

Abstract
The constant threat of viral disease can be combated by the development of novel vaccines and therapeutics designed to disrupt key features of virus structure or infection cycle processes. Such development relies on high-resolution characterization of viruses and their dynamical behaviors, which are often challenging to obtain solely by experiment. In response, all-atom molecular dynamics simulations are widely leveraged to study the structural components of viruses, leading to some of the largest simulation endeavors undertaken to date. The present work reviews exemplary all-atom simulation work on viruses, as well as progress toward simulating entire virions.
Graphical Abstract:
Introduction
Viruses are a constant threat to human health, as well as the health of our domesticated animals and agriculture. Often, viral infections are challenging to treat due to limited therapeutic options. Although vaccines are effective at preventing or reducing the severity of viral disease, protective vaccines against fewer than 20 different viruses are currently available (WHO). Further, the high mutation rates of some viruses necessitate that vaccines be updated annually, or that new antiviral treatments be constantly realized to continue disease management in chronically infected individuals.
The rational development of novel prophylactic and therapeutic interventions against viral pathogens depends on detailed knowledge of virus structure, infection cycle processes, and the conformational dynamics that link structures to their functional roles in these processes. All-atom molecular dynamics (MD) simulations, often referred to as the “computational microscope,” remain the only method capable of elucidating the dynamical properties of biomolecules within their physiological environments at full chemical resolution [1]. Modern supercomputers and MD simulation codes enable researchers to investigate the nuances of virus structure and dynamics, even for very large components of virus structure [2].
Most all-atom MD simulation studies of viruses have focused on their protein-based components, particularly envelope proteins, viroporins, capsids, and accessory proteins or enzymes. Some work has leveraged MD simulations to construct complete atomistic models of viral proteins or their assemblies by integrating experimental data. Other work has taken advantage of MD simulations to characterize, under native conditions, the chemical-physical properties of such structures through analysis of their dynamics and influence on surrounding solvent. The overarching goals of virus simulation endeavors are to reveal critical aspects of virus structure or mechanistic insights into their function that can be targeted for novel disease treatments. Here, exemplary all-atom simulations of viruses are briefly reviewed, and progress toward simulating entire virions at atomistic detail is discussed.
Envelope Proteins
The first stage of viral infection is cell entry. Often, this process begins with adhesion of the virus to the host cell via receptor binding. In enveloped viruses, receptor binding is mediated by membrane-embedded surface proteins, which typically also represent the major antigens of the virus and may participate in virus-host membrane fusion. Other envelope proteins play a role in viral egress. The motivation to elucidate the molecular determinants of receptor recognition, as well as the antigenic and fusogenic properties of envelope proteins, have rendered them of great interest for study with MD simulations.
A broadly-studied viral envelope protein is the influenza hemagglutinin (HA) trimer (Fig. 1a), which is responsible for cellular adhesion and membrane fusion. Much work on HA has been aimed at understanding its specificity for host cell receptor glycans, which is an important factor in the avian-human transmission barrier [6, 7]. Characterization of receptor binding may support the development of adhesion-inhibiting drugs.
Figure 1:
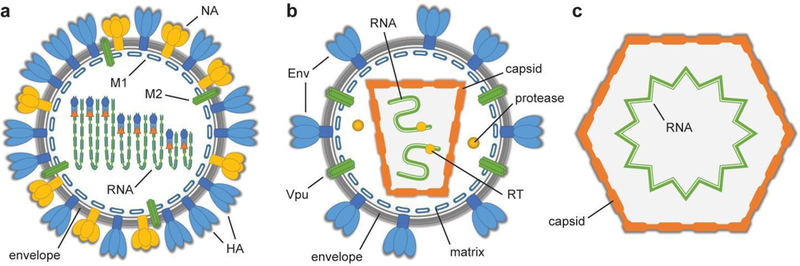
Schematic illustrations of viruses whose protein components have been heavily studied using all-atom MD simulations. a. Influenza A virus virion, average diameter of 120 nm [3]. Hemagglutinin (HA), neuraminidase (NA), and the matrix protein 2 viroporin (M2) are embedded within the viral envelope, which surrounds the matrix protein 1 (M1) assembly and encloses the RNA. b. Mature HIV-1 virion, average diameter of 145 nm [4]. The envelope protein (Env) and viroporin (Vpu) are embedded within the viral envelope, which surrounds the matrix protein assembly and encloses the RNA-containing capsid. The virus also carries copies of a protease and reverse transcriptase (RT). c. STMV virion, diameter of 17 nm [5]. STMV is a non-enveloped virus, composed only of the RNA-containing capsid.
All-atom MD simulations have been applied to numerous crystal complexes of HAs with receptor analogues to characterize binding mode dynamics, as well as to study recognition and specificity based on bound receptor conformations and interaction profiles [8, 9, 10, 11, 12]. Recent modeling and simulation work has also revealed that HA receptor specificity may depend on extended glycan structure and the ability to accommodate bidentate binding to biantennary glycans [13, 14]. MD simulations have been further employed to evaluate the effects of HA mutations that alter receptor binding and specificity [15, 16] and to study HA-antibody recognition and escape [17, 18].
Notably, HA is glycosylated, with N -glycans contributing around 20% of its molecular weight [6]. All-atom simulations of glycosylated HAs have been used to probe the influence of glycosylation on receptor binding [19], to provide insight into accessibility of glycan-remodeling enzymes [20], and to characterize interactions between HA and lung surfactant protein D, an innate immune lectin that neutralizes influenza A via attachment to HA glycans [21].
HA initiates fusion of the viral envelope and host membranes following cell adhesion and endocytosis, when endosomal acidification triggers it to undergo a conformational change. The mechanism of HA-mediated membrane fusion has been extensively studied with simulation approaches, including characterization of fusion peptide dynamics, interactions with the host membrane, and structural effects of reduced pH [22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32].
The secondary surface antigen of the influenza A virus is the neuraminidase (NA) tetramer (Fig. 1a), which cleaves host cell glycans to allow escape of progeny virions. Several compounds that mimic the native substrate of NA and bind to inhibit its action have been licensed for use as antiviral drugs for the treatment of influenza A (i.e., tamivir and zanamivir) [33]. The majority of simulation work on NA has focused on exploiting it as a drug target. In some cases, all-atom simulations have been employed to characterize the conformational dynamics of NA from the perspective of designing novel drug compounds or elucidating the determinants of ligand recognition and inhibition [34, 35, 36, 37]. In other cases, simulations of NA have been used as part of computational screening protocols to discover new druggable hot spots or inhibitor leads [38, 39, 40]. Additional studies have provided insights into the mechanisms of drug resistance [41, 42, 43], the role of calcium in NA stability and drug binding [44], and the structural basis for increased virulence of NA stalk-deletion mutants [45].
Another well-studied envelope protein is the human immunodeficiency virus type 1 (HIV-1) envelope (Env) trimer (Fig. 1b and 2d), which mediates cellular adhesion and membrane fusion. All-atom MD simulations have been employed to characterize the conformational properties of Env, particularly its V3 variable domain, a critical determinant of CD4 receptor binding [46, 47]. Other simulation work on Env has focused on identifying correlated motions that may underlie allosteric communication networks within the protein [48, 47, 49, 50]. The transmembrane domain and fusion peptide of Env have also been extensively investigated with MD simulations to explore conformational variability and membrane interactions, providing insights into the mechanism of membrane fusion [51, 52, 53, 54, 55, 56].
Figure 2:

Graphical size comparison of viral protein components. a. HIV-1 viroporin (Vpu), transmembrane domain. b. HIV-1 protease. c. HIV-1 reverse transcriptase (RT). d. HIV-1 envelope protein (Env) ectodomain. e. HIV-1 capsid. g. HIV-1 envelope membrane. g. STMV capsid.
Importantly, Env is heavily glycosylated, with N -glycans accounting for roughly 50% of its molecular weight [57]. These glycans form a shield around Env, cloaking it from the host immune system, and may also influence the binding affinity of CD4. The glycan shield is of great interest as a target for broadly-neutralizing antibody and vaccine design [57], and can only be accurately described with simulations that encompass chemical detail. Several all-atom MD studies have investigated the effects of glycosylation on the conformation and dynamics of Env variable domains V1, V2, and V3, as well as implications for receptor and antibody binding [58, 59, 60]. More recent work has used computational modeling and simulation to produce fully glycosylated Env trimers, with the goals of characterizing the dynamical behavior of the glycan shield and determining its impact on antibody elicitation, recognition, and binding [50, 61, 62, 63].
