

Abstract
The APOBEC3 family of cytosine deaminases catalyzes the conversion of cytosines-to-uracils in single-stranded DNA. Traditionally, these enzymes are associated with antiviral immunity and restriction of DNA-based pathogens. However, a role for these enzymes in tumor evolution and metastatic disease has also become evident. The primary APOBEC3 candidate in cancer mutagenesis is APOBEC3B (A3B) for three reasons: 1) A3B mRNA is upregulated in several different cancers, 2) A3B expression and mutational loads correlate with poor clinical outcomes, and 3) A3B is the only family member known to be constitutively nuclear. Previous studies have mapped non-canonical A3B nuclear localization determinants to a single surface-exposed patch within the N-terminal domain (NTD). Here, we show that A3B has an additional, distinct, surface-exposed NTD region that contributes to nuclear localization. Disruption of residues within the first 30 amino acids of A3B (import surface 1) or loop 5/α-helix 3 (import surface 2) completely abolish nuclear localization. These import determinants also graft into NTDs of related family members and mediate re-localization from cell-wide-to-nucleus or cytoplasm-to-nucleus. These findings demonstrate that both sets of residues are required for non-canonical A3B nuclear localization and describe unique surfaces that may serve as novel therapeutic targets.
Keywords: APOBEC3B, cancer genomic DNA deaminase, retrovirus restriction factor, nuclear import, subcellular localization
INTRODUCTION
The APOBEC3 (apolipoprotein B mRNA-editing enzyme, catalytic polypeptide-like 3) enzymes are a family of single-stranded (ss)DNA cytosine deaminases that provide overlapping protection against diverse DNA-based pathogens, such as transposable retroelements and retroviruses like HTLV-1, HIV-1, and HIV-2 (reviewed by [1–4]). APOBEC3D (A3D), APOBEC3F (A3F), APOBEC3G (A3G), and APOBEC3H (A3H) are capable of suppressing HIV-1 replication by packaging into nascent viral particles and mutating cytosines-to-uracils in newly synthesized viral cDNA [5–15]. APOBEC3B (A3B) has been shown to be a restrictor of specific retroviral families [16, 17], and can potently restrict HIV-1 when virus is produced from adherent cells (293T and HeLa) but not from T-lymphocyte derived cell lines [7, 18]. Additionally, A3B is capable of restricting intracellular elements, such as LINE-1 retrotransposons and foreign DNA [19–24]. Overall, these studies support the idea that the APOBEC3 enzymes, including A3B, exist as an overlapping innate defense mechanism that serves to block the spread of a wide variety of DNA-based parasites.
A3B has also been implicated as a major source of mutation in primary breast tumors (comprising 20% of base substitutions) and metastasis (mutational contributions greater than 50%), as well as contributing to the mutational landscapes of several other cancer types including cervical, lung, head/neck, and bladder cancers ([25–31] and reviewed by [32–35]). Additionally, recent studies have shown that A3B contributes to drug resistance in breast cancer and correlates with poor clinical outcomes [36–44]. Furthermore, A3B mRNA levels become highly upregulated in over 80% of estrogen receptor-positive breast tumors with cervical, bladder, and head/neck cancers among the tumor types displaying the highest A3B expression levels and cytosine mutational loads [25–27, 45]. Notably, the upregulation of A3B in head/neck and cervical cancers, many of which are due to human papilloma virus infection, is most likely attributable to collateral damage from responding to the virus infection [46–49].
While it is becoming clear that A3B is a major contributor to cancer mutagenesis and tumor evolution, a major question for both innate immunity and cancer mutagenesis is: how does A3B enter the nuclear compartment? Previous studies have reported the localization patterns for several of the APOBEC3 family members and shown that A3B is the only member that is constitutively nuclear [20, 22, 50–53]. These observations raise the questions of 1) what is the mechanism of nuclear import, and 2) what are the protein determinants required? It has been suggested that present day A3B obtained both its mechanism of nuclear import and the required protein determinants from the ancestral antibody gene diversification enzyme AID (activation-induced cytidine deaminase) during the expansion of the APOBEC3 locus in the primate lineage [51]. Support for this model comes from A3B’s ability to interact with the same subset of IMPORTINα proteins utilized by AID, and from evidence that substitution of an NTD amino acid, V54D, which is positioned within an analogous region in AID, abates nuclear localization [51]. However, only two studies have examined the protein determinant of A3B required for nuclear import. One study narrowed down the nuclear localization determinant to the first 60 residues of the N-terminal domain, and the other suggested that a single surface including amino acids D19 and E24 was the sole localization determinant [52, 53].
Our studies here began with the goal of fine-mapping A3B nuclear localization determinants with a long-term aim of investigating the prevention of nuclear localization as a strategy to slow rates of tumor evolution. Chimeric constructs between A3B and non-nuclear family members yielded unexpected results and a comprehensive definition of nuclear import requirements. First, we show that residues D19 and E24 (import surface 1) are required for nuclear import of full-length A3B. Second, through the use of additional chimeric constructs, we define a new region within loop 5/α-helix 3 (import surface 2) and demonstrate its involvement in nuclear localization. Finally, we demonstrate that these two regions are sufficient to drive the nuclear import of normally cytoplasmic APOBEC3 enzymes.
RESULTS
Characterization of A3B NTD import surface 1 using A3G/B chimeras and single amino acid substitutions
To gain a better understanding of which residues in A3B contribute to nuclear import, we revisited the use of A3G/B chimeras described previously [52, 53] to see whether additional localization determinates could be identified. To exacerbate localization phenotypes, a C-terminal eGFP tag was added to generate a larger protein complex that needs to be actively imported into the nuclear compartment. Our previous studies using similar chimeras indicated that a nuclear localization determinant was located within the first 60 residues of A3B NTD [52]. Therefore, we generated chimeric junctions at regularly spaced intervals (each exchange at ~10 amino acid residues) that avoided any major structural changes based on a recently solved crystal structure of an A3B NTD variant [54]. As shown in Fig. 1a, grafting the first 21 residues of A3Gi-eGFP into full-length A3Bi-eGFP results in whole-cell localization in both 293T and HeLa cells (see Materials and Methods for details on intron-disrupted constructs; representative images in Fig. 1b and Fig. S1, plus quantification in Fig. 1c). Importantly, A3B localization could be almost fully redirected to the cytoplasmic compartment by grafting an additional 11 or more residues from A3G into A3Bi-eGFP (i.e., residues 1–32 from A3Gi-eGFP into A3Bi-eGFP; Fig. 1b and 1c).
Figure 1. A3G/B chimeras reveal that a key nuclear localization determinant of A3B resides in the first 30 amino acids.

