Medicine 1992;117(Suppl 2):36. [CRS: 6800131000045702; EMBASE: 1992360747] [Google Scholar]
- Ramond MJ, Poynard T, Rueff B, Mathurin P, Theodore C, Chaput JC, et al. A randomized trial of prednisolone in patients with severe alcoholic hepatitis. New England Journal of Medicine 1992;326(8):507‐12. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Ramond MJ, Poynard T, Rueff B, Theodore C, Matthurin P, Chaput JC, et al. Prednisolone markedly improves short‐term survival of patients with severe biopsy‐proven alcoholic hepatitis A double blind randomized bicenter trial. Journal of Hepatology 1991;13(Suppl 2):S63. [CENTRAL: 222028; CRS: 6800100000005449; CN‐00222028] [Google Scholar]
Richardet 1993 {published data only}
- Mal F, Pham Huu T, Richardet JP, Roulot D, Labadie H, Pol S, et al. Corticosteroids (Cs) does not influence plasma tumor necrosis factor‐alpha (pTNF) and interleukin 6 (pIL6) concentrations in severe alcoholic hepatitis (AH): a cross over study. Hepatology (Baltimore, Md.) 1992;16:234A. [Google Scholar]
- Richardet JP, Dehoux M, Mal F, Roulot D, Labadie H, Pol S, et al. Influence of corticosteroids (CS) on plasma cytokines concentrations in patients with severe alcoholic hepatitis (HA): results of a randomized study. Journal of Hepatology 1993;18:S75. [Google Scholar]
Shumaker 1978 {published data only}
- Conn HO. Steroid treatment of alcoholic hepatitis. The yeas and the nays. Gastroenterology 1978;74(2 (Pt 1)):319‐22. [PUBMED: 620902] [PubMed] [Google Scholar]
- Galambos JT. Alcoholic liver disease, new aspects of an old problem. Schweizerische Medizinische Wochenschrift 1978;108(28):1050‐2. [CENTRAL: 221034; CRS: 6800100000004689; EMBASE: 0978366839; JC‐‐NLM: Journal ID:uei, 0404401; PUBMED: 78228296] [PubMed] [Google Scholar]
- Shumaker JB, Resnick RH, Galambos JT, Makopour H, Iber FL. A controlled trial of 6‐methylprednisolone in acute alcoholic hepatitis. With a note on published results in encephalopathic patients. American Journal of Gastroenterology 1978;69(4):443‐9. [MEDLINE: ] [PubMed] [Google Scholar]
Theodossi 1982 {published data only}
- Theodossi A, Eddleston AL, Williams R. Controlled trial of methylprednisolone therapy in severe acute alcoholic hepatitis. Gut 1982;23(1):75‐9. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Thursz 2015 {published data only}
- Forrest E, Mellor J, Stanton L, Bowers M, Ryder P, Austin A, et al. STeroids Or Pentoxifylline for Alcoholic Hepatitis (STOPAH): study protocol for a randomised controlled trial. Trials 2013;14(1):262. [CRS: 6800131000045566; EMBASE: 2013572163; CN‐00917636] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petts G, Lloyd K, Vergis N, Kudo H, Quaglia A, Forrest E, et al. The liver biopsy in alcoholic hepatitis: data from the STeroids Or Pentoxifylline in Alcoholic Hepatitis (STOPAH) clinical trial. Gut 2015;64(Suppl 1):A262. [CRS: 6800131000059987; EMBASE: 72009796] [Google Scholar]
- Petts G, Lloyd K, Vergis N, Kudo H, Quaglia A, Forrest E, et al. The liver biopsy in alcoholic hepatitis: data from the STeroids Or Pentoxifylline in Alcoholic Hepatitis (STOPAH) clinical trial. Journal of Pathology 2015;237(Suppl 1):S23. [CRS: 6800131000075013; EMBASE: 72082064] [Google Scholar]
- Petts G, Lloyd K, Vergis N, Kudo H, Quaglia A, Forrest E, et al. Utility of liver biopsy in alcoholic hepatitis: data from the STeroids Or Pentoxyfilline in Alcoholic Hepatitis (STOPAH) clinical trial. Journal of Hepatology 2015;62(Suppl 2):S776‐7. [CRS: 6800131000059992; EMBASE: 71937847] [Google Scholar]
- Thursz M, Forrest E, Roderick P, Day C, Austin A, O'Grady J, et al. The clinical effectiveness and cost‐effectiveness of STeroids Or Pentoxifylline for Alcoholic Hepatitis (STOPAH): a 2 x 2 factorial randomised controlled trial. Health Technology Assessment 2015;19(102):1‐138. [CRS: 6800131000075049] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thursz MR, Richardson P, Allison M, Austin A, Bowers M, Day CP, et al. Prednisolone or pentoxifylline for alcoholic hepatitis. New England Journal of Medicine 2015;372(17):1619‐28. [CRS: 6800131000075005; JC‐‐NLM: Journal ID:0255562, now; PUBMED: 25901427] [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Alvarez 2004 {published data only}
- Alvarez MA, Cabre E, Lorenzo‐Zuniga V, Montoliu S, Planas R, Gassull MA. Combining steroids with enteral nutrition: a better therapeutic strategy for severe alcoholic hepatitis? Results of a pilot study. European Journal of Gastroenterology & Hepatology 2004;16:1375‐80. [DOI] [PubMed] [Google Scholar]
Cabré 2000 {published data only}
