

Abstract
Rationale:
Cardiac amyloidosis is a rare condition that is difficult to diagnose, because the clinical manifestations are often varied and nonspecific. The presence and degree of cardiac involvement are the main prognosis determinants, with a median survival of 6 months expected when presented with heart failure. Moreover, the optimal treatment for cardiac amyloidosis is still unclear.
Patient concerns:
We report a case of a 50-year-old man who was admitted with evolution of progressive dyspnea. Two months before the present admission, the patient was diagnosed with bacterial pneumonia complicated by bilateral parapneumonic effusion that required drainage.
Diagnosis:
Electrocardiography demonstrated poor R-wave progression in leads V1-V3 with right axis deviation and low voltage criteria. Echocardiography revealed diffuse left ventricular hypertrophy with normal ventricular cavity size, severe diastolic dysfunction, and sparkling and granular texture of the ventricle wall. Serum free light-chain analysis showed an altered kappa/lambda ratio of 0.01 with lambda light chains greatly elevated. A periumbilical fat aspirate sample confirmed amyloidosis. Bone marrow examination confirmed benign monoclonal gammopathy with 8.5% plasma cells, and biopsy stained for Congo red was negative.
Intervention:
A combination of bortezomib with cyclophosphamide and dexamethasone treatment was initiated.
Outcome:
Unfortunately, 5 days after the second therapy with bortezomib, the patient died.
Lessons:
Cardiac amyloidosis should be seriously considered in any adult with signs or nonspecific symptoms of cardiac distress, most notably congestive heart failure due to underlying restrictive cardiomyopathy.
Keywords: amyloidosis, cardiac amyloidosis, heart failure, heart involvement
1. Introduction
Amyloidosis is a rare systemic disease caused by extracellular deposition of proteinaceous fibrils with a characteristic β-pleated sheet structure that typically produces apple-green birefringence under polarized light after Congo red staining. There are several types of systemic amyloidosis, including immunoglobulin light chain (AL), serum amyloid A protein, familial hereditary mainly transthyretin mutation, and senile systemic amyloid.[1,2] The most common form of amyloidosis is the AL type.[3] Although almost every amyloidogenic protein has been associated with cases of cardiac amyloidosis, the actual prevalence of heart involvement varies among each type of amyloidosis.
Diagnosis of systemic amyloidosis is frequently delayed because the clinical manifestations are often varied and nonspecific. The presence and degree of cardiac involvement are the main prognosis determinants. For example, the median survival of individuals with AL amyloidosis who present with heart failure is 6 months. In this study, we report a 50-year-old man diagnosed with AL amyloidosis involving the heart, and aim to illustrate rapid progression of the disease and complicated and poor effects of treatment.
2. Case report
A 50-year-old man was admitted to our hospital for evolution of progressive dyspnea, paroxysmal nocturnal dyspnea, orthopnea and fatigue, and abdominal distension. These symptoms began 2 months earlier, when he was diagnosed at a different hospital with bacterial pneumonia complicated by bilateral parapneumonic effusion, which required drainage. His past medical history included bilateral lipoma, which was surgically treated 1 year prior to admission, and thyroiditis 3 years earlier. He reported regular moderate alcohol intake (100 g/wk) and that he had smoked 20 cigarettes a day for 30 years. The study protocol was approved by the ethics review board of the First Hospital of Jilin University (No. 2016-263). Informed written consent was obtained from the patient for publication of this case report and accompanying images.
Upon admission, his respiratory rate was 20 breaths/min, pulse was 102 beats/min, and blood pressure was 98/65 mm Hg. Bleeding points were scattered in the abdominal skin and hemorrhagic purpura were around the eye orbit (Fig. 1). Physical examination revealed jugular venous distention and abolition of bilateral basilar breath sounds based on pulmonary auscultation. There was no cardiac murmur; however, a third heart sound was heard.
Figure 1.
