

Abstract
Neuronal calcium sensor-1 (NCS-1) protein has been implicated in multiple neuronal functions by binding partners mostly through a largely exposed hydrophobic crevice (HC). In the absence of a ligand, the C-terminal tail (loop L3, residues D176 to V190) binds directly to the HC pocket as a ligand mimetic, occupying the HC and regulating its conformational stability. A recent experimental study reported that L3 deletion resulted in global structure destabilization. However, the influence of C-terminal tail on the conformations of NCS-1 protein is unclear at the atomic level. In this study, we investigated the structural properties and the conformational dynamics of wild type NCS-1 and L3 truncation variant by extensive all-atom molecular dynamics (MD) simulations. Our cumulative 2 μs MD simulations demonstrated that L3 deletion increased the structural flexibility of the C-domain and the distant N-domain. The community network analysis illustrated that C-terminal tail truncation weakened the interdomain correlation. Moreover, our data showed that the variant significantly disrupted the salt bridges network and expanded simultaneously the global structure and HC. These conformational changes caused by C-terminal tail truncation may affect the regulation of target interactions. Our study provides atomic details of the conformational dynamics effects of the C-terminal tail on human wild type NCS-1.

1. INTRODUCTION
Neuronal calcium sensor 1 (NCS-1) protein is one of the members of the neuronal calcium sensor (NCS) family, which consists of NCS-1, hippocalcin, neurocalcin-δ, VILIP1-3, recoverin, GCAP1-3, and KChIP1-4.1 NCS proteins have been implicated in multiple neuronal functions. However, genetic studies have showed that these functions do not overlap,2 and NCS-1 is able to interact with and regulate different target proteins.1 Structural studies have showed that NCS-1, recoverin, and GCAP1 have similar main chain topologies.3 Some NCS proteins, such as NCS-1, are N-terminally myristoylated.4 The solvent exposed myristoyl group drives constitutive membrane association5 dynamically cycling between the membrane and cytosol.6
Human NCS-1 plays key roles in central neuronal system disorders and is associated with psychiatric diseases.7 For example, a rare missense, substituting R102Qwas identified in an autistic patient.8 The dysfunctions of NCS-1 are poorly characterized on the molecular level.9 NCS-1 specificity of target protein recognition requires that the hydrophobic crevice (HC) consisting of conserved hydrophobic residues is exposed.1 The specificity of the interactions with binding partners is due to the varying size and shape of the hydrophobic groove, with varied patterns of surrounding charged residues.1,3 In complexes, HC is occupied by a Pik1 peptide fragment10–12 or poly(ethylene glycol) (PEG) molecules13 which stabilize the conformation.14 In addition, NCS-1 can mediate desensitization of D2/D3 dopamine receptors through interaction with the short cytoplasmic C-terminus of the receptor.15 A synthetic dopamine D2 receptor peptide can bind to the large, solvent-exposed HC.16,17 The movement of the C-terminal helix 11 of NCS-1 which fully exposed the HC was shown to be important for this D2 peptide binding.17 A NMR study showed that the C-terminal tail (loop L3, residues D176 to V190) of NCS-1 protein bound directly to the HC pocket as a ligand mimetic in the absence of binding ligand and regulated the conformational stability of the activated state.14 L3 can initially bind to the binding site in HC when there are no binding ligands, while it is released by incoming interacting proteins.3,14,28 Our recent MD simulation study showed that in the R102Q variant of the human NCS-1, L3 was in a more extended state, largely occupying the hydrophobic crevice,18 suggesting that it may block the binding of NCS-1 to its receptors. The detailed mechanism of the C-terminal tail of NCS-1 is currently not understood. Kragelund et al. found that the C-terminal tail deletion reduced the global stability of NCS-1.14 However, the effect of L3 deletion on the conformational dynamics of NCS-1 is unclear at the atomic level.
In this study, we investigated the role of C-terminal tail on the structural properties by conducting two independent 500 ns molecular dynamics (MD) simulations with explicit solvent for both WT NCS-1 and its L3 deletion type (DT) NCS-1 (NCS- 1Δ176–190), each starting from two different initial structures. We found that the secondary structure content of NCS-1Δ176–190 variant was quiet similar to that of WT NCS-1, consistent with a recent experimental study.14 The structural flexibilities of the C-domain and the N-domain distal to C-terminal tail were increased, indicating local and global perturbations of C- terminal tail on the conformation of NCS-1 protein. The global protein structure and HC were simultaneously expanded, inducing the HC to be fully exposed to the solvent. The salt bridges, especially in the C-domain, were significantly disrupted. The community network analysis illustrated that the C-terminal tail truncation weakened the interdomain correlation. These results reveal that L3 plays a critical role in the NCS-1 conformation dynamics, and these structural changes caused by C-terminal tail truncation may affect the regulation of target interactions.
2. MATERIALS AND METHODS
Human WT NCS-1 Protein and the NCS-1Δ176–190 Variant.
The solution NMR structures of the unmyristoylated calcium-bound human NCS-1 (PDB id: 2LCP) contain 20 structure models.14 The pairwise backbone RMSD values (see Table S1) spanned from 0.13 to 0.37 nm for the core structure (E11 to K174) and from 0.21 to 0.64 nm for the full-length NCS-1 protein (M1 to V190). The first two minimum-energy models of these solution NMR structures were utilized as the initial states of two independent molecular dynamic (MD) simulations of WT NCS-1. The two initial conformations of the NCS-1㤄176–190 variant were modeled by taking the two WT initial states as the starting point, with the last 15 residues from D176 to V190 deleted, followed by energy minimization. The four initial states are labeled as iWT-1, iWT-2, iDT-1, and iDT-2. The root-mean-square deviations (RMSDs) evaluated for the backbone of the residues E11 to K174 of the NMR NCS-1 protein structure were both 0.18 nm for the iWT-1–iWT-2 pair and for the iDT-1–iDT-2 pair. In this study, to mimic the experimental neutral pH condition,14 the N-terminus and the side chains of Lys and Arg were protonated (NH3+, Arg+, Lys+), and the C-terminus and the side chains of Glu and Asp were deprotonated (COO−, Glu−, Asp−).