- Cabré E, Rodríguez‐Iglesias P, Caballería J, Quer JC, Sánchez‐Lombraña JL, Parés A, et al. Short‐ and long‐term outcome of severe alcohol‐induced hepatitis treated with steroids or enteral nutrition: a multicenter randomized trial. Hepatology (Baltimore, Md.) 2000;32:36‐42. [DOI] [PubMed] [Google Scholar]
- Cabré E, on behalf of the Spanish Group for the Study of Alcoholic Hepatitis. Treatment of severe alcoholic hepatitis with steroids or total enteral nutrition: interim results of a prospective, randomized, multicentric trial. Gastroenterology 1998;114:A868‐9. [Google Scholar]
- Gassull M, Cabré E. Short and long‐term outcome in severe alcoholic hepatitis (SAH) treated with steroids or total enteral nutrition (TEN). A multicentric randomized controlled trial by the Spanish Group for the Study of Alcoholic Hepatitis. Journal of Parenteral and Enteral Nutrition 2000;24:S5. [Google Scholar]
Christensen 1981 {published data only}
- Christensen E, Fauerholdt L, Schlichting P, Juhl E, Poulsen H, Tygstrup N. Aspects of the natural history of gastrointestinal bleeding in cirrhosis and the effect of prednisone. Gastroenterology 1981;81(5):944‐52. [PubMed] [Google Scholar]
Copenhagen 1969 {published data only}
- Copenhagen Study Group for Liver Diseases. Effect of prednisone on the survival of patients with cirrhosis of the liver. A report from the Copenhagen Study Group for Liver Diseases. Lancet 1969;1(586):119‐21. [CENTRAL: 844264; CRS: 6800100000024872; PUBMED: 69076529] [PubMed] [Google Scholar]
Daures 1991 {published data only}
- Daures J‐P, Peray P, Bories P, Blanc P, Yousfi A, Michel H, et al. Steroid therapy in acute alcoholic hepatitis: a pooled estimate based on published randomized controlled trials [Place de la corticotherapie dans le traitement des hépatites alcooliques aiguës. Résultants d'une méta‐analyse]. Gastroenterologie Clinique et Biologique 1991;51:223‐8. [PubMed] [Google Scholar]
Dhanda 2016 {published data only}
- Dhanda A, Collins P. Infection is common in patients with severe alcoholic hepatitis treated with steroids but not associated with poor outcome. Journal of Hepatology 2016;64:S228. [Google Scholar]
Galambos 1984 {published data only}
- Galambos JT, Riepe SP. Use of colchicine and steroids in the treatment of alcoholic liver disease. Recent Developments in Alcoholism 1984;2:181‐94. [CENTRAL: 34655; CRS: 6800100000000618; EMBASE: 6374780; JC‐‐NLM: Journal ID:rda, 8301996; PUBMED: 84222884] [DOI] [PubMed] [Google Scholar]
Gill 1984 {published data only}
- Gill R, Zieve L, Logan G. Severe alcoholic hepatitis improved by combined treatment with prednisolone, testosterone and an amino acid supplement. Hepatology (Baltimore, Md.) 1984;4:2894. [Google Scholar]
Goldis 2000 {published data only}
- Goldis A, Matei R, Vernic C, Strain R. Treatment of acute alcoholic hepatitis with glucocorticosteroids‐prognostic factors. Steatohepatitis (NASH and ASH). Falk Symposium 121 2000;123:25. [Google Scholar]
Hozo 1996 {published data only}
- Hozo I, Mise S, Rumboldt Z, Bagatin J, Tonkic A. A controlled clinical trial of methylprednisolone in patients with the cholestatic form of alcoholic liver cirrhosis [Kontrolirano klinicko ispitivanje metilprednisolona u bolesnika sa kolestatskim oblikom alkoholne ciroze jetre]. Medicinski Arhiv 1996;50:81‐3. [PMID: 9601759] [PubMed] [Google Scholar]
- Hozo I, Mise S, Rumboldt Z, Bagatin J, Tonkic A. Controlled clinical trial of methylprednisolone in patients with the cholestatic form of alcoholic liver cirrhosis. Gastroenterology International 1997;10:137‐9. [PubMed] [Google Scholar]
Imperiale 1990 {published data only}
- Imperiale TF, McCullough AJ. Do corticosteroids reduce mortality from alcoholic hepatitis? A meta‐analysis of the randomized trials. Annals of Internal Medicine 1990;113(4):299‐307. [DOI] [PubMed] [Google Scholar]
Lee 2016 {published data only}
- Lee Y‐S, Kim HJ, Kim JH, Yoo YJ, Kim TS, Kang SH, et al. Systematic review: steroid, pentoxifylline or combined therapy for acute alcoholic hepatitis. Hepatology International 2016;10(Suppl 1):424. [Google Scholar]
Lesesne 1978 {published data only}
- Lesesne HR, Bozymski EM, Fallon HJ. Treatment of alcoholic hepatitis with encephalopathy. Comparison of prednisolone with caloric supplements. Gastroenterology 1978;74(2 Pt 1):169‐73. [MEDLINE: ] [PubMed] [Google Scholar]
Mal 1991 {published data only}
- Mal F, Huu TR, Roulot D, Pol S, Labadie H, Ricardet JP, et al. In severe alcoholic hepatitis (AH) plasma tumor necrosis factor (pTNF) is inconstantly elevated and is not influenced by corticosteroid (Cs) therapy. Hepatology (Baltimore, Md.) 1991;14(4 (Pt 2)):232A. [CENTRAL: 221590; CRS: 6800100000005112; CN‐00221590] [Google Scholar]
Mathurin 2018 {published data only}