Beyond influenza A and HIV-1, all-atom MD simulations have also been applied to study the envelope proteins of several other viruses. Such work has included investigations of the effects of varying pH on the dengue virus [64, 65, 66, 67, 68, 69] and vesicular stomatitis virus (VSV) [70] envelope proteins, structure and host membrane interactions for the Ebola virus [71, 72, 73] and herpes simplex virus (HSV) [74] envelope protein fusion peptides, solution dynamics of human parainfluenza virus type 3 (HPIV-3) hemagglutinin-neuraminidase [75], and conformational flexibility in the hepatitis C virus (HCV) E2 envelope protein [76, 77].
Viroporins
Enveloped viruses can encode another class of membrane-embedded proteins, called viroporins, that play diverse roles in their respective viral infection cycles. Viroporins are small, hydrophobic proteins that oligomerize within the envelope to form hydrophilic channels, capable of transporting ionic species and small molecules [78]. The essential activities of these channels, their druggability, and potential as models to understand transport mechanisms within the human cell, have rendered them of long-standing interest for study with MD simulations.
The most widely-studied viroporin, from a computational perspective, is the influenza A matrix protein 2 (M2) ion channel (Fig. 1a), which transports protons across the viral envelope following endosomal acidification to prepare the particle for host membrane fusion and subsequent genome release [79]. All-atom MD simulations have been broadly employed to study M2 channel activation, gating, and proton permeation [80, 81, 82, 83, 84, 85, 86], as well as investigate the relationship between pH and channel conductance [87, 88] and internal water structure [89]. Adamantane drug compounds (i.e., amantadine and rimantidine) are known to inhibit M2 channel activity [79]. Additional all-atom MD simulation work on M2 has been aimed at characterizing drug binding and channel inhibition [84, 85, 90], probing the mechanisms of drug resistance [91, 92], and designing new inhibitors against drug-resistant mutants [93, 94].
Other viroporins that have been examined using all-atom MD simulations include the viral protein U (Vpu) of HIV-1 (Fig. 1b and 2a). Various early modeling and simulation studies of Vpu [95] led to evaluation of its preferred oligomeric state and all-atom investigation of its dynamics and channel activity [96]. All-atom simulations were also applied to develop a model for the Paramecium bursaria chlorella virus type 1 (PBCV-1) Kcv potassium channel to probe structure-function relationships [97, 98] and elucidate its mechanism of ion transport [99]. More recently, all-atom simulations were employed to determine the structure of the hepatitis C virus (HCV) p7 viroporin monomer [100] and to model various channel oligomer states, enabling characterization of their dynamics and conductance properties [101, 102, 103, 104, 105].
Capsids
Many viruses package their genome within a remarkable protein shell called a capsid. Often, capsids play key functional roles in delivering the viral genome to the host cell nucleus, rendering them of great interest as drug targets. Capsids generally represent the largest protein components of virus structures, and simulations of intact capsids have accounted for the most substantial all-atom virus simulations yet published [106, 2]. Work by Perilla et al. has clearly demonstrated the need for chemical detail to facilitate accurate simulation studies of virus capsids as drug targets [107].
The first all-atom simulation of a complete virus capsid was based on satellite tobacco mosaic virus (STMV, Fig. 1c and 2g) and encompassed a landmark particle count of 1 million atoms with solvent [108]. Both an empty and genome -containing capsid were investigated over 10–13 ns. With packaged RNA, the simulated capsid maintained structural integrity and exhibited only minor deviations from icosahedral symmetry. Without RNA, the simulated capsid rapidly broke symmetry and began to implode. The results of the work strongly suggest that RNA is responsible for nucleating assembly of STMV virions.
Prior to work on STMV, simulations of complete capsids were approximated using rotational symmetry boundary conditions, which exploit the icosahedral symmetry of capsid structures to model, at least in effect, a complete capsid using only the capsid asymmetric unit [106]. As time has progressed, all-atom simulations of intact capsids have become increasingly accessible. Notably, such work has included characterization of the stress-response of the southern bean mosaic virus (SBMV) capsid [109], validation of multi-scale modeling approaches based on the Sesbania mosaic virus (SeMV) capsid [110], derivation of a complete atomic model for the rabbit hemorrhagic disease virus (RHDV) capsid [111], and examination of the interplay between solvent and ions and the porcine circovirus type 2 (PCV2) capsid [112, 113].
The first all-atom simulation of a virus capsid to explore the microsecond timescale was based on an empty model of satellite tobacco necrosis virus (STNV), which encompassed 1.2 million atoms with solvent [114]. Like many non-enveloped plant viruses, STNV exhibits specific binding sites for divalent ions, likely calcium, and has been shown experimentally to undergo structural expansion unless these ions are bound. Simulations with and without bound calcium confirmed that the former maintain a smaller diameter and the latter swell. The results of the work produced an atomistic model for the swollen STNV capsid, which was not previously available, as well as an atomistic description of the dynamics underlying the swelling process.
The empty poliovirus capsid was studied over a timescale of 200 ns, based on a system of 6 million atoms with solvent [115]. Simulation results revealed that the capsid functions as a semipermeable membrane structure, translocating water molecules, but not ions, in equilibrium across its surface. Further, the solution pressure on the interior of the capsid was found to be negative, owing to electrostatic interaction of solution electrolytes with the charged capsid surface. The results of the work demonstrate that the capsid plays an essential role in maintaining an environment conducive to the stable accommodation of its RNA genome and associated counterions.
The empty hepatitis B virus (HBV) capsid was simulated for 1.1 µs based on a model of the complete assembly domain, derived from crystallography and molecular modeling [116]. The simulation encompassed 6 million atoms with solvent and revealed that the capsid undergoes notable asymmetric distortion at equilibrium. Further, the capsid exhibited a fivefold preference to translocate sodium over chloride through its triangular pores, and induced the localization of sodium along its interior surface. The results of the work implicate the triangular pores as the extrusion site of the capsid’s carboxy-terminal domain tails, which contain cellular signals, and suggest a mechanism by which the capsid could modulate signal exposure based on tail phosphorylation. Simulations have also been used to investigate the effects of small-molecule drugs on HBV capsid morphology [107] and to determine the structural basis for enhanced assembly and drug resistance in HBV capsid mutants [117].
An all-atom structure of the human immunodeficiency virus type 1 (HIV-1) capsid (Fig. 1b and 2e) was derived by combining results from cryo-EM and NMR experiments with computational modeling and data-guided MD simulations [118]. This landmark simulation endeavor, encompassing 64 million atoms with solvent, overcame the challenges of HIV-1 capsid size and asymmetric architecture to generate the first chemically complete model. The results of the study revealed key structural elements defining pentamer-hexamer and hexamer-hexamer interfaces within the capsid and produced a much-needed platform to enable investigation of capsid function and targeted drug development. Notably, the all-atom model has been used to study capsid interactions with the host cell factors cyclophilin A [119, 120] and MxB [121], which regulate and restrict HIV-1 viral infectivity, respectively.
In a subsequent study, the empty HIV-1 capsid model was simulated for 1.2 µs, setting the current published record for the most substantial all-atom simulation ever performed [122]. The simulation revealed that the capsid translocates chloride across its surface at twice the rate of sodium through ion-specific channels, which may relate to the translocation of nucleotides during reverse transcription. Further, the capsid exhibited complex dynamics, including collective motions that divide the structure into two hemispheres and suggest a mechanism for capsid uncoating, as well as oscillatory surface waves at four different frequencies that may underlie emergent properties of the capsid.
Immature Particle Lattices
Retroviruses such as HIV-1, human T-cell leukemia virus type 1 (HTLV-1), and Rous sarcoma virus (RSV) initially assemble as immature particles from Group-specific antigen (Gag) polyproteins. During maturation, proteolytic processing of Gag yields cleavage products that reassemble to produce mature virions [123]. All-atom simulations proved invaluable in determining the structures of the immature HTLV-1 [124] and RSV [125] Gag lattices, as well as the HIV-1 capsid protein-SP1 peptide maturation intermediate (CA-SP1) [126]. Notably, simulations revealed that the CA-SP1 six-helix bundle exists in a dynamic helix-coil equilibrium, and that maturation-inhibiting drugs and mutations act by stabilizing a helical form of the bundle [126]. MD studies of CA-SP1 bound to inositol phosphates showed that these small molecules facilitate the formation of the six-helix bundle and, thus, promote assembly of the Gag lattice [127]. Simulations of the RSV lattice model were used to determine the effects of charge distribution and mutations on Gag assembly [128].
Viral Enzymes
Some viruses encode and package enzymes necessary to carry out the processes that drive their infection cycles. All-atom MD simulations of the HIV-1 protease (Fig. 1b and 2b), which is responsible for proteolytic cleavage of Gag prior to maturation, revealed the motion of the enzyme’s active-site flaps, whose opening and closing are considered essential to function [129, 130, 131, 132, 133, 134]. Further, studies of the West Nile virus (WNV) NS3 protease identified a conformational selection mechanism used to identify its substrate [135]. Additionally, all-atom simulations were instrumental in elucidating the mechanism by which non-nucleoside reverse transcription inhibitors (NNRTIs), an important class of therapeutics against HIV-1, interfere with the virus’ reverse transcripase [136] (Fig. 1b and 2c), which synthesizes DNA from the RNA template.
