

Abstract
An essential advantage during eukaryotic cell evolution was the acquisition of a network of mitochondria as a source of energy for cell metabolism and contrary to conventional wisdom, functional mitochondria are essential for the cancer cell. Multiple aspects of mitochondrial biology beyond bioenergetics support transformation including mitochondrial biogenesis, fission and fusion dynamics, cell death susceptibility, oxidative stress regulation, metabolism, and signaling. In cancer, the metabolism of cells is reprogrammed for energy generation from oxidative phosphorylation to aerobic glycolysis and impacts cancer mitochondrial function. Furthermore cancer cells can also modulate energy metabolism within the cancer microenvironment including immune cells and induce “metabolic anergy” of antitumor immune response. Classical approaches targeting the mitochondria of cancer cells usually aim at inducing changing energy metabolism or directly affecting functions of mitochondrial antiapoptotic proteins but most of such approaches miss the required specificity of action and carry important side effects. Several types of cancers harbor somatic mitochondrial DNA mutations and specific immune response to mutated mitochondrial proteins has been observed. An attractive alternative way to target the mitochondria in cancer cells is the induction of an adaptive immune response against mutated mitochondrial proteins. Here, we review the cancer cell-intrinsic and cell-extrinsic mechanisms through which mitochondria influence all steps of oncogenesis, with a focus on the therapeutic potential of targeting mitochondrial DNA mutations or Tumor Associated Mitochondria Antigens using the immune system. (Translational Research 2018; 202:35—51)
MITOCHONDRIA: THE POWERHOUSE OF THE CELL
Mitochondria are essential organelles derived from endosymbiotic bacteria, necessary for cellular activity. They are an exceptional example of natural selection, as the host allowed their coevolution since most of the mitochondrial proteins are encoded by the nuclear genome. Mitochondria, however, retain a small 16 Kb DNA genome that encodes tRNAs, rRNAs, and proteins essential for respiration.1 Indeed, they are the powerhouse of the cell. These organelles are maternally inherited with 1 cell containing hundreds of mitochondria that can be wild-type (a state referred to as homoplasmy) or exist in mixtures of wild-type and mutant forms (heteroplasmy) dependently on the mtDNA.2 The system regulating turnover of mitochondria is known as mitophagy, a mechanism by which damaged or excess mitochondria are selectively eliminated. Mitophagy is accompanied by the balance of fission (the separation of long, tubular mitochondria into 2 or more smaller parts) and fusion (the combination of two mitochondria into a single organelle).3
As the powerhouse of the cell, mitochondria are essential bioenergetic and biosynthetic factories critical for normal cell function. They use substrates from cytoplasm to drive fatty acid oxidation (FAO), the tricarboxylic acid cycle or the Krebs cycle, the electron transport chain (ETC), and respiration, to synthesize the molecules essential for the construction of macromolecules including amino acids, lipids, nucleotides, heme, and iron-sulfur clusters, and to regenerate reduced nicotinamide adenine dinucleotide phosphate for antioxidant defense.2 Reducing agents NADH and hydroquinone form of flavin adenine dinucleotide (FADH2), produced by Krebs cycle, are indispensable and allow, by the ETC, generation of a proton gradient throughout the mitochondrial inner membrane (cristae) that generates adenosine triphosphate (ATP) by way of the H+-ATP synthase enzyme. This enzyme allows protons to cross the membrane in a single direction, according to the process of chemiosmosis.4 This metabolic pathway is called oxidative phosphorylation (OXPHOS), in which cells oxidize nutrients to produce ATP. During this mechanism, electrons are transferred from electron donors to electron acceptors, such as molecular oxygen, in the redox reaction. The reduction of oxygen can potentially produce harmful intermediates called reactive oxygen species (ROS), like superoxide or peroxide anions. Cytochrome c oxidase, complex IV, can, however, ameliorate these by-products by reducing oxygen to water.5 The OXPHOS mechanism is highly efficient with 36 ATP molecules as the maximum yield from an initial glucose molecule.6
MITOCHONDRIA AND CANCER
Tumor cell phenotypes are characterized by genetic alterations driving the expression of 10 main characteristics: genetic instability, sustaining proliferative signaling, evading growth suppressors, avoiding immune destruction, sustain promoting inflammation resisting cell death, enabling replicative immortality, inducing angiogenesis, deregulating cellular energetics, activating invasion, and metastasis.7 Warburg observed that cancer cells can reprogram their metabolism by switching from oxidative phosphorylation to glycolysis and consequently to lactic acid fermentation, even in the presence of oxygen, leading to a state that has been termed “aerobic glycolysis.”8 Unlike the high energy yield of OXPHOS, the conversion of a glucose molecule into lactate leads to the low-energy yield with the formation of only 2 ADP molecules.6 However, although the energetic yield per molecule of glucose is much lower for aerobic glycolysis compared with OXPHOS, when glucose is in excess and flux through the pathway high, glycolysis has the potential to produce ATP in greater quantities and at a faster rate.9
Some cancer cells operate in this manner because glycolysis allows the production of intermediates that can be used in various biosynthetic pathways, such as the genesis of nucleotides and amino acids, necessary during cell proliferation. The so-called Warburg effect, indeed, is also observed in embryonic tissues cells in the proliferative phase. Moreover, embryonic tissues, as well as proliferating tumor cells are not deficient in nutrients, so they can “waste” glucose molecules with a respiratory mechanism that is not as efficient as that of OXPHOS.10 Therefore, Warburg’s hypothesis that cancer cells change their metabolism due to a mitochondrial defect8 cannot be a general rule. Functional mitochondria are essential for the development of tumor cells, and this has been confirmed by the creation of ρo cancer cells, deficient in mtDNA or knocking down mitochondria transcription factor (Tfam). Indeed, these cells have reduced tumor formation in nude mice.2
Mutations that damage bioenergetic system enzymes have been found in cancer cells, as well as mutations on mtDNA. Mitochondrial mutations have been described for some tumor cells: mtDNA control region variants are associated with endometrial11 and cervical cancer12; mutation in NADH dehydrogenase subunit 3 complex I (ND3) gene has been associated with breast cancer risk13; a mtDNA cytochrome c oxidase subunit 1 (CO1) nucleotide variant has been linked with epithelial ovarian cancer.14
We can divide carcinogenic mutations that involve energy system into two classes: (1) mitochondrial enzyme mutations that have a negative effect on the OXPHOS mechanism that leads to the tumor phenotype development; (2) mutations that facilitate the adaptation of tumor cell to the microenvironment constantly changing over the disease progression.
Altered OXPHOS in cancer cells.
Most of mitochondrial enzyme mutations lead to loss of functionality of enzymes with consequent stabilization of hypoxia-inducible factor 1α (HIF-1α). HIF-1α is a transcription factor that, when stabilized, is translocated into the nucleus and causes a shift in energy metabolism from oxidative to glycolytic.15 Inhibition of SDH (Succinate Dehydrogenase, an integral mitochondrial inner membrane protein complex that oxidizes succinate to fumarate and transfers 2 electrons to coenzyme Q10) increases mitochondrial and cytosolic succinate levels, which inhibits α-ketoglutarate-dependent prolyl hydroxylases, inducing stabilization of HIF1α.16 Mutations that lead to loss of SDH functionality have been found in paragangliomas and pheochromocytomas.17 Mutations in fumarate hydratase (FH) gene can stabilize HIF-1α transcription factor as well. FH converts fumarate to malate and lack of function of FH results in increased levels of fumarate and succinate.18 Fumarate, like succinate has also been hypothesized to inhibit prolyl hydroxylases and stabilize HIF-1α.19 Homozygous null mutations in FH gene are associated with multiple cutaneous and uterine leiomyomatas and aggressive forms of renal cell cancer.20
Mutations in another family of mitochondrial enzymes, isocitrate dehydrogenase (IDH), results in an energy shift in tumor cells and has been characterized as mutations that result in the generation of IDH1 and IDH2 variants. The wild-type form of these enzymes allows the conversion of isocitrate in α-ketoglutarate while mutant variations reduce α-ketoglutarate to R(−)-2-hydroxyglutarate (R)-2HG). Consequently, (R)-2HG has been shown to cause epigenetic methylation, know to alter expression of target genes.21–23 Among the genes that are targeted is WNT gene.24 Wnt signaling activates mitochondrial biogenesis and oxidative phosphorylation (OXPHOS) gene expression.25 In conclusion, it is evident that mutations in mtDNA lead to mitochondrial dysfunction that consequently causes changes in nuclear gene expression, a mechanism called retrograde signaling.2 This mechanism likely permits tumor cells to adapt to stringent metabolic requirements and foster tumor progression.
IMPACT OF CANCER ON STROMA METABOLISM, “THE REVERSE WARBURG EFFECT”
Cancer cells can be viewed to adapt to unfavorable conditions such as altered and reduced energetic substrate, reduced oxygen tension, variation in environmental temperature, increased ROS toxicity, and exposure to conditions that induce apoptosis.26 The development of these adverse conditions can be attributable to the presence of the cancer cells and their impaired metabolism. An example that illustrates this phenomenon is observed in breast cancer cells. Stromal fibroblasts associated in breast cancer epithelial cells are glycolytic; this metabolism switch is induced by the H2O2 released by adjacent cancer cells. This results in loss of caveolin 1 by fibroblasts, which in turn causes induction of mitophagy in these stromal fibroblasts: this self-cannibalism reduces mitochondrial function and switches the fibroblast metabolism over to glycolysis. The lactate and ketones that are produced are utilized by cancer cells to feed mitochondrial energy production. This intercellular cooperation has been designated “reverse Warburg effect.”27 This study demonstrates that the metabolic alteration in the tumor cell is not always caused by a mitochondrial defect, but it can be born from mutations that favor the adaptation to a new environment. Paraphrasing Brandon et al26 “the tumorigenic mutations are analogous to human pathogenic mutations while the tumor adaptive mutations are analogous to the human regional adaptive variants.”
MITOCHONDRIA AND METABOLISM IN IMMUNE SYSTEM
There is no doubt that the cellular energy system, complex and finely regulated, is responsible not only for forming the fuel necessary for cells’ survival but also for other cellular processes, specific according to the cell type.28 Therefore, events that guide the plasticity of the mitochondria can cause consequences not only in terms of energy metabolism but also favoring a tumor microenvironment. In the following sections we will give some examples of how mitochondrial biogenesis, bioenergetic reactions, mitochondrial structures, and energy metabolites can cause changes in the immune system cellular activity and how the cells are affected in case they are defective.
DEFECT IN T CELL MITOCHONDRIA BIOGENESIS REPRESSES ANTICANCER RESPONSES
The presence of T cells within tumors, in a landmark study, was shown to correlate with improved clinical outcomes.29 Later the correlation was confirmed for many other tumor types including, for example, melanoma30 and breast cancer.31 T cells play a pivotal role in the antitumor immune response and as all other cells, needs a functional energy source to carry out its cellular functions. Therefore, it is necessary that T cells have an abundant supply of nutrients and the efficient machinery to generate energy. T cells, once activated, have an enormous demand for energy as they exponentially proliferate and synthesize inflammatory cytokines among other activities.