- Mathurin P, Dufour JF, Bzowej NH, Shiffman ML, Arterburn S, Nguyen T, et al. Selonsertib in combination with prednisolone for the treatment of severe alcoholic hepatitis: a phase 2 randomized controlled trial. Hepatology 2018;68(1):8A‐9A. [Google Scholar]
Mendenhall 1993 {published data only}
- Mendenhall CL. Alcoholic hepatitis. In: Schiff L, Schiff ER editor(s). Diseases of the Liver. 7th Edition. Vol. 2, Philadelphia (PA): JB Lippincott Co, 1993:856‐74. [Google Scholar]
Moreno 2014 {published data only}
- Moreno C, Deltenre P, Senterre C, Louvet A, Gustot T, Bastens B, et al. Intensive enteral nutrition is ineffective for patients with severe alcoholic hepatitis treated with corticosteroids. Gastroenterology 2016;150:903‐10. [DOI] [PubMed] [Google Scholar]
- Moreno C, Trepo E, Louvet A, Degre D, Bastens B, Hittelet A, et al. Impact of intensive enteral nutrition in association with corticosteroids in the treatment of severe alcoholic hepatitis: a multicenter randomized controlled trial. Hepatology (Baltimore, Md.) 2014;60(Suppl 1):269A‐70A. [CRS: 6800131000024411; EMBASE: 71638823; CN‐01023754] [Google Scholar]
Morris 2005 {published data only}
- Morris JM, Forrest EH. Bilirubin response to corticosteroids in severe alcoholic hepatitis. European Journal of Gastroenterology & Hepatology 2005;17:759‐62. [PMID: 15947554] [DOI] [PubMed] [Google Scholar]
Naganuma 2014 {published data only}
- Naganuma A, Hoshino T, Ogashiwa T, Hayashi E, Uehara S, Miyamae N, et al. Pilot study of granulocytapheresis and leukocytapheresis for the treatment of severe alcoholic hepatitis. Hepatology International 2014;8(1 Suppl 1):S8. [CRS: 6800131000045556; CN‐01011068] [Google Scholar]
Naveau 2004 {published data only}
- Naveau S, Chollet‐Martin S, Dharancy S, Mathurin P, Jouet P, Piquet MA, et al. A double‐blind randomized controlled trial of infliximab associated with prednisolone in acute alcoholic hepatitis. Hepatology (Baltimore, Md.) 2004;39:1390‐7. [PMID: 15122768] [DOI] [PubMed] [Google Scholar]
Phillips 2001 {published data only}
- Philips M, Curtis H, Portmann B, Donaldson N, Bomford A, O'Grady J. Antioxidants versus corticosteroids in the treatment of severe alcoholic hepatitis: a randomized trial. Hepatology (Baltimore, Md.) 2001;34:250A. [DOI] [PubMed] [Google Scholar]
Poynard 1991 {published data only}
- Poynard T, Ramond MJ, Rueff B, Mathurin P, Chaput JC, Benhamou JP. Corticosteroids reduce mortality from alcoholic hepatitis in patients without encephalopathy. A meta‐analysis of the randomized trials (RCTs) including French trials. Hepatology (Baltimore, Md.) 1991;14:234A. [Google Scholar]
Reynolds 1989 {published data only}
- Reynolds TB, Benhamou JP, Blake J, Naccarato R, Orrego H. Treatment of acute alcoholic hepatitis. Gastroenterology International 1989;2(4):208‐16. [Google Scholar]
Schlichting 1976 {published data only}
- Schlichting P, Juhl E, Poulsen H, Winkel P. Alcoholic hepatitis superimposed on cirrhosis. Clinical significance and effect of long‐term prednisone treatment. Scandinavian Journal of Gastroenterology 1976;11:305‐12. [MEDLINE: ] [PubMed] [Google Scholar]
Singal 2018 {published data only}
- Singal AK, Bataller R, Ahn J, Kamath PS, Shah VH. ACG clinical guideline: alcoholic liver disease. American Journal of Gastroenterology 2018;113:175‐94. [DOI] [PMC free article] [PubMed] [Google Scholar]
Spahr 2002 {published data only}
- Spahr L, Rubbia‐Brandt L, Giostra E, Rougemont A‐L, Pugin J, Fischer M, et al. Combination of steroids with anti‐TNFa (INFLIXIMAB) or placebo in severe alcoholic hepatitis: early changes in ICAM‐1, histology and biochemical parameters. A pilot study. Journal of Hepatology 2002;37:448‐55. [PMID: 12217597] [DOI] [PubMed] [Google Scholar]
Stewart 2002 {published data only}
- Stewart S, Prince M, Bassendine M, Hudson M, James O, Jones D, et al. A trial of antioxidant therapy alone or with corticosteroids in acute alcoholic hepatitis. Journal of Hepatology 2002;36(Suppl 1):42. [DOI] [PubMed] [Google Scholar]
Szabo 2018 {published data only}
- Szabo G, Mitchell MC, McClain CJ, Dasarathy S, McCullough AJ, Nagy L, et al. IL‐1 receptor antagonist in combination with pentoxifylline and zinc for severe alcoholic hepatitis: a multicenter randomized double‐bind placebo‐controlled clinical trial. Hepatology 2018;68(1):1445A. [Google Scholar]
Tygstrup 1979 {published data only}
- Tygstrup N, Christensen E, Juhl E. Randomised clinical trials in hepatology [Randomisierte klinische therapiestudien in der hepatologie]. Internist 1979;20:565‐70. [PubMed] [Google Scholar]
References to ongoing studies
NCT03160651 {published data only}
- Corticosteroids in severe alcoholic hepatitis patients with early spontaneous improvement. Ongoing study Estimated study start date: June 2017.
