

Abstract
Recent studies suggested that p53 aggregation can lead to loss-of-function (LoF), dominant-negative (DN) and gain-of-function (GoF) effects, with adverse cancer consequences. The p53 aggregation-nucleating 251ILTIITL257 fragment is a key segment in wild-type p53 aggregation; however, an I254R mutation can prevent it. It was suggested that self-assembly of wild-type p53 and its cross-interaction with mutants differ from the classical amyloid nucleation-growth mechanism. Here, using replica exchange molecular dynamics (REMD) simulations, we studied the cross-interactions of this p53 core fragment and its aggregation rescue I254R mutant. We found that the core fragment displays strong aggregation propensity, whereas the gatekeeper I254R mutant tends to be disordered, consistent with experiments. Our cross-interaction results reveal that the wild-type p53 fragment promotes β-sheet formation of the I254R mutant by shifting the disordered mutant peptides into aggregating states. As a result, the system has similar oligomeric structures, inter-peptide interactions and free energy landscape as the wild type fragment does, revealing a prion-like process. We also found that in the cross-interaction system, the wild-type species has a higher tendency to interact with the mutant than with itself. This phenomenon illustrates synergistic effects between the p53 251ILTIITL257 fragment and the mutant resembling prion cross-species propagation, cautioning against exploiting it in drug discovery.
1. Introduction
Many neurodegenerative and metabolic diseases are associated with amyloidogenic proteins which have an intrinsic propensity to self-assemble into toxic oligomers and linear fibrils.1–6 Such proteins includes β-amyloid peptide (Aβ) in Alzheimer’s disease,1,7 α-synuclein in Parkinson’s disease8 and islet amyloid polypeptide (IAPP) in type 2 diabetes mellitus2,9. Recently, amyloid deposits of tumor suppressor protein p53 observed in cancer cells revealed surprising related pathogenic mechanisms between cancer and these neurodegenerative diseases.10,11
p53 plays a central role in cellular life and death by controlling many pathways related to apoptosis, cell arrest, and DNA repair in response to stress.12,13 Besides its critical role as a tumor suppressor, p53 regulates hundreds of genes and is guardian maintaining genome stability.12,14 p53 monomer has three domains: the N-terminal activation domain (residues 1–92), the C-terminal domain (residues 313–393), and the DNA-binding core domain (residues 93–312). In about half of all human tumors, p53 is mutated mainly in the DNA-binding core domain, which leads to either loss of its tumor suppressor function or gain of tumor promoting functions15–20. All three p53 domains are able to aggregate, leading to loss of function for p53.15,21,22 Recently, it was realized that prion-like aggregation of mutant p53 may be related to dysfunction of p53 in cancer.10,11,23,24 Mutant p53 aggregates induce not only the aggregation of wild type p5311 but also aggregation of its paralogs p63 and p73.25,26 The heterotetramers (mutant and wild type) show dominant-negative (DN) effects.27,28 Co-aggregation of p53 with its paralogs p63 and p73 might lead to interaction with new binding sites in the DNA and explain gain-of-function effects23, which may increase cancer aggressiveness and progression.24 The destabilized, oncogenic p53 mutants are likely to increase the exposure of the hydrophobic core region29, thus making it prone to aggregation via self-assembly of the aggregation-nucleating stretch into an intermolecular β-sheet-like structure11. Recent experiments show that the strong hydrophobic region 251ILTIITL257 plays an important role in the aggregation of p53. The segment not only drives full-length p53 aggregation in vivo30 but also facilitates mutant p53 coaggregation with and inactivation of p63 and p7324,31.