(a) Alignment of A3B and A3G with known and predicted secondary structures (α-helices and β-strands) depicted above amino acid sequences. Arrows indicate chimeric junction locations with residue numbers annotated above and below the junctions, and shading indicates identical residues. (b) Representative images of 293T and HeLa cells transiently expressing the indicated full length C-terminally eGFP tagged chimeric proteins (10 𝜇m scale bar). (c) Quantification of localization patterns for the indicated constructs in 293T cells. Individual cells expressing the indicated chimeric proteins were scored and grouped into three categories based on their overall patterns of cellular fluorescence (n=25 per condition; Nuc., nuclear localization; WC, whole cell localization; Cyto., cytoplasmic localization; ns, indicates no significance; ***, p < 0.001 by unpaired student’s t-test).
Closer examination of this region identified 8 amino acid differences between residues 11 and 32 (Fig. 2a). The double mutant of Y18S, D19Y within full-length A3Bi-eGFP recapitulated the whole-cell localization phenotype (Fig. 2b), while the exchange of residues 22–32 in A3Bi-eGFP to those of A3G resulted in complete cytoplasmic localization (Fig. 2c). A refined analysis of these regions utilizing single amino acid substitutions revealed that D19Y confers the whole-cell localization phenotype while E24R alone can abolish nuclear import (Fig. 2b and 2c). These results support the findings of a previous study that also identified these residues as having a role in nuclear import [53]. However, we observe differential localization from only single amino acid substitutions opposed to making the double, or quadruple, mutations reported previously.
Figure 2. Mutational analysis of amino acid resides within a single surface exposed region contribute to A3B nuclear localization.
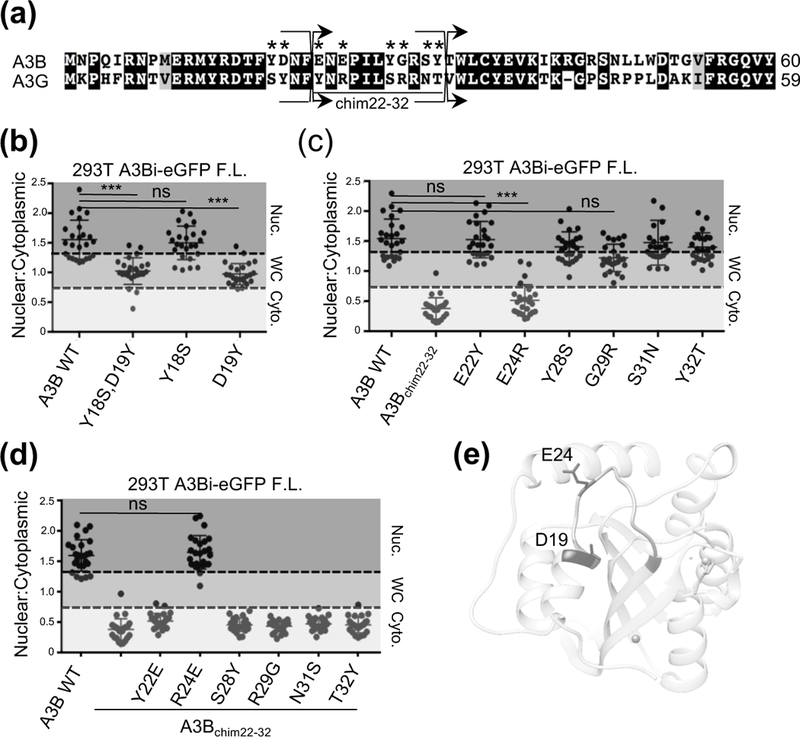
(a) Alignment of relevant regions of A3B and A3G amino acid sequences (labeled as in Fig. 1). (b-d) Quantification of localization patterns for the indicated amino acid substitution mutants (n=25 per condition; image quantification and statistical analysis as described in Fig. 1). (e) Structural representation of wild-type A3B NTD with key localization determinants labeled (see text for details).
To further explore the role of position E24 in nuclear import, we examined the full-length A3Bi-eGFP chimera with residues 22–32 exchanged for those of A3G, which relocalizes fully to the cytoplasm (Fig. 2c). Single amino acid substitutions were made between residues 22–32 to determine which, if any, could revert A3B localization from cytoplasm to nucleus. We observed that reversion of R24E alone is sufficient to restore an A3B-like nuclear localization phenotype (Fig. 2d).
Double-domain APOBEC3 proteins display differential localization patterns regardless of sequence conservation
We next examined the location of D19 and E24 on the 3-dimensional structure of A3B NTD to better understand how these residues might contribute to nuclear import. Computational approaches were used to construct a model for wild-type A3B NTD based on the recently published 6 amino acid substitution mutant structure (PDB 5TKM [54]). The resulting wild-type model indicated that these residues are located on the same surface of the protein (Fig. 2e), which is interesting as this region is completely conserved within the NTD of A3D. We therefore speculated that expression of A3D NTD alone would also display nuclear localization, even though the full-length protein is cytoplasmic [7, 50, 55].
To test this idea, we assessed the localization patterns of all of the double-domain APOBEC3 proteins in comparison to those of the respective NTDs or C-terminal domains (CTDs) alone. Consistent with previous reports, we observed that all of the double-domain APOBEC3s use a single domain for sub-cellular localization [50, 52, 56] (representative images in Fig. 3a, 3c, and Fig. S1, plus quantification in Fig. 3b and 3d). Interestingly, even though import surface 1 is completely conserved between A3B NTD and A3D NTD, the localization of these NTDs were either nuclear or whole-cell, respectively. Furthermore, we noticed that A3B, A3D, and A3F NTDs have ~70% amino acid identity with roughly 85% similarity (all classified as Z2 domains [57]), yet they display three different localization patterns: nuclear, whole-cell, and cytoplasmic, respectively. These observations raised the possibility that these domains may have overlapping localization determinants with subtle variations to independently regulate the sub-cellular localization of each APOBEC3.
Figure 3. Localization of A3B, A3D, A3F, and A3G full-length and single domain variants.
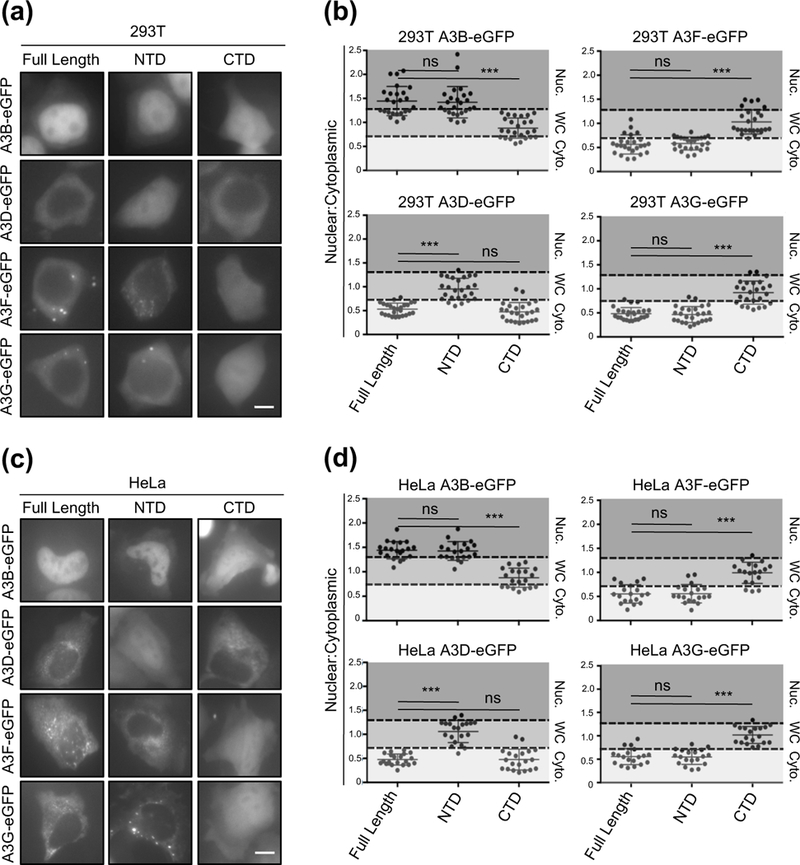
(a and c) Representative images of 293T and HeLa cells transiently expressing indicated C-terminally eGFP-tagged A3 full-length, NTD, or CTD constructs (10 𝜇m scale bar). (b and d) Quantification of localization patterns for the indicated amino acid substitution mutants (n=25 for 293T cells and n=20 for HeLa cells; image quantification and statistical analysis as described in Fig. 1).