Additional references
AASLD 2010
- O'Shea RS, Dasarathy S, McCullough AJ. Alcoholic liver disease. Hepatology (Baltimore, Md.) 2010;51:307‐28. [DOI] [PubMed] [Google Scholar]
Balshem 2011
- Balshem H, Helfand M, Schünemann HJ, Oxman AD, Kunz R, Brozek J, et al. GRADE guidelines: 3. Rating the quality of evidence. Journal of Clinical Epidemiology 2011;64(4):401‐6. [PUBMED: 21208779] [DOI] [PubMed] [Google Scholar]
Becker 1996
- Becker U, Deis A, Sørensen TI, Grønbaek M, Borch‐Johnsen K, Müller CF, et al. Prediction of risk of liver disease by alcohol intake, sex, and age: a prospective population study. Hepatology (Baltimore, Md.) 1996;23:1025‐9. [DOI] [PubMed] [Google Scholar]
Beckett 1961
- Beckett AG, Livingstone AV, Hill KR. Acute alcoholic hepatitis. British Medical Journal 1961;2:1113‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Begg 1994
- Begg CB, Mazumdar M. Operating characteristics of a rank correlation test for publication bias. Biometrics 1994;50(4):1088‐101. [PUBMED: 7786990] [PubMed] [Google Scholar]
Brok 2008
- Brok J, Thorlund K, Gluud C, Wetterslev J. Trial Sequential Analysis reveals insufficient information size and potentially false positive results in many meta‐analyses. Journal of Clinical Epidemiology 2008;61:763‐9. [DOI] [PubMed] [Google Scholar]
Brok 2009
- Brok J, Thorlund K, Wetterslev J, Gluud C. Apparently conclusive meta‐analyses may be inconclusive – Trial Sequential Analysis adjustment of random error risk due to repetitive testing of accumulating data in apparently conclusive neonatal meta‐analyses. International Journal of Epidemiology 2009;38(1):287‐98. [DOI] [PubMed] [Google Scholar]
Buzzetti 2017
- Buzzetti E, Kalafateli M, Thorburn D, Davidson BR, Thiele M, Gluud LL, et al. Pharmacological interventions for alcoholic liver disease (alcohol‐related liver disease). Cochrane Database of Systematic Reviews 2017, Issue 3. [DOI: 10.1002/14651858.CD011646.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Castellini 2017
- Castellini G, Nielsen E, Gluud, C. Comment on: "Cell therapy for heart disease: Trial Sequential Analyses of two Cochrane Reviews". Clinical Pharmacology and Therapeutics 2017;102:21‐4. [DOI] [PubMed] [Google Scholar]
Christensen 1995
- Christensen E, Gluud C. Glucocorticoids are ineffective in alcoholic hepatitis: a meta‐analysis adjusting for confounding variables. Gut 1995;37(1):113‐8. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Christensen 1999
- Christensen E, Gluud C. Glucocorticosteroids are not effective in alcoholic hepatitis. American Journal of Gastroenterology 1999;94(10):3065‐6. [DOI] [PubMed] [Google Scholar]
Deeks 2011
- Deeks JJ, Higgins JP, Altman DG. Chapter 9: Analysing data and undertaking meta‐analyses. In: Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
DeMets 1987
- DeMets DL. Methods for combining randomized clinical trials: strengths and limitations. Statistics in Medicine 1987;6(3):341‐50. [DOI] [PubMed] [Google Scholar]
DerSimonian 1986
- DerSimonian R, Laird N. Meta‐analysis in clinical trials. Controlled Clinical Trials 1986;7(3):177‐88. [DOI] [PubMed] [Google Scholar]
Dominguez 2008
- Dominguez M, Rincon D, Abraldes JG, Miquel R, Colmenero J, Bellot P, et al. A new scoring system for prognostic stratification of patients with alcoholic hepatitis. American Journal of Gastroenterology 2008;103:2747‐56. [DOI] [PubMed] [Google Scholar]
Dunn 2005
- Dunn W, Jamil LH, Brown LS, Wiesner RH, Kim WR, Menon KV, et al. MELD accurately predicts mortality in patients with alcoholic hepatitis. Hepatology (Baltimore, Md.) 2005;41:353‐8. [DOI] [PubMed] [Google Scholar]
EASL 2012
- European Association for the Study of the Liver. EASL Clinical Practical Guidelines: management of alcoholic liver disease. Journal of Hepatology 2012;57:399‐420. [DOI] [PubMed] [Google Scholar]
EASL 2018
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: management of alcohol‐related liver disease. Journal of Hepatology 2018;69:154‐81. [DOI] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Davey SG, Schneider M, Minder C. Bias in meta‐analysis detected by a simple, graphical test. BMJ (Clinical Research Ed.) 1997;315(7109):629‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ellis 2012
- Ellis EL, Mann DA. Clinical evidence for the regression of liver fibrosis. Journal of Hepatology 2012;56(5):1171‐80. [DOI] [PubMed] [Google Scholar]