Scharping et al observed that tumor-infiltrating T cells (TILs) display a phenotype of metabolic insufficiency, prominently characterized by persistent loss of mitochondrial function and mass.32 The hypothesis is that chronic activation, associated with the anticancer response, represses oxidative metabolism. Inflammatory-mediated activation in cancerous state and chronic viral infections lead to an augmented energy demand that T cells are unable to satisfy.33,34,35 Chronic activation may drive the T cells on sustained reliance on glycolysis in tissue sites where most of the available glucose is consumed by tumor cells.36 Scharping et al propose that sustained activation of T cells in cancer promotes a defect in mitochondrial biogenesis due to a defect in PPAR-gamma coactivator 1α (PGC1α)-programmed mitochondrial biogenesis.32 Indeed, metabolic reprogramming of T cells through enforced PGC1α expression rescued mitochondrial function and induced superior antitumor responses characterized by cytokine production and tumor control. PGC1a is repressed by protein kinase B (Akt)37 and Sharping et al showed increased AKT activation in CD8+ TILs.32 Activation of the phosphatidylinositol 3-kinase (PI3K)—phosphatase PTEN—AKT signal transduction pathway drives changes in metabolism to promote cancer cell growth and proliferation.38 Indeed, AKT pharmacologic inhibition enables TIL expansion.39 Growth factor-dependent activation of PI3K leads to the activation of downstream effectors including Akt and the mammalian target of rapamycin (mTOR) that coordinates metabolic activities that support cellular biosynthesis. In normal cells, PI3K activation is tightly controlled by dephosphorylation of PIP3 by the phosphatase PTEN, a potent tumor suppressor.40 Together, mutations in PI3K signaling constitute one of the most common mutations in human tumors.38 Enhanced PI3K/Akt signaling selectively gives tumors an advantage through several pathways, including (1) increased surface expression of nutrient transporters resulting in increased uptake of glucose, (2) Akt-dependent stimulation of hexokinase and phosphofructokinase to drive glycolysis, (3) enhanced transcription of genes involved in glycolysis, and (4) enhanced protein translation through Akt-dependent mTOR activation.40
Exhausted T cells fail to secrete cytokines and express high levels of coinhibitory molecules like Programmed cell death protein 1 (PD-1) and cytotoxic T-lymphocyte associated antigen 4 (CTLA-4).41 PD-1 and CTLA-4 signaling inhibit glycolysis, mechanism necessary to delineate an active phenotype for T cells. Distinctly, unlike CTLA-4 signaling, PD-1 signaling can promote fatty acid oxidation, commonly observed in memory T cells.42 Additionally, CTLA-4 interacts with protein phosphatase 2 a key enzyme that inhibits mTOR signaling, a crucial signaling cascade for T cell differentiation and function.43
THE IMPACT OF MITOCHONDRIAL MASS IN T CELL PHENOTYPE
Since efficient metabolism is fundamental for normal cellular function, it is not surprising that an inadequate antitumor response is also caused by glitches in the gears of the cellular drivetrain, such as the reduction of the number of mitochondria as discussed. Not only in the number of mitochondria, but also the mitochondrial shape is involved in lymphocyte activity. Mitochondria are not inert during metabolic changes in T cells. Indeed, T lymphocytes can be differentiated into T naive, T effectors, and T memory based on the mitochondrial mass analysis. Naïve T cell have fragmented and round mitochondria44 and during activation in effector T cells (Teff) there is an increase in the fission rate and, therefore, the increase in the number of mitochondria, with looser cristae.45 Looser cristae may lead to the dissociation of ETC supercomplexes46,47 that may result in less efficient OXPHOS and promoting aerobic glycolysis. This could assist T cell activation and proliferation.45 Without tight cristae and efficient ETC activity, electrons may linger in the complexes resulting in the generation of ROS.48 In contrast, memory T cells (Tm cells) have greater numbers of mitochondria than Teff and are elongated as a result of decreased fission rate.45 Fused, elongated mitochondria have efficient ETC super complex formation and OXPHOS, which may assist in cell survival and functions.49
METABOLIC STATUS IMPACT T CELLS AND MACROPHAGES DIFFERENTIATION
As we have mentioned, OXPHOS has been an intensely studied mechanism in recent years in the oncology field: OXPHOS inefficiency in favor of glycolysis in tumor cells is considered a hallmark characteristic. The metabolic switch toward glycolysis from OXPHOS leads to polarization toward a proinflammatory tumor microenvironment. Accumulation of the metabolite succinate leads to the stabilization of the HIF-1α and, subsequently, to the expression of a set of genes including genes that permit the switch toward a glycolytic metabolism. In the previous paragraph, we reviewed that, for Teff cells, the glycolytic metabolism is influenced by the mitochondrial conformation. Additional evidence includes the stabilization of the HIF-1α factor, which not only allows glycolytic metabolism, but also allows differentiation of cells with proinflammatory activity. HIF-1α enhances Th17 development through direct transcriptional activation of the RAR-related orphan receptor γT which upon upregulation attenuates regulatory genes involved in regulatory T cells development by binding forkhead box P3 (Foxp3) and targeting it for proteasomal degradation.50 HIF-1α expression in T cells can be induced both by hypoxic and nonhypoxic stimuli, including T cell receptor-triggered stimuli.51
In addition to Teff cells, HIF-1 also plays a role in the inflammatory response in macrophages.52 Macrophages exposed to hypoxia accumulate both HIF-1α and HIF-2α, and overexpression of HIF-2α in tumor-associated macrophages (TAMs) is specifically correlated with high-grade human tumors and poor prognosis. HIF-1α stabilization by succinate occurs in lipopolysaccharide-activated macrophages, a process driven by signaling from Toll-like receptors TLR4.53 Stabilization of HIF-1α followed by upregulation of genes including IL-1B, which encodes IL-1β, allows the metabolic shift toward glycolysis.54 Although succinate and SDH activity are essential for the generation of a proinflammatory response in lipopolysaccharide-activated macrophages, prolonged SDH activity and ROS production result in hyperinflammation state. To control this inflammatory response, macrophages use the metabolite itaconate, which can inhibit SDH function and the subsequent proinflammatory response.28
While M1 cells use aerobic glycolysis, M2 macro-phages use FAO for growth and proliferation.55 M2 macrophages have roles in the regulation of inflammation and tissue repair52 and FAO has been directly linked to their antiinflammatory phenotype. Indeed, inhibition of carnitine palmitoyltransferase 1 (CPT1), a rate-limiting enzyme of FAO, limits M2 activation in response to IL-4.56 CPT1 catalyzes the transfer of long-chain acyl group of the acyl-CoA ester to carnitine, thereby shuttling fatty acids into the mitochondrial matrix for β-oxidation. Induced changes in macrophage metabolism are IL-4-dependent due to the signal transducer and activator of transcription 6 factor and the expression of PGC1β, associated with mitochondrial biogenesis.32,57 The fuel for the additional mitochondria is recovered from a futile cycle of CD36-dependent uptake of triglycerides, which increases the spare respiratory capacity (SRC). SRC is the extra capacity available in cells to produce energy in response to increased stress or work.56 With these examples, we suggest that glycolysis mechanism is observed during development of an inflammatory environment, with polarization toward M1, while the FAO mechanism dominates during development of an anti-inflammatory environment through polarization toward M2 macrophages (Fig 1). However, inhibition of CPT1 has little negative effect on IL-4-induced M2 polarization in humans, suggesting that there is an alternative pathway for M2 development.58
Fig 1.
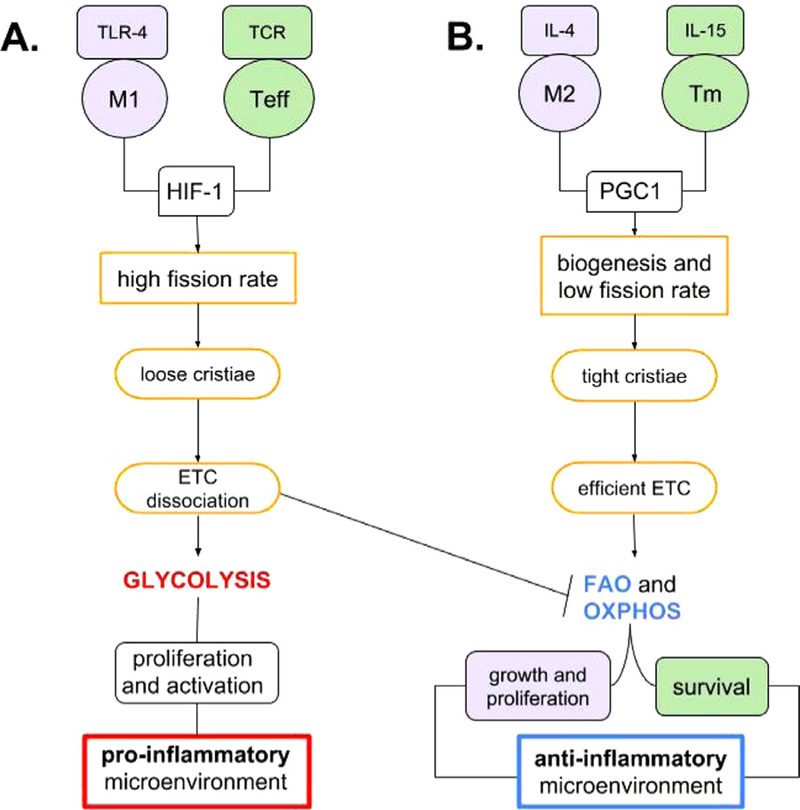
Glycolysis/FAO/OXPHOS metabolic pathways influence pro-or anti-inflammatory microenvironment. (A) Stabilization of the HIF-1 factor allows glycolytic metabolism and polarization toward the pro-inflammatory environment. (B) Production of PGC1 (Peroxisome proliferator-activated receptor gamma coactivator 1) boosts the fatty acids metabolism, OXPHOS, and promotes anti-inflammatory microenvironment. IL-4 and IL-15 increase the production of PGC1, efficient ETC supercomplex formation and OXPHOS. This promotes the survival of Tm cells and polarization of the macrophages toward anti-inflammatory M2. OXPHOS, oxidative phosphorylation.
Fatty acid metabolism is similarly vital for Tm functions and T regulatory differentiation.59 IL-15, a cytokine that promotes CD8+ Tm cell development and maintenance, promotes mitochondrial biogenesis and the expression of CPT1a.60 Thus, as well as for IL-4 in M2 macrophages, IL-15 increases SRC and oxidative metabolism of CD8 + Tm cells.