Reciprocal A3B/G chimeras identify a second surface region required for nuclear localization
The observations above led us to hypothesize that A3B might require an additional import surface to drive it into the nucleus. To test this possibility, we used reciprocal A3B/G chimeras to determine the minimum A3B region required to promote A3G nuclear localization. Based on observations above, and prior reports [52, 53], we reasoned that minimally the first 32 residues (import surface 1) and likely another region would be required to fully relocalize A3G to the nuclear compartment. We generated these reciprocal chimeras to mirror the junctions shown in Fig. 1a to minimize the possibility of structural perturbations and help ensure that any differences in localization patterns are due to the exchanged residues (Fig. 4a).
Figure 4. Reciprocal chimeras implicate a second surface-exposed region in A3B nuclear localization.

(a) Alignment of A3B and A3G amino acid sequences as described in Figure 1. (b) Representative images of 293T and HeLa cells transiently expressing the indicated full length C-terminally eGFP tagged chimeric proteins (10 𝜇m scale bar). (c) Quantification of localization patterns for the indicated amino acid substitution mutants (n=25 per condition; image quantification and statistical analysis as described in Fig. 1).
Interestingly, exchanging residues 1–32 (import surface 1) of A3B with the corresponding residues of A3G only re-localizes A3G from cytoplasm to whole-cell, but not to the nucleus. Moreover, strong nuclear localization could not be achieved until the first 190 residues of A3B were exchanged with those of A3G. Chimeras that were generated from position 32–66, which encompassed 4 separate variants, could only re-localize A3G to whole-cell, with weak nuclear signal (Fig. 4b, 4c, and Fig. S1). Taken together, these results supported the possibility that A3B requires a second set of residues for proper nuclear import, located between positions 66–190 within the NTD.
To determine which additional residues contribute to A3B nuclear import, we focused again on comparisons with A3D and looked for clustered NTD amino acid differences between positions 66–190 (Fig. 5a). We identified two regions that fit this criterion and generated A3B/D NTD chimeras to further test the possibility of a second nuclear import determinant in A3B. In this case, A3D NTD was used as a chimeric partner because it contains an intact import surface 1, unlike A3G NTD. Therefore, any chimeric exchanges (downstream from import surface 1) that result in relocalization of A3D NTD to the nuclear compartment can be attributed directly to the addition of another set of import determinants. In both 293T and HeLa cells, we observed that chimeras just upstream of the first region of interest (first-third chimeras) resulted in a phenotypic switch from nucleus-to-whole-cell, and vice versa, for A3B/D and A3D/B chimeras, respectively (Fig. 5b and Fig. S2). These observations became more evident with quantification of these localization patterns for both cell types (Fig. 5c). In contrast, generating chimeras downstream of the second region of interest (second-third chimeras) had no impact on localization of either A3B/D or A3D/B chimeras (Fig. 5b, 5c, and Fig. S2). These results indicated that a second set of residues is required for nuclear import of A3B and that these residues are located between positions 77 and 126.
Figure 5. A3B/D and A3D/B chimeras identify a second determinant of A3B nuclear localization within loop 5 and α-helix 3.

(a) Alignment of A3B and A3D NTD amino acid sequences as described in Fig. 1. (b and d) Representative images of 293T and HeLa cells transiently expressing the indicated wild-type or chimeric C-terminally eGFP tagged proteins (10 𝜇m scale bar). (c and e) Quantification of localization patterns for the indicated amino acid substitution mutants (n=25 for 293T cells and n=20 for HeLa cells; image quantification and statistical analysis as described in Fig. 1).
Residues within loop 5 and αhelix 3 of A3B NTD encompass a second localization determinant
To further explore the region identified between residues 77 and 126, we made additional chimeric substitutions within A3B and A3D NTDs exchanging loop 4 in combination with loop 5/αhelix 3 (hereafter referred to as αhelix 3+), loop 4 alone, or αhelix3+ alone to identify the residues required for nuclear import (Fig. 5a). Chimeras exchanging the entire region (loop 4 and αhelix 3+) or αhelix 3+ alone displayed differential localization from nucleus-to-whole-cell and vice versa in A3B/D and A3D/B chimeras, respectively, for both 293T and HeLa cells (Fig. 5d, 5e, and Fig. S2). Exchanging loop 4 alone had no effect on localization of either set of chimeras; however, when αhelix 3+ was exchanged, relocalization for both sets of chimeras could be observed (Fig. 5d, 5e, and Fig. S2).
To identify the exact residues within αhelix 3+ required for nuclear localization of A3B, we exchanged the residues within this region with those of A3D targeting two different amino acid segments T95-A102 (H3–1) and K103-S109 (H3–2) (Fig. 6a). Exchanging either the αhelix 3+ or H3–1 regions in the NTD of A3B with those of A3D resulted in abrogated nuclear import in both 293T and HeLa cells, and exchanging H3–2 alone had no effect (Fig. 6b, 6c, and Fig. S2).
Figure 6. Amino acid residues T95-A102 are important for A3B nuclear localization.