Details of Molecular Dynamics Simulations.
MD simulations have been widely used in the conformational studies of biomolecules.19–25 In this work, explicit solvent MD simulations were carried out using the GROMACS 4.5.3 software package26 and the CHARMM27 force field with CMAP corrections,27 in accordance with recent MD studies by Bellucci et al.28 and by us.18 The results from these two MD studies are in line with a recent NMR study,14 indicating that the force field used in these studies is reliable for NCS-1 protein. Two independent 500 ns MD simulations were performed for both WT NCS-1 and NCS-1Δ176–190 variant, using model 1 and model 2 with three calcium ions in PDB structure (PDB id: 2LCP) as starting points.14 In this study, the parameters used for calcium ions were q = +2.0e, σ = 0.2436 nm, and ε = 0.5021 kJ/mol. The TIP3P water model was used. Additional Na+ and Cl− were added to the systems with a NaCl concentration of 0.1 M. The simulation box type is rhombic dodecahedron. In GROMACS, the simulation box is defined by three vectors a (d, 0, 0), b (0, d, 0), and c (d/2, d/2, √2d/2) and written as (d, d, √2d/2, d/2, d/2), where d is the edge length of the rhombic dodecahedron. In our simulations, the d values for model 1 and model 2 were respectively 9.30 and 9.16 nm. Thus, the simulation boxes were described as (9.30, 9.30, 6.58, 4.65, 4.65) nm for model 1 (corresponding to MD runs: WT-1 and DT-1) and (9.16, 9.16, 6.47, 4.58, 4.58) nm for model 2 (corresponding to MD runs: WT-2 and DT-2). The boxes were big enough that the protein has no atomic contact with its own periodic image. Bond lengths within protein and water molecules were respectively constrained by the LINCS29 and SETTLE algorithms,30 allowing an integration time step of 2 fs. The particle mesh Ewald (PME) method was used to calculate the electrostatic interaction with a real space cutoff of 1.0 nm, and the van der Waals interactions were calculated using a cutoff of 1.4 nm. The simulations were performed in isothermal–isobaric (NPT) ensemble using periodic boundary condition. The solute and solvent were separately coupled to external temperature bath using velocity rescaling method31 and pressure bath using the Parrinello–Rahman method.32 The temperature and pressure were maintained at 310 K and 1 bar using coupling constants of 0.1 and 1.0 ps, respectively.
Analysis Methods.
Analysis of the trajectories was performed using the tools implemented in the GROMACS 4.5.3 software package.26 The RMSDs were calculated following structural alignment. The RMSD was respectively the backbone RMSD of the core structure (residues E11 to K174), the N-domain (residues E11 to S93), and the C-domain (residues D98 to K174) of NCS-1 protein. The structural stability of the NCS-1 protein was examined by the time evolution of the number of backbone H-bonds. A hydrogen bond was considered to be formed if the distance between N and O is ≤3.5 Å and the angle of N–H…O is ≥ 150°. The DSSP program was used to determine the secondary structure.33 Root-mean-square fluctuation (RMSF) was calculated for each residue with respect to the MD generated average structure in the last 400 ns. The free energy landscape was constructed using the relation –RT ln H(x,y), where H(x,y) was the histogram of two selected reaction coordinates, x and y. A salt bridge between a pair of oppositely charged residues was considered to be formed if the centroids of the side-chain charged groups in two oppositely charged residues lie within 0.4 nm of each other.34 The distances between the centroids of the side-chain charged groups were calculated to show the change of the salt bridges. The visual molecular dynamics (VMD 1.9.1) package was used for graphical structure analysis.35 Interaction networks of WT NCS-1 and of the NCS-1Δ176–190 variant were analyzed and displayed by Network View36 implemented in VMD.
3. RESULTS AND DISCUSSION
NCS-1Δ176-190 Variant Maintained the Overall Secondary Structure Content; However, the Dynamics of the C-domain and Distant N-Domain Changed.
A cartoon representation of the solution structure of unmyristoylated calcium-bound human NCS-1 protein with Ca2+ ions14 and the amino acid sequence of the C-terminal tail L3 are given in Figure 1. The NCS-1 structure primarily consists of nine α-helices and three loops. The helices arrange in four EF hands, namely EF1, EF2, EF3, and EF4. Three of the four EF hands (EF2, EF3, and EF4) can bind Ca2+ ions in corresponding sites (Figure 1a). EF1 is not able to bind Ca2+ ions due to a conserved Cys/Pro substitution in the calcium binding motif.13,37 According to Heidarsson et al.,14 the nine helices are helix 1 (H1) (residues E11 to R18), H2 (E24 to F34), H3 (A45 to Q54), H4 (T62 to F72), H5 (F82 to S93), H6 (D98 to Y108), H7 (R118 to V132), H8 (E146 to M155), and H9 (L166 to K174). The three loops are L1 (F56 to P61), L2 (G133 to P145), and L3 (D176 to V190). Residues E24 to K174 shape a hydrophobic crevice (HC), of which helices H4, H5, and H6 form the floor of the crevice, H3 and H7 on one side and H9 on the other side of the HC, and helices H2 and H8 close the HC at the opposite edges. L3 partly occupies the HC as a ligand mimic in the absence of a binding partner. Two domains which are connected by the hinge loop R94 to L97 are defined in the NCS-1 protein:14 N-domain consisting of residues M1 to S93 and C-domain consisting of residues D98 to V190.
Figure 1.
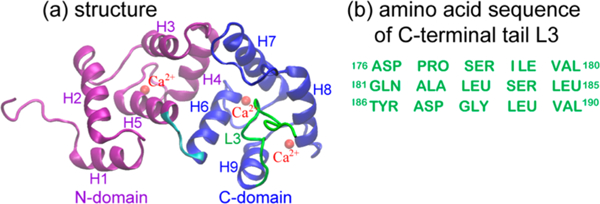
Cartoon diagram of the NMR structure of unmyristoylated calcium-bound human NCS-1 (PDB: 2LCP, model 1) (a) and the amino acid sequence of the C-terminal tail L3 (b). The N-domain and C-domain segments are shown respectively in purple and blue, and the L3 segment is shown using the same color as the amino acid sequence. Three Ca2+ ions (shown in red) bind only to EF2, EF3, and EF4.