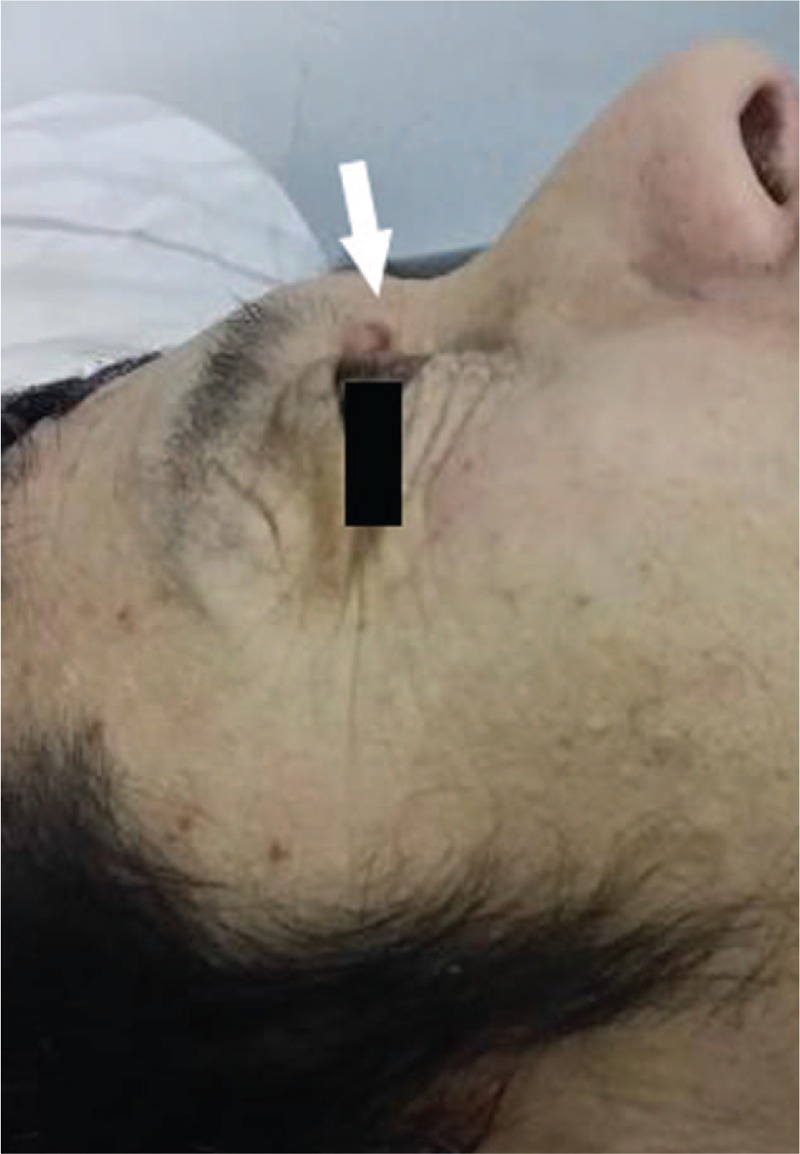
Hemorrhagic purpura was around the eye orbit.
An echocardiography test of the chest demonstrated small bilateral pleural effusions, 42 mm under the left 7th rib and 54 mm under the right 7th rib. Electrocardiography (ECG) demonstrated a sinus rhythm with a heart rate of 93 beats/min, poor R-wave progression in leads V1–V3 with right axis deviation, and low voltage criteria (Fig. 2). Echocardiography revealed diffuse left ventricular hypertrophy (septal and free wall thickness of 17 and 14 mm, respectively) with normal ventricular cavity size (diastolic and systolic chamber diameters of 45 and 23 mm, respectively). Enlarged atria (35 × 50 × 45 mm), and mild mitral and tricuspid regurgitation were observed. The left ventricle was interpreted to have severe diastolic dysfunction (mitral E wave velocity = 0.8 m/s, A wave = 0.27 m/s, medial E/e′ = 20), as well as mildly impaired systolic function with an impaired ejection fraction of 54% based on 2-dimensional echocardiography. The early diastolic transmitral filling velocity to atrial filling velocity (E/A) ratio increased to 4.25. The ventricle wall was characteristic sparkling and of granular texture (Fig. 3). The echocardiographic appearance of thickened ventricle accompanied with low QRS voltage in ECG was characteristic. These abnormalities were considered suggestive of amyloidosis. To further evaluate both myocardial and pericardial pathologies, cardiac magnetic resonance imaging (MRI) was performed on a 3.0-T MR scan system. The cardiac MRI showed diffuse, subendocardial delayed gadolinium enhancement (Fig. 4).
Figure 2.