In contrast, in the plasmacytoid dendritic cells (pDC), FAO is observed during the antiviral response. pDCs are a specialized subset of DCs that release high levels of Type I interferons (IFN-I) in response to infection.61 Stimulation of pDCs with CpG or recombinant IFN-α upregulates FAO and OXPHOS. Inhibition of CPT1 decreases CpG-induced OXPHOS and the production of IFN-α, TNF, and IL-6 in Pdc.62
DAMAGE OF MTDNA BY OXPHOS/ROS AND MUTATIONS
Under normal conditions, the main source of mtDNA damage is generated by the OXPHOS process that generates ROS. Excess ROS such as superoxide, hydrogen peroxide and hydroxyl radicals can damage mtDNA directly or serve as precursors of oxidants (such as 4-hydroxynonenal, malondialdehyde, acrolein, and crotonaldehyde) that also induce mtDNA aberrations.63 ROS and oxidants not only affect DNA structures but also modify essential mitochondrial proteins and lipid membranes (Fig 2). Without external sources of oxidative stress, normal level of OXPHOS and ROS/oxidant generation is counteracted by enzymatic and nonenzymatic antioxidants. The antioxidant system, consisting of a set enzymes and metabolites converting O2●‒ into H2O2 and H2O, is regulated by transcription factors sensitive to the redox state, therefore cells can quickly adapt to moderate oxidative stress.64
Fig 2.
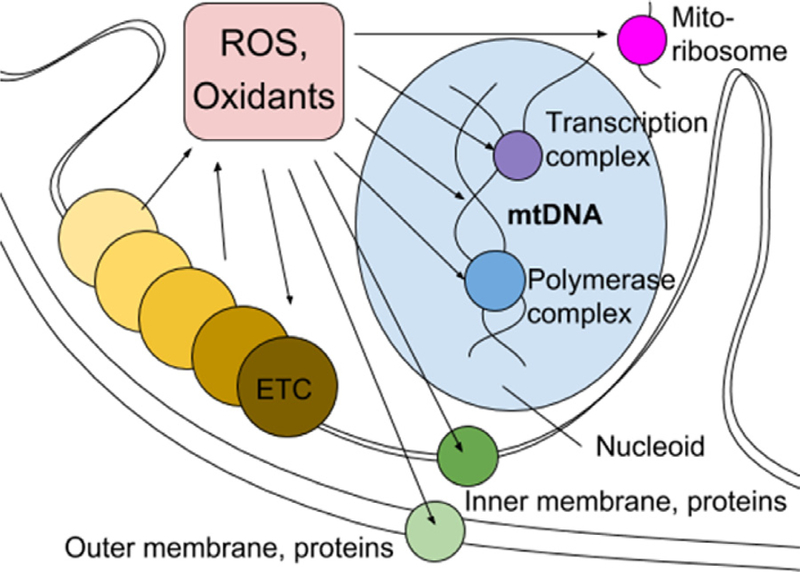
Electron transfer chain (ETC) generates reactive oxygen species (ROS). In excess ROS and oxidants, their metabolites, cause mutations in mtDNA and proteins with “virtual opportunity” of damaging all essential systems in the mitochondria.
Types of mtDNA damage include single-strain breaks, abasic sites, oxidative base damage, and DNA cross-links. Repairing of abasic sites and oxidized/altered bases may lead to generation of SSBs that lead to stalled or blocked replication and transcription. Blocked replication/transcription processes as well as the double-strand breaks can induce mtDNA degradation. In turn, degradation of the aberrant mtDNA due to the presence of multiple functional mtDNA copies within the cell is a way of preserving mitochondrial genetic material from mutations, in addition to the repair tools.63,65–68 Cells survive and maintain the OXPHOS function with a high mtDNA mutation load: up to 80%−90% of the mtDNA can be mutant,69 up to 40% can be deleted70; when the mutation load is above the threshold mitochondria undergo fragmentation (multiple fission events) and dysfunctional parts are eliminated. The mechanism of mtDNA degradation after severe damage with double-stranded breaks is not mediated by mitochondrial nucleases and is not yet known.68
Though both mtDNA damage and repair can be sources of mutations, most of the mtDNA mutations that accumulate with age are most likely the result of mitochondrial polymerase POLγ nucleotide misinsertion errors that occur just once per 500,000 nucleotides inserted.71,72 Indeed, the ROS/oxidants mostly induce transversion mutations G to T that do not significantly increase with aging, while older people accumulate the transition mtDNA mutations demonstrated by ultrasensitive sequencing analysis of point mutations frequencies in young (<1 yr) and old (>75 yr) individuals.73 The very high fidelity of POLγ and a small mitochondrial genome size (16,569 base pairs) make mtDNA mutations rare events. Accumulation of exclusively point mutations that cannot affect mitochondrial functions is explained by so-called “purifying selection” through autophagy of dysfunctional mitochondria.74
TURNOVER OF MITOCHONDRIA
Mitochondria biogenesis and quality control.
The “quality control of mitochondria” is the balance between several processes that aim to provide the cell with a functional mitochondrial system: mitochondria biogenesis, lysis of dysfunctional proteins, vesicular transport to the lysosomes, mtDNA degradation, fusion and fission, and mitophagy (autophagy of the mitochondria). To accomplish this quality control communication between the mitochondria and nucleus is essential.
Mitochondrial biogenesis begins with the activation of signaling cascades and transcriptional complexes that promote the de novo synthesis of mitochondrial proteins and replication of mtDNA, recruitment and transport of the new proteins to mitochondrial compartments, and assembly of expanding mitochondrial structures. Therefore, growth of mitochondrial mass starts from enlargement of mitochondria, which later undergo fission (or division) events increasing the number of mitochondria.
Known events that activate mitochondrial biogenesis include the lack of energy sensed as a high ADP/ATP ratio and cell functions activation sensed as the presence of growth factors, increase of cAMP or cytoplasmic Ca2+ ions. All these events are shown to lead to increased activity of PGC1a, with consequent increase of mitochondrial biogenesis. Thus, low levels of ATP lead to increased activity of 5׳-adenosine monophosphate-activated protein kinase AMPK that phosphorylates and activates PGC1α.75–77 Though AMPK downregulate functions of mechanistic target of rapamycin (mTOR),78 independent activation of mTOR by growth factors can also increase PGC1α activity. High levels of cAMP activate cAMP response element-binding protein, that inhibits the mitophagy-related LC3-II molecule and upregulates the expression of PGC1α. PGC1α activity also can be upregulated through p38 mitogen-activated kinase and calcium/calmodulin-dependent kinase activation upon increase of Ca2+ levels.76,77
PGC1α regulates major transcription factors regulating biogenesis of mitochondria: nuclear respiratory factors NRF1 and NRF2 (also known as NFE2L2). Both of them upregulate mitochondria-related genes, including proteins providing respiratory function, heme synthesis, mitochondrial transport, tRNA synthesis, ribosomal proteins, etc. Mitochondrial biogenesis is necessarily associated with proper functioning of protein transport complexes, given that more than 90% of these mitochondrial proteins are encoded in the nucleus of the cell.76
For effective protein turnover mitochondria use their separate proteolytic system: protein quality control proteases present in the mitochondrial compartments. Excess unfolded proteins in mitochondria induce the mitochondrial unfolded protein response mtUPR and includes upregulation of the chaperones HSP60, HSP10 and mtDnaJ, and protease ClpP expression in the nucleus, together with upregulation of mitochondrial proteases YME1L1, PMPCB, and other proteins.79 Outer membrane proteins can be degraded by the proteasome, dysfunctional proteins aggregates on the focal sides of outer membrane can form mitochondria-derived vesicles (MDV) that fuse with lysosomes.80 Damaged mitochondria can be removed through the acquisition of lysosomal markers or enveloped by autophagosomes via mitophagy.81
Vesicular transport of the MDVs to the lysosomes is a complimentary tool to the mechanisms of fission and mitophagy, allowing the cell to recycle small portions of mitochondria.82 The Parkinson’s disease-associated proteins Vps35, Parkin, and putative kinase protein 1 (PINK1) are involved in the biogenesis of a subset of these MDVs, linking this novel trafficking pathway to human disease. A working hypothesis of PINK1/Parkin-mediated MDV formation suggests that unfolded, oxidized proteins within the matrix (triggered by ROS or failure of the protein complex assembly) leads to protein aggregation. As an example, local oxidation of cardiolipin affects import channels and PINK1 accumulates at these failed channels to induce recruitment and activation of Parkin. After that, the ubiquitination activity of Parkin is required for the generation and release of MDVs. Another example of vesicular transport from the mitochondria is the recycling of the protein dynamin-related protein 1 (Drp1) after the mitochondria fission/division process.83
The mitochondria of a cell comprise a dynamic system that undergoes a series of fusion events between individual mitochondria and fission events—separation of mitochondria to smaller organelles. Fusion can help mutated mitochondria to recover the loss of function by utilizing the genetic material of fusion partner. Fission allows the cell to detect dysfunctional parts of mitochondria with low membrane potential, encapsulate, and eliminate them by mitophagy.84,85
In mammals mitochondrial fusion is mediated by proteins mitofusin 1 (Mfn1), Mfn2, and optic atrophy 1 (Opa1); Mfn1 and Mfn2 are involved in the merger of the outer membranes, Opa1 is responsible for the merger of inner membranes. Degradation of Mfn1/2, modified by ubiquitination, and cleavage of Opa1 are the factors that prevent fusion. Mitochondrial fission is a result of constriction of the site dividing mitochondria in 2 parts. Initial constriction is mediated by inverted formin 2 inducing polymerization of actin on the endoplasmic reticulum and later ER-mitochondrial interaction.86 Drp1, a cytosolic protein, is recruited to the mitochondrial membrane in which its conformational change promotes constriction and fission. Recently another protein shown to be a key player in the final fission is dynamin 2.87 Mitochondrial fission is an intricate process regulating both cellular and organ dynamics, including development, apoptosis, acute organ injury, and various diseases.88 Inhibition of mitochondrial fission prevents mitophagy, therefore both of these processes are essential for mitochondria quality control.89
Mitophagy.
Mitophagy allows for the constant turn-over of mitochondria and helps to maintain their optimal number or to eliminate them (such as during red blood cell differentiation) and is associated with the elimination of paternal mitochondria after the fusion of a spermatozoon with the oocyte. Mitophagy can be activated via different pathways. One of the best characterized pathways is mediated by 2 key regulators: PTEN-induced PINK1 and Parkin, E3 ubiquitin ligase.
Parkin-dependent mitophagy activation.
PINK1 is constantly transferred to mitochondria and is constantly transported in the inner membrane by the TOM complex and degraded there by presenilin-associated rhomboid-like protein in functional organelles.90 In unhealthy mitochondria, the inner mitochondrial membrane becomes depolarized. This membrane potential is necessary for the TOM complex mediated protein import and in depolarized mitochondria PINK1 is no longer imported to the inner membrane. PINK1 concentration in the outer mitochondrial membrane increases which lead to autophosphorilation91,92 and recruitment of cytosolic Parkin to the mitochondria, where PINK1 phosphorylates the Ubl domain of Parkin which activates its E3 ligase activity. Active Parkin ligates the ubiquitin chains to the mitochondrial outer membrane proteins (tethering itself also), PINK1 phosphorylates them, and this event can cause recruitment of new Parkin molecules (Fig 3).93 Parkin interacts with the adaptor protein, activating molecule in Beclin-1-regulated autophagy (Ambra1), which activates PI3K next to the mitochondria to assist in mitophagy,89 but the main role of Parkin in mitophagy activation, remain is formation of Lys63-linked ubiquitin chains that promote substrates degradation, amplify the mitophagy activation by serving as the new substrates for PINK1, and together with PINK1, play major effector roles in mitophagy.85,94
Fig 3.