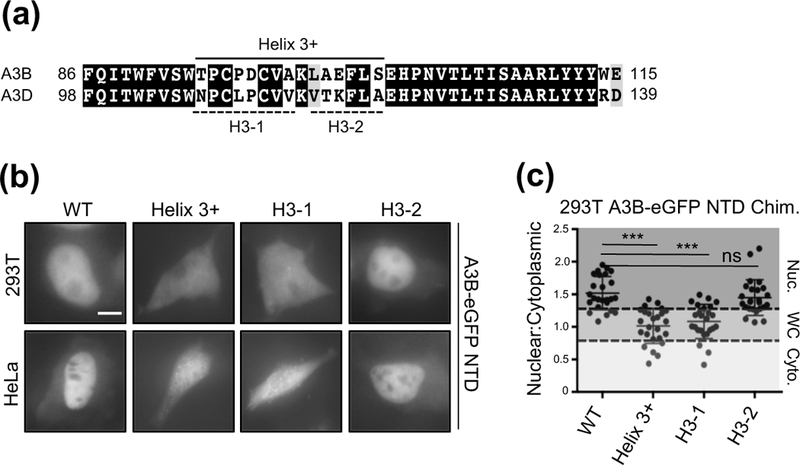
(a) Alignment of A3B and A3D protein sequences (as described in Fig. 1). The solid line indicates the full α−Helix 3+ region exchanged in A3B for the corresponding region of A3D. The dashed lines indicate the sub-regions of α−Helix 3+ that were exchanged individually in A3B. (b) Representative images of 293T and HeLa cells transiently expressing the indicated A3B NTD C-terminally eGFP tagged chimeric proteins (10 𝜇m scale bar). (c) Quantification of localization patterns for the indicated amino acid substitution mutants (n=25 per condition; image quantification and statistical analysis as described in Figure 1).
Cytoplasmic A3F becomes nuclear by exchanging residues 1–32 with A3B
As shown above, the high degree of sequence identity between A3B and A3D NTDs led to the discovery of a region within loop 5/αhelix 3 of A3B required for nuclear import (Fig. 5 and 6). With this in mind, we asked why A3F NTD is cytoplasmic, even though it has ~85% sequence identity (~92% similarity) to A3B NTD (Fig. 3a and 3c). Upon closer inspection, a sequence alignment between A3B and A3F NTDs revealed that these domains have an identical loop 5/αhelix 3 region (import surface 2) but that the first 32 residues of A3F are identical to those of A3G (missing import surface 1) (Fig. 7a). Therefore, we reasoned that exchanging the first 32 residues of A3F for those of A3B (reconstituting import surface 1) would result in nuclear import. As expected, exchanging these residues resulted in strong nuclear localization of the A3F NTD chimera in both 293T and HeLa cells (Fig. 7b, 7c, and Fig. S2).
Figure 7. A3F NTD relocalization from cytoplasmic to nucleus by A3B residues 1–32.
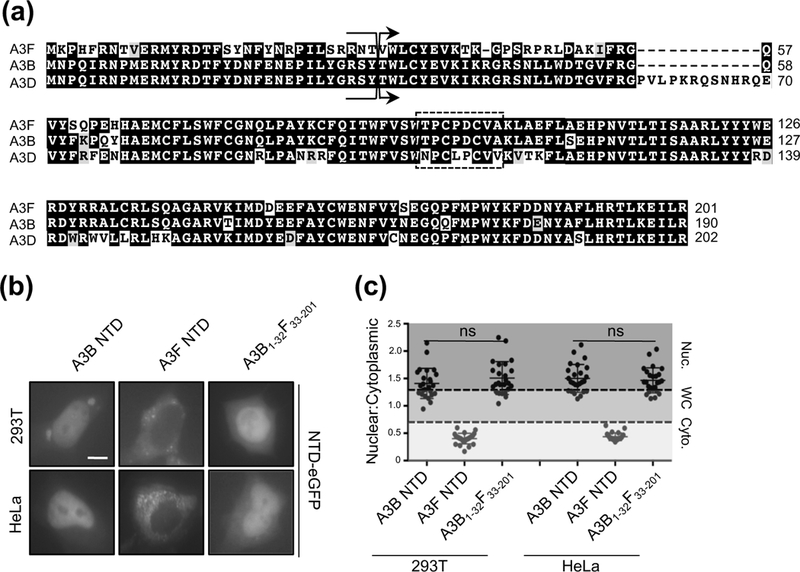
(a) Alignment of A3F, A3B, and A3D NTD protein sequences (as described in Fig. 1). (b) Representative images of 293T and HeLa cells transiently expressing the indicated wild-type and chimeric NTD C-terminally eGFP tagged proteins (10 𝜇m scale bar). (c) Quantification of localization patterns for the indicated amino acid substitution mutants (n=25 for 293T cells and n=20 for HeLa cells; image quantification and statistical analysis as described in Fig. 1).
DISCUSSION
A3B is unique amongst the human APOBEC3 family of DNA cytosine deaminase enzymes in that it exhibits constitutive nuclear localization and is upregulated in several different cancer types. Here, we demonstrate that the nuclear import of A3B is dictated by two distinct sets of residues within the NTD, and that grafting either of these surfaces onto related but non-nuclear APOBEC3 family members – A3D and A3F – results in relocalization to the nuclear compartment. Import surface 1, which has been previously reported on and further explored here, encompasses residues D19 and E24 (Fig. 1 and 2). Import surface 2 is comprised of a segment of loop 5 and αhelix 3 and it is also necessary for proper nuclear localization (Figs. 4-6). A previous residue implicated in nuclear import of A3B, V54 [51], was not identified here as being a strong contributor to the nuclear import mechanism. Analysis of the recently solved structure of A3B NTD (PDB: 5TKM) and a model here for wild-type NTD combine to indicate that this residue is buried within the hydrophobic interior of the protein. Therefore, it is possible that substitution of V54 to a negatively charged, hydrophilic amino acid (aspartate) in the previous study may have disrupted the conformation of “import surface 1” and indirectly disrupted nuclear import.
In the case of A3D NTD, import surface 1 (D19 and E24) is conserved but import surface 2 within loop 5/αhelix 3 (T95-A102) is not, and thus localization is whole-cell (Fig. 3a and 3c). However, when the A3D NTD loop 5/αhelix 3 region is replaced with that of A3B, nuclear localization becomes evident in both 293T and HeLa cells (Fig. 5). Our model also applies to the NTD of A3F, except that import surface 2 is conserved but import surface 1 is not. When A3F residues 1–32 are replaced with those of A3B, thus re-constituting import surface 1, the NTD of A3F becomes strongly nuclear (Fig. 7). In the case of A3G, neither set of residues is conserved and nuclear import only occurs when both import surfaces are replaced (A3B/G chimera data shown in Fig. 4). Additionally, A3G has been shown to have an active cytoplasmic retention property encompassing residues R24, F126, and W127 [52, 58]. An alternative explanation for the chimera localization phenotypes depicted in Fig. 4b is that this mechanism has been partially disrupted in the A3B/G chimeras, resulting in whole cell localization patterns. Therefore, nuclear import is not observed until all cytoplasmic retention residues of A3G have been replaced by A3B nuclear localization determinants. This is particularly noteworthy in two regards: 1) the R24 position in A3G structurally overlaps with “import surface 1” of A3B, and 2) the F126 and W127 positions in A3G are adjacent to “import surface 2” residues in A3B, A3D, and A3F. These data combine to indicate that the distinct localization activities of A3B (nucleus) and A3G (cytoplasm) may be mediated by similar solvent-exposed surface areas.