We first examined the conformational dynamics of the WT species of the two NMR conformers during the two simulations (WT1 and WT2). Figure 2 (black curve) presents the time evolution of the backbone RMSDs of the core structure (residues E11 to IK174), N-domain (residues E11 to S93), and C-domain (residues D98 to K174) of WT NCS-1 with respect to the corresponding initial states. As shown in Figure 2 (black curve in a, d), the backbone RMSDs of the core structure of the WT increased quickly within the first 50 ns, then changed gradually, and stabilized respectively around 0.32 and 0.43 nm after t = 150 ns for WT1 and WT2. The backbone RMSDs of the N-domain and C-domain showed a similar tendency with those of the core structure. The RMSDs of the N-domain in WT1 and WT2 during the last 400 ns were stabilized respectively around 0.29 and 0.40 nm, while those of C-domain changed gradually and finally maintained respectively around 0.32 and 0.35 nm.
Figure 2.
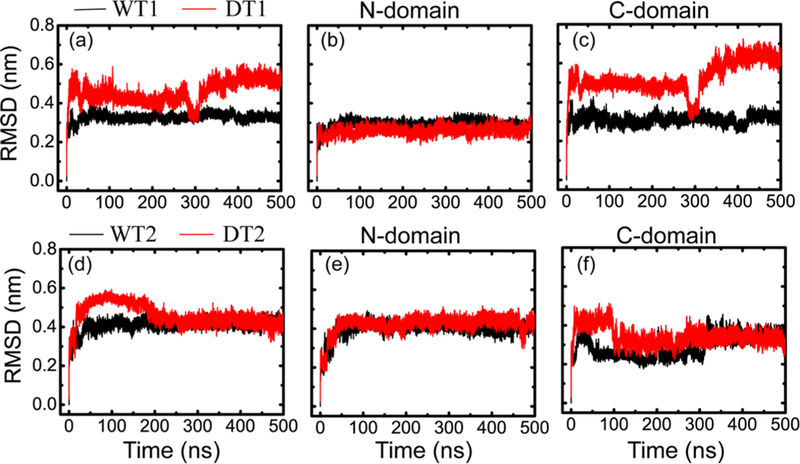
Time evolution of the backbone RMSDs for WT NCS-1 (black curve) and its L3 deletion type DT NCS-1 (red curve) in two different MD trajectories (WT1, WT2 for the WT species and DT1, DT2 for the variant). The backbone RMSDs include the core structure (E11 to K174), N-domain (residues E11 to S93), and C-domain (residues D98 to K174) with respect to the corresponding energy-minimized initial structures (iWT-1, iWT-2) of WT NCS-1 (aligned on all backbone atoms from residues E11 to K174, E11 to S93, and D98 to K174).
We then probed the structural properties of the NCS-1Δ176–190 variant using two independent MD runs (DT1, DT2) initiated from two different initial states (iDT-1, iDT-2). The two initial conformations were modeled using the two energy- minimized states (iWT-1, iWT-2) of the WT species with the C-terminal tail (loop L3) removed. The backbone RMSDs of the core structure, N-domain, and C-domain for the variant with respect to the corresponding initial states (iWT-1, iWT-2) of the WT NCS-1 are shown in Figure 2 (red curve). The backbone RMSDs of the core structure of NCS-1Δ176–190 variant in Figure 2a,d changed largely respectively during the time period of t = 0–320 and t = 0–250 ns, then changed gradually, and stabilized respectively around 0.52 and 0.43 nm. To examine the origin of the large RMSDs, we calculated the backbone RMSD values for the N-domain and the C-domain. The RMSDs of the N-domain in DT1 and DT2 during the last 400 ns were respectively stabilized around 0.28 and 0.43 nm, while the RMSD values of C-domain presented a similar tendency with those of the core structure during the whole simulation time, and they were finally respectively stabilized around 0.64 and 0.35 nm during the last 150 and 200 ns. The data showed that the large RMSD values of the core structure resulted from the C-domain. This result is consistent with a recent NMR study by Kragelund et al. showing that the more severe perturbations of L3 deletion on the conformation of WTNCS-1 were found mostly in the C-domain.14
The probability distributions of the backbone RMSD values of the core structure spanning residues E11 to K174 were also calculated for the WT and DT species (see Figure S1). The distribution curves of WT species in the two MD trajectories displayed one dominant peak centered at respectively 0.34 (WT1) and 0.41 nm (WT2), while two dominant peaks were seen in the distribution curves of DT species, which were located at respectively 0.44 and 0.51 nm for DT1 and 0.44 and 0.54 nm for DT2. These data show that obviously the overall RMSD values of DT variant are larger than those of the WT species, indicating that the tail truncation reduced the stability of the NCS-1 protein.
The local conformational dynamics was monitored by calculating the Cα root-mean-square fluctuation (RMSF) for the MD-generated average structure in the time duration of 100−500 ns. The Cα -RMSF curves of the WT species and its NCS-1Δ176–190 variant from the two independent MD runs are shown in Figure 3. Except for few regions, the NCS-1Δ176–190 variant exhibited overall larger fluctuations than WT NCS-1, indicating that it has larger flexibility. The results are consistent with a NMR study by Kragelund et al. reporting that the tail truncation variant resulted in drastic global destabilization.14
Figure 3.

Cα -RMSF of each residue for WT NCS-1 and NCS-1Δ176–190 variant in the two independent MD runs (a, b). Cα-RMSF values were calculated using the last 400 ns data of each MD trajectory for both systems.
The variant displayed larger RMSF values in helix H9 (L166 to K174) region (red curves) than the WT species, indicating an increased local flexibility/mobility of the variant in the region. This may explain the larger RMSD values in the C- domain. However, unexpectedly, the N-domain of the variant in both simulations presented a spectrum with larger RMSF values than those of WT NCS-1, albeit with a much smaller magnitude than the C-domain. A previous study14 reported that despite being positioned most distal to the N-domain, L3 had a significant stabilizing effect on the structural integrity of this domain. Our observation of an increasing of RMSF values of N-domain is consistent with the above-mentioned experimental study,14 confirming that C-terminal tail deletion affects the distant N-domain. A recent optical tweezer experiment study reported that the unfolding was based on the stable packing of the helix H9.38 Our simulations showed that the deletion of C-terminal tail resulted in an increased flexibility of helix H9 (see Figure 3). On the basis of these findings, we propose that L3-truncation may facilitate the unfolding process of NCS-1 protein.