A bioinformatics study showed that protein sequences often encode gatekeeper residues (charged residues and proline) against aggregation.32 Mutations of these gatekeeper residues, especially Arg, cause a significant rise in aggregation and polymorphism.33 Consistently, it has been shown that introducing a gatekeeper residue (I254R) into p53 aggregation core region 251ILTIITL257 can rescue p53 from aggregation23. A study of a related short peptide 250PILTIITL257 indicated that multiple arginine substitutions (PIRTIITR and PIRTRRTL) also abolish peptide aggregation. Introducing various rescue residues into wild type or mutant p53 might be an intriguing potential strategy in p53 gene therapy, even though it may also be toxic. It has been shown that the loss of the aggregation propensity caused by the I254R mutation alleviated the interaction of aggregating mutants with wild-type p5323; however, it is unclear how the I254R mutation changes the aggregation behavior of p53 at the atomic level.
Exploring the cross interactions between two co-existing proteins with similar or different aggregation propensities is of paramount importance in understanding prion-like propagation in general3,4,34 and the triggering p53 dysfunction in particular. It has been suggested that the mechanism of wild type p53 aggregation and its cross-interaction with mutant p53 is different from the classical nucleation-growth mechanism of amyloid fibril formation31. On the one hand, it appears that small seeds from already polymerized molecules are the initiators of aggregation35,36, followed by a relatively rapid spread37. On the other hand, the mechanism corresponds more to trapping by cross-reaction and co-aggregation31 than to classical seeding and growth.38,39 In this study, we use all-atom replica exchange molecular dynamics (REMD) simulations to investigate the self-assembly of p53 aggregation-nucleating segment 251ILTIITL257and its cross interactions with its aggregation rescue I254R mutant. We found that the nucleation core fragment displays strong aggregation propensity, whereas its gatekeeper I254R mutant tends to form disordered structure, consistent with experiments23. However, the co-aggregation of the aggregation prone fragment with the gatekeeper I254R mutant still displays a significant prion-like behavior. Wild type 251ILTIITL257 has a higher propensity to form β-sheet and interact with I254R mutant, resulting in trapping the disordered oligomers on to the ordered β-sheet-rich oligomeric structures. This observation questions the efficacy of such a mutant fragment as a drug in p53 cancers.
2. Materials and method
Peptide systems
We studied the self-aggregation of p53 wild type (WT) aggregation-nucleating fragment 251ILTIITL257 and its I254R gatekeeper mutant (MT), and the cross-interaction (CI) between the WT and MT fragments. The simulated WT, MT and CI systems respectively consist of six 251ILTIITL257 chains, six I254R mutant chains, and 3WT+3MT chains. Selection of hexamer as amyloid size in the simulation is common. Among these, studies of hexamers of amyloid-beta peptide (16–35) and its mutants revealed the influence of charge states on amyloid formation40.
The 251ILTIITL257 peptide and its I254R mutant were both capped by the ACE (CH3CO) group at the N-terminus and the NH2 group at the C-terminus. The mutation of Ile254 to Arg (amyloid gatekeeper amino acid)32 introduces a positive charge.
Simulation Methods
Three 250 ns REMD41 simulations were performed using the GROMACS-4.5.3 software package42. We chose the AMBER99SB-ILDN force field43 and carried out REMD simulations in the NPT ensemble at a pressure of 1 bar. The parameters used for our REMD simulations are widely used in numerous REMD studies44–47. There are 48 replicas, each of 250 ns duration, at temperatures exponentially spaced between 307.86 and 421.82 K.48 Thus the second replica has the physiological temperature of 310 K, which helps to ensure more exchange chances with its neighboring replicas, thus accelerate sampling at the physiological temperature of 310 K.