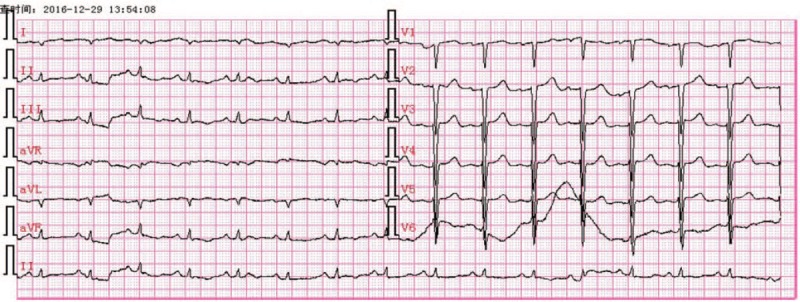
Electrocardiography demonstrated low voltage QRS predominantly in limb leads.
Figure 3.

Two-dimensional echocardiography revealed diffuse left ventricular hypertrophy (septal and free wall) with normal ventricular cavity size. The wall of ventricle was characteristic sparkling and granular texture. IVS = interventricular septum, LA = left atria, LV = left ventricle, PWLV = posterior wall.
Figure 4.
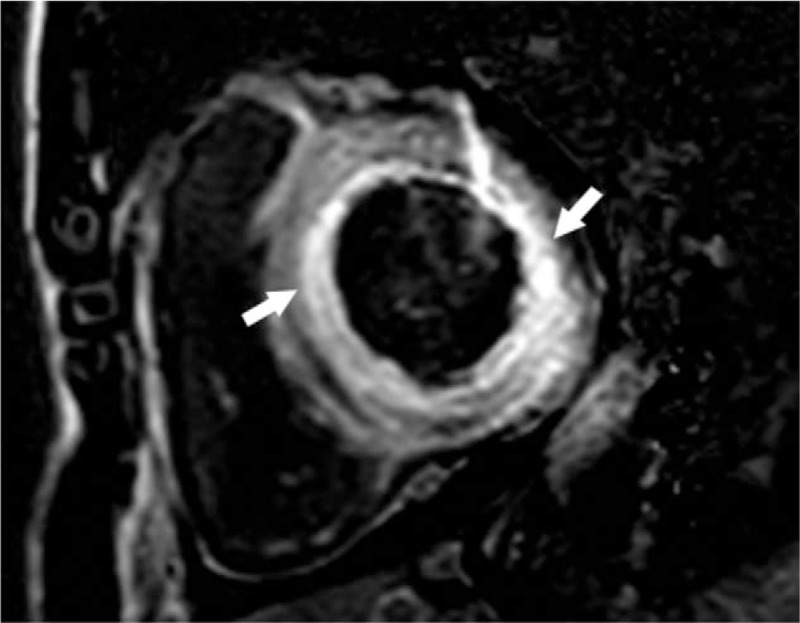
Cardiac magnetic resonance images acquired 15 minutes after administration of intravenous gadolinium contrast material showed diffuse subendocardial (arrowhead) enhancement of the left ventricular myocardium.
Additional testing revealed troponin I was mildly elevated at 0.14 ng/mL (normal 0–0.034 ng/mL). Plasma N-terminal brain natriuretic peptide (NT-proBNP) was elevated at 15,000 pg/mL (normal 0–400 pg/mL). Serum electrophoresis failed to detect monoclonal gammopathy in the serum, but immunoelectrophoresis revealed free lambda light chains. Serum free light–chain (FLC) analysis showed an altered kappa/lambda ratio of 0.01, with lambda light chains increased at 1240 mg/L (normal 8.3–27.0 mg/L). Bence-Jones protein was not detected in the urine. Bone marrow examination confirmed benign monoclonal gammopathy with 8.5% plasma cells, and biopsy stained for Congo red was negative. There was no evidence of multiple myeloma on skeletal survey. Autoantibody screening was normal. The glomerular filtration ratios were normal with daily proteinuria. A periumbilical fat aspirate sample confirmed amyloidosis. The patient was finally diagnosed with AL amyloidosis with cardiac and skin involvement, and in stage III using the Mayo 2012 staging system.