Toward All-Atom Simulations of Complete Virions
As experimental methods, particularly cryo-EM and cryo-ET, continue to provide greater detail on virus composition and architecture, and as supercomputers and simulation codes continue to offer greater performance for the study of large biological systems, the computational community becomes poised to study the dynamics of complete virions within physiological environments. Nonetheless, the paucity of accurate, full-length, and fully atomistic models for some major components of virus structure presents a significant challenge to the derivation and investigation of all-atom virions.
Importantly, the lack of accurate all-atom models for encapsidated genome structures is the foremost limiting factor for the construction of complete virions. While the work of Freddolino et al. on genome-containing STMV (Fig. 1c) represents the only all-atom simulation of a complete virion performed to date [108], the genome structure employed was not based on the actual STMV RNA sequence. Similarly, a complete pariacoto virus (PaV) virion derived using molecular modeling included an RNA model based on an artificial RNA sequence [137]. For both of these virions, limitations in determining genome organization from crystal structures, which typically rely on icosahedral averaging, led to inaccuracies in the resulting RNA models [138, 139]. Recently, Zeng et al. used computational methods to generate the first all-atom model for the complete structure of any virus using the natural genome sequence, that of STMV containing realistic RNA [5].
Moving forward, modeling and simulation will continue to play an essential role in the determination of all-atom structures for genome-containing capsids, particularly through approaches that integrate experimental data from techniques such as NMR and cryo-EM [140, 141]. Although simulation studies of the intact HBV capsid have shown that the inherent flexibility of some virus structures may limit the ability to obtain true atomic (1–2 ˚A) resolution with cryo-EM [116], advances in image processing are enabling high-resolution asymmetric reconstructions capable of producing m o r e authentic descriptions of encapsidated genome, such as those obtained for the immature HBV particle and bacteriophage MS2 [142, 143]. The availability of more complete structural information, along with improved understanding of genome organization within capsids [144] and integrative modeling approaches [1] that employ increasingly accurate protein and nucleic acid force fields [145, 146], will support new simulation studies of all-atom virions in the near future, particularly for non-enveloped viruses.
All-atom simulations of enveloped viruses will face the additional challenge of treating the envelope membrane lipid bilayer. Due to size, complex lipid compositions, and the inclusion of membrane-embedded surface proteins, viral envelopes require careful consideration to model and equilibrate [147]. Further, enclosing a virus capsid in a fully atomistic envelope will significantly increase system size and simulation expense, particularly given the inclusion of solvent. Figure 2 emphasizes the dramatic size disparity between several protein-based components of HIV-1 that have been previously investigated with all-atom simulations and a model of the intact envelope membrane (Fig. 2f). The envelope necessarily dwarfs the HIV-1 capsid, the simulations of which currently represent the most far-reaching accomplishment of computational virology reported for structures of atomic detail [118, 122]. Following construction of a complete HIV-1 virion, it is clear that substantial supercomputing power will be required to simulate such a colossal biological assembly at chemical resolution, especially over timescales sufficient to allow meaningful study.
In the meantime, three viral envelopes have been investigated using coarse-grained simulation methods, including that of an immature HIV-1 particle [148], influenza A virus [149], and dengue virus [150] While the immature HIV-1 particle included its viral matrix component, the influenza A virion did not explicitly include matrix protein 1 (M1, Fig. 1a). The general lack of high-resolution structures for viral matrix assemblies represents another limiting factor in the construction of realistic all-atom virions.
In addition to suitable numbers and distributions of envelope proteins and viroporins embedded in the viral surface, truly complete and realistic virion models will also incorporate appropriate glycosylation on proteins and glycolipid species, as well as stoichiometric numbers of viral enzymes and accessory proteins encoded and packaged by the respective virus. The availability of in situ structures for intact virions, such as those recently obtained for HIV-1 using cryo-ET [151, 152], will dramatically support future all-atom modeling and simulation efforts. Additionally, experimental characterization of viral envelope lipidomes and improved software and force fields for building and simulating large membrane assemblies [147] are essential advancements that will soon enable all-atom simulations of complete enveloped virions. Undoubtedly, all-atom virions will be among the first large-scale biological systems to be investigated on upcoming exascale supercomputing platforms.
Highlights:
Viral protein simulations are among the largest all-atom simulations ever published.
All-atom simulations reveal the dynamics of viral proteins at chemical resolution.
All-atom detail is required for accurate simulations, particularly with drugs.
Data-guided simulations help determine viral structures inaccessible to experiment.
In situ structures and integrative modeling will enable all-atom simulations of virions.
Acknowledgments
This work was supported by the National Institutes of Health grants P50GM082251 and P30GM110758– 04 and the University of Delaware.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Jodi A. Hadden, Department of Chemistry and Biochemistry, University of Delaware, Newark, Delaware 19716.
Juan R. Perilla, Department of Chemistry and Biochemistry, University of Delaware, Newark, Delaware 19716
References
- [1].Goh Boon Chong, Hadden Jodi A., Bernardi Rafael C., Singharoy Abhishek, McGreevy Ryan, Rudack Till, Cassidy C. Keith, and Schulten Klaus. Computational methodologies for real-space structural refinement of large macromolecular complexes. Annual Review of Biophysics, 45:253–278, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.A thorough review of integrative modeling and simulationapproachescurrentlyappliedtodetermine and refine atomistic structures of large biological systems, including virus capsids.
- [2].Perilla Juan R., Goh Boon Chong, Cassidy C. Keith, Liu Bo, Bernardi Rafael C., Rudack Till, Yu Hang, Wu Zhe, and Schulten Klaus. Molecular dynamics simulations of large macromolecular complexes. Current Opinion in Structural Biology, 31:64–74, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Harris Audray, Cardone Giovanni, Winkler Dennis, Heymann J Bernard, Brecher Matthew, White Judith M, and Steven Alasdair C. Influenza virus pleiomorphy characterized by cryo-electron tomography. Proceedings of the National cademy of Sciences, 103(50):19123–19127, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Briggs JohnAG,Wilk Thomas,Welker Reinhold, ausslich Hans-Georg Kr, and Fuller Stephen D. Structural organization of authentic, mature HIV-1 virions and cores. The EMBO journal, 22(7):1707–1715, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Zeng Yingying, Larson Steven B, Heitsch Christine E, McPherson Alexander, and Harvey Stephen C. A model for the structure of satellite tobacco mosaic virus. Journal of Structural Biology, 180(1):110–116, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.All-atom modeling of STMV with its encapsidated RNA genome, producing the first realistic and chemically-complete structure of any virus.