Six 251ILTIITL257 chains, six I254R mutant chains, or 3WT+3MT chains, with random conformations for each chain, were initially placed randomly in a 6.1 × 6.1 × 6.1 nm3 box filled with TIP3P water molecules. There are 7322, 7296, 7301 water molecules in the WT, MT and WT+MT systems, respectively. The peptide concentration in the three systems is 43.9 mM. The attempt swap time between two neighboring replicas is 2 ps. The acceptance ratio is ~ 24%, as shown in the supporting information (Fig. S1). A large number of REMD simulation studies demonstrate that an acceptance ratio of 20~30% is good34,44,49–53. Periodic boundary conditions were applied in all three directions. Constraints were applied to all-bond lengths using the SETTLE algorithm and the LINCS method for the peptides, allowing an integration time step of 2 fs. The protein and non-protein (water and counterions) groups were separately coupled to an external heat bath with a relaxation time of 0.1 ps using a velocity rescaling coupling method54. The pressure was kept at 1 bar using the Parrinello-Rahman method55 with a coupling time constant of 1.0 ps. A cutoff of 1.4 nm was used for van der Waals interaction. The Particle Mesh Ewald (PME) method56 with a real space cutoff of 1.0 nm was used for electrostatics interactions. The coordinates were saved every 2 ps.
Analysis Methods
Trajectory analysis was performed with our in-house-developed codes and the facilities implemented in GROMACS-4.5.3 software package42. We discarded the first 150 ns data of each REMD run to remove the bias of the initial states. Therefore, the structural properties of each system were based on the simulation data generated in the last 100 ns. The secondary structure of the peptide was identified using the DSSP program. The tertiary structure analysis was performed by combining a Cα root-mean-square deviation (RMSD) cluster analysis method with the percentages of various sizes of the β-sheet. In this study, we performed a chain-independent RMSD calculation because all the chains are topologically identical, as done previously by Li et al57. In this study, for a single pdb file generated from the REMD trajectory, we calculated its RMSD using 6! (6×5×4×3×2×1) different coordinate files (the x, y, z values of all the atoms are unchanged, only the numbering order of the atoms are changed). That is to say, we got 6! RMSDs. The smallest RMSD, i.e. the chain-independent RMSD, was taken for structure clustering.
The size of a β-sheet is the number of β-strand in an n-stranded β-sheet, e.g., the β-sheet size of a three-stranded β-sheet is three. Two peptide chains are considered to form a β-sheet if (i) at least two consecutive residues in each chain visit the β-sheet state and (ii) the two chains form at least two backbone hydrogen bonds (H-bonds). One H-bond is taken as formed if the N···O distance is less than 0.35 nm and the N-H···O angle is greater than 150°O angle is greater than 150°. We used a topological parameter, connectivity length (CL)58, to describe the orderness of the peptide aggregates. CL is defined as the sum over the square root of the β-sheet size and the number of disordered chains in each conformation. For example, the CL of a hexamer consisting of a 4-stranded β-sheet and two random chains is sqrt(4) + sqrt(1) + sqrt(1) = 4. Thus, the larger the connectivity length, the more disordered the hexamer is. The VMD program59 was used for graphical structure analysis
The solvation extent of the peptide backbone was estimated by the number of water molecules that are within 0.35 nm from the peptide backbone. The interpeptide interactions were analyzed by the residue-residue (including main-chain-main-chain (MC-MC) and side-chain-side-chain (SC-SC)) contact probabilities. Here, a contact is defined when the aliphatic carbon atoms of two nonsequential side chains (or main chains) come within 0.54 nm or any other atoms of two nonsequential side chains (or main chains) lie within 0.46 nm. The free energy surface of each system was constructed using - RT ln H(x, y), where H(x, y) is the histogram of two selected reaction coordinates, H-bond number and Rg. Here, H-bond number and Rg denote the total number of hydrogen bonds (including intra- and intermolecular H-bonds) and the radius of gyration of the hexamer, respectively. The value of the minimum free energy is defined as zero. We also estimated the total energy and binding free energy using the Molecular Mechanics/linear Poisson-Boltzmann Surface Area (MM/PBSA) method implemented in the AMBER package60. In MM/PBSA, the total energy is calculated as: Etotal = Eangle + Edih + Eelec + Evdw + Epolar + Enonpolar. Here, Eangle, Edih, Evdw and Eelec are respectively angle, dihedral angle, the van der Waals (vdw) and the electrostatic interaction energies in vacuum. The Gpolar + Gnonpolar is the solvation free energy that is required to transfer a solute from vacuum into the solvent, where, Gpolar and Gnonpolar are the electrostatic and non-electrostatic contributions to the solvation free energy, respectively. Gpolar is calculated by the PB model and Gnonpolar is estimated by the solvent accessible surface area (SASA). The binding free energy (ΔGbinding) between a ligand and a receptor also is calculated as: ΔGbinding = ΔEvdw + ΔEelec + ΔGpolar + ΔGnonpolar.