Our model of A3B requiring two distinct import surfaces, containing non-positively charged residues, differs from classical nuclear import motifs that are typically clusters of basic amino acids [59, 60]. These “classical” positively charged import sequences can be categorized as either monopartite, as in the case of the SV40 Large T-antigen [61], or bipartite, as in the case of nucleoplasmin [59, 62], BPM1 [63], or PRP20 [64]. However, “non-classical” nuclear localization sequences (NLS) are emerging that deviate from this dogma and utilize distinctly different sequences which can contain acidic residues, such as the case of hnRNP-A1 [65], or even proline-tyrosine based motifs in the case of yeast PAB2 [66]. Our findings here strongly suggest that the NLS of A3B, with two distinct surface-exposed regions, should be categorized as both non-classical and bipartite (Fig. 8).
Figure 8. Structural depiction of A3B NTD surfaces required for nuclear import.

(a) Structural representation of wild-type A3B NTD that highlights both import surfaces. Import surface 1 is defined by D19 and E24 (blue). Import surface 2 is defined by T95-A102 (green, this study). (b) Surface-filled representation of the ribbon structure in panel (a).
Our previous studies examining the molecular mechanism of A3B import showed that exogenously expressed IMPORTINα1, α3, and α5 could immunoprecipitate wild-type A3B, but not mutant V54D [51]. These data suggested that A3B might utilize the canonical IMPORTINα-dependent pathway to translocate into the nucleus. When we examined the two sets of residues on the 3-dimensional structural model of A3B NTD (Fig. 8), we observed that these surfaces are on the same molecular plane, in the shape of an arc. This is noteworthy since the solved crystal structure of IMPORTINα forms a horseshoe shape [67, 68]. Therefore, it is possible that IMPORTINα can bind both surfaces simultaneously, even in the absence of a canonical NLS, to engage the molecular import pathway. Interestingly, one of the greatest differences between A3B and A3D NTDs is a 12-residue longer loop 3 region in the latter protein (Fig. 5a). When this extension is mapped to the 3-dimensional structure, it resides directly between both sets of nuclear import surfaces. To determine if this extended loop 3 sequence could disrupt nuclear import of A3B, we introduced an additional 12 residues, and an even longer 23-residue piece, into the loop 3 region of A3B and assessed nuclear import. Neither of these long loop 3 extensions had any effect on A3B nuclear import (data not shown), suggesting that IMPORTINα may not be engaging both sets of residues simultaneously. If it were, then the presence of this extra 23-amino acid loop sequence would have been expected to attenuate import.
A more likely possibility is that A3B binds to an as yet unidentified import partner, and upon binding, engages the nuclear import machinery. This would be similar to the import mechanism utilized by CDK4/CyclinD1 [69]. In this model, the import partner may exhibit a canonical NLS, or an A3B/binding-partner complex could form a shared structural NLS for import. This is an appealing model since it would raise the possibility of a more specific way to chemically inhibit A3B nuclear import and could serve as a novel approach to develop inhibitors to counteract A3B mutagenesis observed different cancer types. Several efforts are underway to discover strategies to repress A3B function and exploiting this new set of import residues may serve as an alternative approach that could potentially prove both effective and specific.
MATERIALS AND METHODS
Cell Lines and Culture Conditions
293T cells were maintained in DMEM (Hyclone) supplemented with 10% FBS (Gibco) and 0.5% penicillin/streptomycin (50 units). HeLa were maintained in RPMI (Hyclone) supplemented with 10% FBS (Gibco) and 0.5% penicillin/streptomycin (50 units). 293T and HeLa cells were transfected with TransIT LTI (Mirus) according to the manufacture’s protocol.
Plasmids and Cloning
All A3-eGFP expression plasmids used in this study were cloned into a pQCXIH retroviral expression vector. Point mutants were generated by PCR amplification using Phusion high fidelity DNA polymerase (NEB) and overlapping PCR to introduce the indicated mutation. A similar approach was taken to generate chimeric A3 proteins, except that the indicated A3s were used as individual templates and then overlapping PCR was used to combine the two amplicons. For all NTD and CTD constructs, including mutants and chimeras, PCR products were digested with NotI and AgeI restriction enzymes and ligated into pQCXIH that contained an in-frame c-terminal eGFP tag. For cloning mutants and chimeras into the full-length A3Bi and A3Gi intron disrupted constructs, NotI and XhoI (within the intron sequence) were used to digest the PCR amplicons for cloning. Digested products were cloned into vectors that contain the respective intron-CTD sequence with a C-terminal eGFP tag. All primer sequences used to clone the point mutant and chimeric proteins are listed in Supplementary Table 1. Full-length A3Bi-eGFP and A3Gi-eGFP constructs used in Figs. 1-4 contain an intron within the A3 CTD coding sequence. This intron prevents A3B-mediated self-inactivation of the vector during amplification in E. coli, which is genotoxic as reported previously [70].
Fluorescence Microscopy Experiments
Approximately 10,000 293T cells, or, 6,000 HeLa cells were plated in growth media described above into a 96-well CellBIND microplate (Corning) and allowed to adhere overnight. The next day, cells were transfected with 100 ng of the indicated A3-eGFP construct and imaged 48 h post transfection. Images were collected at 40x magnification using an EVOS FL Color microscope (ThermoFisher), and quantified using ImageJ software. Individual cells were scored and grouped into three categories (nuclear, whole cell, or cytoplasmic). Subcellular compartments were defined based on DAPI staining (nucleus). The anti-A3 signal was calculated by dividing the nuclear anti-A3 staining intensity by the cytoplasmic A3 intensity, and then graphed with Prism 6.0 (GraphPad Software). All constructs were tested in biological triplicate with respective quantifications being calculated from representative images taken from a single set of experiments.
Generation of A3B NTD Structural Model
The wild-type A3B NTD system was generated based on the A3B-CD1m A3B (Y13D/Y28S/Y83D/W127S/Y162D/Y191H) crystal structure, PDB ID: 5TKM [54]. Amino acid substitutions introduced for protein solubility and crystallization were reverted in silico using MOE software [71].
Statistical Analysis
GraphPad Prism 6.0 was used for statistical analysis (GraphPad). Quantitative data were represented as mean +/−SEM. Statistical significance was determined by an unpaired student’s t-test.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Gabriel Starrett and Christopher Richards for intellectual discussion. DJS received salary support from the University of Minnesota Craniofacial Research Training (MinnCResT) program (NIH T90DE022732). This work was supported by NIAID R37 AI064046. Support from NIH P41 GM103426 to REA is gratefully acknowledged. RSH is the Margaret Harvey Schering Land Grant Chair for Cancer Research, a Distinguished University McKnight Professor, and an Investigator of the Howard Hughes Medical Institute.
ABBREVIATIONS:
- APOBEC3
apolipoprotein B mRNA-editing enzyme, catalytic polypeptide-like 3
- A3B
APOBEC3B
- A3D
APOBEC3D
- A3G
APOBEC3G
- A3F
APOBEC3F
- NTD
N-terminal domain
- CTD
C-terminal domain
- ssDNA
single-stranded DNA
- AID
activation-induced cytidine deaminase
Footnotes
COMPETING FINANCIAL INTEREST
R.S.H. is a co-founder, shareholder, and consultant of ApoGen Biotechnologies Inc. R.E.A. is a co-founder, is on the Scientific Advisory Board, and has equity interest in Actavalon, Inc. The other authors declare no competing financial interests.