- [6].Kasson Peter M. Receptor binding by influenza virus: using computational techniques to extend structural data. Biochemistry, 51(12):2359–2365, 2012. [DOI] [PubMed] [Google Scholar]
- [7].Ji Ye, White Yohanna JB, Hadden Jodi A, Grant Oliver C, and Woods Robert J. New insights into influenza A specificity: an evolution of paradigms. Current Opinion in Structural Biology, 44:219–231, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Newhouse E Irene, Xu Dong, Markwick Phineus RL, Amaro Rommie E, Pao Hsing C, Wu Kevin J, Alam Maqsudul, McCammon J Andrew, and Li Wilfred W. Mechanism of glycan re-ceptor recognition and specificity switch for avian, swine, and human adapted influenza virus hemagglutinins: a molecular dynamics perspective. Journal of the American Chemical Society, 131(47):17430–17442, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Dong Xu E Newhouse Irene, Amaro Rommie E., Pao Hsing C., Cheng Lily S., Markwick Phineus R L, McCammon J. Andrew, Li Wilfred W., and Arzberger Peter W.. Distinct Glycan Topology for Avian and Human Sialopentasaccharide Receptor Analogues upon Binding Different Hemagglutinins: A Molecular Dynamics Perspective. JournalofMolecularBiology,387:465–491, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sieben Christian, Kappel Christian, Zhu Rong, Wozniak Anna, Rankl Christian, Hinter- dorfer Peter, Grubmu¨ller Helmut, and Herrmann Andreas. Influenza virus binds its host cell using multiple dynamic interactions. Proceedings of the National Academy of Sciences, 109(34):13626– 13631, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Elli Stefano, Macchi Eleonora, Rudd Timothy, Raman Rahul, Sassaki Guillherme, Viswanathan Karthik, Yates Edwin A, Shriver Zachary, Naggi Annamaria, Torri Giangiacomo, Sasisekharan Ram, and Guerrini Marco. Insights into the human glycan receptor conformation of 1918 pandemic hemagglutinin–glycan complexes derived from nuclear magnetic resonance and molecular dynamicsstudies. Biochemistry, 53(25):4122–4135, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Macchi Eleonora, Rudd Timothy R, Raman Rahul, Sasisekharan Ram, Yates Edwin A, Naggi An-namaria, Guerrini Marco, and Elli Stefano. Nuclear magnetic resonance and molecular dynamics simulation of the interaction between recognition protein H7 of the novel influenza virus H7N9 and glycan cell surface receptors. Biochemistry, 55(48):6605–6616, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Peng Wenjie, Vries Robert de, Grant Oliver C, Thompson Andrew J, McBride Ryan, Tsogtbaatar Buyankhishig, Lee Peter S, Razi Nahid, Wilson Ian A, Woods Robert J, et al. Recent H3N2 viruses have evolved specificity for extended, branched human-type receptors, con-ferring potential for increased avidity. Cell Host & Microbe, 21(1):23–34, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Vries Robert P De, Peng Wenjie, Grant Oliver C, Thompson Andrew J, Zhu Xueyong, Bouwman Kim M, Torrents de la Pena Alba T, van Breemen Marielle J, Wickra-masinghe Iresha N Ambepitiya, de Haan Cornelis AM, et al. Three mutations switch H7N9 influenza to human-type receptor specificity. PLoS Pathogens, 13(6):e1006390, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kasson Peter M., Ensign Daniel L., and Pande Vijay S.. Combining molecular dynamics with Bayesian analysis to predict and evaluate ligand-binding mutations in influenza hemagglutinin. Journal of the American Chemical Society, 131:11338–11340, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Das Payel, Li Jingyuan, Royyuru Ajay K, and Zhou Ruhong. Free energy simulations reveal a double mutant avian H5N1 virus hemagglutinin with altered receptor binding specificity. Journal of Computational Chemistry, 30(11):1654–1663,2009. [DOI] [PubMed] [Google Scholar]
- [17].Zhou Ruhong, Das Payel, and Royyuru Ajay K. Single mutation induced H3N2 hemagglutinin antibody neutralization: a free energy perturbation study. The Journal of Physical Chemistry B, 112(49):15813–15820, 2008. [DOI] [PubMed] [Google Scholar]
- [18].Ieong Pek, Amaro Rommie E, and Li Wilfred W. Molecular dynamics analysis of antibody recog-nition and escape by human H1N1 influenza hemagglutinin. Biophysical Journal, 108(11):2704– 2712, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kasson Peter M and Pande Vijay S. Structural basis for influence of viral glycans on ligand binding by influenza hemagglutinin. Biophysical Journal, 95(7):L48–L50, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Khatri Kshitij,Klein Joshua A,White Mitchell, Grant Oliver C, Leymarie Nancy, Woods Robert J, Hartshorn Kevan L, and Zaia Joseph. Integrated omics and computational glycobiology reveal structural basis for influenza a virus glycan microheterogeneity and host interactions. Molecular & Cellular Proteomics, 15(6):1895–1912, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Goh Boon Chong, Rynkiewicz Michael J., Cafarella Tanya R., White Mitchell R., Hartshorn Kevan L., Allen Kimberly, Crouch Erika C., Calin Oliviana, Seeberger Peter H., Schulten Klaus, and Seaton Barbara A.. Molecular mechanisms of inhibition of influenza by surfactant protein D revealed by large-scale molecular dynamics simulation. Biochemistry, 52:8527–8538, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.All-atom simulations of intact influenza HA complexed with an innate immune lectin, investigating the effects of lectin mutations on viral glycan attachment and inhibition of viral adhesion.
- [22].Huang Qiang, Chen Cheng-Lung, and Herrmann Andreas. Bilayer conformation of fusion peptide of influenza virus hemagglutin: A molecular dynamics simulation study. Biophysical Journal, 87:14–22, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Vaccaro Loredana, Cross Karen J., Kleinjung Jens, Straus Suzana K., Thomas David J., Wharton Stephen A., Skehel John J., and Franternali Franca. Plasticity of influenza haemag- glutinin fusion peptides and their interaction with lipid bilayers. Biophysical Journal, 88:25–36, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lagu¨e Patrick, Roux Benoˆıt, and Pastor Richard W.. Molecular dynamics simulations of the in-fluenza hemagglutinin fusion peptide in micelles and bilayers: Conformational analysis of peptide and lipids. Journal of Molecular Biology, 354(5):1129–1141, 2005. [DOI] [PubMed] [Google Scholar]
- [25].Jang Hyunbum, Michaud-Agrawal Naveen, Johnston Jennifer M.,and Woolf Thomas B.. How to lose a kink and gain a helix: pH independent conformational changes of the fusion domains from influenza hemagglutinin in heterogeneous lipid bilayers. Proteins: Structure, Function, and Bioinformatics, 72(1):299–312. [DOI] [PubMed] [Google Scholar]
- [26].Kasson Peter M., Lindahl Erik, and Pande Vijay S.. Atomic-resolution simulations predict a transition state for vesicle fusion defined by contact of a few lipid tails. PLoS Comput. Biol, 6(6):e1000829, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Legare Sebastien and Lagu¨e Patrick. The influenza fusion peptide adopts a flexible flat V conformation in membranes. Biophysical Journal, 102:2270–2278, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kalani Mohamad R., Moradi Abdulvahab, Moradi Mahmoud, and Tajkhorshid Emad. Char-acterizing a histidine switch controlling pH-dependent conformational changes of the influenza virus hemagglutinin. Biophysical Journal, 105(4):993–1003, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Larsson Per and Kasson Peter M.. Lipid tail protrusion in simulations predicts fusogenic activity of influenza fusion peptide mutants and conformational models. PLoS Computational Biology, 9:e1002950, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].L´egar´e S and Lagu¨e P. The influenza fusion peptide promotes lipid polar head intrusion through hydrogen bonding with phosphates and N-terminal membrane insertion depth. PROTEINS: Structure, Function, and Bioinformatics, 82:2118–2127, 2014. [DOI] [PubMed] [Google Scholar]
- [31].Zhou Yu, Wu Chao, Zhao Lifeng, and Huang Niu. Exploring the early stages of the pH-induced conformational change of influenza hemagglutinin. Proteins: Structure, Function, and Bioinfor-matics, 82(10):2412–2428, 2014. [DOI] [PubMed] [Google Scholar]
- [32].Brice Allyn R. and Lazaridis Themis. Structure and dynamics of a fusion peptide helical hairpin on the membrane surface: Comparison of molecular simulations and NMR. Journal of Physical Chemistry B, 118(17):4461–4470, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Beck Charles R, Sokal Rachel, Arunachalam Nachiappan, Puleston Richard, Cichowska Anna, Kessel Anthony, Zambon Maria, and Nguyen-Van-Tam Jonathan S. Neuraminidase inhibitors for influenza: a review and public healthMANUSCRIPTperspectiveintheaftermathofthe2009pandemic. Influenza and Other Respiratory Viruses, 7(s1):14–24, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Amaro Rommie E., Minh David D. L., Cheng Lily S. Jr. and Olson Arthur J. Lind-strom William M., Lin Jung-Hsin, and Liand Wilfred W. McCammon J. Andrew. Remarkable loop flexibility in avian influenza N1 and its implications for antiviral drug design. Journal of the American Chemical Society, 129:7764–7765, 2007. [DOI] [PubMed] [Google Scholar]
- [35].Amaro RE, Cheng X, Ivanov I, Xu D, and McCammon JA. Characterizing loop dynamics and ligand recognition in human- and avian-type influenza neuraminidases via generalized born molecular dynamics and end-point free energy calculations. Journal of the American Chemical Society, 131:4702–4709, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lawrenz M,Baron R,and McCammon JA. Independent-trajectories thermodynamic-integration free-energy changes for biomolecular systems: Determinants of H5N1 avian influenza virus neuraminidase inhibition by peramivir. Journal of Chemical Theory and Computation, 5:1106–1116, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Amaro Rommie E, Swift Robert V, Votapka Lane, Li Wilfred W, Walker Ross C, and Bush Robin M. Mechanism of 150-cavity formation in influenza neuraminidase. Nature Communications, 2:388, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Cheng LS, Amaro RE, Xu D, Li WW, Arzberger PW, and McCammon JA. Ensemble-based virtual screening reveals potential novel antiviral compounds for avian influenza neu-raminidase. Journal of Medicinal Chemistry, 51:3878–3894, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Landon Melissa R., Amaro Rommie E., Baron Riccardo, Ho-Ngan Chi, Ozonoff David, McCammon J. Andrew, and Vajda Sandor. Novel druggable hot spots in avian influenza neuraminidase H5N1 revealed by computational solvent mapping of a reduced and representative receptor en-semble. Chemical Biology and Drug Design, 71:106–116, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Durrant Jacob D and McCammon J Andrew. Potential drug-like inhibitors of group 1 in-fluenza neuraminidase identified through computer-aided drug design. Computational Biology and Chemistry, 34(2):97–105, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Le Ly, Lee Eric H., Schulten Klaus, and Truong Thahn.Molecularmodelingofswineinfluenza A/H1N1, Spanish H1N1, and avian H5N1 flu N1 neuraminidases bound to Tamiflu and Relenza. PLoS Currents: Influenza, 2009. August 27:RRN1015, 2010. (9 pages). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Le Ly, Lee Eric H., Hardy David J., Truong Thanh N., and Schulten Klaus. Molecular dy-namics simulations suggest that electrostatic funnel directs binding of Tamiflu to influenza N1 neuraminidases. PLoS Computational Biology, 6:e1000939, 2010. (13 pages). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Vergara-Jaque Ariela, Poblete Horacio, Lee Eric, chulten Klaus, Gonza´lez-Nilo Fernando, and Chipot Christophe. Molecular basis of drug resistance in A/H1N1 virus. Journal of Chemical Information and Modeling, 52:2650–2656, 2012. (DNA P41). [DOI] [PubMed] [Google Scholar]
- [44].Lawrenz Morgan, Wereszczynski Jeff, Amaro Rommie, Walker Ross, Roitberg Adrian, and McCammon J An-drew. Impact of calcium on N1 influenza neuraminidase dynamics and binding free energy. Proteins: Structure, Function, and Bioinformatics, 78(11):2523–2532, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Durrant Jacob, Bush Robin M, and Amaro Rommie E. Microsecond molecular dynamics simulations of influenza neuraminidase suggest a mechanism for the increased virulence of stalk-deletion mutants. The Journal of Physical Chemistry B, 120(33):8590–8599, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.All-atom simulations of intact influenza NA embedded in a realistic envelope bilayer, investigating druggability and the relationship between stalk length and virulence.