The stability of helices was explored by calculating the number of backbone hydrogen bonds (H-bonds) and the percentage of helix for residues M1 to A175. The number of backbone H-bonds in WT NCS-1 (black curve in Figure S2a,b) fluctuated respectively around 75 and 80, and the helix percentage (black curve in c, d) remained around 57% during 500 ns of the simulations. The fluctuations in the number of backbone H-bonds correspond to disruption and re-formation of H-bonds. A similar extent of fluctuation was observed in the percentage of helix, corresponding to partial unfolding-refolding of the helices, which could be seen from the secondary structure profile in Figure S3. As shown in trajectories of WT1 and WT2 (Figure S3a,b), helices H2 (E24 to F34), H4 (T62 to F72), H7 (R118 to V132), and H8 (E146 to M155) showed partially unfolding-refolding processes during the simulation. For example, helix H4 in WT1 simultaneously unfolded and refolded during the time period of t = 0–500 ns. Especially during the time period of t = 320–400 ns, helix H4 partially unfolded and changed into turn (in yellow) and then refolded into a helix after t = 400 ns. Similar behavior was also seen in helices H2, H7, and H8. Thus, as expected, the breaking and re-formation of the backbone H-bonds were accompanied by partial unfolding-refolding of helices.
The curves of the number of backbone H-bonds and of the percentage of helix as a function of time for NCS-1Δ176–190 variant (red curve in Figure S2) overlapped well with those of the WT species, revealing similar global conformational dynamics between the variant and WT NCS-1 protein. The secondary structure profiles in Figure S3 also showed that the variant largely maintained its secondary structure. Thus, the larger RMSD values of >0.5/0.4 nm of the variant in Figure 2 (red curve) indicated that the tertiary structure might undergo certain conformational rearrangements with time while the secondary structure content kept mostly unchanged. These data indicate that even though protein flexibility are increased significantly by the L3 deletion, the overall secondary structure content of NCS-1 is largely maintained. These results are consistent with a recent study showing that the far-UV circular dichroism (CD) spectrum of NCS-1Δ176–190 variant was closely similar to that of WT NCS-1.14
The probability distributions of the number of H-bonds and of the helix percentage are plotted in Figure S4 for the WT species (black curve) and the variant (red curve). It can be seen that the average numbers of H-bonds (a, b) of WT species in the two MD trajectories were respectively 77 and 79, while those of the variant were respectively 73 and 79, slightly less than those of the WT species. Similar curves were also seen in the distribution of the helix percentage (c, d). The average numbers of helix percentage of WT species in the two MD runs were both 0.58, while those of the variant were respectively 0.56 and 0.57. These data indicate that the tail truncation variant is less stable than the WT NCS-1 species.
Simultaneous Expansion of the Global Protein Structure and the Hydrophobic Crevice of the NCS-1Δ176–190 Variant.
After characterizing the secondary structure of the protein, we further probed its size and the three-dimensional (3D) structure. The 3D cartoon representative structures of the WT species (a) and the NCS-1Δ176–190 variant (b) at 500 ns are shown in Figure S5. At the end of the simulation, the three Ca2+ ions were still located in the native sites for the WT species and the variant. To inspect the structure of the Ca2+ binding sites of WT NCS-1 protein, we plotted in Figure S6 the time evolution of backbone RMSD values of three different WT segments containing amino acid residues coordinating to the Ca2+ ions. The reference structure for the RMSD calculation was the initial energy-minimized NMR structure. The amino acid sequences of these three segments are 73DENKDGRIEFSE84, 109DLDNDGYITRNE120, and 157DKNADGKLTLQE168. It can be seen from Figure S6 that the backbone RMSD values of the three segments were smaller than 0.2 nm during the full 500 ns simulation time, indicative of the structural stability of the Ca2+ binding sites.Recently, Jungwirth and co-workers39 investigated the calcium binding to calmodulin using MD simulations including electronic polarization effect via ionic charge rescaling, and their MD simulation results showed that inclusion of the polarization effect improved the agreements with experiments in terms of strength of calcium binding and structure of calcium binding sites. It is noted that in our study the electronic polarization effect was not considered for Ca2+ ions and their coordinating oxygen atoms. Whether and how the inclusion of effective polarization affect the calcium binding and the structure of calcium ions binding sites of NCS-1 protein remain to be determined.
To highlight the differences between the structures of WT species and the DT variant, we presented in Figure 4 the superimposed cartoon representations of the two conformations generated at t = 500 ns. For clarity, the three Ca2+ ions were not shown. It can be seen from Figure 4 that the size of the variant was obviously larger than that of the WT species. We further calculated the radius of gyration (Rg) for the two species in the four different MD trajectories. Figure S7 showed that the Rg of the WT NCS-1 (black curve) in the two MD runs reduced quickly from the start of MD simulation, then fluctuated slightly, and got stabilized respectively around 1.72 and 1.75 nm. The Rg of the variant also decreased within the first 150 ns, then displayed little fluctuation, and was finally stabilized at 1.80 and 1.83 nm. In the two MD trajectories (red curve), the Rg of the NCS-1Δ176–190 variant was obviously larger than that of the WT species, indicating that the tail deletion induced global structural expansion. The results were further observed from the probability distributions of Rg in Figure S8 for WT and DT NCS-1. It can be seen that the average values (around the curve peak) of Rg for WT species in the two MD runs were respectively 1.72 and 1.74 nm, while those of the DT variant were respectively 1.79 and 1.83 nm, larger than those of the WT NCS-1. Figure 4 also shows that although the tail deletion variant is less compact than the WT species, it is still able to maintain a well-folded structure.
Figure 4.