REFERENCES
- 1.Harris R, and Dudley J (2015) APOBECs and virus restriction. Virol 479:131–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Simon V, Bloch N, and Landau N (2015) Intrinsic host restrictions to HIV-1 and mechanisms of viral escape. Nat. Immunol 16:546–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Malim M, and Beiniasz P (2012) HIV restriction factors and mechanisms of evasion. Cold Spring Harbor Perspectives in Medicine 2:a006940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koito A, and Ikeda T (2013) Intrinsic immunity against retrotransposons by APOBEC cytidine deaminases. Front. Microbiol 4:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Refsland E, Hulquist J, and Harris R (2012) Endogenous origins of HIV-1 G-to-A hypermutation and restriction in the nonpermissive T cell line CEM2n. PLoS Path 8:e1002800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang T, Zhang W, Tian C, Liu B, Yu Y, Ding L, Spearman P, and Yu XF. (2008) Distinct viral determinants for the packagining of human cytidine deaminases APOBEC3G and APOBEC3C. Virol 377:71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hultquist J, Lengyel J, Refsland E, LaRue R, Lackey L, Brown W, and Harris R (2011) Human and rhesus APOBECD, APOBEC3F, APOBEC3G, and APOBEC3H demonstrate a conserved capacity to restrcit Vif-deficient HIV-1. J Virol 85:11220–11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dang Y, Wang X, Esselman W, and Zheng Y (2006) Identification of APOBEC3DE as another antiretroviral factor from the human APOBEC family. J Virol 80:10522–10533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harris R, Bishop K, Sheehy A, Craig H, Petersen-Mahrt S, Watt I, Neuberger M, and Malim M. (2003) DNA deamination mediates innate immunity to retroviral infection. Cell 113:803–809. [DOI] [PubMed] [Google Scholar]
- 10.Mangeat B, Turelli P, Caron G, Friedli M, Perrin L, and Trono D (2003) Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 424:99–103. [DOI] [PubMed] [Google Scholar]
- 11.Zhang H, Yang B, Pomerantz R, Zhang C, Arunachalam S, and Gao L (2003) The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature 424:94–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liddament M, Brown W, Schumacher A, and Harris R (2004) APOBEC3F properties and hypermutation preferences indicate activity against HIV-1 in vivo. Curr. Biol 14:1385–91. [DOI] [PubMed] [Google Scholar]
- 13.Zheng Y, Irwin D, Kurosu T, Tokunaga K, Sata T, and Peterlin B (2004) Human APOBEC3F is another host factor that blocks human immunodeficiency virus type 1 replication. J Virol 78:6073–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wiegand H, Doehle B, Bogerd H, and Cullen B (2004) A second human antiretroviral factor, APOBEC3F, is suppressed by the HIV-1 and HIV-2 Vif proteins. EMBO J 23:2451–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.OhAinle M, Kerns J, Li M, Malik H, and Emerman M (2009) Anti-retroelement activity of APOBEC3H was lost twice in recent human evolution. Cell Host Microbe 4:249–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ooms M, Krikini A, Kress A, Simon V, and Munk C (2012) APOBEC3A, APOBEC3B, and APOBEC3H haplotype 2 restrict human T-lymphotropic virus type 1. J Virol 86:6097–6108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu Q, Chen D, Konig R, Marianai R, and Unutmaz D (2004) APOBEC3B and APOBEC3C are potent inhibitors of simian immunodeficiency virus replication. J Virol 279:53379–53386. [DOI] [PubMed] [Google Scholar]
- 18.Doehle B, Schafer A, and Cullen B (2005) Human APOBEC3B is a potent inhibitor of HIV-1 infectivity and is resistant to HIV-1 Vif. Virol 339:281–288. [DOI] [PubMed] [Google Scholar]
- 19.Wissing S, Montano M, Garcia-Perez J, Moran J, and Greene W (2011) Endogenous APOBEC3B restricts LINE-1 retrotransposition in transformed cells and human embryonic stem cells. J Biol. Chem 266:36427–36437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stenglein M, and Harris R (2006) APOBEC3B and APOBEC3F inhibit L1 retrotransposition by a DNA deamination-independent mechanism. J Biol. Chem 281:16837–41. [DOI] [PubMed] [Google Scholar]
- 21.Stenglein M, Burns M, Li M, Lengyel J and Harris R (2010) APOBEC3 proteins mediate the clearance of foreign DNA from human cells. Nat. Struct. Mol. Biol 17:222–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bogerd H, Wiegand H, Hulme A, Garcia-Perez J, O’Shea K, Moran J, and Cullen B (2006) Cellular inhibitors of long interspersed element 1 and Alu retrotransposition. PNAS 103:8780–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lovsin N, and Peterlin B (2009) APOBEC3 proteins inhibit LINE-1 retrotransposition in the absence of ORF1p binding. Ann. N. Y. Acad. Sci 1178:268–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen H, Lilley C, Lee D, Chou J, Narvaiza I, Landau N, and Weitzman M (2006) APOBEC3A is a potent inhibitor of adeno-associated virus and retrotransposons. Curr. Biol 16:480–5. [DOI] [PubMed] [Google Scholar]
- 25.Burns M, Lackey L, Carpenter M, Rathore A, Land A, Leonard B, Refsland E, Kotndeniya D, Tretyakova N, Nikas J, Yee D, Temiz N, Donohue D, McDougle R, Brown W, Law E, and Harris R (2013) APOBEC3B is an enzymatic source of mutation in breast cancer. Nature 494:366–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burns M, Temiz N, and Harris R (2013) Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat. Gen 45:977–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roberts S, Lawrence M, Mlimczak L, Grimm S, Fargo D, Stojanov P Kiezun A, Kryukov G, Carter S, Saksena G, Harris S, Shah R, Resnick M, Getz G, and Gordenin D. (2013) An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Gen 45:970–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lefebvre C, Bachelot T, Filleron T, Pedrero M, Campone M, Soria JC, Massard C, Levy C, Arnedos M, Lacroix-Triki M, Garrabey J, Boursin Y, Deloger M, Fu Y, Commo F, Scott V, Lacroix L, Dieci MV, Kamal M, Dieras V, Goncalves A, Ferrerro JM, Romieu G, Vanlemmens L, Mouret Reynier MA, Thery JC, Le Du F, Guiu S, Dalenc F, Clapisson G, Bonnefoi H, Jimenez M, Le Tourneau C and Andre F (2016) Mutational profile of metastatic breast cancers: a retrospective analysis. PLoS Med 13:e1002201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, Boyault S, Burkhardt B, Butler AP, Caldas C, Davies HR, Desmedt C, Eils R, Eyfjord JE, Foekens JA, Greaves M, Hosoda F, Hutter B, Ilicic T, Imbeaud S, Imielinski M, Jager N, Jones