Feinberg 2017
- Feinberg J, Nielsen EE, Korang SK, Halberg Engell K, Nielsen MS, Zhang K, et al. Nutrition support in hospitalised adults at nutritional risk. Cochrane Database of Systematic Reviews 2017, Issue 5. [DOI: 10.1002/14651858.CD011598.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Fisher 1922
- Fisher RA. On the interpretation of X^2 from contingency tables, and the calculation of P. Journal of the Royal Statistical Society 1922;85(1):87‐94. [Google Scholar]
Forrest 2005
- Forrest EH, Evans CD, Stewart S, Phillips M, Oo YH, McAvoy NC, et al. Analysis of factors predictive of mortality in alcoholic hepatitis and derivation and validation of the Glasgow alcoholic hepatitis score. Gut 2005;54:1174‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
French 1912
- French H. An index of differential diagnosis of main symptoms. Wright.Bristol 1912:368. [Google Scholar]
Garattini 2016
- Garattini S, Jakobsen JC, Wetterslev J, Bertelé V, Banzi R, Rath A, et al. Evidence‐based clinical practice: overview of threats to the validity of evidence and how to minimise them. European Journal of Internal Medicine 2016;32:13‐21. [DOI] [PubMed] [Google Scholar]
Gerber 1973
- Gerber MA, Orr W, Denk H, Schaffner F, Popper H. Hepatocellular hyalin in cholestasis and cirrhosis: its diagnostic significance. Gastroenterology 1973;64(1):89‐98. [PubMed] [Google Scholar]
Gluud 2017
- Gluud C, Nikolova D, Klingenberg SL. Cochrane Hepato‐Biliary Group. About Cochrane (Cochrane Review Groups (CRGs)) 2017, Issue 2. Art. No.: LIVER 2017.
GRADEpro GDT 2015 [Computer program]
- McMaster University (developed by Evidence Prime). GRADEpro GDT. Hamilton (ON): McMaster University (developed by Evidence Prime), 2015.
Guyatt 2011a
- Guyatt G, Oxman AD, Akl EA, Kunz R, Vist G, Brozek J, et al. GRADE guidelines: 1. Introduction – GRADE evidence profiles and summary of findings tables. Journal of Clinical Epidemiology 2011;64(4):383‐94. [PUBMED: 21195583] [DOI] [PubMed] [Google Scholar]
Guyatt 2011b
- Guyatt GH, Oxman AD, Kunz R, Atkins D, Brozek J, Vist G, et al. GRADE guidelines: 2. Framing the question and deciding on important outcomes. Journal of Clinical Epidemiology 2011;64(4):395‐400. [PUBMED: 21194891] [DOI] [PubMed] [Google Scholar]
Guyatt 2011c
- Guyatt GH, Oxman AD, Vist G, Kunz R, Brozek J, Alonso‐Coello P, et al. GRADE guidelines: 4. Rating the quality of evidence – study limitations (risk of bias). Journal of Clinical Epidemiology 2011;64(4):407‐15. [PUBMED: 21247734] [DOI] [PubMed] [Google Scholar]
Guyatt 2011d
- Guyatt GH, Oxman AD, Montori V, Vist G, Kunz R, Brozek J, et al. GRADE guidelines: 5. Rating the quality of evidence – publication bias. Journal of Clinical Epidemiology 2011;64(12):1277‐82. [PUBMED: 21802904] [DOI] [PubMed] [Google Scholar]
Guyatt 2011e
- Guyatt GH, Oxman AD, Kunz R, Brozek J, Alonso‐Coello P, Rind D, et al. GRADE guidelines 6. Rating the quality of evidence – imprecision. Journal of Clinical Epidemiology 2011;64(12):1283‐93. [PUBMED: 21839614] [DOI] [PubMed] [Google Scholar]
Guyatt 2011f
- Guyatt GH, Oxman AD, Kunz R, Woodcock J, Brozek J, Helfand M, et al. GRADE guidelines: 7. Rating the quality of evidence – inconsistency. Journal of Clinical Epidemiology 2011;64(12):1294‐302. [PUBMED: 21803546] [DOI] [PubMed] [Google Scholar]
Guyatt 2011g
- Guyatt GH, Oxman AD, Kunz R, Woodcock J, Brozek J, Helfand M, et al. GRADE guidelines: 8. Rating the quality of evidence – indirectness. Journal of Clinical Epidemiology 2011;64(12):1303‐10. [PUBMED: 21802903] [DOI] [PubMed] [Google Scholar]
Guyatt 2011h
- Guyatt GH, Oxman AD, Sultan S, Glasziou P, Akl EA, Alonso‐Coello P, et al. GRADE guidelines: 9. Rating up the quality of evidence. Journal of Clinical Epidemiology 2011;64(12):1311‐6. [PUBMED: 21802902] [DOI] [PubMed] [Google Scholar]
Guyatt 2013a
- Guyatt G, Oxman AD, Sultan S, Brozek J, Glasziou P, Alonso‐Coello P, et al. GRADE guidelines: 11. Making an overall rating of confidence in effect estimates for a single outcome and for all outcomes. Journal of Clinical Epidemiology 2013;66(2):151‐7. [PUBMED: 22542023] [DOI] [PubMed] [Google Scholar]
Guyatt 2013b