- [46].Yokoyama Masaru, Naganawa Satoshi, Yoshimura Kazuhisa, Matsushita Shuzo, and Sato Hironori. Structural dynamics of HIV-1 envelope Gp120 outer domain with V3 loop. PLoS One, 7(5):e37530, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Lo´pez Aliana Victoria de, Tamamis Phanourios, Kieslich Chris A, and Morikis Dimitrios. Insights into the structure, correlated motions, and electrostatic properties of two HIV-1 gp120 V3 loops. PLoS One, 7(11):e49925, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Shrivastava Indira and LaLonde Judith M. Fluctuation dynamics analysis of gp120 envelope protein reveals a topologically based communication network. Proteins: Structure, Function, and Bioinformatics, 78(14):2935–2949, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Sethi Anurag, Tian Jianhui, Derdeyn Cynthia A, Korber Bette, and Gnanakaran S. A mecha-nistic understanding of allosteric immune escape pathways in the HIV-1 envelope glycoprotein. PLoS Computational Biology, 9(5):e1003046, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Lemmin Thomas, Soto Cinque, Stuckey Jonathan, and Kwong Peter D. Microsecond dynamics and network analysis of the HIV-1 SOSIP Env trimer reveal collective behavior and conserved microdomains of the glycan shield. Structure, 25(10):1631–1639, 2017. [DOI] [PubMed] [Google Scholar]
- [51].Kamath Shantaram and Wong Tuck C. Membrane structure of the human immunodeficiency virus gp41 fusion domain by molecular dynamics simulation. Biophysical Journal, 83(1):135–143, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Wong Tuck C. Membrane structure of the human immunodeficiency virus gp41 fusion peptide by molecular dynamics simulation: II. the glycine mutants. Biochimica et Biophysica Acta (BBA)-Biomembranes,1609(1):45–54, 2003. [DOI] [PubMed] [Google Scholar]
- [53].Kim Jong Hwa, Hartley Taryn L, Curran A Rachael, and Engelman Donald M. Molecular dynamics studies of the transmembrane domain of gp41 from hiv-1. Biochimica et Biophysica Acta (BBA)-Biomembranes, 1788(9):1804–1812, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Gangupomu Vamshi K and Abrams Cameron F. All-atom models of the membrane-spanning domain of HIV-1 gp41 from metadynamics. Biophysical Journal, 99(10):3438–3444, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].pell´aniz Beatriz, Rujas Edurne, Carravilla Pablo, Requejo-Isidro Jos´e, Huarte Nerea, Domene Car-men, and Nieva Jos´e L. Cholesterol-dependent membrane fusion induced by the gp41 membrane-proximal external region–transmembrane domain connection suggests a mechanism for broad HIV-1 neutralization. Journal of Virology, 88(22):13367–13377, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Baker Michelle K, Gangupomu Vamshi K, and Abrams Cameron F. Characterization of the water defect at the HIV-1 gp41 membrane spanning domain in bilayers with and without cholesterol us-ing molecular simulations. Biochimica et Biophysica Acta (BBA)-Biomembranes, 1838(5):1396– 1405, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Doores Katie J. The HIV glycan shield as a target for broadly neutralizing antibodies. The FEBS Journal, 282(24):4679–4691, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Wood Natasha T, Fadda Elisa, Davis Robert, Grant Oliver C, Martin Joanne C, Woods Robert J, and Travers Simon A. The influence of N-linked glycans on the molecular dynamics of the HIV-1 gp120 V3 loop. PloS One, 8(11):e80301, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Qi Yifei, Jo Sunhwan, and Im Wonpil. Roles of glycans in interactions between gp120 and HIV broadly neutralizing antibodies. Glycobiology, 26(3):251–260, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Tian Jianhui, Lo´pez Cesar A, Derdeyn Cynthia, Jones Morris S, Pinter Abraham, Korber Bette, and Gnanakaran Sandrasegaram. Effect of glycosylation on an immunodominant region in the V1V2 variable domain of the HIV-1 envelope gp120 protein. PLoS Computational Biology, 12(10):e1005094, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Stewart-Jones Guillaume BE, Soto Cinque, Lemmin Thomas, Chuang Gwo-Yu, Druz Aliaksandr, Kong Rui, Thomas Paul V, Wagh Kshitij, Zhou Tongqing, Behrens Anna-Janina, By-lund Tatsiana, Choi Chang W, Davison Jack R, Georgiev Ivelin S, Joyce M Gordon, Kwon Young Do, Pancera Marie, Taft Justin, Yang Yongping, Zhang Baoshan, Shivatare Sachin S, Shi-vatare Vidya S, Lee Chang-Chun D, Wu Chung-Yi, Bewley Carole A, Burton Dennis R, Koff Wayne C, Connors Mark, Crispin Max, Baxa Ulrich, Korber Bette T, Wong Chi-Huey, Mascola John R, and Kwong Peter. Trimeric HIV-1-Env structures define glycan shields from clades A, B, and G. Cell, 165(4):813–826, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.All-atom simulations of multiple glycoforms of intact HIV-1 Env, investigating the dynamics of the glycan shield, as well as glycan involvement in antibody recognition.