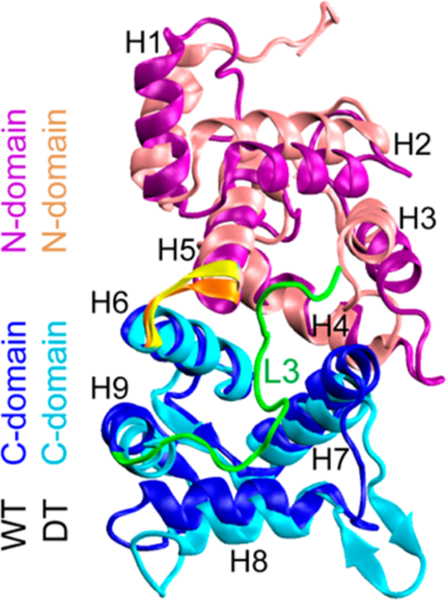
Cartoon representations of WT NCS-1 and its DT variant superimposed on each other. The WT NCS-1 is more compact than the DT variant. The two structures were taken respectively from the MD trajectories of WT1 and DT1 at t = 500 ns. The N-domain and C-domain segments are shown respectively in purple and blue for WT species and in pink and cyan for DT NCS-1. The hinge loops are showed in orange and yellow for WT and DT protein. Loop L3 segment is shown in green for WT NCS-1.
We next compared the free energy landscape of the WT species and that of the DT variant. The free energy landscape was projected onto two reaction coordinates: the radius of gyration and the number of H-bonds (see Figure 5). The free energy landscapes plotted in Figure 5 are similar: there was one global minimum-energy basin. However, differences are also seen for the plots of the two species. For WT NCS-1 species, the basin (in blue) was located at (Rg, number of H-bonds) values of (1.72 nm, 76) (Figure 5a) and (1.74 nm, 79) (Figure 5b), while the basin shifted to respectively (Rg, number of H- bonds) values of (1.79 nm, 72) (Figure 5c) and (1.83 nm, 79) (Figure 5d) for the DT variant. The whole basin of free energy landscape moved to the right and down in the variant. Comparison of the free energy landscapes in Figure 5 demonstrates that the tail deletion changed the free energy surface of the NCS-1 protein.
Figure 5.
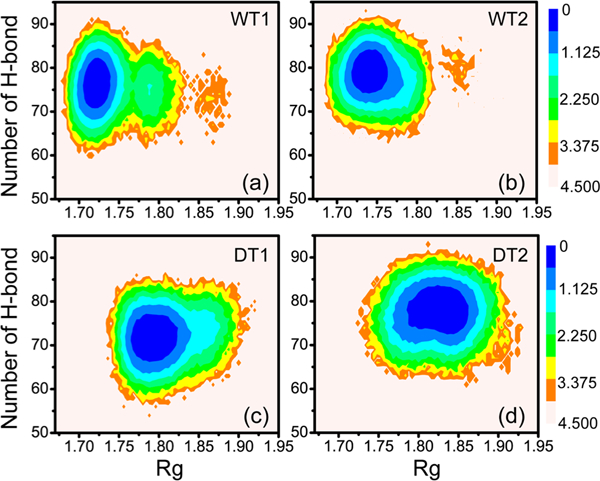
Free energy landscapes (in kcal/mol) for WT (a, b) and L3 deletion type (DT) (c, d) NCS-1 protein, plotted as a function of the radius of gyration (Rg) and the number of backbone H-bonds using the data from 0 to 500 ns.
We next estimated the size of the hydrophobic crevice (HC) by calculating the solvent accessible surface area (SASA) of the hydrophobic residues located within the HC (SASA_HC). Figure 6 gives the probability distribution function (PDF) of the SASA_HC. It can be seen from the figures that the SASA_HC PDF peaks were centered at 15.4 and 15.5 nm2 for the WT (black curve). In contrast, the SASA_HC PDF peaks were centered at 15.8 and 16.1 nm2 for the NCS-1Δ176–190 variant (red curve). The results demonstrate that with the global structural expansion of the variant its hydrophobic crevice becomes larger. The largely opened hydrophobic crevice makes it possible for the incoming ligand to bind, indicating a functionally active NCS-1Δ176–190 variant.
Figure 6.

Probability distribution function (PDF) of solvent accessible surface area (SASA) for the hydrophobic crevice (HC) formed by the hydrophobic residue located inside the HC for WT and DT NCS-1.
L3 Deletion Altered Significantly the Salt Bridge Network in the C-Domain.
NCS-1 contains 24 positively charged residues and 33 negatively charged residues out of total 190 residues. Whether the salt bridges are buried40,41 or solvent exposed,42–45 they play an important role in stabilizing the proteins.46,47 To probe whether the L3 deletion affects the salt bridges, we calculated their formation probabilities between all positively and negatively charged residues in the WT and the DT variant. The salt bridge probability maps are given in Figure 7 for the WT species and the NCS-1Δ176–190 variant. Figure 7a shows that most of the salt bridges in WT NCS-1 were intradomain salt bridges, such as E15-R18, E15-K19, D44-R79, and E26-R94 salt bridges in N-domain and E99-R102, R118- D150, E140-R148, and E142-R151 salt bridges in C-domain, but interdomain salt bridges also existed, such as K63-D123, K63-D126, and E74-R102. These intradomain and interdomain salt bridges were also reported in the recent MD studies by Bellucci et al.28 and by us.18 The salt bridge probability map of NCS-1Δ176–190 variant in Figure 7b is different from that of WT species, especially in the C-domain. It can be seen from Figure 7 that the C-terminal tail deletion significantly removed the K25-D187 and R151-D176 salt bridges and changed the overall salt bridge network. The mostly affected salt bridges were D37- K50 in N-domain and D98-R102, E137-R148, E140-R148, E142-R148, and E140-R151 in C-domain, among which salt bridges E137-R148, E140-R148, and E142-R148 disappeared while D98-R102 and E140-R151 were newly formed salt bridges. The disruption of the three salt bridges in the C- domain provides an explanation for the larger RMSD values (see Figure 2) and the increased flexibility (see Figure 3) of the C-domain. Other salt bridges were also affected, including E15- R18 and E15-K19 in H1, E99-R102 in H6, and R118-D150 between H7 and H8, salt bridges E142-K147 and E142-R151 connecting helix H8 and loop L2. These perturbed salt bridges are mostly located in the C-domain, revealing that the C- terminal tail truncation changes dramatically the salt bridge network in the C-domain.