3. Results and discussion
To examine the convergence of the REMD simulations, we first checked the time evolution of the replica initiated from 310 K. Figure S2A shows that this replica visited sufficiently the whole temperature space within a 250 ns REMD simulation, indicating the replica was not trapped in one single temperature. Other replicas display similar sampling behavior (data not shown). The convergences of the three REMD runs were further verified by comparing the β-sheet probability of each residue and the probability density function (PDF) of end-to-end distance of each chain within two different time intervals using the 150–200 ns and 200–250 ns data. As shown in Fig. S2B, the residue-based β-sheet probabilities are almost the same between the two time periods. The distributions of end-to-end distance within the two independent time intervals overlap very well for the three systems (Fig. S2C). These data suggest that our REMD simulations for the three systems are reasonably converged within 250 ns. Unless specified, all the REMD simulation results presented below are based on the last 100 ns (t = 150–250 ns) simulation data generated at 310 K.
I254R gatekeeper mutant decreases the β-sheet propensity of the P53 251ILTIITL257 peptide.
We first examined the secondary structure properties of the wild type 251ILTIITL257 peptide. As can be seen in Fig. 1A, the hydrogen bonding counts steadily increase with the simulation time. Our highly efficient REMD sampling provides much better results than a previous 65 ns simulation of a system consisting of 24 250PILTIITL257 peptide chains, where only about 20 hydrogen bonds were obtained.30 Fig. 1B shows the percentage of secondary structure, including coil, β-sheet, β-bridge, bend, turn, α-helix and 3-helix. The equilibrated 251ILTIITL257 hexamer contains 36.0% β-sheet. When the two terminal residues I251 and L257 were not considered, the β-sheet content of the peptide reaches as high as 40–60%. As expected, introducing I254R gatekeeper mutation changes the aggregation propensity of the 251ILTIITL257 peptide. Compared with the aggregation prone wild type sequence, the substitution of the hydrophobic isoleucine residue with a positively charged arginine considerably decreases the average β-sheet content from 36.0% to 25.7% and increases the average coil content from 48.0% to 54.7%. The residue-based β-sheet probability (Fig. 1C) shows that the β-sheet probability for all residues drops from 36%−61% for the wild type to 24%−47% for the mutant. While it seems that the 47% β-sheet probability is still not too low, the size of the β-sheet decreases and the dimer is dominant (Fig. 2A), indicating that the mutation suppresses the aggregation of the peptide.
Fig 1.
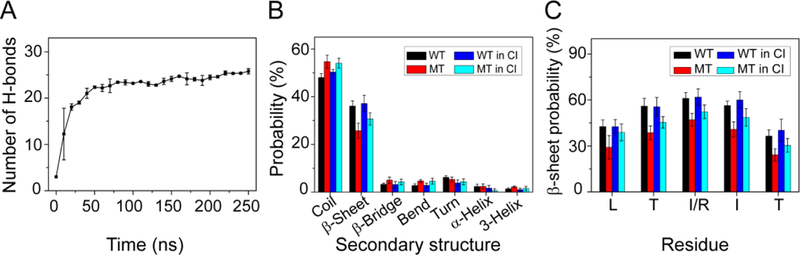
Secondary structure analyses. (A) The number of hydrogen bonds as a function of simulation time in WT hexameric systems. Hydrogen bonding counts increase to 24 after 150 ns, showing highly efficient REMD sampling. (B, C) Analysis of the secondary structure of the WT, MT, and CI hexameric systems. The positively charged arginine considerably decreases the average β-sheet content and increases the average coil content. The average β-sheet probabilities of the WT increase slightly and MT increase significantly in the CI system.