- Guyatt GH, Oxman AD, Santesso N, Helfand M, Vist G, Kunz R, et al. GRADE guidelines: 12. Preparing summary of findings tables – binary outcomes. Journal of Clinical Epidemiology 2013;66(2):158‐72. [PUBMED: 22609141] [DOI] [PubMed] [Google Scholar]
Guyatt 2013c
- Guyatt GH, Thorlund K, Oxman AD, Walter SD, Patrick D, Furukawa TA, et al. GRADE guidelines: 13. Preparing summary of findings tables and evidence profiles – continuous outcomes. Journal of Clinical Epidemiology 2013;66(2):173‐83. [PUBMED: 23116689] [DOI] [PubMed] [Google Scholar]
Guyatt 2017
- Guyatt GH, Ebrahim S, Alonso‐Coello P, Johnston BC, Mathioudakis AG, Briel M, et al. GRADE guidelines 17: assessing the risk of bias associated with missing participant outcome data in a body of evidence. Journal of Clinical Epidemiology 2017;87:14‐22. [PUBMED: 28529188] [DOI] [PubMed] [Google Scholar]
Harbord 2006
- Harbord RM, Egger M, Sterne JA. A modified test for small‐study effects in meta‐analyses of controlled trials with binary endpoints. Statistics in Medicine 2006;25(20):3443‐57. [DOI] [PubMed] [Google Scholar]
Higgins 2002
- Higgins JP, Thompson SG. Quantifying heterogeneity in a meta‐analysis. Statistics in Medicine 2002;21(11):1539‐58. [DOI] [PubMed] [Google Scholar]
Higgins 2011a
- Higgins JP, Deeks JJ. Chapter 7: Selecting studies and collecting data. In: Higgins JP, Green S, editor(s), Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Higgins 2011b
- Higgins JP, Altman DG, Sterne JA. Chapter 8: Assessing risk of bias in included studies. In: Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Higgins 2011c
- Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Houghton 2009
- Houghton M. Discovery of the hepatitis C virus. Liver International 2009;29(Suppl 1):82‐8. [DOI] [PubMed] [Google Scholar]
ICH‐GCP 1997
- International Conference on Harmonisation Expert Working Group. International conference on harmonisation of technical requirements for registration of pharmaceuticals for human use. ICH harmonised tripartite guideline. Guideline for good clinical practice CFR & ICH Guidelines. Vol. 1, Philadelphia (PA): Barnett International/PAREXEL, 1997. [Google Scholar]
Ioannidis 2009
- Ioannidis JP. Adverse events in randomized trials: neglected, restricted, distorted, and silenced. Archives of Internal Medicine 2009;169(19):1737‐9. [DOI] [PubMed] [Google Scholar]
Jakobsen 2014
- Jakobsen J, Wetterslev J, Winkel P, Lange T, Gluud C. Thresholds for statistical and clinical significance in systematic reviews with meta‐analytic methods. BMC Medical Research Methodology 2014;14:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
Jensen 1994
- Jensen K, Gluud C. The Mallory body: morphological, clinical and experimental studies (Part 1 of a literature survey). Hepatology (Baltimore, Md.) 1994;20:1061‐77. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Kjaergard 2001
- Kjaergard LL, Villumsen J, Gluud C. Reported methodologic quality and discrepancies between large and small randomized trials in meta‐analyses. Annals of Internal Medicine 2001;135(11):982‐9. [DOI] [PubMed] [Google Scholar]
Lefkowitch 2005
- Lefkowitch JH. Morphology of alcoholic liver disease. Clinics in Liver Disease 2005;9(1):37‐53. [DOI] [PubMed] [Google Scholar]
Lieber 1999
- Lieber CS. Role of S‐adenosyl‐L‐methionine in the treatment of liver diseases. Journal of Hepatology 1999;30:1155‐9. [DOI] [PubMed] [Google Scholar]
Louvet 2007
- Louvet A, Naveau S, Abdelnour M, Ramond MJ, Diaz E, Fartoux L. The Lille model: a new tool for therapeutic strategy in patients with severe alcoholic hepatitis treated with steroids. Hepatology (Baltimore, Md.) 2007;45(6):1348‐54. [DOI] [PubMed] [Google Scholar]
Louvet 2018
- Louvet A, Thursz M, Kim DJ, Labreuche J, Atkinson SR, Sidhu SS, et al. Corticosteroids reduce risk of death within 28 days for patients with severe alcoholic hepatitis, compared with pentoxifylline or placebo – a meta‐analysis of individual data from controlled trials. Gastroenterology 2018;155:458‐68. [DOI] [PubMed] [Google Scholar]
Lundh 2017
- Lundh A, Lexchin J, Mintzes B, Schroll JB, Bero L. Industry sponsorship and research outcome. Cochrane Database of Systematic Reviews 2017, Issue 2. [DOI: 10.1002/14651858.MR000033.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Mathurin 2011