Figure 7.
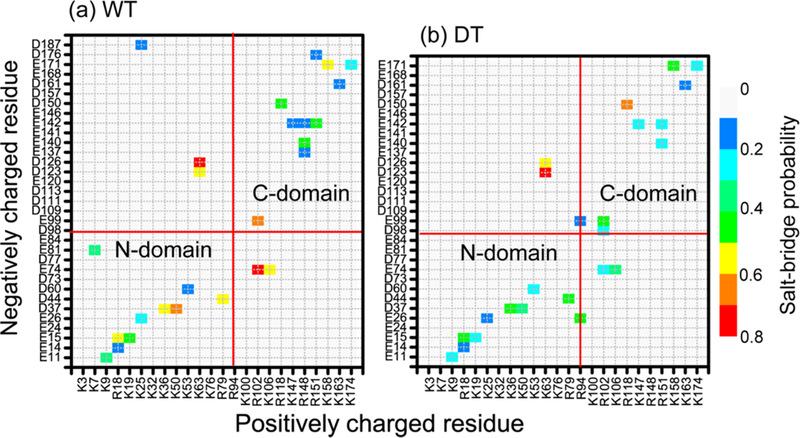
L3 deletion shifts the salt bridge network of WT NCS-1. The salt bridge probability maps are given in (a) for the WT species and in (b) for the NCS-1Δ176–190 variant.
Of particular interest are the interdomain salt bridges: K25- D187, E26-R94, R94-E99, K63-D123, K63-D126, E74-R102, and E74-K106. L3 deletion removed the K25-D187 salt bridge and reduced the probability of E74-R102 and E74-K106 salt bridges. These changes could weaken the interaction between the N-domain and the C-domain. However, the interdomain interactions could be partly compensated by the newly forming salt bridges E26-R94 and R94-E99 in the variant.
The folding mechanism of human NCS-1 protein has been studied recently by Heidarsson et al. by optical tweezer experiment.38 They reported that the complete folding of the C-domain (containing helices H6, H7, H8, and H9) was crucial for the subsequent folding of the N-domain (containing helices H1, H2, H3, H4, and H5) of NCS-1, indicating interdomain dependence. Consistent with this experimental study, our multiple MD simulations demonstrate that L3 deletion not only alters the salt bridge network in the C-domain but also affects the interdomain salt bridges, revealing interdomain correlation.
C-Terminal Tail Deletion Weakened the Interdomain Correlation and Strengthened the Intradomain Relevance.
The conformational dynamics of the wild type NCS-1 and its NCS-1Δ176–190 variant were also studied using the community network analysis based on the MD trajectories.48 In this analysis, each residue was assigned a node centered on its Cα atom, and edges were defined to connect pairs of residues (nodes) those interacted at least 75% of the time (within 4.5 Å) throughout the time duration of 0–500 ns simulations. The time-averaged connectivity of the nodes is used to identify the disjoint subnetworks called community in the network.49 Nodes belonging to the same community have more and stronger connections to one another and have weaker connections to nodes outside the community. The thickness of the edges between two nodes indicates the strength of their dynamic correlation.
The Girvan-Newman algorithm split the network of WT NCS-1 into 9 communities (Figure 8a) and the variant into 11 communities (Figure 8b). It can be seen from Figure 8 that the community networks are different. The connections between the N-domain and C-domain were dense in WT NCS-1, especially between helices H5 and H6, illustrating the stronger interdomain coupling which might be important for the global stability of the protein. In contrast to the wild type, the intracommunities connections of the variant became denser and stronger than those of the WT species; meanwhile, the coupling between the two domains became weak, implying weakened interdomain cross-talk. The data show that even though the secondary structure content of the variant was largely maintained, the residue-residue connections and the dynamic communities were altered. In the variant, although the interactions within some communities such as helices H2, H6, and H8 were enhanced, the coupling between different communities decreased. These results illustrate that L3 deletion weakens the global connections of NCS-1 protein.
Figure 8.

Community networks formed in the WT (a) and DT (b) NCS-1 protein with edge widths corresponding to their weights based on MD simulations. The corresponding cartoon representations at t = 0 ns are shown in (c) and (d). Each community has its own color, superimposed on the initial MD simulation structure.
4. CONCLUSIONS
Functional neuronal calcium sensor-1 protein binds partners mostly through a large exposed hydrophobic crevice. In the absence of the ligand, the C-terminal tail (loop L3) of NCS-1 binds to the HC pocket as a ligand mimetic and regulates the conformational stability of the activated state.14 It was reported that full length of the HC was required for the regulatory function, whereas the C-terminal tail of NCS-1 was not essential in C. elegans.50 In order to delineate the role of the C- terminal tail L3 in human NCS-1 protein, we simulated NCS-1Δ176–190 variant with the last 15 residues removed from the C- terminal tail. We analyzed the structural flexibility, the secondary structure population, the salt bridge network, and the community network of the wild type human NCS-1 protein and NCS-1Δ176–190 variant. Our multiple simulations starting from two different initial structures showed that the C-terminal tail deletion increased the structural flexibility of the C-domain and the distant N-domain and induced simultaneous expansion of the whole structure and HC. The community network analysis illustrated that the C-terminal tail truncation weakened the interdomain correlation and altered the residue-residue connections. Analysis of the salt bridge population in WT NCS- 1 and in the variant demonstrated that the tail truncation changed the overall salt bridge network, especially those in the C-domain. The L3-deletion-induced salt bridge alterations resulted in a reduced conformational stability, which provided an atomic-level explanation for the conformational changes. However, the NCS-1Δ176–190 variant is still able to maintain a well-organized structure, albeit with a higher flexibility; the largely opened hydrophobic crevice may make binding of incoming ligand possible, indicating a functionally active NCS-1Δ176–190 variant.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Cong Guo and Dr. Yin Luo for helpful discussions. This project has been funded from Graduate Student Education Innovation Plan of Shanghai University of Sport (Grant yjscx2015003). Simulations were performed at the High Performance Computing Server (PowerEdge T710) of Shanghai University of Sport. This project has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, under Contract HHSN261200800001E. This research was supported (in part) by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpcb.5b07962.