Fig 2.
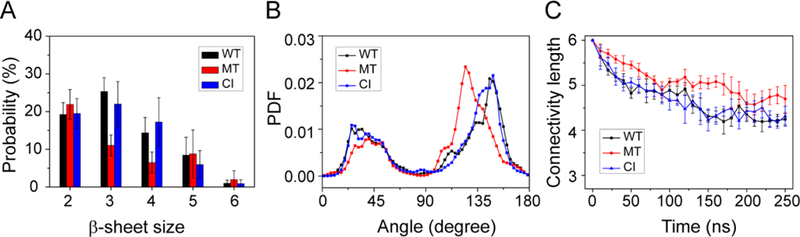
Characteristic analyses of interpeptide β-sheets. (A) Analysis of the β-sheet size distribution. The majority of β-sheets are three β-stranded β-sheets in WT and CI systems, while the size of β-sheet decrease and the dimer is dominant in MT system. (B) The distribution of the angle between two β-sheet-forming chains in WT, MT, and CI systems. Two peaks located at 25° and 150° indicate that the antiparallel alignment is dominant for three systems. PDF is the probability density function. (C) The time evolution of the connectivity length for the WT, MT, and CI hexamers. The smaller CLs values of the WT and CI systems than those of the MT systems reveal the much more ordered nature of the WT and CI hexamers.
The majority of the β-sheets are three β-stranded β-sheets, but large sizes of β-sheets consisting of 4–6 β-strands were also observed (Fig. 2A). Figure 2B shows the distribution of the orientations of two neighboring β-strands in all β-sheet sizes. For the WT system, the two peaks located at 25° and 150° correspond to respectively parallel and antiparallel alignment of β-strands, with the antiparallel alignment being dominant. Similar results are seen for the MT and CI systems. These results indicate that the peptide chains in the three systems have a preference to adopt antiparallel β-stranded sheets.
The ordering of the WT, MT and CI hexamers is examined by monitoring the time evolution of the connectivity length (CL) (Fig. 2C). The CLs of the hexamers of the WT, MT and CI systems decrease rapidly from the initial value of 6.0 respectively to 4.2, 5.0, and 4.2 within the first 150 ns of the simulations and fluctuate around these three values during t = 150–250 ns. The CLs of CI hexamers during the last 100 ns is very close to those of the WT hexamer, indicating that the CI system displays similar aggregation properties as the WT system. The larger CL value of the MT system as compared to those of the WT and CI systems reveals the much more disordered nature of MT hexamers.
Experiments show that high temperature can lead wild type p53 to lose its function61–63. Thus, we further investigated the β-sheet probability as a function of temperature for WT, MT, and CI hexamers (Fig. 3). For both MT and CI systems, the probability of β sheet decreases monotonically with increasing temperature, whereas the β-sheet probability of the WT increases slightly form 36.1% (308K) to 37.1% (325 K) and then decreases, also revealing that the WT sequence prefers to aggregate at higher temperature.
Fig 3.