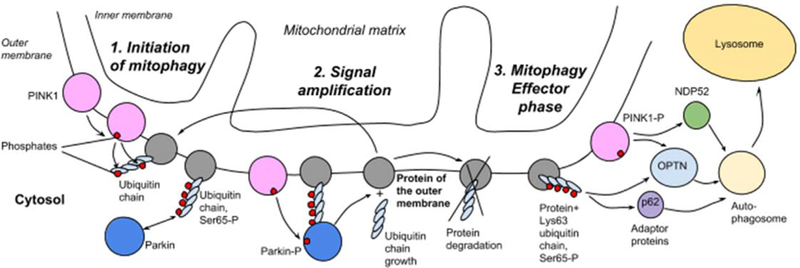
Mitophagy initiation, amplification and effector stages. 1. Initiation. In a functional mitochondria PINK1 is short-lived, but in the dysfunctional ones it accumulates and autophosphorylates and recruit Parkin. 2. Amplification. Phosphorylated by PINK1, Parkin ubiquitinates the mitochondrial outer membrane’s proteins: new substrates for PINK1-Parkin signal amplification for mitophagy activation. 3. Effector stage. Phosphorylated PINK1 and Lys63 ubiquitin chains serve as receptors for phagophore recruitment and mitophagy. PINK1, putative kinase protein 1.
Mitophagy effector stage.
Ubiquitination of mitochondrial proteins promote the recruitment of Ub-binding autophagy receptors such as p62 and optineurin, which in turn elicit the targeting of damaged mitochondria to LC3-positive phagophores for clearance in the lysosomes.95,96 PINK1 is bound by NDP52 and optineurin that not only interact with LC3, but also recruit autophagy factors ULK1, DFCP1, and WIPI1 to the mitochondria’s focal spots94 (Fig 3).
Parking-independent mitophagy activation.
Damaged mitochondria have elevated levels of Fundc1 and Nix on the surface; these proteins can directly recruit LC3-positive phagophores to the mitochondria. Mitochondrial depolarization also triggers ubiquitin ligases Smurf1 and Mul1 that promote mitophagy, presumably ubiquitinating the mitochondrial proteins. Other less studied mechanisms of mitophagy are: Atg-independent mitophagy, Ulk1-dependent and Atg5-independent macroautophagy, autophagy via the 15-lipoxygenase pathway and lysosomal invagination/direct interaction with mitochondria (microautophagy).89
Interestingly, growing body of evidence has shown the inactivation of Parkin in a broad panel of cancers. Indeed, Parkin has been shown to be downregulated in multiple cancer cell lines and primary tumors.97–99 Moreover, Parkin-deficient mice are more susceptible to tumorigenesis100 and the ectopic expression of Parkin restores normal phenotype suggesting a tumor-suppressive role for Parkin101 (for detailed review, see102). As Parkin is often downregulated in tumors, recently Villa et al identified an E3 ubiquitin ligase (ARIH1/HHARI) that triggers mitophagy in cancer cells in a PINK1-dependent manner. They found that ARIH1/HHARI polyubiquitinates damaged mitochondria, leading to their removal via autophagy. Importantly, ARIH1 is widely expressed in cancer cells, notably in breast and lung adenocarcinomas; ARIH1 expression protects against chemotherapy-induced death.
Mitochondria DAMPs and mtDNA mutations in innate and adaptive immunity
Immune system constantly detects and destroys the cells that are mutated or transformed, this is what is termed cancer immuno-surveillance.103 Damage and mutations accumulated in mitochondria can drive both innate104,105 and acquired immune responses,106 thus contributing to immune surveillance. Additionally, mitochondria produce many of the danger-associated molecular patterns, DAMPs, (including mtDNA) therefore mitochondrial functions are linked to regulation of immune responses.107,108
Innate immunity.
Mitochondrial but not genomic DNA of the tumor cells is found in abundance in the cytosol of tumor-infiltrating DCs. There it can upregulate the cross-priming function of DCs by activating the STING pathway.109 Lymphocytes (B cells, T cells, and natural killer cells) activated by CpG and non-CpG oligodeoxynucleotides of class C can release extracellular mtDNA; extracellular mtDNA can activate TLR9 or cGAS/STING pathways in the leukocytes and especially neutrophils, inducing proinflammatory and type I IFN responses.110 Extracellular mtDNA also binds IL-26 and this complex can be taken up by myeloid cells where it activates proinflammatory STING and P2Y1 receptor-cAMP pathways111 Inside the cells mtDNA released from the mitochondria can activate immune responses interacting with cGAS, TLR9 and inflammasome proteins NOD, LRR, Pyrin domain-containing protein 3 (NLRP3) and absent in melanoma 2 (AIM2).105
Adaptive immunity.
Cells with damaged/mutated mitochondria have increased expression of MHC class I that may facilitate exposure of the neoantigens to immune system and clearance of the affected cells.112 Indeed the mutations of mtDNA are shown to be a possible source of immunogenic self-peptides initiating the T cell-mediated immune responses.113,114 Autophagy can produce the antigenic peptides conjugated with both MHCI and MHCII molecules,115,116 but antigen presentation mechanisms activated in mitophagy are different. Mitophagy activators PINK1 and Parkin do not activate, but in contrary, suppress mitochondrial antigens presentation (MitAP). This can be explained by PINK1/Par-kin-dependent suppression of MDVs production: the latter provide mitochondrial antigens transport to the lysosomes with consequent presentation of the antigenic peptides in MHC class I complex.106
Targeting mitochondria for cancer therapy.
Mitochondria are responsible for cell energy metabolism, ROS production and induced cell death thus it is not surprising that functional defects can contributes to development and progression of cancer.117 The recent understanding of the crucial role played by the mitochondria in cancer has led to the development of different therapeutic approaches focused on this organelle. Agents that target this organelle has been extensively studied in the context of cancer.118–121 In this section, we discuss some of the most relevant mitochondrial targets in cancer therapy and give a few examples of drugs that are currently being tested in clinical trials.
Targeting mitochondria metabolism.
Mitochondria are defined as the powerhouse of the cell; they are the major source of ATP, control redox homeostasis, they are involved in oncogenic signaling, innate immunity, and cell death.1 Under physiological conditions, ATP is synthesized through oxidative phosphorylation by the mitochondria. However, cancer cells switch their energy metabolism into aerobic glycolysis, a phenomenon known as the “Warburg effect.”2 Warburg hypothesized that this metabolic switch was due to defects in mitochondria, however, recent studies show that depleting mitochondria from cancer cells decreases tumor progression.122 Glucose is one of the main key players in cancer progression and is the promoter of tumor invasion and metastasis and glycolysis is a rapid way to produce ATP supplying ample energy for proliferation of malignant cells. This is likely why in both hypoxic and nonhypoxic conditions; tumor cells utilize glycolysis.123 Therefore, reversing the hyperglycolytic state of cancer cells in order to induce cell death is a possible therapeutic approach.
The first attempt to reduce glycolysis is to block glucose uptake in malignant cells. For example, WZB117 a bis-hydroxybenzoate compound that acts as a fastacting, irreversible blocker of glucose transport by GLUT1, is a drug able to block Glucose Transporter 1 (GLUT1) activity reducing glucose transport inside the cell lowering glycolysis metabolism in the tumor. This effect is mediated by the reduction of ATP levels in the cell and induction of endoplasmic reticulum stress and eventually cell-cycle arrest.124 This drug has been tested in combination therapy with either paclitaxel or cisplatin displaying a synergistic effect in vitro and in vivo against lung and breast cancer cell lines.125 Another drug that is able to decrease glycolysis metabolism in cancer cells is 2-deoxy-d-glucose (2DG). 2-DG is a glucose analog that competes with glucose-6-phosphate to inhibit glucose metabolism. However, it has been observed that its 2-DG activity is not sufficiently potent to significantly reduce glucose uptake and thus induce cell death.118 2-DG showed a significant increase in cytotoxicity of cisplatin in human head and neck cancer by enhancing ROS levels.126 Two different clinical trials have been performed to test 2-DG anticancer activity. However, there are concerns that 2-DG might compromise the glycolytic metabolism of the brain and of the heart.121 Another example is Dichloroacetate (DCA) which is a small molecule that inhibits pyruvate dehydrogenase kinase. DCA is able to revert the hyperglycolytic state of cancer cells to normal glucose oxidation metabolism, in this manner, tumor cells become more sensitive to apoptotic signals and their proliferative capacity is impaired. DCA has been tested as a monotherapy in a Phase I study in patients with recurrent malignant brain tumors.127
Metformin is a biguanide hypoglycemic oral drug that is currently used to treat type 2 diabetes. Metformin also acts as an inhibitor of the mitochondrial electron transfer chain Complex I and activator of AMP-activated protein kinase, therefore metformin can reduce ATP production.128,129 Recent epidemiological studies have shown that patients with type 2 diabetes treated with metformin to control their blood glucose levels have a reduced risk to develop cancer.130 Later this drug has been proven effective therapy against cancer in numerous cancer clinical trials on diabetic and nondiabetic cancer patients.131 Two mechanisms can explain the antitumor activity of metformin.132 Decreasing blood glucose levels lead to decreased levels of insulin, serving as a mitogen in some cancers.133 Another mechanism is inhibition of the respiratory complex I by metformin and its analogs.134 In low glucose availability metformin decreases ATP production and induces cell death; when glucose concentration is high metformin inhibits ATP levels and induces AMPK activation that reduces activity of mTOR necessary for proliferation.135 Despite the number of clinical trials assessing the positive effect of this drug, results are still to be considered inconclusive.136
Targeting ROS.
Reactive oxygen species are important for the cell to perform normal functions; however, when mitochondrial metabolism is accelerated the result is a dangerous accumulation of ROS that can lead to neoplastic transformation.137 Mutations in nuclear (nDNA) and mtDNA in cancer cells may affect components of the ETC, leading to an increase of ROS levels.138 Moreover, malignant cells are able to escape apoptotic signals, thus there is an over-accumulation of ROS that triggers genomic instability and cancer proliferation.139 Not surprisingly, overproduction of ROS is also considered a hallmark of cancer.
Elesclomol sodium (STA-4783) is an example of a drug targeting ROS levels in cancer cells. Its role is to inhibit electron transport flux and enhance ROS production inducing oxidative stress in both transformed and healthy cells. However, as tumor cells have already elevated levels of ROS, this drug will be able to induce cytotoxicity selectively in malignant cells, resulting in activation of apoptotic cell death.140 STA-4783 alone and in combination with paclitaxel showed promising data in phase I/II clinical studies in patients with refractory solid tumors.141,142 However, a phase III study in patients with melanoma has been discontinued for safety reasons.121 Lastly, β-phenylethylisothiocyanates (PEITC) is a thiol modifier that interacts with ROS regulators. PEITC targets CIII complex decreasing OXPHOS and inducing ROS production to trigger cancer cell apoptosis.143 PEITC has also been tested in vitro in combination with metformin in ovarian cancer tumors and significant anti-proliferative and cytotoxic effect have been observed.144 Recently a Phase II clinical trial has been completed in which PEITC was used as a lung cancer-preventing agent in smoking subjects.