- Mathurin P, O'Grady J, Carithers RL, Phillips M, Louvet A, Mendenhall CL, et al. Corticosteroids improve short‐term survival in patients with severe alcoholic hepatitis: meta‐analysis of individual patient data. Gut 2011;60(2):255‐60. [DOI] [PubMed] [Google Scholar]
Moher 1998
- Moher D, Pham B, Jones A, Cook DJ, Jadad AR, Moher M, et al. Does quality of reports of randomised trials affect estimates of intervention efficacy reported in meta‐analyses?. Lancet 1998;352(9128):609‐13. [DOI] [PubMed] [Google Scholar]
Mustafa 2013
- Mustafa RA, Santesso N, Brozek J, Akl EA, Walter SD, Norman G, et al. The GRADE approach is reproducible in assessing the quality of evidence of quantitative evidence syntheses. Journal of Clinical Epidemiology 2013;66(7):736‐42; quiz 742.e1‐5. [PUBMED: 23623694] [DOI] [PubMed] [Google Scholar]
NTAWG 2015
- Nordic Trial Alliance Working Group on Transparency and Registration. Transparency and registration in clinical research in the Nordic countries (Report), 2015. nta.nordforsk.org/projects/nta_transparency_report.pdf (accessed 21 June 2017).
Petrasek 2013
- Petrasek J, Iracheta‐Vellve A, Csak T, Satishchandran A, Kodys K, Kurt‐Jones EA, et al. STING‐IRF3 pathway links endoplasmic reticulum stress with hepatocyte apoptosis in early alcoholic liver disease. Proceedings of the National Academy of Sciences of the United States of America 2013;110(41):16544‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Phillips 2006
- Phillips M, Curtis H, Portmann B, Donaldson N, Bomford A, O'Grady J. Antioxidants versus corticosteroids in the treatment of severe alcoholic hepatitis – a randomised clinical trial. Journal of Hepatology 2006;44:784‐90. [DOI] [PubMed] [Google Scholar]
Pugh 1973
- Pugh RN, Murray‐Lyon IM, Dawson JL, Pietroni MC, Williams R. Transection of the oesophagus for bleeding oesophageal varices. British Journal of Surgery 1973;60:646‐9. [DOI] [PubMed] [Google Scholar]
Rambaldi 2006
- Rambaldi A, Gluud C. Anabolic‐androgenic steroids for alcoholic liver disease. Cochrane Database of Systematic Reviews 2006, Issue 4. [DOI: 10.1002/14651858.CD003045.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Rambaldi 2008
- Rambaldi A, Saconato HH, Christensen E, Thorlund K, Wetterslev J, Gluud C. Systematic review: glucocorticosteroids for alcoholic hepatitis – a Cochrane Hepato‐Biliary Group systematic review with meta‐analyses and trial sequential analyses of randomized clinical trials. Alimentary Pharmacology & Therapeutics 2008;27(12):1167‐78. [CRS: 6800131000005200; EMBASE: 2008243224; JC‐‐NLM: Journal ID:a5d, 8707234; PUBMED: 18363896] [DOI] [PubMed] [Google Scholar]
Review Manager 2014 [Computer program]
- Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager 5 (RevMan 5). Version 5.3. Copenhagen: Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Rhen 2005
- Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids – new mechanisms for old drugs. New England Journal of Medicine 2005;353(16):1711‐23. [DOI] [PubMed] [Google Scholar]
Royle 2003
- Royle P, Milne R. Literature searching for randomized controlled trials used in Cochrane Reviews: rapid versus exhaustive searches. International Journal of Technology Assessment in Health Care 2003;19(4):591‐603. [DOI] [PubMed] [Google Scholar]
Rücker 2008
- Rücker G, Schwarzer G, Carpenter J. Arcsine test for publication bias in meta‐analyses with binary outcomes. Statistics in Medicine 2008;27:746‐63. [DOI] [PubMed] [Google Scholar]
Savović 2012a
- Savović J, Jones HE, Altman DG, Harris RJ, Jüni P, Pildal J, et al. Influence of reported study design characteristics on intervention effect estimates from randomized, controlled trials. Health Technology Assessment 2012;16(35):1‐82. [DOI] [PubMed] [Google Scholar]
Savović 2012b
- Savović J, Jones HE, Altman DG, Harris RJ, Jüni P, Pildal J, et al. Influence of reported study design characteristics on intervention effect estimates from randomized, controlled trials. Annals of Internal Medicine 2012;157(6):429‐38. [DOI] [PubMed] [Google Scholar]
Schulz 1995
- Schulz KF, Chalmers I, Hayes, R, Altman DG. Empirical evidence of bias. Dimensions of methodological quality associated with estimates of treatment in controlled trials. JAMA 1995;273:408‐12. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Schäcke 2002
- Schäcke H, Döcke WD, Asadullah K. Mechanisms involved in the side effects of glucocorticoids. Pharmacology & Therapeutics 2002;96(1):23‐43. [DOI] [PubMed] [Google Scholar]