- [62].Li Xiaoyan, Grant Oliver C, Ito Keigo, Wallace Aaron, Wang Shixia, Zhao Peng, Wells Lance, Lu Shan, Robert J Woods, and Joshua S Sharp. Structural analysis of the glycosylated intact HIV-1 gp120–b12 antibody complex using hydroxyl radical protein footprinting. Biochemistry, 56(7):957–970, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Zhou Tongqing, Doria-Rose Nicole A, Cheng Cheng, Stewart-Jones Guillaume BE, Chuang Gwo-Yu, Chambers Michael, Druz Aliaksandr, Geng Hui, McKee Krisha, Kwon Young Do, et al. Quantification of the impact of the HIV-1-glycan shield on antibody elicitation. Cell Reports, 19(4):719–732, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Kampmann T, Mueller DS, Mark AE, Young PR, and Kobe B. The role of histidine residues in low-pH-mediated viral membrane fusion. Structure, 14:1481–1487, 2006. [DOI] [PubMed] [Google Scholar]
- [65].Mueller DS, Kampmann T, Yennamalli R, Young PR, Kobe B, and Mark AE. Histidine protonation and the activation of viral fusion proteins. Biochemistry, 36:43–45, 2008. [DOI] [PubMed] [Google Scholar]
- [66].Prakash MK, Barducci A, and Parrinello M. Probing the mechanism of pH-induced large scale conformational change in the dengue virus envelope protein using atomistic simulations. Biophysics, 99:588–594, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Dubey Kshatresh Dutta, Chaubey Amit Kumar, and Ojha Rajendra Prasad. Role of ph on dimeric interactions for DENV envelope protein: An insight from molecular dynamics study. Biochimica et Biophysica Acta (BBA)-Proteins and Proteomics, 1814(12):1796–1801, 2011 [DOI] [PubMed] [Google Scholar]
- [68].Fuzo Carlos A and Degr`eve L´eo. New pockets in dengue virus 2 surface identified by molecular dynamics simulation. Journal of Molecular odeling, 19(3):1369–1377, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Degr´eve L, Fuzo CA, et al. Structure and dynamics of the monomer of protein E of dengue virus type 2 with unprotonated histidine residues. Genetics and Molecular Research, 12(1):348–359,2013. [DOI] [PubMed] [Google Scholar]
- [70].Ru¨cker Pia, Wieninger Silke A, Ullmann G Matthias, and Sticht Heinrich. pH-dependent molec-ular dynamics of vesicular stomatitis virus glycoprotein G. Proteins: Structure, Function, and Bioinformatics, 80(11):2601–2613, 2012. [DOI] [PubMed] [Google Scholar]
- [71].Freitas MonicaˆS, Gaspar Luciane, Lorenzoni Marcos, Almeida Fabio CL, Tinoco Luzineide W, Almeida Marcius S, Maia Lenize F, Degr`eve L´eo, Valente Ana Paula, and Silva Jerson L. Structure of the ebola fusion peptide in a membrane-mimetic environment and the interaction with lipid rafts. Journal of Biological Chemistry, 282(37):27306–27314, 2007. [DOI] [PubMed] [Google Scholar]
- [72].Jaskierny Adam J, Panahi Afra, and Feig Michael. Effect of flanking residues on the conforma-tional sampling of the internal fusion peptide from ebola virus. Proteins: Structure, Function, and Bioinformatics, 79(4):1109–1117, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Gregory Sonia M, Larsson Per, Nelson Elizabeth A, Kasson Peter M, White Judith M, and Tamm Lukas K. Ebolavirus entry requires a compact hydrophobic fist at the tip of the fusion loop. Journal of Virology, 88(12):6636–6649,2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Vitiello Giuseppe, Falanga Annarita, Petruk Ariel Alcides, Merlino Antonello, Frag-neto Giovanna, Paduano Luigi, Galdiero Stefania, and D’Errico Gerardino. Fusion of raft-like lipid bilayers operated by a membranotropic domain of the HSV-type I glycoprotein gH occurs through a cholesterol-dependent mechanism. Soft Matter, 11(15):3003–3016, 2015. [DOI] [PubMed] [Google Scholar]
- [75].Winger Moritz and Itzstein Mark von. Exposing the flexibility of human parainfluenza virus hemagglutinin-neuraminidase. Journal of the American Chemical Society, 134(44):18447–18452, 2012. [DOI] [PubMed] [Google Scholar]
- [76].Kong Leopold, Lee David E, Kadam Rameshwar, Liu Tong, Giang Erick, Nieusma Travis, Garces Fernando, Tzarum Netanel,Woods Virgil L, Ward Andrew B, et al. Structural flexibility at a major conserved antibody target on hepatitis C virus e2 antigen. Proceedings of the National Academy of Sciences, 113(45):12768–12773, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Barone Daniela, Balasco Nicole, Autiero Ida, and Vitagliano Luigi. The dynamic properties of the hepatitis C virus e2 envelope protein unraveled by molecular dynamics. Journal of Biomolecular Structure and Dynamics, 35(4):805–816, 2017. [DOI] [PubMed] [Google Scholar]
- [78].Nieva Jos´e Luis, Madan Vanesa, and Carrasco Luis. Viroporins: structure and biological func-tions. Nature Reviews Microbiology, 10(8):563, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Cady Sarah, Luo Wenbin, Hu Fanghao, and Hong Mei. Structure and function of the influenza A M2 proton channel. Biochemistry, 48(31):7356–7364, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Schweighofer Karl J and Pohorille Andrew. Computer simulation of ion channel gating: the M2 channel of influenza A virus in a lipid bilayer. Biophysical Journal, 78(1):150–163, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Smondyrev AM and Voth GA. Molecular dynamics simulation of proton transport through the influenza A virus M2 channel. Biophysical Journal, (4):1987–1996, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Wu Yujie and Voth Gregory A. A computational study of the closed and open states of the influenza a M2 proton channel. Biophysical Journal, 89(4):2402–2411, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Chen Hanning, Wu Yujie, and Voth Gregory A. Protontransportbehaviorthroughtheinfluenza a M2 channel: insights from molecular simulation. Biophysical Journal, 93(10):3470–3479, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Yi Myunggi, Cross Timothy A., and Zhou Huan-Xiang. A secondary gate as a mechanism for inhibition of the M2 proton channel by amantadine. Journal of Physical Chemistry B, 112(27):7977–7979, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Yi Myunggi, Cross Timothy A, and Zhou Huan-Xiang. Conformational heterogeneity of the M2 proton channel and a structural model for channel activation. Proceedings of the National Academy of Sciences, 106(32):13311–13316, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Liang Ruibin, Li Hui, Swanson Jessica MJ, and Voth Gregory. Multiscale simulation reveals a multifaceted mechanism of proton permeation through the influenza a M2 proton channel. ProceedingsoftheNationalAcademyof Sciences, 111(26):9396–9401, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Zhong Qingfeng, Newns Dennis M, Pattnaik Pratap, Lear James D, and Klein Michael L. Two possible conducting states of the influenza A virus M2 ion channel. FEBS Letters, 473(2):195– 198, 2000. [DOI] [PubMed] [Google Scholar]
- [88].Khurana Ekta, Peraro Matteo Dal, DeVane Russell, Vemparala Satyavani, DeGrado William F., and Klein Michael L.. Molecular dynamics calculations suggest a conduction mechanism for the M2 proton channel from influenza virus. Proceedings of the National Academy of Sciences, USA, 106(4):1069–1074, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Thomaston Jessica L, Alfonso-Prieto Mercedes, Woldeyes Rahel A, Fraser James S, Klein Michael L, Fiorin Giacomo, and DeGrado William F. High-resolution structures of the M2 chan-nel from influenza A virus reveal dynamic pathways for proton stabilization and transduction. Proceedings of the National Academy of Sciences, 112(46):14260–14265, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Khurana Ekta, DeVane Russell H., Peraro Matteo Dal, and Klein Michael L.. Computational study of drug binding to the membrane-bound tetrameric M2 peptide bundle from influenza A virus. Biochimica et Biophysica Acta, 1808:530–537, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Leonov Hadas, Astrahan Peleg, Krugliak Miriam, and Arkin Isaiah T. How do aminoadaman- tanes block the influenza M2 channel, and how does resistance develop? Journal of the American Chemical Society, 133(25):9903–9911, 2011 [DOI] [PubMed] [Google Scholar]
- [92].Gu Ruo-Xu, Liu Limin Angela, Wang Yong-Hua, Xu Qin, and Wei Dong-Qing. Structural comparison of the wild-type and drug-resistant mutants of the influenza M2 proton channel by molecular dynamics simulations. The Journal of Physical Chemistry B, 117(20):6042–6051, 2013. [DOI] [PubMed] [Google Scholar]
- [93].Wang Jun, Ma Chunlong, Fiorin Giacomo, Carnevale Vincenzo, Wang Tuo, Hu Fanghao, Lamb Robert A, Pinto Lawrence H, Hong Mei, Klein Michael L, et al. Molecular dynamics simulation directed rational design of inhibitors targeting drug-resistant mutants of influenza A virus M2. Journal of the American Chemical Society, 133(32):12834–12841, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Wang Jun,Wu Yibing,Ma Chunlong, Fiorin Giacomo, Wang Jizhou, Pinto Lawrence H, Lamb Robert, Klein Michael L, and DeGrado William F. Structure and inhibition of the drug-resistant S31N mutant of the M2 ion channel of influenza A virus. Proceedings of the National Academy of Sciences, 110(4):1315–1320, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Fischer WB. Vpu from HIV-1 on an atomic scale: experiments and computer simulations. FEBS Letters, 552(1):39–46, 2003. [DOI] [PubMed] [Google Scholar]
- [96].Lopez Carlos F, Montal Mauricio, Blasie J Kent, Klein Michael L, and Moore Preston B. Molec-ular dynamics investigation of membrane-bound bundles of the channel-forming transmembrane domain of viral protein U from the human immunodeficiency virus HIV-1. Biophysical Journal, 83(3):1259–1267, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Hertel Brigitte, Tayefeh Sascha, Kloss Thomas, Hewing Jennifer, Gebhardt Manuela, Baumeister Dirk, Moroni Anna, Thiel Gerhard, and Kast Stefan M. Salt bridges in the miniature viral channel Kcv are important for function. European Biophysics Journal, 39(7):1057–1068, 2010. [DOI] [PubMed] [Google Scholar]
- [98].Gebhardt Manuela, Hoffgaard Franziska, Hamacher Kay, Kast Stefan M, Moroni Anna, and Thiel Gerhard. Membrane anchoring and interaction between transmembrane domains are crucial for K+ channel function. Journal of Biological Chemistry, 286(13):11299–11306, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Tayefeh Sascha, Kloss Thomas, Kreim Michael,Gebhardt Manuela,Baumeister Dirk,Hertel Brigitte, Richter Christian, Schwalbe Harald, Moroni Anna, Thiel Gerhard, et al. Model develop-ment for the viral Kcv potassium channel. Biophysical Journal, 96(2):485–498, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Montserret Roland, Saint Nathalie, Vanbelle Christophe, Salvay Andr´es Gerardo, Simorre Jean-Pierre, Ebel Christine, Sapay Nicolas, Renisio Jean-Guillaume, B¨ockmann Anja, Stein-mann Eike, Pietschmann Thomas, Dubuisson Jean, Chipot hristophe, and Penin Franc¸ois. NMR structure and ion channel activity of the p7 protein from hepatitis C virus. Journal of Biological Chemistry, 285(41):31446–31461, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Chandler Danielle E., Penin Francois, Schulten Klaus, and Chipot Christophe. The p7 protein ofhepatitisCvirusformsstructurallyplastic, minimalist ion channels. PLoS Computational Biology, 8:e1002702, 2012. (10 pages). [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.All-atom simulations of HCV p7 viroporin embedded in membrane bilayers, elucidating likely oligomeric states and functional ion channel characteristics.