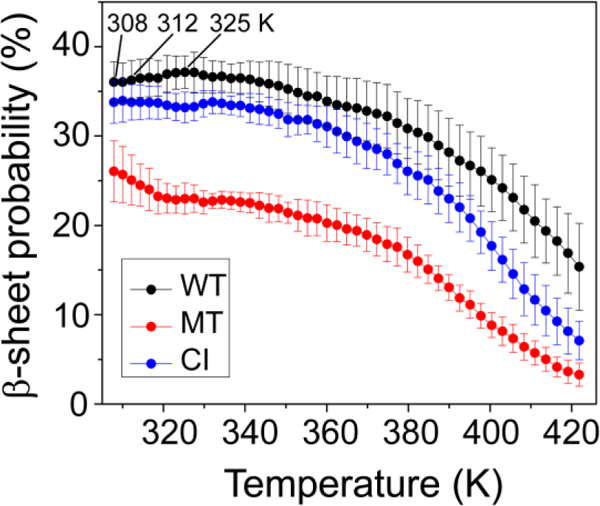
β-sheet probability as a function of temperature for WT, MT, and CI hexamers. β-sheet probability is 36.1% at 308 K, 36.2% at 312 K and 37.1% at 325 K, revealing that the WT sequence prefers to aggregate at higher temperature.
We then calculated the average number of water molecules within 0.35 nm from the backbone atoms of each residue to monitor the extent of solvation of the peptide backbone (Fig. 4). In the wild type system, the number of water molecules drops successively from 2.1 to 1.1. The backbone atoms of residues T256 (~2.0) and L257 (~2.7) are more solvent-exposed than other residues. The three residues (TII) in the middle of the amino acid sequence of the WT peptide are well protected from the solvent. Due to the smaller oligomer size and lower β-sheet content, the residue has higher solvation (Figure 4, red bar). Compared with the wild type system, the backbone of all residues of the mutant hexamer are more solvent-exposed, except I251, indicating that the I254R mutation affects not only itself, but also the entire peptide.
Fig 4.
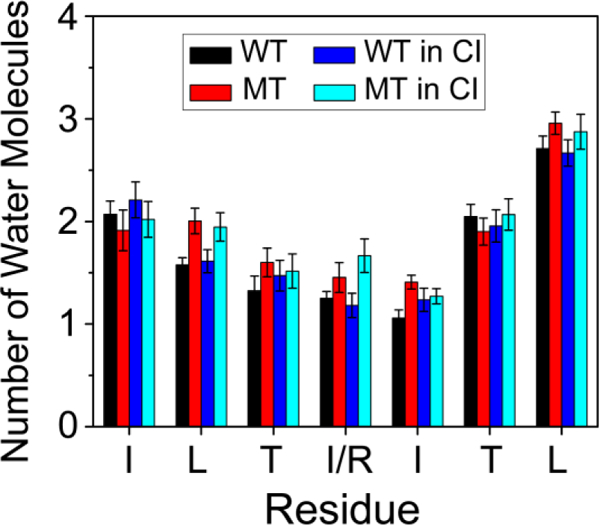
The average number of water molecules within 0.35 nm of the mainchain atom of each residue. The figure shows that C-termini are more solvent-exposed and the backbone of all residues of the mutant hexamer are more solvent-exposed.
We also calculated the interpeptide MC-MC and SC-SC contact probabilities between all pairs of residues for WT, MT, and CI hexamers. As seen from Fig. 5, while the wild type hexamers have balanced both MC-MC interaction and SC-SC interactions, the I254R gatekeeper mutant hexamers lost most of its MC-MC interactions. Interestingly, the nearby Ile255 residue now plays an important role in peptide associations. For the wild type hexamer system (Fig. 5A), compared to other residue pairs, the T253-T253 (with a contact probability of 13.1%), T253-I254 (13.3%), I254-I254 (12.6%) and T253-I255 (13.9%) pairs display high MC-MC contact probabilities. The relatively high MC-MC contact probabilities along the left diagonal of the MC-MC contact map in Fig. 5A indicate that WT peptides are aligned predominantly in antiparallel orientation. For SC-SC interactions, the isoleucine-isoleucine pairs have highest contact probabilities of 20.9% (I254-I254 pair), 20.1% (I254-I255 pair) and 16.5% (I255-I255 pair), reflecting strong hydrophobic interaction. When I254 is substituted by proline, the MC-MC contact probabilities (see Fig. 5B) of T253-T253, T253-I254, I254-I254 and T253-I255 pairs dramatically drop to 9.3%, 11.0%, 10.5% and 9.8%, respectively. This reduced MC-MC interaction is associated with the increasing interactions between MC atoms and water molecules. For SC-SC contact probabilities (see Fig. 5E), arginine shows weak interactions with other residues because of its positive charged side chain, leading to high contact probability of I255-I255 (32.1%) and T253-I255 (21.6%) pairs. The error bars of MC-MC contact probabilities in each probability map were calculated using the 150–200 ns and 200–250 ns data. Our calculations show that all the error bars are very small compared to the contact probabilities (Table S1-S6). The small errors reflect the reliability of the REMD simulations.