The patient's therapeutic plan was to perform 3 cycles of therapy with bortezomib (1.3 mg/sqm/d) and dexamethasone (20 mg/d) administered on the 1st, 8th, 15th, and 22nd days of each 3-week course. Cyclophosphamide was subsequently administered on the 2nd, 9th, 16th, and 23rd days (300 mg/d). Unfortunately, 5 days after the second cycle of therapy with bortezomib, the NT-proBNP level increased from 15,000 to 31,100 pg/mL, and troponin I increased from 0.14 to 0.463 ng/mL. The patient died because of a fulminant syndrome characterized by infiltrative pulmonary disease with hemorrhage, severe pulmonary infection, and heart failure.
3. Discussion
Cardiac amyloidosis is a rare condition that is difficult to diagnose, but should be seriously considered in any adult with signs or s nonspecific symptoms of cardiac problems, most notably congestive heart failure due to underlying restrictive cardiomyopathy. Our patient presented with initial symptoms of dyspnea and rapidly progressed to severe diastolic heart failure, which demonstrated systemic AL amyloidosis with cardiac and skin involvement that was refractory to combinatory drug treatment using bortezomib, dexamethasone, and cyclophosphamide.
The most common findings in ECG are low precordial and limb lead voltage and a pseudo infarction pattern. Although echocardiography usually reveals increased left and right ventricle wall thickness with greater echogenicity (granular sparking), the granular appearance of myocardium is not specific. However, low voltage in ECG and interventricular septal thickness of > 19.8 mm in echo, together, have a sensitivity of 72% and specificity of 91% for cardiac amyloidosis.[4] Also, ECG and echocardiography are very useful first-line examinations which are commonly used for heart, and also provide us with clues that lead to the diagnosis of amyloidosis in our patient.
Further cardiac MRI examination was made to recognize the granular appearance of myocardium and for differential diagnosis. The patient's cardiac MRI revealed a characteristic diffuse, subendocardial enhancement in delayed gadolinium enhancement images. Although cardiac MRI has advantages in noninvasive diagnosis of cardiomyopathy, it currently has a limited role in identifying early cardiac involvement in amyloidosis.
The diagnosis of suspected AL amyloidosis is made without consideration of specific organ involvement, and involves identification of amyloid typing and clonal disease.[5] Serum FLC analysis and serum and urine immunoelectrophoresis should be performed for all patients. The concentration of serum FLC can also be used to determine treatment strategies to achieve the fastest improvement.[6] After the above-indicated initial screening tests, bone marrow biopsy should be performed to determine whether plasma cell dyscrasia exists, and the percentage and type of λ- or κ-producing plasma cells.[7] In our case, urine sample immunofixation revealed a monoclonal peak of λ-FLC proteins, which is a hallmark of AL amyloidosis. Plasma cell dyscrasia was excluded by bone marrow biopsy.
Cardiac biomarkers are the most powerful predictors of prognosis and therapeutic response in AL amyloidosis,[8,9] and this has led to the current Mayo 2012 staging system[10] for this disease. The system is based on serum levels of NT-proBNP and cardiac troponin T and the concentration of circulating amyloidogenic FLC proteins.[10] This staging system assigns 1 point for each of the following: NT-proBNP ≥ 1800 pg/mL, troponin T ≥ 0.025 ng/mL, and a difference in serum FLC proteins ≥ 18 ng/dL; defining stages I (no points) to IV (3 points) with increasing risk of mortality. Our patient was in stage III, which hinted at a poor prognosis and therapeutic response.
Biopsy is the gold standard for diagnosis, and organ or other tissue biopsy specimens from patients with amyloidosis should demonstrate positive staining with Congo red dye and characteristic apple-green birefringence observed with polarized light.[11] Cardiac biopsy conclusively diagnoses cardiac AL amyloidosis; however, the invasive endocardial biopsy procedure is difficult to implement clinically. Biopsies of rectum and periumbilical fat aspiration are frequently used methods in the clinic for patients with suspected cardiac AL amyloidosis for less invasiveness and relatively high sensitivity and specificity.[12]
Therapeutic interventions for cardiac AL amyloidosis remain controversial. Initial management typically involves diuretics for symptomatic relief. Angiotensin-converting enzyme inhibitors and angiotensin II inhibitors are noted to be intolerant for profound hypotension. Definitive treatment is to stop production of the paraprotein responsible for amyloid formation. Bortezomib, which is a reversible proteasome inhibitor, is effective at killing clonal plasma cells and prompt reduction of circulating amyloidogenic monoclonal proteins in a majority of patients. Bortezomib combinations emerged as a front-line therapy in AL amyloidosis in phase II clinical trials.[13] Although no therapy has yet been proven successful for removing existing amyloid deposits, a combination of bortezomib with cyclophosphamide and dexamethasone (CyBorD) is associated with very high response rates and is the best studied in the literature. A recent study showed that approximately 60% of patients achieved a complete response or very good partial response after 230 patients were treated with CyBorD as a front-line therapy; however, just 17% achieved a cardiac response.[14] Response to treatment is predictive of outcome in cardiac AL amyloidosis and is measured both by hematologic parameters and cardiac parameters, primarily using NT-proBNP. Our patient, whose NT-proBNP level increased from 15,000 to 31,100 pg/mL during treatment, had a poor prognosis. Vigilantly monitoring the disease is, therefore, essential. Further studies are needed for effective strategies to remove existing amyloid deposits and develop targeted therapies to effectively improve prognosis.