One table shows the backbone RMSD values between each pair of the 20 solution NMR structure models for wild type NCS-1 protein; eight figures present the probability of the backbone RMSDs of the core structure for WT/DT NCS-1 with respect to the energy-minimized initial structures, time evolution and distribution of the number of backbone H-bonds, the helix percentage and the radius of gyration, the secondary structure profiles of the conformations, cartoon representative structures of WT/DT NCS-1 from the MD trajectories at 500 ns, backbone RMSDs for three WT segments containing amino acid residues coordinating to the Ca2+ ions as a function of simulation time (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Burgoyne RD; Haynes LP Understanding the physiological roles of the neuronal calcium sensor proteins. Mol. Brain 2012, 5, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Burgoyne RD Neuronal calcium sensor proteins: generating diversity in neuronal Ca2+ signalling. Nat. Rev. Neurosci. 2007, 8, 182–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Ames JB; Lim S Molecular structure and target recognition of neuronal calcium sensor proteins. Biochim. Biophys. Acta, Gen. Subj. 2012, 1820, 1205–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Haynes LP; Burgoyne RD Unexpected tails of a Ca2+ sensor. Nat. Chem. Biol. 2008, 4, 90–91. [DOI] [PubMed] [Google Scholar]
- (5).O’Callaghan DW; Ivings L; Weiss JL; Ashby MC; Tepikin AV; Burgoyne RD Differential use of myristoyl groups on neuronal calcium sensor proteins as a determinant of spatio-temporal aspects of Ca2+ signal transduction. J. Biol. Chem. 2002, 277, 14227–14237. [DOI] [PubMed] [Google Scholar]
- (6).Handley MT; Lian LY; Haynes LP; Burgoyne RD Structural and functional deficits in a neuronal calcium sensor-1 mutant identified in a case of autistic spectrum disorder. PLoS One 2010, 5, e10534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Braunewell KH The darker side of Ca2+ signaling by neuronal Ca2+-sensor proteins: from Alzheimer’s disease to cancer. Trends Pharmacol. Sci. 2005, 26, 345–351. [DOI] [PubMed] [Google Scholar]
- (8).Piton A; Michaud JL; Peng H; Aradhya S; Gauthier J; Mottron L; Champagne N; Lafreniere RG; Hamdan FF; Joober R; et al. Mutations in the calcium-related gene IL1RAPL1 are associated with autism. Hum. Mol. Genet. 2008, 17, 3965–3974. [DOI] [PubMed] [Google Scholar]
- (9).Heidarsson PO; Naqvi MM; Otazo MR; Mossa A; Kragelund BB; Cecconi C Direct single-molecule observation of calcium-dependent misfolding in human neuronal calcium sensor-1. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 13069–13074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Ames JB; Hendricks KB; Strahl T; Huttner IG; Hamasaki N; Thorner J Structure and calcium-binding properties of Frq1, a novel calcium sensor in the yeast Saccharomyces cerevisiae. Biochemistry 2000, 39, 12149–12161. [DOI] [PubMed] [Google Scholar]
- (11).Lim S; Strahl T; Thorner J; Ames JB Structure of a Ca2+- myristoyl switch protein that controls activation of a phosphatidyli- nositol 4-kinase in fission yeast. J. Biol. Chem. 2011, 286, 12565–12577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Strahl T; Huttner IG; Lusin JD; Osawa M; King D; Thorner J; Ames JB Structural insights into activation of phosphatidylinositol 4-kinase (Pik1) by yeast frequenin (Frq1). J. Biol. Chem. 2007, 282, 30949–30959. [DOI] [PubMed] [Google Scholar]
- (13).Bourne Y; Dannenberg J; Pollmann V; Marchot P; Pongs, Immunocytochemical localization and crystal structure of human frequenin (neuronal calcium sensor 1). J. Biol. Chem. 2001, 276, 11949–11955. [DOI] [PubMed] [Google Scholar]
- (14).Heidarsson PO; Bjerrum-Bohr IJ; Jensen GA; Pongs O; Finn BE; Poulsen FM; Kragelund BB The C-terminal tail of human neuronal calcium sensor 1 regulates the conformational stability of the Ca 2+- activated state. J. Mol. Biol. 2012, 417, 51–64. [DOI] [PubMed] [Google Scholar]
- (15).Kabbani N; Negyessy L; Lin R; Goldman-Rakic P; Levenson R Interaction with neuronal calcium sensor NCS-1 mediates desensitization of the D2 dopamine receptor. J. Neurosci. 2002, 22, 8476–8486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Woll MP; De Cotiis DA; Bewley MC; Tacelosky DM; Levenson R; Flanagan JM Interaction between the D2 dopamine receptor and neuronal calcium sensor-1 analyzed by fluorescence anisotropy. Biochemistry 2011, 50, 8780–8791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Lian LY; Pandalaneni SR; Patel P; McCue HV; Haynes LP; Burgoyne RD Characterisation of the interaction of the C- terminus of the dopamine D2 receptor with neuronal calcium sensor-PLoS One 2011, 6, e27779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Zhu YZ; Wu Y; Luo Y; Zou Y; Ma BY; Zhang QW R102Q mutation shifts the salt-bridge network and reduces the structural flexibility of human neuronal calcium sensor-1 protein. J. Phys. Chem. B 2014, 118, 13112–13122. [DOI] [PubMed] [Google Scholar]
- (19).Liu P; Huang XH; Zhou RH; Berne BJ Observation of a dewetting transition in the collapse of the melittin tetramer. Nature 2005, 437, 159–162. [DOI] [PubMed] [Google Scholar]
- (20).Zhou RH; Huang XH; Margulis CJ; Berne BJ Hydrophobic collapse in multidomain protein folding. Science 2004, 305, 1605–1609. [DOI] [PubMed] [Google Scholar]
- (21).Garcia AE; Paschek D Simulation of the pressure and temperature folding/unfolding equilibrium of a small RNA hairpin. J. Am. Chem. Soc. 2008, 130, 815–817. [DOI] [PubMed] [Google Scholar]