Targeting the permeability transition pore complex.
The permeability transition pore complex (PTPC) is a highly dynamic supramolecular complex found on the mitochondria membrane and is responsible for mitochondria membrane permeabilization. PTPC is composed of different molecular components; voltage-dependent anion channel in the outer membrane, the adenine nucleotide translocase (ANT) in the mitochondrial inner membrane and cyclophilin D in the mitochondrial matrix. Additional PTPC-interacting proteins include the peripheral benzodiazepine receptor in the outer membrane and hexokinase (HK), which makes contact with the mitochondrial outer surface from the cytosol.145 HK is an inhibitor of the mitochondria membrane permeabilization. PTP opening normally occur in dysfunctional mitochondria and its occurrence starts a process that leads to cessation of ATP synthesis, Ca2+ release, inhibition of respiration and, in vitro at least, pyridine nucleotide depletion, matrix swelling, mitochondrial depolarization; mobilization of cytochrome c and eventually release of proapoptotic proteins such as cytochrome c itself, endonuclease G and AIF.146 To induce mitochondrial membrane permeabilization, 2 strategies can be utilized: a direct strategy that targets different components of the PTPC using drugs that either increase cytosolic levels of Ca2+ or trigger ROS production; or indirect permeabilization by depletion of endogenous inhibitors of PTCP like glucose, phosphocreatine, and glutathione.121
The most promising drug targeting ANT that efficiently triggers apoptosis is lonidamine or 1-(2,4-dichlorobenzyl)-1-H-indazole-3-carboxylic acid.147 Lonidamine is an ANT ligand that acts as a HK and induces a conformational change in the ANT leading to mitochondrial channel formation. It has been reported that lonidamine is able to target the respiratory activity of complex II, suppressing the formation of fumarate and malate leading to the accumulation of succinate in the treated cells.148 Lonidamine enhances the apoptotic response to cisplatin, cyclophosphamide, doxorubicin, paclitaxel, melphalan, and γ-irradiation both in vivo and in vitro. This drug has been tested in the early phase clinical trials but the study was terminated due to the lack of benefits for the patients.149 4-(N-(S-glutathionylacetyl)amino) phenylarsenoxide (GSAO), a glutathione-coupled trivalent arsenical compound, has the ability to cross-link critical cysteine residues of ANT to inhibit the antiporter activity of ATP/ADP, ROS accumulation and cytosolic ATP depletion, mitochondrial depolarization, and apoptosis. GSAO targets preferentially proliferating cells due to the high Ca2+ levels and elevated respiration rates in mitochondria, which make them more susceptible to PTPC. However, a clinical trial on patients with solid tumors did not show any significant effect.150 PENAO (4-[N-[S-penicillaminylacetyl]amino] phenylarsonous acid) is a second generation ANT inhibitor that accumulates in the tumor cells more rapidly than GSAO, resulting in enhanced antiproliferative activity and increased anti-tumor efficacy in mice.151 There is currently a Phase Ia clinical trial ongoing using a continuous intravenous infusion of PENAO in patients with solid tumors.152
Targeting mitochondrial outer membrane permeabilization.
Mitochondrial outer membrane permeabilization (MOMP) is the process that mediates intrinsic apoptotic pathway in cells. Apoptosis impairment serves a critical feature of the tumor cells and makes them more chemotherapy resistant. MOMP is one of the most important mechanisms of apoptosis. Restoring normal apoptosis mechanisms in the tumor cells, including mitochondria-dependent apoptosis, is a logical approach to treat cancer. MOMP is under control of the Bcl2 family proteins, this family consists of BH3-only proapoptotic proteins and prosurvival proteins. Major proapoptotic players are Bax and Bak able to form the pores, they can be activated by other proapoptotic proteins of Bcl2 family; prosurvival family members are able to inhibit Bax and Bak.153,154 In cancer cells, Bcl-2 and Bcl-XL antiapoptotic proteins are overexpressed and able to sequester BAX and BAK, inhibiting cell death.155
One of the best characterized drugs used to restore MOMP in cancer cells is ABT-737. This molecule binds to BCl-2, BCl-Xl, and BCl-w mimicking BH3-only proteins, antagonists of cell death.156 The drug induces cell death through the intrinsic pathway of apoptosis and depends on expression of the BCl-2 family proteins.157 ABT-737 has been shown to enhance conventional chemotherapy and radiotherapy treatment in hematological and solid tumors.158,159 However, ABT-737 has presented delivery problems and moreover, when Mcl-1, another prosurvival relative, is expressed in cancer cells, this drug encounters resistance due to its inability to bind Mcl-1.157,160 An orally available derivative is known as ABT-263 (Navitoclax). Its activity has been observed in SCLC xenografts and in combination with chemotherapy agents this drug is in preclinical studies and phase I studies in B cell lymphoma, multiple myeloma, and SCLC.161–163 However, Phase II clinical trials showed a limited efficacy of this drug against advanced and recurrent SCLC.164 Gossypol (AT-101) is a natural phenolic compound that can simultaneously inhibit BCl-2, BCl-Xl, BlC-w, and MCl1.165 It has been reported to be active against prostate cancer in a Phase I/II clinical trials.166 Phase II clinical trial has just been completed to treat patients with progressive or recurrent glioblastoma multiforme.121 Apogossypol (NSC736630) is a semisynthetic advanced version of gossypol with superior antitumor activity and reduced toxicity both in vitro and in vivo.167
Mitochondria drug delivery strategy.
The challenge that comes with the development of mitochondria-targeting drugs involves the necessity to generate an appropriate way to deliver the desired molecule to its intended target. Different strategies have already been assessed; delocalized lipophilic cations are attracted by the negatively charged mitochondria matrix and thus are able to cross the membrane and accumulate within the mitochondria.168 The only concern when this delocalized lipophilic cations are used to deliver drugs to the mitochondria is that they can be toxic for the organelle when accumulated in high concentration also in normal tissues.169,170 Another strategy that has been considered is mitochondria targeting sequence (MTS)-containing peptides. They are nuclear-encoded mitochondrial proteins that contain an MTS of 20–40 amino acids recognized by receptors on the mitochondrial membrane. This delivery approach has been successfully used to transport chemical cargos to the mitochondria,171,172 however, the size and the solubility of MTS is a major limitation for this approach.173,174 Synthetic peptides and amino-acid-based transporters have been recently evaluated as an alternative drug delivery method.173,175 Lastly, vesicle-based carriers have been considered to bring large or impermeable cargos to the mitochondria. These strategies and many others have already been extensively discussed in the literature.173,176
Mitochondria and new approaches to cancer immunotherapy.
The ability of the adaptive immune system to distinguish self from nonself is established during central tolerance development, during which time self-reactive clones of T and B cells are either eliminated or develop as immune regulatory cells. Other positively selected surviving cells recognize non‒self-entities, notably infectious agents and cancerous tissues that display mutated proteins, promising active immunotherapy approaches including cancer vaccines such as dendritic cell therapy and peptide vaccination, are as effective as the Tumor Associated Antigens (TAA) these vaccines target. Potential TAA selection for active or passive immunotherapy strategies such as vaccine and chimeric antigen receptor T cell therapy respectively, must meet a number of criteria. They must be immunogenic, abundantly expressed by tumor cells, not be expressed in normal tissue (to limit off-target effects) and (preferably) functionally important for the tumor (so that expression is unlikely to be lost).
Chen et al suggested that somatically mutated self-proteins are involved in autoimmune disease development.113 To demonstrate their hypothesis, PBMCs were collected from healthy donors and autoimmune patients with systemic lupus erythematosus or ankylosing spondylitis, T cells were stimulated with either mitochondrial self-peptides or mitochondria-mutated peptides; T cells activated by the mutated self-peptide were cross-reactive with the self-peptide both in healthy and autoimmune patients as measured by IFNγ levels.113 This study suggests that somatic mutations may participate in the induction of autoimmune diseases by antigen presentation. Interestingly, many different types of cancers harbor somatic mitochondrial DNA mutations117,177,178 and consequently an increased immune response has been observed against mutated mitochondrial peptides; for example, mitochondrial cytochrome b peptide derived from melanoma cells of patients is recognized by CD4+ T cells.179
Mitochondrial antigen presentation has been proposed as a possible cause for autoimmune disease development. In 2016, Matheoud et al published a study in which they show how Pink1 and Parkin proteins are involved in adaptive immune responses and suggested the possibility of autoimmune mechanisms involved in Parkinson disease (PD) antigen presentation.106 Parkin is one of the most important players in the development of PD; this protein is an E3 ubiquitinase that is able to mediate mitophagy in conditions of stress or damage of the mitochondria. Parkin is activated by phosphorylation by Pink1, an outer membrane mitochondrial surface protein. In PD, Parkin is mutated and subsequently, Pink1 is impaired in the activation of Parkin ubiquitinase functions.180 The loss of Pink1/Parkin activity leads to a significant reduction of mitophagy.181 Interestingly, Matheoud et al observed that mitAP is enhanced in PD, likely as a response to the downregulation of mitophagy. MitAP relies on mitochondrial-derived vesicles which are able to transport the damaged mitochondria to late endosomes where peptides are loaded on MHC I molecule and presented on the surface of the cell. The conclusion that was proposed is that Parkin and Pink1 are able to inhibit the immune response in PD.106 This new finding may be the first step to link a neurodegenerative disease like PD to autoimmunity.
Moreover, Voo et al provided the first example of a mitochondrial immune target for CD4+ T cells expanded from a melanoma patient. The tumor infiltrating T cells expanded, by high dose IL-2 from such patient, demonstrated recognition of a peptide translated from an alternative open reading frame of the mitochondrial cytochrome b (cyt b).182
With these studies providing the sound rationale that cancer patients bearing mutations in mitochondrial DNA and tumor-associated mitochondrial antigens (TAMAs) meet all of the criteria of an ideal TAA. Pierini et al developed a cancer vaccine based on using aberrant mitochondrial protein, isolated from the tumor, as an immunotherapeutic strategy.182,183 Syngeneic dendritic cells were pulsed with enriched mitochondrial protein extract derived from the RENCA cancer cell line or solid tumors and then administered in vivo to determine the vaccine immunogenicity and efficacy. Upon vaccine administration followed by tumor challenge (prophylactically), the vaccine rejected tumors in 70%−80% of the animals and delayed tumor growth in the remaining in a CD8+ cytotoxic T cell-specific manner. In a therapeutic setting, mice were challenged initially with tumor followed by administration of the TAMAs-based vaccine and similarly, tumor delay was observed. Subsequently, sequencing of the RENCA mtDNA found 2 mutated proteins (COX1 and ND5) which were then used to produce a tumor mitochondria vaccine. COX1 mutated peptide used as a vaccine resulted in delayed tumor growth and induction of T cell specific immune response.183 To the best of our knowledge, this is the first vaccine based on mtDNA-mutated peptides derived from tumor cells that elicit an immune response specifically against the tumor.