DT, Jones D, Knappskog S, Kool M, Lakhani SR, Lopez-Otin C, Martin S, Munshi NC, Nakamura H, Northcott PA, Pajic M, Papaemmanuil E, Paradiso A, Pearson JV, Puente XS, Raine K, Ramakrishna M, Richardson AL, Richter J, Rosenstiel P, Schlesner M, Schumacher TN, Span PN, Teague JW, Totoki Y, Tutt AN, Valdes-Mas R, van Buuren MM, van ‘t Veer L, Vincent-Salomon A, Waddell N, Yates LR, Australian Pancreatic Cancer Genome I, Consortium IBC, Consortium IM-S, PedBrain I, Zucman-Rossi J, Futreal PA, McDermott U, Lichter P, Meyerson M, Grimmond SM, Siebert R, Campo E, Shibata T, Pfister SM, Campbell PJ and Stratton MR (2013) Signatures of mutational processes in human cancer. Nature 500:415–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nik-Zainal S, Alexandrov LB, Wedge DC, Van Loo P, Greenman CD, Raine K, Jones D, Hinton J, Marshall J, Stebbings LA, Menzies A, Martin S, Leung K, Chen L, Leroy C, Ramakrishna M, Rance R, Lau KW, Mudie LJ, Varela I, McBride DJ, Bignell GR, Cooke SL, Shlien A, Gamble J, Whitmore I, Maddison M, Tarpey PS, Davies HR, Papaemmanuil E, Stephens PJ, McLaren S, Butler AP, Teague JW, Jonsson G, Garber JE, Silver D, Miron P, Fatima A, Boyault S, Langerod A, Tutt A, Martens JW, Aparicio SA, Borg A, Salomon AV, Thomas G, Borresen-Dale AL, Richardson AL, Neuberger MS, Futreal PA, Campbell PJ, Stratton MR and Breast Cancer Working Group of the International Cancer Genome C (2012) Mutational processes molding the genomes of 21 breast cancers. Cell 149:979–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ullah I, Karthik GM, Alkodsi A, Kjallquist U, Stalhammar G, Lovrot J, Martinez NF, Lagergren J, Hautaniemi S, Hartman J and Bergh J (2018) Evolutionary history of metastatic breast cancer reveals minimal seeding from axillary lymph nodes. J Clin. Invest 128:1355–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Swanton C, McGranahan N, Starrett G, and Harris R (2015) APOBEC enzymes: mutagenic fuel for cancer evolution and heterogeneity. Cancer Discovery 5:704–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roberts S, and Gordenin D (2014) Hypermutation in human cancer genomes: footprints and mechanisms. Nat. Rev. Cancer 14:786–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Helleday T, Eshtad S and Nik-Zainal S (2014) Mechanisms underlying mutational signatures in human cancers. Nat. Rev. Genet 15:585–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Venkatesan S, Rosenthal R, Kanu N, McGranahan N, Bartek J, Quezada SA, Hare J, Harris RS and Swanton C (2018) Perspective: APOBEC mutagenesis in drug resistance and immune escape in HIV and cancer evolution. Ann. Oncol 29:563–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Law E, Sieuwerts A, LaPara K, Leonard B, Starrett G, Molan A, Temiz N, Vogel R, Gelder M, Sweep F, Span P, Foekens J, Martens J, Yee D, and Harris R (2016) The DNA cytosine deaminase APOBEC3B promotes tamoxifen resistance in ER-positive breast cancer. Sci. Advan 2:e1601737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sieuwerts A, Willis S, Burns M, Loop M, Meijer-Van Gelder M Schlicker A, Heideman M, Jacobs H, Wessels L, Leyland-Jones B, Gray KP, Foekens JA, Harris RS, and Mertens JW. (2014) Elevated APOBEC3B correlates with poor outcomes for estrogen receptor-positive breast cancers. Hormones and Cancer 5:405–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsuboi M, Yamane A, Horiguchi J, Yokobori T, Kawabata-Iwakawa R, Yoshiyama S, Rokudai S, Odawara H, Tokiniwa H, Oyama T, Takeyoshi I, and Nishiyama M (2016) APOBEC3B high expression status is associated with aggressive phenotype in Japanese breast cancers. Breast Cancer 23:780–8. [DOI] [PubMed] [Google Scholar]
- 39.Cescon D, Haibe-Kains B, and Mak T (2015) APOBEC3B expression in breast cancer reflects cellular proliferation, while a deletion polymorphism is associated with immune activation. PNAS 112:2841–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Walker B, Wardell C, Murison A, Boyle E, Begum D Dahir N, Proszek PZ, Melchor L, Pawlyn C, Kaiser MF, Johnson DC, Qiang YW, Jones JR, Cairns DA, Gregory WM, Owen RG, Cook G, Drayson MT, Jackson GH, Davies FE, and Morgan GJ. (2015) APOBEC family mutational signatures are associated with poor prognosis translocations in multiple myeloma. Nat. Comm 6:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nik-Zainal S, Wedge D, Alexandrov L, Petljak M, Butler A, Bolli A, Davies HR, Knappskog S, Martin S, Papaemmanuil E, Ramakrishna M, Shlien A, Simonic I, Xue Y, Tyler-Smith C, Campbell PJ, and Stratton MR. (2014) Association of a germline copy number polymorphism of APOBEC3A and APOBEC3B with burden of putative APOBEC-dependent mutations in breast cancer. Nat. Genet 46:487–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tokunaga E, Yamashita N, Tanaka K, Inoue Y, Akiyoshi S, Saeki H, Oki E, Kitao H, and Maehara Y (2016) Expression of APOBEC3B mRNA in primary breast cancer of Japanese women. PLoS One 11:e0168090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fanourakis GT,K, Papanikolaou N, Chatzistamou I, Xydous M, Tseleni-Balafouta S, Skiavounou A, Voutsinas GE, and Vastardis H. (2016) Evidence for APOBEC3B mRNA and protein expression in oral squamous cell carcinomas. Exp. Mol. Pathol 101:314–319. [DOI] [PubMed] [Google Scholar]
- 44.Middlebrooks C, Banday A, Matsuda K, Udquim K, Onabajo O, Paquin A, Figueroa JD, Zhu B, Koutros S, Kubo M, Shuin T, Freedman ND, Kogevinas M, Malats N, Chanock SJ, Garcia-Closas M, Siverman DT, Rothman N, and Prokunina-Olsson L (2016) Association of germline variants in the APOBEC3 region with cancer risk and enrichment with APOBEC-signature mutations in tumors. Nat. Genet 48:1330–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alexandrov L, Kin-Zainal S, Wedge D, Aparicio S, Behjati S, Blankin A, Bignell G, Bolli N, Borg A, Borresen-Dale A, Boyauly S, Burkhardt B, Butler A, Caldas C, Davies H, Desmedt C, Eils R, Eyfjord J, Foekens J, Greaves M, Hosoda F, Hutter B, Ilicic T, Imbeaud S, Imielinski M, Jager N, Jones D, Knappskog S, Kool M, Lakhani S, Lopez-Otin C, Martin S, Munchi N, Nakamura H, Northcott P, Pajic M, Papaemmanuli E, Paradiso A, Pearson J, Puente X, Raine K, Ramakrisna M, Richardson A, Richter J, Rosentiel P, Schlesner M, Schumacher T, Span P, Teague J, Totoki Y, Tutt A, Valdes-Mas R, van Buuren M, van ‘t Veer L, Vincent-Salomon A, Waddell N, Yates L, Austrailian Pancreatic Cancer Genome Initiative, ICGC BReast Cancer Consortium, ICGC MMML-Seq Consortium, ICGC