Fig 5.
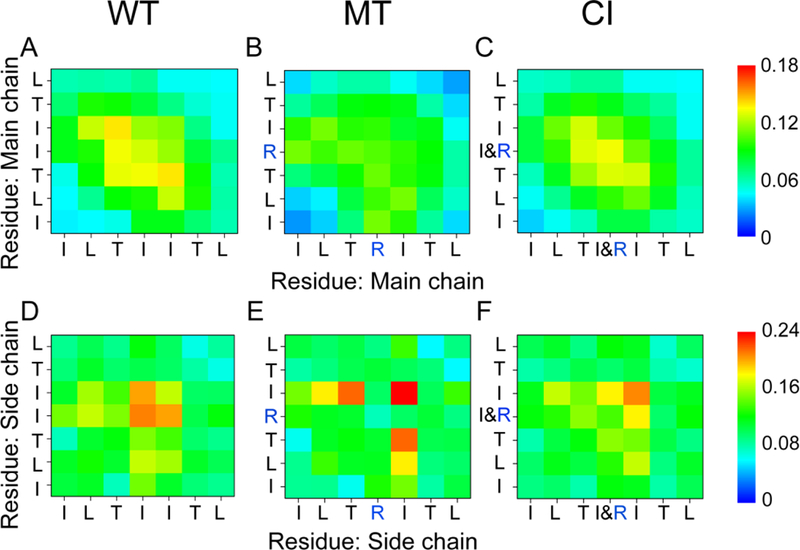
Contact probability map. Main-chain-main-chain (MC-MC) and side-chain -side-chain (SC-SC) contact probabilities averaged over the 150–250 ns REMD generated conformations for the hexamers of wild type (A, D), I254R mutant (B, E), and the CI (C, F) systems at 310 K. Strong hydrophobic interactions between the isoleucines essentially stabilize oligomeric structures in the WT and CI systems.
Synergistic mutual β-sheet promotion between the p53 251ILTIITL257 fragment and its I254R gatekeeper mutant.
In order to study the cross-interaction between the aggregation prone 251ILTIITL257 fragment and its I254R gatekeeper mutant, we simulated the system with a mixture of three wild type and three mutant peptides. Surprisingly, we found that there is synergistic effect of the cross-interaction between the wild type and its I254R mutant. The β-sheet probability of the mutant in the CI hetero-hexamer is promoted to 30.6%, which is about 4.9% higher than that of MT homo-hexamer. Even the average β-sheet probability of the WT species in the CI system is about 1.1% higher than that in the WT homo-hexamer (Fig. 1B). Comparing the WT trimer in the CI system with the WT homo-hexamer in the WT system, we found that the average β-sheet probabilities of L255 and T256 increase slightly (~3.6% and ~3.7%). Meanwhile, the probabilities of turn, α-helix and 3-helix of both WT and MT peptides in the CI system slightly decrease. These results indicate that the interaction between the WT and MT peptides in the CI system can induce the secondary structure transition from turn, α-helix and 3-helix to β-sheet structure for the I254R mutant. In the CI system (Fig. 1C), β-sheet probabilities of all residues, except terminal residues of the MT peptide, distinctly increase (5.25%−9.7%). These data show that wild type species can significantly promote β-sheet formation of its I254R mutant. In the CI system, even though the I254R mutation rendered higher contact numbers with water molecules (Fig. 4), the hydrophobic I254 in the wild type peptide still provides effective protection of backbone atoms from solvation. This trend can also be seen from the contact probability map (Fig. 5). In the CI system, the MC-MC contact probabilities of T253-T253 (11.6%), T253-I254 (12.9%), I254-I254 (13.4%) and T253-I255 (12.6%) pairs show similarly strong MC-MC interactions as the wild type system (Fig. 5C). The SC-SC contact probability of I/R254- I/R254 (13.6%), I/R254-I255 (18.2%) and I255-I255 (20.7%) also show strong hydrophobic interactions between the isoleucines (Fig. 5F). The maps show that the wild type peptides weaken the negative I254R mutational effect on the SC-SC interactions in the CI system. These data reveal that the oligomeric structures in the WT and CI systems are essentially stabilized by MC-MC interactions between threonine and isoleucine residues and SC-SC interactions between isoleucine and isoleucine residues.