- (22).Miyashita N; Straub JE; Thirumalai D Structures of beta-amyloid peptide 1–40, 1–42, and 1–55-the 672–726 fragment of APP-in a membrane environment with implications for interactions with gamma-secretase. J. Am. Chem. Soc. 2009, 131, 17843–17852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Gao YQ; Yang W; Karplus M A structure-based model for the synthesis and hydrolysis of ATP by F1-ATPase. Cell 2005, 123, 195–205. [DOI] [PubMed] [Google Scholar]
- (24).Krone MG; Hua L; Soto P; Zhou RH; Berne BJ; Shea JE Role of water in mediating the assembly of Alzheimer amyloid-beta Abeta16–22 protofilaments. J. Am. Chem. Soc. 2008, 130, 11066–11072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Zuo GH; Huang Q; Wei GH; Zhou RH; Fang HP Plugging into proteins: poisoning protein function by a hydrophobic nanoparticle. ACS Nano 2010, 4, 7508–7514. [DOI] [PubMed] [Google Scholar]
- (26).Hess B; Kutzner C; van der Spoel D; Lindahl E GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [DOI] [PubMed] [Google Scholar]
- (27).Bjelkmar P; Larsson P; Cuendet MA; Hess B; Lindahl E Implementation of the CHARMM force field in GROMACS: analysis of protein stability effects from correction maps, virtual interaction sites, and water models. J. Chem. Theory Comput. 2010, 6, 459–466. [DOI] [PubMed] [Google Scholar]
- (28).Bellucci L; Corni S; Di Felice R; Paci E The structure of neuronal calcium sensor-1 in solution revealed by molecular dynamics simulations. PLoS One 2013, 8, e74383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Hess B; Bekker H; Berendsen HJC; Fraaije JGEM LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar]
- (30).Miyamoto S; Kollman PA Settle- an analytical version of the shake and rattle algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar]
- (31).Bussi G; Donadio D; Parrinello M Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [DOI] [PubMed] [Google Scholar]
- (32).Parrinello M; Rahman A Polymotphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar]
- (33).Kabsch W; Sander C Dictionary of potein secondary structure: Pattern recognition of hydrogen -bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [DOI] [PubMed] [Google Scholar]
- (34).Ma BY; Kumar S; Tsai CJ; Nussinov R Folding funnels and binding mechanisms. Protein Eng., Des. Sel 1999, 12, 713–720. [DOI] [PubMed] [Google Scholar]
- (35).Humphrey W; Dalke A; Schulten K VMD: visual molecular dynamics. J. Mol. Graphics 1996, 14 (33–38), 33–38. [DOI] [PubMed] [Google Scholar]
- (36).Eargle J; Luthey-Schulten Z NetworkView: 3D display and analysis of protein.RNA interaction networks. Bioinformatics 2012, 28, 3000–3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Burgoyne RD The neuronal calcium-sensor proteins. Biochim. Biophys. Acta, Mol. Cell Res. 2004, 1742, 59–68. [DOI] [PubMed] [Google Scholar]
- (38).Heidarsson PO; Otazo MR; Bellucci L; Mossa A; Imparato A; Paci E; Corni S; Di Felice R; Kragelund BB; Cecconi C Single-molecule folding mechanism of an EF-hand neuronal calcium sensor. Structure 2013, 21, 1812–1821. [DOI] [PubMed] [Google Scholar]
- (39).Kohagen M; Lepsik M; Jungwirth P Calcium binding to calmodulin by molecular dynamics with effective Polarization. J. Phys. Chem. Lett. 2014, 5, 3964–3969. [DOI] [PubMed] [Google Scholar]
- (40).Waldburger CD; Schildbach J. f.; Sauer RT Are buried salt bridges important for protein stability and conformational specificity? Nat. Struct. Biol. 1995, 2, 122–128. [DOI] [PubMed] [Google Scholar]
- (41).Tissot AC; Vuilleumier S; Fersht AR Importance of two buried salt bridges in the stability and folding pathway of barnase. Biochemistry 1996, 35, 6786–6794. [DOI] [PubMed] [Google Scholar]
- (42).Makhatadze GI; Loladze VV; Ermolenko DN; Chen X; Thomas ST Contribution of surface salt bridges to protein stability: guidelines for protein engineering. J. Mol. Biol. 2003, 327, 1135–1148. [DOI] [PubMed] [Google Scholar]
- (43).Ibarra-Molero B; Zitzewitz JA; Matthews CR Salt-bridges can stabilize but do not accelerate the folding of the homodimeric coiled-coil peptide GCN4-p1. J. Mol. Biol. 2004, 336, 989–996. [DOI] [PubMed] [Google Scholar]
- (44).Williams DV; Byrne A; Stewart J; Andersen NH Optimal salt bridge for Trp-cage stabilization. Biochemistry 2011, 50, 1143–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Andersson HS; Figueredo SM; Haugaard-Kedstrom LM; Bengtsson E; Daly NL; Qu X; Craik DJ; Ouellette AJ; Rosengren KJ The alpha-defensin salt-bridge induces backbone stability to facilitate folding and confer proteolytic resistance. Amino Acids 2012, 43, 1471–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Kumar S; Tsai CJ; Ma BY; Nussinov R Contribution of salt bridges toward protein thermostability. J. Biomol. Struct. Dyn. 2000, 17, 79–85. [DOI] [PubMed] [Google Scholar]
- (47).Kumar S; Ma BY; Tsai CJ; Nussinov R Electrostatic strengths of salt bridges in thermophilic and mesophilic glutamate dehydrogenase monomers. Proteins: Struct. Funct.., Genet. 2000, 38, 368–383. [DOI] [PubMed] [Google Scholar]
- (48).Sethi A; Eargle J; Black AA; Luthey-Schulten Z Dynamical networks in tRNA: protein complexes. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 6620–6625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Girvan M; Newman ME Community structure in social and biological networks. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 7821–7826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Martin VM; Johnson JR; Haynes LP; Barclay JW; Burgoyne RD Identification of key structural elements for neuronal calcium sensor-1 function in the regulation of the temperature- dependency of locomotion in C. elegans. Mol. Brain 2013, 6, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.