Spahr 2001
- Spahr L, Rubbia‐Brandt L, Pugin J, Giostra E, Frossard JL, Borisch B, et al. Rapid changes in alcoholic hepatitis histology under steroids: correlation with soluble intercellular adhesion molecule‐1 in hepatic venous blood. Journal of Hepatology 2001;35(5):582‐9. [DOI] [PubMed] [Google Scholar]
Sterne 2011
- Sterne JA, Egger M, Moher D. Chapter 10: Addressing reporting biases. In: Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions. Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Student 1908
- Student. The probable error of a mean. Biometrika 1908;6(1):1‐25. [Google Scholar]
Taïeb 2000
- Taïeb J, Mathurin P, Elbim C, Cluzel P, Arce‐Vicioso M, Bernard B, et al. Blood neutrophil functions and cytokine release in severe alcoholic hepatitis: effect of corticosteroids. Journal of Hepatology 2000;32(4):579‐86. [DOI] [PubMed] [Google Scholar]
Thorlund 2009
- Thorlund K, Devereaux PJ, Wetterslev J, Guyatt G, Ioannidis JP, Thabane L, et al. Can trial sequential monitoring boundaries reduce spurious inferences from meta‐analyses. International Journal of Epidemiology 2009;38(1):276‐86. [DOI] [PubMed] [Google Scholar]
Thorlund 2010
- Thorlund K, Anema A, Mills E. Interpreting meta‐analysis according to the adequacy of sample size. An example using isoniazid chemoprophylaxis for tuberculosis in purified protein derivative negative HIV‐infected individuals. Clinical Epidemiology 2010;2:57‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
Thorlund 2011
- Thorlund K, Engstrøm J, Wetterslev J, Brok J, Imberger G, Gluud C. User manual for Trial Sequential Analysis (TSA), 2011. ctu.dk/tsa/files/tsa_manual.pdf (accessed 21 June 2017).
TSA 2011 [Computer program]
- Copenhagen Trial Unit. TSA – Trial Sequential Analysis. Version 0.9 Beta. Copenhagen: Copenhagen Trial Unit, 2011.
Wetterslev 2008
- Wetterslev J, Thorlund K, Brok J, Gluud C. Trial Sequential Analysis may establish when firm evidence is reached in cumulative meta‐analysis. Journal of Clinical Epidemiology 2008;61(1):64‐75. [DOI] [PubMed] [Google Scholar]
Wetterslev 2009
- Wetterslev J, Thorlund K, Brok J, Gluud C. Estimating required information size by quantifying diversity in a random‐effects meta‐analysis. BMC Medical Research Methodology 2009;9:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wetterslev 2017
- Wetterslev J, Jakobsen JC, Gluud C. Trial Sequential Analysis in systematic reviews with meta‐analysis. BMC Medical Research Methodology 2017;17(1):39. [DOI] [PMC free article] [PubMed] [Google Scholar]
WHO 2010
- World Health Organization. Global strategy to reduce the harmful use of alcohol, 2010. www.who.int/substance_abuse/msbalcstragegy.pdf (accessed 21 June 2017).
WHO 2013
- Sauced RS. Update on background paper 6.14. Harmful use of alcohol, alcohol use, disorders, and alcoholic liver diseases, 2013. www.who.int/medicines/areas/priority_medicines/BP6_14Alcohol.pdf (accessed 21 June 2017).
Wood 2008
- Wood L, Egger M, Gluud LL, Schulz KF, Jüni P, Altman GD, et al. Empirical evidence of bias in treatment effect estimates in controlled trials with different interventions and outcomes: meta‐epidemiological study. BMJ (Clinical Research Ed.) 2008;336:601‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wu 1999
- Wu D, Cederbaum AI. Ethanol‐induced apoptosis to stable HepG2 cell lines expressing human cytochrome P‐4502E1. Alcoholism: Clinical and Experimental Research 1999;23:67‐76. [PubMed] [Google Scholar]
References to other published versions of this review
Pavlov 2016
- Pavlov CS, Tsochatzis E, Casazza G, Nikolova D, Volcek E, Gluud C. Glucocorticosteroids for people with alcoholic hepatitis. Cochrane Database of Systematic Reviews 2016, Issue 6. [DOI: 10.1002/14651858.CD001511.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Pavlov 2017
- Pavlov CS, Varganova DL, Casazza G, Tsochatzis E, Nikolova D, Gluud C. Glucocorticosteroids for people with alcoholic hepatitis. Cochrane Database of Systematic Reviews 2017, Issue 11. [DOI: 10.1002/14651858.CD001511.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Saconato 1999
- Saconato H, Gluud C, Christensen E, Atallah ÁN. Glucocorticosteroids for alcoholic hepatitis. Cochrane Database of Systematic Reviews 1999, Issue 1. [DOI: 10.1002/14651858.CD001511] [DOI] [Google Scholar]