CONCLUSIONS
Mitochondria are essential organelles derived from symbiotic bacteria, necessary for cellular activity. The emergence, in recent decades, of a whole new field of “Mitochondrial Medicine” is the recognition to the important role that mitochondria play in both human health and disease. Targeting mitochondrial molecules in the development of cancer therapy requires selective action on the target and selective accumulation at the target site. It has to be noted that the chances of finding a molecule with selective effect in diseases like cancer is very small given the subtle differences in cancer cell-specific targets from normal cells. Even lower is the chance to discover the molecules with both selective accumulation and target specificity. Therefore, the implementation of the immune system as the mechanism for targeting unhealthy mitochondria within cancerous cells becomes of extreme interest, as the adaptive immune system can specifically target cancer cells bearing TAMAs, sparing normal tissues from the TAMAs-specific CTL-mediated response.
ACKNOWLEDGMENTS
This work is supported by NIH-1R01CA206012–01 and Basser Research Center for BRCA Award. We would like to thank John Facciponte for his assistance in manuscript preparation. All authors have read the journal’s policy on disclosure of potential conflicts of interest. All authors have read the journal’s authorship statement, and the manuscript has been reviewed by and approved by all named authors.
REFERENCES
- 1.Zong W-X, Rabinowitz JD, White E. Mitochondria and cancer. Mol Cell 2016;61:667–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wallace DC. Mitochondria and cancer. Nat Rev Cancer 2012;12:685–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arduíno DM, Esteves AR, Cardoso SM. Mitochondrial fusion/fission, transport and autophagy in Parkinson’s disease: when mitochondria get nasty. Parkinsons Dis 2011;2011:767230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mitchell P, Moyle J. Acid-base titration across the membrane system of rat-liver mitochondria. Catalysis by uncouplers. Bio-chem J 1967;104:588–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature 2000;408:239–47. [DOI] [PubMed] [Google Scholar]
- 6.Lehninger AL, Nelson DL, Cox MM (1993) Principles of bio-chemistry 241–242
- 7.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646–74. [DOI] [PubMed] [Google Scholar]
- 8.Warburg O On the origin of cancer cells. Science 1956;123:309–14. [DOI] [PubMed] [Google Scholar]
- 9.Guppy M, Greiner E, Brand K. The role of the Crabtree effect and an endogenous fuel in the energy metabolism of resting and proliferating thymocytes. Eur J Biochem 1993;212:95–9. [DOI] [PubMed] [Google Scholar]
- 10.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009;324:1029–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu VWS, Wang Y, Yang H-J, et al. Mitochondrial DNA variant 16189T>C is associated with susceptibility to endometrial cancer. Hum Mutat 2003;22:173–4. [DOI] [PubMed] [Google Scholar]
- 12.Zhai K, Chang L, Zhang Q, Liu B, Wu Y. Mitochondrial C150T polymorphism increases the risk of cervical cancer and HPV infection. Mitochondrion 2011;11:559–63. [DOI] [PubMed] [Google Scholar]
- 13.Canter JA, Kallianpur AR, Parl FF, Millikan RC. Mitochondrial DNA G10398A polymorphism and invasive breast cancer in African-American women. Cancer Res 2005;65:8028–33. [DOI] [PubMed] [Google Scholar]
- 14.Permuth-Wey J, Chen YA, Tsai Y-Y, et al. Inherited variants in mitochondrial biogenesis genes may influence epithelial ovarian cancer risk. Cancer Epidemiol Biomarkers Prev 2011;20:1131–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wallace DC, Fan W, Procaccio V. Mitochondrial energetics and therapeutics. Annu Rev Pathol 2010;5:297–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kurelac I, Romeo G, Gasparre G. Mitochondrial metabolism and cancer. Mitochondrion 2011;11:635–7. [DOI] [PubMed] [Google Scholar]
- 17.Bardella C, Pollard PJ, Tomlinson I. SDH mutations in cancer. Biochim Biophys Acta 2011;1807:1432–43. [DOI] [PubMed] [Google Scholar]
- 18.Frezza C, Zheng L, Folger O, et al. Haem oxygenase is synthetically lethal with the tumour suppressor fumarate hydratase. Nature 2011;477:225–8. [DOI] [PubMed] [Google Scholar]
- 19.Adam J, Hatipoglu E, O’Flaherty L, et al. Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell 2011;20:524–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Picaud S, Kavanagh KL, Yue WW, et al. Structural basis of fumarate hydratase deficiency. J Inherit Metab Dis 2011;34:671–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koivunen P, Lee S, Duncan CG, et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 2012;483:484–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Figueroa ME, Lugthart S, Li Y, et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell 2010;17:13–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chowdhury R, Yeoh KK, Tian Y-M, et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep 2011;12:463–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Turcan S, Rohle D, Goenka A, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012;483:479–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yoon JC, Ng A, Kim BH, Bianco A, Xavier RJ, Elledge SJ. Wnt signaling regulates mitochondrial physiology and insulin sensitivity. Genes Dev 2010;24:1507–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brandon M, Baldi P, Wallace DC. Mitochondrial mutations in cancer. Oncogene 2006;25:4647–62. [DOI] [PubMed] [Google Scholar]
- 27.Pavlides S, Whitaker-Menezes D, Castello-Cros R, et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009;8:3984–4001. [DOI] [PubMed] [Google Scholar]
- 28.Mills EL, Kelly B, O’Neill LAJ. Mitochondria are the power-houses of immunity. Nat Immunol 2017;18:488–98. [DOI] [PubMed] [Google Scholar]
- 29.Zhang L, Conejo-Garcia JR, Katsaros D, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med 2003;348:203–13. [DOI] [PubMed] [Google Scholar]
- 30.Erdag G, Schaefer JT, Smolkin ME, et al. Immunotype and immunohistologic characteristics of tumor-infiltrating immune cells are associated with clinical outcome in metastatic melanoma. Cancer Res 2012;72:1070–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gu-Trantien C, Loi S, Garaud S, et al. CD4+ follicular helper T cell infiltration predicts breast cancer survival. J Clin Invest 2013;123:2873–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scharping NE, Menk AV, Moreci RS, et al. The tumor micro-environment represses T cell mitochondrial biogenesis to drive intratumoral T cell metabolic insufficiency and dysfunction. Immunity 2016;45:701–3. [DOI] [PubMed] [Google Scholar]
- 33.Schietinger A, Greenberg PD. Tolerance and exhaustion: defining mechanisms of T cell dysfunction. Trends Immunol 2014;35:51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 2015;15:486–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang Y, Li Y, Zhu B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis 2015;6:e1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Siska PJ, Rathmell JC. T cell metabolic fitness in antitumor immunity. Trends Immunol 2015;36:257–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fernandez-Marcos PJ, Auwerx J. Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am J Clin Nutr 2011;93:884S–90S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 2006;441:424–30. [DOI] [PubMed] [Google Scholar]
- 39.Crompton JG, Sukumar M, Roychoudhuri R, et al. Akt inhibition enhances expansion of potent tumor-specific lymphocytes with memory cell characteristics. Cancer Res 2015;75:296–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jones RG, Thompson CB. Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev 2009;23:537–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pauken KE, Wherry EJ. Overcoming T cell exhaustion in infection and cancer. Trends Immunol 2015;36:265–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Patsoukis N, Bardhan K, Chatterjee P, et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun 2015;6:6692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chuang E, Fisher TS, Morgan RW, et al. The CD28 and CTLA-4 receptors associate with the serine/threonine phosphatase PP2A. Immunity 2000;13:313–22. [DOI] [PubMed] [Google Scholar]
- 44.Ron-Harel N, Santos D, Ghergurovich JM, et al. Mitochondrial biogenesis and proteome remodeling promote one-carbon metabolism for T cell activation. Cell Metab 2016;24:104–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Buck MD, O’Sullivan D, Klein Geltink RI, et al. Mitochondrial dynamics controls T cell fate through metabolic programming. Cell 2016;166:63–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Frank M, Duvezin-Caubet S, Koob S, et al. Mitophagy is triggered by mild oxidative stress in a mitochondrial fission dependent manner. Biochim Biophys Acta 2012;1823:2297–310. [DOI] [PubMed] [Google Scholar]
- 47.Bird L T cells: Mitochondrial shape shifters. Nat Rev Immunol 2016;16:402–3. [DOI] [PubMed] [Google Scholar]
- 48.Sena LA, Li S, Jairaman A, et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 2013;38:225–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol 2011;13:589–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dang EV, Barbi J, Yang HY, et al. Control of T H 17/T reg balance by hypoxia-inducible factor 1. Cell 2011;146: 772–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lukashev D, Klebanov B, Kojima H, et al. Cutting edge: hypoxia-inducible factor 1alpha and its activation-inducible short isoform I.1 negatively regulate functions of CD4+ and CD8+ T lymphocytes. J Immunol 2006;177:4962–5. [DOI] [PubMed] [Google Scholar]
- 52.Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity 2013;38:633–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tannahill GM, Curtis AM, Adamik J, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013;496:238–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mills EL, Kelly B, Logan A, et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell 2016;167:457–470.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tan Z, Xie N, Cui H, et al. Pyruvate dehydrogenase kinase 1 participates in macrophage polarization via regulating glucose metabolism. J Immunol 2015;194:6082–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huang SC-C, Everts B, Ivanova Y, et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol 2014;15:846–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vats D, Mukundan L, Odegaard JI, et al. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab 2006;4:13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Namgaladze D, Lips S, Leiker TJ, et al. Inhibition of macrophage fatty acid β-oxidation exacerbates palmitate-induced inflammatory and endoplasmic reticulum stress responses. Diabetologia 2014;57:1067–77. [DOI] [PubMed] [Google Scholar]
- 59.Michalek RD, Gerriets VA, Jacobs SR, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol 2011;186:3299–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.van der Windt GJW, Everts B, Chang C-H, et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity 2012;36:68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chistiakov DA, Orekhov AN, Sobenin IA, Bobryshev YV. Plasmacytoid dendritic cells: development, functions, and role in atherosclerotic inflammation. Front Physiol 2014;5:279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Benn CS, Netea MG, Selin LK, Aaby P. A small jab - a big effect: nonspecific immunomodulation by vaccines. Trends Immunol 2013;34:431–9. [DOI] [PubMed] [Google Scholar]