PedBrain, Zucman-Rossi J, Futreal A, McDermott U, Lichter P, Meyerson M, Grimmond S, Siebert R, Campo E, Shibata T, Pfister S, Campbell P, and Stratton M. (2013) Signatures of mutational processes in human cancer. Nature 500:415–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Verhalen B, Starrett G, Harris R, and Jiang M (2016) Functional upregulation of the DNA cytosine deaminase APOBEC3B by polyomaviruses. J Virol 90:6379–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vieira V, Leonard B, White E, Starrett G, Temiz N, Lorenz L, Lee D, Soares MA, Lambert PF, Howley PM, and Harris RS. (2014) Human papillomavirus E6 triggers upregulation of the antiviral and cancer genomic DNA deaminase APOBEC3B. mBio 5:e02234–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mori S, Takeuchi T, Ishii Y, Yugawa T, Kiyono T, Nishina H and Kukimoto I (2017) Human papillomavirus 16 E6 up-reguates APOBEC3B via the TEAD transcription factor. J Virol 91:e02413–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Starrett G, Marcelus C, Cantalupo P, Katz J, Cheng J, Akagi K, Thakuria M, Rabinowits G, Wang L, Symer D, Pipas J, Harris RS, and DeCaprio J (2017) Merkel cell polyomavirus exhibits dominant control of the tumor genome and transcriptome in virus-associated Merkel cell carcinoma. MBio 8:e02079–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lackey L, Law E, Brown W, and Harris R (2013) Subcellular localization of the APOBEC3 proeins during mitosis and implications for genomic DNA deamination. Cell Cycle 12:762–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lackey L, Demorest Z, Land A, Hultquist J, Brown W, and Harris R (2012) APOBEC3B and AID have similar nuclear import mechanisms. J Mol. Biol 419:301–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stenglein M, Matsuo H, and Harris R (2008) Two regions within the amino-terminal half of APOBEC3G cooperate to determine cytoplasmic localization. J Virol 82:9591–9599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pak V, Heidecker G, Pathak V, and Derse D (2011) The role of amino-terminal sequences in cellular localization and antiviral activity of APOBEC3B. J Virol 85:8538–8547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xiao X, Yang H, Arutiunian V, Besse G, Morimoto C, Zirkle B, and Chen X (2017) Structural detminants of APOBEC3B non-catalytic domain for molecular assembly and catalytic regulation. Nucleic Acids Res 45:7494–7506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liang W, Xu J, Yuan W, Song X, Zhang J, Wei W Yu X, and Yang Y. (2016) APOBEC3DE inhibits LINE-1 retrotransposition by interacting with ORF1p and influencing LINE reverse transcriptase activity. PLoS One 11:e0157220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wichroski M, Robb G, and Rana T (2006) Human retroviral host restriction factors APOBEC3G and APOBEC3F localize to mRNA processing bodies. PLoS Path 2:e41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.LaRue R, Jonsson S, Silverstein K, Lajoie M, Bertrand D, El-Mabrouk N, Hotzel I, Andresdottir V, Smith T, and Harris R (2008) The artiodactyl APOBEC3 innate immune repertoire shows evidence for a multi-functional domain organization that existed in the ancestor of placental mammals. BMC Mol. Biol 9:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bennett RP, Presnyak V, Wedekind JE and Smith HC (2008) Nuclear exclusion of the HIV-1 host defense factor APOBEC3G requires a novel cytoplasmic retention signal and is not dependent on RNA binding. J Biol. Chem 283:7320–7. [DOI] [PubMed] [Google Scholar]
- 59.Dingwall C, Sharnick S, and Laskey R (1982) A polypeptide domain that specifies migration of nucleoplasmin into the nucleus. Cell 30:449–58. [DOI] [PubMed] [Google Scholar]
- 60.Lange A, Mills R, Lange C, Stewart M, Devine S, and Corbett A (2007) Classical nuclear localization signal: definition, function, and interaction with importin. J Biol. Chem 282:5101–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kalderon D, Roberts B, Richardson W, and Smith A (1984) A short amino acid sequence able to specify nuclear localization. Cell 39:499–509. [DOI] [PubMed] [Google Scholar]
- 62.Dingwall C, Robbins J, Dilworth S, Roberts B, and Richardson W (1988) The nucleoplasmin nuclear localization sequence is larger and more complex than that of SV-40 T antigen. J Cell Biol 107:841–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Levanic D, Horvat T, Martincic J, and Bauer N (2012) A novel bipartite nuclear localization signal guides BPM1 protein to nucleolus suggesting its CULLIN3 independent function. PLoS One 7:e51184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Roman N, Christie M, Swarbrick C, Kobe B, and Forwood J (2013) Structural characterisation of the nuclear import receptor importin alpha in complex with the bipartite NLS of Prp20. PLoS One 8:e82038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Makkerh J, Dingwall C, and Laskey R (1996) Comparative mutagenesis of nuclear localization signals reveals the importance of neutral and acidic amino acids. Curr. Biol 6:1025–7. [DOI] [PubMed] [Google Scholar]
- 66.Mallet P, and Bachand F (2013) A proline-tyrosine nuclear localization signal (PY-NLS) is required for the nuclear import of fission yeast PAB2, but not of human PABN1. Traffic 14:282–94. [DOI] [PubMed] [Google Scholar]
- 67.Mtnott A, Harrop S, Brown L, Breit S, Boke B, and Curmi P (2011) Crystal structure of importin-alpha bound to a peptide bearing the nuclear localization signal from chloride intracellular channel protein 4. FEBS J 278:1662–75. [DOI] [PubMed] [Google Scholar]
- 68.Nakada R, Hirano H and Matsuura Y (2015) Structure of importin-alpha bound to a non-classical nuclear localization signal of the influenza A virus nucleoprotein. Sci. Rep 5:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bockstaele L, Coulonval K, Kooken H, Paternot S, and Roger Pierre (2006) Regulation of CDK4. Cell Division 1:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hulquist J, Lengyel J, Refsland E, LaRue R, Lackey L, Brown W, and Harris R (2011) Human and rhesus APOBECD, APOBEC3F, APOBEC3G, and APOBEC3H demonstrate a conserved capacity to restrict Vif-deficient HIV-1. J Virol 85:11220–11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Molecular Operating Environment (MOE), 2014.09; Chemical Computing Group ULC, 1010 Sherbrooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7 (2018) [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.