- [102].Wang Yi-Ting, Schilling Roman, Fink Rainer HA, and Fischer Wolfgang B. Ion-dynamics in hepatitisvirus p7 helical transmembrane domainsa molecular dynamics simulation study. Biophysical Chemistry, 192:33–40, 2014. [DOI] [PubMed] [Google Scholar]
- [103].Cook Gabriel, Dawson Lindsay A, Tian Ye, and Opella Stanley J. Three-dimensional structure and interaction studies of hepatitis C virus p7 in 1, 2-dihexanoyl-sn-glycero-3-phosphocholine by solution nuclear magnetic resonance. Biochemistry, 52(31):5295–5303, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Kalita Monoj Mon, Griffin Stephen, Chou James J, and Fischer Wolfgang B. Genotype-specific differences in structural features of hepatitis C virus (HCV) p7 membrane protein. Biochimica et Biophysica Acta (BBA)-Biomembranes, 1848(6):1383–1392, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Kalita Monoj Mon and Fischer Wolfgang B. Asymmetric dynamics of ion channel forming pro-teinshepatitis C virus (HCV) p7 bundles. Biochimica et Biophysica Acta (BBA)-Biomembranes, 1858(7):1462–1470, 2016. [DOI] [PubMed] [Google Scholar]
- [106].May Eric R. Recent developments in molecular simulation approaches to study spherical virus capsids. Molecular Simulation, 40(10–11):878–888, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Perilla Juan R., Hadden Jodi A., Goh Boon Chong, Mayne Christopher G., and Schulten Klaus. All-atom molecular dynamics of virus capsids as drug targets. Journal of Physical Chemistry Letters, 7:1836–1844, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **.A perspective on applying all-atom simulations to study drug-bound capsids, demonstrating that chemical resolution is required to achieve accurate characterization of protein-ligand interactions.
- [108].Freddolino Peter L., Arkhipov Anton S., Larson Steven B., McPherson Alexander, and Schulten Klaus. Molecular dynamics simulations of the complete satellite tobacco mosaic virus. Structure, 14:437–449, 2006. [DOI] [PubMed] [Google Scholar]
- *.The first all-atom simulations of an intact capsid and complete virion, demonstrating the role of packagedgenomeinthestructuralstabilityof STMV.
- [109].Zink Mareike and Grubmu¨ller Helmut. Mechanical properties of the icosahedral shell of southern bean mosaic virus: molecular dynamics study. Biophysical Journal, 96:1350–1363, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].May Eric R and Brooks Charles L III. Determination of viral capsid elastic properties from equilibrium thermal fluctuations. Physical Review Letters, 106(18):188101, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Wang Xue, Xu Fengting, Liu Jiasen, Gao Bingquan, Liu Yanxin, Zhai Yujia, Ma Jun, Zhang Kai, Baker Timothy S., Schulten Klaus, Zheng Dong, Pang Hai, and Sun Fei. Atomic model of rabbit hemorrhagic disease virus by cryo-electron microscopy and crystallography. PLoS Pathogens, 9:e1003132, 2013. (14 pages). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Tarasova Elvira, Farafonov Vladimir, Khayat Reza, Okimoto Noriaki, Komatsu Teruhisa S, Taiji Makoto, and Nerukh Dmitry. All-atom molecular dynamics simulations of entire virus capsid reveal the role of ion distribution in capsid’s stability. The Journal of Physical Chemistry Letters, 8(4):779–784, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Tarasova Elvira, Korotkin Ivan, Farafonov Vladimir, Karabasov Sergey, and Nerukh Dmitry. Complete virus capsid at all-atom resolution: Simulations using molecular dynamics and hybrid molecular dynamics/hydrodynamics methods reveal semipermeable membrane function. Journal of Molecular Liquids, 245:109–114, 2017. [Google Scholar]
- [114].Larsson Daniel S D, Liljas Lars, and Spoel David van der. Virus capsid dissolution studied by microsecond molecular dynamics simulations. PLoS Computational Biology, 8:e1002502, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.The first all-atom simulations of an intact capsid to reach the microsecond timescale, revealing the mechanistic pathway for STNV swelling in the absence of structural calcium ions.
- [115].Andoh Y, Yoshii N, Yamada A, Fujimoto K, Kojima H, Mizutani K, Nakagawa A, Nomoto A, and Okazaki S. All-atom molecular dynamics calculation study of entire poliovirus empty capsids in solution. Journal of Chemical Physics, 141(165101):165101, 2014. [DOI] [PubMed] [Google Scholar]
- [116].Hadden Jodi A, Perilla Juan R, Schlicksup Christopher John, Venkatakrish-nan Balasubramanian, Zlotnick Adam, and Schulten Klaus. All-atom molecular dynamics of the HBV capsid reveals insights into biological function and cryo-EM resolution limits. eLife, 7, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **.All-atom simulations of the HBV capsid over one microsecond, revealing flexibility and asymmetry in an icosahedral structure, with implications for resolution of single-particle image reconstructions.
- [117].Ruan Lu, Hadden Jodi A, and Zlotnick Adam. ssembly properties of Hepatitis B Virus core protein mutants correlate with their resistance to assembly-directed antivirals. Journal of Virol-ogy, page in press, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Zhao Gongpu, Perilla Juan R., Yufenyuy Ernest L., Meng Xin, Chen Bo, Ning Jiying, Ahn Jinwoo, Gronenborn Angela M., Schulten Klaus, Aiken Christopher, and Zhang Peijun. Mature HIV-1 capsid structure by cryo-electron microscopy and all-atom molecular dynamics. Nature, 497:643–646, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **.A landmark publication for computational virology, presenting an all-atom structure of the HIV-1 capsid derived from data-guided modeling and simulations totaling 64 million particles.
- [119].Liu Chuang, Perilla Juan R., Ning Jiying, Lu Manman, Hou Guangjin, Ramalho Ruben, Bedwell Gre-gory, Byeon In-Ja, Ahn Jinwoo, Shi Jiong, Gronenborn Angela, Prevelige Peter, Rousso Itay, Aiken Christopher, Polenova Tatyana, Schulten Klaus, and Zhang Peijun. Cyclophilin A stabilizes HIV-1 capsid through a novel non-canonical binding site. Nature Communications, 7:10714:(10 pages), 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Lu Manman, Hou Guangjin, Zhang Huilan, Suiter Christopher L., Ahn Jinwoo, Byeon In-Ja L., Perilla Juan R., Langmead Christopher J., Hung Ivan, Gor’kov Peter L., Gan Zhehong, Brey William, Aiken Christopher, Zhang Peijun, Schulten Klaus, Gronenborn Angela M., and Polenova Tatyana. Dynamic allostery governs cyclophylin A-HIV capsid interplay. Proceedings of the National Academy of Sciences, USA, 112:14617–14622, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Alvarez Frances JD, He Shaoda, Perilla Juan R, Jang Sooin, Schulten Klaus, Engelman Alan N, Scheres Sjors HW, and Zhang Peijun. CryoEM structure of MxB reveals a novel oligomerization interface critical for HIV restriction. Science Advances, 3(9):e1701264, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Perilla Juan R and Schulten Klaus. Physical properties of the HIV-1 capsid from all-atom molecular dynamics simulations. Nature Communications, 8:15959, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **.The most substantial all-atom MD study ever reported,employingsimulationsoftheHIV-1capsid system of 64 million particles over one microsecond to investigate its biophysical properties.