- 63.Cline SD. Mitochondrial DNA damage and its consequences for mitochondrial gene expression. Biochim Biophys Acta 2012;1819:979–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Birben E, Sahiner UM, Sackesen C, Erzurum S, Kalayci O. Oxidative stress and antioxidant defense. World Allergy Organiz J 2012;5:9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Alexeyev M, Shokolenko I, Wilson G, LeDoux S. The maintenance of mitochondrial DNA integrity—critical analysis and update. Cold Spring Harb Perspect Biol 2013;5:a012641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Akhmedov AT, Marín-García J. Mitochondrial DNA maintenance: an appraisal. Mol Cell Biochem 2015;409:283–305. [DOI] [PubMed] [Google Scholar]
- 67.McKinnon PJ, Caldecott KW. DNA strand break repair and human genetic disease. Annu Rev Genomics Hum Genet 2007;8:37–55. [DOI] [PubMed] [Google Scholar]
- 68.Moretton A, Morel F, Macao B, et al. Selective mitochondrial DNA degradation following double-strand breaks. PLoS One 2017;12:e0176795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jones E, Gaytan N, Garcia I, et al. A threshold of transmembrane potential is required for mitochondrial dynamic balance mediated by DRP1 and OMA1. Cell Mol Life Sci 2017;74: 1347–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rossignol R, Faustin B, Rocher C, Malgat M, Mazat J-P, Letellier T. Mitochondrial threshold effects. Biochem J 2003;370: 751–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Szczepanowska K, Trifunovic A. Origins of mtDNA mutations in ageing. Essays Biochem 2017;61:325–37. [DOI] [PubMed] [Google Scholar]
- 72.Pinto M, Moraes CT. Mechanisms linking mtDNA damage and aging. Free Radic Biol Med 2015;85:250–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kennedy SR, Salk JJ, Schmitt MW, Loeb LA. Ultra-sensitive sequencing reveals an age-related increase in somatic mitochondrial mutations that are inconsistent with oxidative damage. PLoS Genet 2013;9:e1003794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Copeland WC, Longley MJ. Mitochondrial genome maintenance in health and disease. DNA Repair 2014;19:190–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jäger S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci USA 2007;104:12017–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ploumi C, Daskalaki I, Tavernarakis N. Mitochondrial biogenesis and clearance: a balancing act. FEBS J 2017;284:183–95. [DOI] [PubMed] [Google Scholar]
- 77.Palikaras K, Tavernarakis N. Mitochondrial homeostasis: the interplay between mitophagy and mitochondrial biogenesis. Exp Gerontol 2014;56:182–8. [DOI] [PubMed] [Google Scholar]
- 78.Xu J, Ji J, Yan X-H. Cross-talk between AMPK and mTOR in regulating energy balance. Crit Rev Food Sci Nutr 2012;52:373–81. [DOI] [PubMed] [Google Scholar]
- 79.Jovaisaite V, Mouchiroud L, Auwerx J. The mitochondrial unfolded protein response, a conserved stress response pathway with implications in health and disease. J Exp Biol 2014;217: 137–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Burman JL, Pickles S, Wang C, et al. Mitochondrial fission facilitates the selective mitophagy of protein aggregates. J Cell Biol 2017;216:3231–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ni H-M, Williams JA, Ding W-X. Mitochondrial dynamics and mitochondrial quality control. Redox Biol 2015;4:6–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Soubannier V, McLelland G-L, Zunino R, et al. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr Biol 2012;22:135–41. [DOI] [PubMed] [Google Scholar]
- 83.McLelland GL, Fon EA. Principles of mitochondrial vesicle transport. Curr Opin Physiol 2018;3:25–33. [Google Scholar]
- 84.Westermann B Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol 2010;11:872–84. [DOI] [PubMed] [Google Scholar]
- 85.Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol 2011;12:9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Steffen J, Koehler CM. ER-mitochondria contacts: actin dynamics at the ER control mitochondrial fission via calcium release. J Cell Biol 2018;217:15–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lee JE, Westrate LM, Wu H, Page C, Voeltz GK. Multiple dynamin family members collaborate to drive mitochondrial division. Nature 2016;540:139–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hu C, Huang Y, Li L. Drp1-dependent mitochondrial fission plays critical roles in physiological and pathological progresses in mammals. Int J Mol Sci 2017. 10.3390/ijms18010144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Williams JA, Ding W-X. Mechanisms, pathophysiological roles and methods for analyzing mitophagy - recent insights. Biol Chem 2018;399:147–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol 2010;191: 933–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015;85:257–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rasool S, Soya N, Truong L, Croteau N, Lukacs GL, Trempe J-F. PINK1 autophosphorylation is required for ubiquitin recognition. EMBO Rep 2018. 10.15252/embr.201744981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Okatsu K, Koyano F, Kimura M, et al. Phosphorylated ubiquitin chain is the genuine Parkin receptor. J Cell Biol 2015;209:111–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lazarou M, Sliter DA, Kane LA, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015;524:309–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kwon YT, Ciechanover A. The ubiquitin code in the ubiquitin-proteasome system and autophagy. Trends Biochem Sci 2017;42:873–86. [DOI] [PubMed] [Google Scholar]
- 96.Tanida I, Ueno T, Kominami E. LC3 and autophagy. Methods Mol Biol 2008;445:77–88. [DOI] [PubMed] [Google Scholar]
- 97.Gong Y, Zack TI, Morris LGT, et al. Pan-cancer genetic analysis identifies PARK2 as a master regulator of G1/S cyclins. Nat Genet 2014;46:588–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Veeriah S, Taylor BS, Meng S, et al. Somatic mutations of the Parkinson’s disease-associated gene PARK2 in glioblastoma and other human malignancies. Nat Genet 2010;42:77–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Morris LG, Veeriah S, Chan TA. Genetic determinants at the interface of cancer and neurodegenerative disease. Oncogene 2010;29. [DOI] [PMC free article] [PubMed]
- 100.Fujiwara M, Marusawa H, Wang HQ, et al. Parkin as a tumor suppressor gene for hepatocellular carcinoma. Oncogene 2008;27:6002–11. [DOI] [PubMed] [Google Scholar]
- 101.Lin D-C, Xu L, Chen Y, et al. Genomic and functional analysis of the E3 ligase PARK2 in glioma. Cancer Res 2015;75:1815–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Xu L, Lin D, Yin D, Koeffler HP. An emerging role of PARK2 in cancer. J Mol Med 2014;92:31–42. [DOI] [PubMed] [Google Scholar]
- 103.Ribatti D The concept of immune surveillance against tumors. The first theories. Oncotarget 2017;8:7175–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Arnoult D, Soares F, Tattoli I, Girardin SE. Mitochondria in innate immunity. EMBO Rep 2011;12:901–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.West AP, Shadel GS. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat Rev Immunol 2017;17:363–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Matheoud D, Sugiura A, Bellemare-Pelletier A, et al. Parkinson’s disease-related proteins PINK1 and Parkin repress mitochondrial antigen presentation. Cell 2016;166:314–27. [DOI] [PubMed] [Google Scholar]
- 107.Zhang Q, Raoof M, Chen Y, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010;464:104–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Galluzzi L, Kepp O, Kroemer G. Mitochondria: master regulators of danger signalling. Nat Rev Mol Cell Biol 2012;13:780–8. [DOI] [PubMed] [Google Scholar]
- 109.Xu MM, Pu Y, Han D, et al. Dendritic cells but not macrophages sense tumor mitochondrial DNA for cross-priming through signal regulatory protein a signaling. Immunity 2017;47:363–73, e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ingelsson B, Söerberg D, Strid T, et al. Lymphocytes eject interferogenic mitochondrial DNA webs in response to CpG and non-CpG oligodeoxynucleotides of class C. Proc Natl Acad Sci USA 2018;115:E478–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wang L, Liebmen MN, Wang X. Roles of mitochondrial DNA signaling in immune responses. Adv Exp Med Biol 2017;1038:39–53. [DOI] [PubMed] [Google Scholar]
- 112.Gu Y, Wang C, Roifman CM, Cohen A. Role of MHC class I in immune surveillance of mitochondrial DNA integrity. J Immunol 2003;170:3603–7. [DOI] [PubMed] [Google Scholar]
- 113.Chen L, Duvvuri B, Grigull J, Jamnik R, Wither JE, Wu GE. Experimental evidence that mutated-self peptides derived from mitochondrial DNA somatic mutations have the potential to trigger autoimmunity. Hum Immunol 2014;75:873–9. [DOI] [PubMed] [Google Scholar]
- 114.Duvvuri B, Duvvuri VR, Wang C, et al. The human immune system recognizes neopeptides derived from mitochondrial DNA deletions. J Immunol 2014;192:4581–91. [DOI] [PubMed] [Google Scholar]
- 115.Oliveira CC, van Hall T. Alternative antigen processing for MHC class I: multiple roads lead to rome. Front Immunol 2015;6:298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Münz C Autophagy proteins in antigen processing for presentation on MHC molecules. Immunol Rev 2016;272:17–27. [DOI] [PubMed] [Google Scholar]
- 117.Carew JS, Huang P. Mitochondrial defects in cancer. Mol Cancer 2002;1:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ubah OC, Wallace HM. Cancer therapy: Targeting mitochondria and other sub-cellular organelles. Curr Pharm Des 2014;20:201–22. [DOI] [PubMed] [Google Scholar]
- 119.Weinberg SE, Chandel NS. Targeting mitochondria metabolism for cancer therapy. Nat Chem Biol 2015;11:9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Marchetti P, Guerreschi P, Mortier L, Kluza J. Integration of mitochondrial targeting for molecular cancer therapeutics. Int J Cell Biol 2015;2015:283145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Fulda S, Galluzzi L, Kroemer G. Targeting mitochondria for cancer therapy. Nat Rev Drug Discov 2010;9:447–64. [DOI] [PubMed] [Google Scholar]
- 122.Tan AS, Baty JW, Dong L-F, et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab 2015;21:81–94. [DOI] [PubMed] [Google Scholar]
- 123.Ganapathy-Kanniappan S, Geschwind J-FH. Tumor glycolysis as a target for cancer therapy: progress and prospects. Mol Cancer 2013;12:152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zhao Y, Butler EB, Tan M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis 2013;4: e532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Liu Y, Cao Y, Zhang W, et al. A small-molecule inhibitor of glucose transporter 1 downregulates glycolysis, induces cell-cycle arrest, and inhibits cancer cell growth in vitro and in vivo. Mol Cancer Ther 2012;11:1672–82. [DOI] [PubMed] [Google Scholar]