Author contributions
Resources: Quan Liu.
Writing - Original Draft: Ming Gao.
Writing - Review & Editing: Liping Chen.
Footnotes
Abbreviations: AL = immunoglobulin light chain, CyBorD = bortezomib with cyclophosphamide and dexamethasone, ECG = electrocardiography, EF = ejection fraction, FLC = free light–chain, MRI = magnetic resonance imaging, NT-proBNP = N-terminal brain natriuretic peptide.
This work was supported by grant from Outstanding Young Talent Foundation of Jilin Province (No. 20190103085JH).
The authors have no conflicts of interest to disclose.
References
- [1].Blancas-Mejia LM, Ramirez-Alvarado M. Systemic amyloidoses. AnnuRev Biochem 2013;82:745–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Rocken C, Schwotzer EB, Linke RP, et al. The classification of amyloid deposits in clinicopathological practice. Histopathology 1996;29:325–35. [DOI] [PubMed] [Google Scholar]
- [3].Fikrle M, Paleček T, Kuchynka P, et al. Cardiac amyloidosis: a comprehensive review. Cor et Vasa 2013;55:e60–75. [Google Scholar]
- [4].Rahman JE, Helou EF, Gelzer-Bell R, et al. Noninvasive diagnosis of biopsy-proven cardiac amyloidosis. J Am Coll Cardiol 2004;43:410–5. [DOI] [PubMed] [Google Scholar]
- [5].Grogan M, Dispenzieri A, Gertz MA. Light-chain cardiac amyloidosis: strategies to promote early diagnosis and cardiac response. Heart 2017;103:1065–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Larsen J. Serum free light chain ratio as a biomarker for high-risk smoldering multiple myeloma. Leukemia 2013;27:941–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Sanchorawala V. Light-chain (AL) amyloidosis: diagnosis and treatment. Clin J Am Soc Nephrol 2006;1:1331–41. [DOI] [PubMed] [Google Scholar]
- [8].Palladini G, Merlini G. What is new in diagnosis and management of light chain amyloidosis? Blood 2016;128:159–68. [DOI] [PubMed] [Google Scholar]
- [9].Merlini G, Lousada I, Ando Y, et al. Rationale, application and clinical qualification for NT-proBNP as a surrogate end point in pivotal clinical trials in patients with AL amyloidosis. Leukemia 2016;30:1979–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kumar S, Dispenzieri A, Lacy MQ, et al. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J Clin Oncol 2012;30:989–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gameren IIV, Hazenberg BPC, Bijzet J, et al. Diagnostic accuracy of subcutaneous abdominal fat tissue aspiration for detecting systemic amyloidosis and its utility in clinical practice. Arthritis Rheum 2006;54:2015–21. [DOI] [PubMed] [Google Scholar]
- [12].Devata S, Hari P, Markelova N, et al. Detection of amyloid in abdominal fat pad aspirates in early amyloidosis: role of electron microscopy and Congo red stained cell block sections. Cytojournal 2011;8:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Laubach JP, Mitsiades CS, Roccaro AM, et al. Clinical challenges associated with bortezomib therapy in multiple myeloma and Waldenströms Macroglobulinemia. Leuk Lymphoma 2009;50:694–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Palladini G, Sachchithanantham S, Milani P, et al. A European collaborative study of cyclophosphamide, bortezomib, and dexamethasone in upfront treatment of systemic AL amyloidosis. Blood 2015;126:612–5. [DOI] [PubMed] [Google Scholar]