- [123].Perilla Juan R and Gronenborn Angela M. Molecular architecture of the retroviral capsid. Trends in Biochemical Sciences, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Martin Jessica L, Mendonc¸a Luiza, Marusinec Rachel, Zuczek Jennifer, Angert Isaac, Blower Ruth J, Mueller Joachim D, Perilla Juan R, Zhang Wei, and Mansky Louis M. Critical role of the HTLV-1 capsid n-terminal domain for Gag-Gag interactions and virus particle assembly. Journal of virology, pages JVI–00333, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Goh Boon Chong, Perilla Juan R., England Matthew R., Heyrana Katrina J., Craven Rebecca C., and Schulten Klaus. Atomic modeling of an immature retroviral lattice using molecular dynamics and mutagenesis. Structure, 23:1414–1425, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Wang Mingzhang, Quinn Caitlin M, Perilla Juan R, Zhang Huilan, Shirra Randall Jr, Hou Guangjin, Byeon In-Ja, Suiter Christopher L, Ablan Sherimay, Urano Emiko, et al. Quenching protein dynamics interferes with HIV capsid maturation. Nature Communications, 8(1):1779, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.All-atom simulated tempering of the HIV-1 CA-SP1 hexamer on ANTON2, revealing that the structure of the six-helix bundle is characterized by a dynamic helix-coil equilibrium.
- [127].Dick Rob, Zadrozny Kaneil, Xu Chaoyi, Schur Florian, Lyddon Terri, Ricana Clifton, Wagner Jonathan, Perilla Juan, Ganser-Pornillos Barbie, Johnson Marc, Pornillos Owen, and Vogt Volker. Inositol phosphates are assembly co-factors for HIV-1. Nature, page in press, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.All-atom simulations of the HIV-1 CA-SP1 hexamer in complex with inositol phosphates, revealing the mechanism by which they promote assembly of the immature virus particls.
- [128].Heyrana Katrina J, Goh Boon Chong, Perilla Juan R, Nguyen Tam-Linh N, England Matthew R, Bewley Maria C, Schulten Klaus, and Craven Rebecca C. Contributions of charged residues in structurally dynamic capsid surface loops to Rous sarcoma virus assembly. Journal of Virology, 90:5700–5714, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Collins Jack R., Burt Stanley K., and Erickson John W.. Flap opening in HIV-1 protease simulated by ‘activated’ molecular dynamics. Nature Structural Biology, 2:334–338, 1995. [DOI] [PubMed] [Google Scholar]
- [130].Hornak Viktor, Okur Asim, Rizzo Robert C, and Simmerling Carlos. HIV-1 protease flaps spontaneously open and reclose in molecular dynamics simulations. Proceedings of the National Academy of Sciences, USA, 103(4):915–920, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Hornak Viktor, Okur Asim, Rizzo Robert C, and Simmerling Carlos. Hiv-1 protease flaps spontaneously close to the correct structure in simulations following manual placement of an inhibitor into the open state. Journal of the American Chemical Society, 128(9):2812–2813,2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Ding Fangyu, Layten Melinda, and Simmerling Carlos. Solution structure of HIV-1 protease flaps probed by comparison of molecular dynamics simulation ensembles and EPR experiments. Journal of the American Chemical Society, 130(23):7184–7185, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Deng Nan-jie, Zheng Weihua, Gallicchio Emillio, and Levy Ronald. Insights into the dynamics of hiv-1 protease: a kinetic network model constructed from atomistic simulations. J Am Chem Soc, 133(24):9387–9394, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Sadiq S Kashif, No´e Frank, and Fabritiis Gianni De. Kinetic characterization of the critical step in HIV-1 protease maturation. Proceedings of the National Academy of Sciences, USA,109(50):20449–20454, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Ekonomiuk Dariusz and Caflisch Amedeo. Activation of the West Nile virus NS3 protease: molec-ular dynamics evidence for a conformational selection mechanism. Protein Science, 18(5):1003–1011, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Ivetac Anthony and McCammon J Andrew. Elucidating the inhibition mechanism of HIV-1 non-nucleoside reverse transcriptase inhibitors through multicopy molecular dynamics simulations. Journal of molecular biology, 388(3):644–658, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Devkota Batsal, Petrov Anton S., Lemieux S´ebastien, Burak Boz Mustafa, Tang Liang, Schneemann Anette, Johnson John E., and Harvey Stephen C.. Structural and electrostatic characteri-zation of Pariacoto virus: Implications for viral assembly. Biopolymers, 91(7):530–538, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Larson SB and McPherson A. Satellite tobacco mosaic virus RNA: structure and implications for assembly. Current Opinion in Structural Biology, 11:59–65, 2001. [DOI] [PubMed] [Google Scholar]
- [139].Tang Liang, Johnson Karyn N., Ball L. Andrew, Lin Tianwei, Yeager Mark, and Johnson John E.. The structure of pariacoto virus reveals a dodecahedral cage of duplex RNA. Nature Structural Biology, 8(1):77–83, 2001. [DOI] [PubMed] [Google Scholar]
- [140].Zhang Huilan, Hou Guangjin, Lu Manman, Ahn Jinwoo, Byeon In-Ja L., Lang-mead Christopher J., Perilla Juan R., Hung Ivan, Gor’kov Peter L., Gan Zhehong, Brey William W., Case David A., Schulten Klaus, Gronenborn Angela M., and Polenova Tatyana. HIV-1 capsid function is regulated by dynamics: Quantitative atomic-resolution insights by integrating magic-angle-spinning NMR, QM/MM, and MD. Journal of the American Chemical Society, 138:14066–14075, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Perilla Juan R, Zhao Gongpu, Lu Manman, Ning Jiying, Hou Guangjin, Byeon In-Ja L, Gronenborn Angela M, Polenova Tatyana, and Zhang Peijun. CryoEM structure refinement by integrating NMR chemical shifts with molecular dynamics simulations. Journal of Physical Chemistry B, page 10.1021/acs.jpcb.6b13105, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Wang Joseph Che-Yen, Nickens David G, Lentz Thomas B, Loeb Daniel D, and Zlotnick Adam. Encapsidated hepatitis B virus reverse transcriptase is poised on an ordered RNA lattice. Proceedings oftheNationalAcademyof Sciences, 111(31):11329–11334, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Dai Xinghong, Li Zhihai, Lai Mason, Shu Sara, Du Yushen, Zhou Z Hong, and Sun Ren. In situ structures of the genome and genome-delivery apparatus in a single-stranded RNA virus. Nature, 541(7635):112, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Harvey Stephen, Zeng Yingying, and Heitsch Christine E. The icosahedral RNA virus as a grotto: organizing the genome into stalagmites and stalactites. Journal of Biological Physics, 39(2):163–172, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Huang Jing, Rauscher Sarah, Nawrocki Grzegorz, Ran Ting, Feig Michael, de Groot Bert L, Grubmu¨ller Helmut, and MacKerell Alexander D Jr. Charmm36m: an improved force field for folded and intrinsically disordered proteins. Nature Methods, 14(1):71, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Tan Dazhi, Piana Stefano, Dirks Robert M, and Shaw David E. RNA force field with accu-racy comparable to state-of-the-art protein force fields. Proceedings of the National Academy of Sciences, page 201713027, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Reddy Tyler and Sansom Mark SP . Computational virology: From the inside out. Biochimica et Biophysica Acta (BBA)-Biomembranes, page in press, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Ayton Gary S. and Voth Gregory A.. Multiscale computersimulationoftheimmature HIV-1 virion. Biophysical Journal, 99:2757–2765, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Reddy Tyler, Shorthouse David, Parton Daniel L, Jefferys Elizabeth, Fowler Philip W, Chavent Matthieu, Baaden Marc, and Sansom Mark SP. Nothing to sneeze at: dynamic and integrative computational model of an influenza a virion. Structure, 23:584–597, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Reddy Tyler and Sansom Mark SP. The role of the membrane in the structure and biophysical robustness of the dengue virion envelope. Structure, 24(3):375–382, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Schur Florian K M, Hagen Wim J H, Rumlova Michaela, Ruml Tomas, Muller Barbara, Krausslich Hans- Georg, and Briggs John A G. Structure of the immature HIV-1 capsid in intact virus particlesat8.8 resolution. Nature,517:505–508, 2015. [DOI] [PubMed] [Google Scholar]
- [152].Mattei Simone, Glass B¨arbel, Hagen Wim JH, Kr¨ausslich Hans-Georg, and Briggs John AG. The structure and flexibility of conical HIV-1 capsids determined within intact virions. Science, 354(6318):1434–1437, 2016. [DOI] [PubMed] [Google Scholar]