- 126.Simons AL, Ahmad IM, Mattson DM, Dornfeld KJ, Spitz DR. 2-Deoxy-D-glucose combined with cisplatin enhances cytotoxicity via metabolic oxidative stress in human head and neck cancer cells. Cancer Res 2007;67:3364–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Dunbar EM, Coats BS, Shroads AL, et al. Phase 1 trial of dichloroacetate (DCA) in adults with recurrent malignant brain tumors. Invest New Drugs 2014;32:452–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hawley SA, Gadalla AE, Olsen GS, Hardie DG. The antidiabetic drug metformin activates the AMP-activated protein kinase cascade via an adenine nucleotide-independent mechanism. Diabetes 2002;51:2420–5. [DOI] [PubMed] [Google Scholar]
- 129.Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J 2000;348(Pt 3):607–14. [PMC free article] [PubMed] [Google Scholar]
- 130.Evans JMM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ 2005;330:1304–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kasznicki J, Sliwinska A, Drzewoski J. Metformin in cancer prevention and therapy. Ann Transl Med 2014;2:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Birsoy K, Sabatini DM, Possemato R. Untuning the tumor metabolic machine: Targeting cancer metabolism: a bedside lesson. Nat Med 2012;18:1022–3. [DOI] [PubMed] [Google Scholar]
- 133.Pollak M The insulin and insulin-like growth factor receptor family in neoplasia: an update. Nat Rev Cancer 2012;12:159–69. [DOI] [PubMed] [Google Scholar]
- 134.Bridges HR, Jones AJY, Pollak MN, Hirst J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem J 2014;462:475–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hardie DG. AMPK: a target for drugs and natural products with effects on both diabetes and cancer. Diabetes 2013;62:2164–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Mallik R, Chowdhury TA. Metformin in cancer. Diabetes Res Clin Pract 2018. 10.1016/j.diabres.2018.05.023. [DOI] [PubMed] [Google Scholar]
- 137.Nogueira V, Hay N. Molecular pathways: reactive oxygen species homeostasis in cancer cells and implications for cancer therapy. Clin Cancer Res 2013;19:4309–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Weinberg F, Hamanaka R, Wheaton WW, et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci USA 2010;107:8788–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Porporato PE, Filigheddu N, Pedro JMB-S, Kroemer G, Galluzzi L. Mitochondrial metabolism and cancer. Cell Res 2018;28:265–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kirshner JR, He S, Balasubramanyam V, et al. Elesclomol induces cancer cell apoptosis through oxidative stress. Mol Cancer Ther 2008;7:2319–27. [DOI] [PubMed] [Google Scholar]
- 141.Berkenblit A, Eder JP, Ryan DP, et al. Phase I clinical trial of STA-4783 in combination with paclitaxel in patients with refractory solid tumors. Clin Cancer Res 2007;13:584–90. [DOI] [PubMed] [Google Scholar]
- 142.O’Day S, Gonzalez R, Lawson D, et al. Phase II, randomized, controlled, double-blinded trial of weekly elesclomol plus paclitaxel versus paclitaxel alone for stage IV metastatic melanoma. J Clin Oncol 2009;27:5452–8. [DOI] [PubMed] [Google Scholar]
- 143.Xiao D, Powolny AA, Moura MB, et al. Phenethyl isothiocyanate inhibits oxidative phosphorylation to trigger reactive oxygen species-mediated death of human prostate cancer cells. J Biol Chem 2010;285:26558–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Chan DK, Miskimins WK. Metformin and phenethyl isothiocyanate combined treatment in vitro is cytotoxic to ovarian cancer cultures. J Ovarian Res 2012;5:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev 2007;87:99–163. [DOI] [PubMed] [Google Scholar]
- 146.Bernardi P, Di Lisa F. The mitochondrial permeability transition pore: molecular nature and role as a target in cardioprotection. J Mol Cell Cardiol 2015;78:100–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Belzacq AS, El Hamel C, Vieira HL, et al. Adenine nucleotide translocator mediates the mitochondrial membrane permeabilization induced by lonidamine, arsenite and CD437. Oncogene 2001;20:7579–87. [DOI] [PubMed] [Google Scholar]
- 148.Guo L, Shestov AA, Worth AJ, et al. Inhibition of mitochondrial complex II by the anticancer agent lonidamine. J Biol Chem 2016;291:42–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Oudard S, Carpentier A, Banu E, et al. Phase II study of lonidamine and diazepam in the treatment of recurrent glioblastoma multiforme. J Neurooncol 2003;63:81–6. [DOI] [PubMed] [Google Scholar]
- 150.Horsley L, Cummings J, Middleton M, et al. A phase 1 trial of intravenous 4-(N-(S-glutathionylacetyl)amino) phenylarsenoxide (GSAO) in patients with advanced solid tumours. Cancer Chemother Pharmacol 2013;72:1343–52. [DOI] [PubMed] [Google Scholar]
- 151.Park D, Chiu J, Perrone GG, Dilda PJ, Hogg PJ. The tumour metabolism inhibitors GSAO and PENAO react with cysteines 57 and 257 of mitochondrial adenine nucleotide translocase. Cancer Cell Int 2012;12:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.First-in-man trial of 4-(N-(S-penicillaminylacetyl)amino) phenylarsonous acid (PENAO) as a continuous intravenous infusion (CIVI), in patients (pt) with advanced solid tumours. J Clin Oncol: Vol 34, No 15_suppl. [Google Scholar]
- 153.Pavlov EV, Priault M, Pietkiewicz D, et al. A novel, high conductance channel of mitochondria linked to apoptosis in mammalian cells and Bax expression in yeast. J Cell Biol 2001;155:725–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Rooswinkel RW, van de Kooij B, Verheij M, Borst J. Bcl-2 is a better ABT-737 target than Bcl-xL or Bcl-w and only Noxa overcomes resistance mediated by Mcl-1, Bfl-1, or Bcl-B. Cell Death Dis 2012;3:e366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Reshi L, Wang H-V, Hui C-F, Su Y-C, Hong J-R. Anti-apoptotic genes Bcl-2 and Bcl-xL overexpression can block iridovirus serine/threonine kinase-induced Bax/mitochondria-mediated cell death in GF-1 cells. Fish Shellfish Immunol 2017;61:120–9. [DOI] [PubMed] [Google Scholar]
- 156.Oltersdorf T, Elmore SW, Shoemaker AR, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005;435:677–81. [DOI] [PubMed] [Google Scholar]
- 157.Konopleva M, Contractor R, Tsao T, et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell 2006;10: 375–88. [DOI] [PubMed] [Google Scholar]
- 158.Kang MH, Kang YH, Szymanska B, et al. Activity of vincristine, L-ASP, and dexamethasone against acute lymphoblastic leukemia is enhanced by the BH3-mimetic ABT-737 in vitro and in vivo. Blood 2007;110:2057–66. [DOI] [PubMed] [Google Scholar]
- 159.Hann CL, Daniel VC, Sugar EA, et al. Therapeutic efficacy of ABT-737, a selective inhibitor of BCL-2, in small cell lung cancer. Cancer Res 2008;68:2321–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Tagscherer KE, Fassl A, Campos B, et al. Apoptosis-based treatment of glioblastomas with ABT-737, a novel small molecule inhibitor of Bcl-2 family proteins. Oncogene 2008;27:6646–56. [DOI] [PubMed] [Google Scholar]
- 161.Tse C, Shoemaker AR, Adickes J, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res 2008;68:3421–8. [DOI] [PubMed] [Google Scholar]
- 162.Lock R, Carol H, Houghton PJ, et al. Initial testing (stage 1) of the BH3 mimetic ABT-263 by the pediatric preclinical testing program. Pediatr Blood Cancer 2008;50:1181–9. [DOI] [PubMed] [Google Scholar]
- 163.Gandhi L, Camidge DR, Ribeiro de Oliveira M, et al. Phase I study of Navitoclax (ABT-263), a novel Bcl-2 family inhibitor, in patients with small-cell lung cancer and other solid tumors. J Clin Oncol 2011;29:909–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Rudin CM, Hann CL, Garon EB, et al. Phase II study of single-agent navitoclax (ABT-263) and biomarker correlates in patients with relapsed small cell lung cancer. Clin Cancer Res 2012;18:3163–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Azmi AS, Mohammad RM. Non-peptidic small molecule inhibitors against Bcl-2 for cancer therapy. J Cell Physiol 2009;218:13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Liu G, Kelly WK, Wilding G, Leopold L, Brill K, Somer B. An open-label, multicenter, phase I/II study of single-agent AT-101 in men with castrate-resistant prostate cancer. Clin Cancer Res 2009;15:3172–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kitada S, Kress CL, Krajewska M, Jia L, Pellecchia M, Reed JC. Bcl-2 antagonist apogossypol (NSC736630) displays single-agent activity in Bcl-2-transgenic mice and has superior efficacy with less toxicity compared with gossypol (NSC19048). Blood 2008;111:3211–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Murphy MP. Selective targeting of bioactive compounds to mitochondria. Trends Biotechnol 1997;15:326–30. [DOI] [PubMed] [Google Scholar]
- 169.Ross MF, Filipovska A, Smith RAJ, Gait MJ, Murphy MP. Cell-penetrating peptides do not cross mitochondrial mem-branes even when conjugated to a lipophilic cation: evidence against direct passage through phospholipid bilayers. Biochem J 2004;383:457–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Kelso GF, Porteous CM, Coulter CV, et al. Selective targeting of a redox-active ubiquinone to mitochondria within cells: antioxidant and antiapoptotic properties. J Biol Chem 2001;276:4588–96. [DOI] [PubMed] [Google Scholar]
- 171.Mukhopadhyay A, Ni L, Yang CS, Weiner H. Bacterial signal peptide recognizes HeLa cell mitochondrial import receptors and functions as a mitochondrial leader sequence. Cell Mol Life Sci 2005;62:1890–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Vestweber D, Schatz G. DNA-protein conjugates can enter mitochondria via the protein import pathway. Nature 1989;338:170–2. [DOI] [PubMed] [Google Scholar]
- 173.Yousif LF, Stewart KM, Kelley SO. Targeting mitochondria with organelle-specific compounds: strategies and applications. Chembiochem 2009;10:1939–50. [DOI] [PubMed] [Google Scholar]
- 174.Yousif LF, Stewart KM, Horton KL, Kelley SO. Mitochondria-penetrating peptides: sequence effects and model cargo transport. Chembiochem 2009;10:2081–8. [DOI] [PubMed] [Google Scholar]
- 175.Horton KL, Stewart KM, Fonseca SB, Guo Q, Kelley SO. Mitochondria-penetrating peptides. Chem Biol 2008;15:375–82. [DOI] [PubMed] [Google Scholar]
- 176.Lu P, Bruno BJ, Rabenau M, Lim CS. Delivery of drugs and macromolecules to the mitochondria for cancer therapy. J Control Release 2016;240:38–51. [DOI] [PubMed] [Google Scholar]
- 177.Fliss MS, Usadel H, Caballero OL, et al. Facile detection of mitochondrial DNA mutations in tumors and bodily fluids. Science 2000;287:2017–9. [DOI] [PubMed] [Google Scholar]
- 178.Chatterjee A, Mambo E, Sidransky D. Mitochondrial DNA mutations in human cancer. Oncogene 2006;25:4663–74. [DOI] [PubMed] [Google Scholar]
- 179.Voo KS, Zeng G, Mu J-B, Zhou J, Su X-Z, Wang R-F. CD4+ T-cell response to mitochondrial cytochrome B in human melanoma. Cancer Res 2006;66:5919–26. [DOI] [PubMed] [Google Scholar]
- 180.Hernandez DG, Reed X, Singleton AB. Genetics in Parkinson disease: Mendelian versus non-Mendelian inheritance. J Neuro-chem 2016;139(Suppl 1):59–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Gao F, Yang J, Wang D, et al. Mitophagy in parkinson’s disease: pathogenic and therapeutic implications. Front Neurol 2017;8:527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Yang B, Jeang J, Yang A, Wu TC, Hung C-F. DNA vaccine for cancer immunotherapy. Hum Vaccin Immunother 2014;10:3153–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Pierini S, Fang C, Rafail S, et al. A tumor mitochondria vaccine protects against experimental renal cell carcinoma. J Immunol 2015;195:4020–7. [DOI] [PubMed] [Google Scholar]