After characterizing the secondary structure properties of WT, MT and CI hexamers, we investigated their three dimensional conformational states by first performing RMSD-based cluster analysis for each system. With a Cα-RMSD cutoff of 0.3 nm, the conformations of the WT hexamer, MT hexamer and CI hexamer at 310 K were separated into 293 clusters, 443 clusters and 392 clusters, respectively. The representative conformations of the first eight most-populated clusters are shown in Fig. S3. These clusters represent 37.5%, 27.6% and 27.4% of all conformations of the WT, MT and CI hexamers, respectively. The WT homo-hexamer and the CI hetero-hexamer contain more ordered β-sheet rich conformations than the MT homo-hexamer. For example, the fourth and sixth clusters of the WT hexamer contain bi-layer β-sheet structures (three-stranded + two-stranded β-sheets) with mainly antiparallel alignment. In the mutant system, the first cluster (with a probability of 7.6%) and the eighth cluster (1.9%) contain respectively five-stranded and six-stranded open β-barrels in which the side chains of positively charged arginine residues are solvent-exposed. For the CI hexamer, similar to the WT system, bilayer β-sheets structures, such as those in the first cluster (5.3%) and the fifth cluster (3.0%), are populated. Mono-layer and bi-layer β-sheet structures are fibril-competent states, while disordered aggregates and β-barrels are difficult to form fibrils.
Our various analyses have shown that the aggregation prone 251ILTIITL257 fragment effectively recruits the I254R gatekeeper mutant into its aggregation pattern. We found that the oligomeric structures of the WT homo-hexamer and the CI hetero-hexamer share fairly similar characteristics of ordered bilayer antiparallel β-sheet structures (Fig. 2B).
The CI system displays similar free energy landscape as the wild type
To have an overall view of the conformational distribution of hexamers of the three systems, we constructed the 2D free energy surface in Fig. 6 using -RT ln H (H-bond number, Rg) as described in Analysis Methods. The locations of representative structures are labeled on the PMF plot. For the wild type hexamer, the black, light blue and green lines point to the location of the first cluster (monolayer structure, with a probability of 10.1%), the fourth cluster (bilayer β-sheet structure, 4.1%) and the sixth cluster (bilayer structure, 2.8%). For the I254R mutant system, we highlight the location of the first cluster (five-stranded open β-barrel, 7.6%), the fourth cluster (disordered coil-rich aggregate, 3.5%) and the fifth cluster (disordered coil-rich hexamer, 2.2%). For the CI hexamer, the location of the first cluster (bilayer β-sheet structure 5.3%), the fourth cluster (four-stranded mono-layer structure 3.0%) and the fifth cluster (monolayer structure, 3.0%) are labeled. The distributions of the conformations of the WT and CI hexamers are quite concentrated in the free energy surface, while they are scattered in the mutant system. The global minimum energy basins of the WT, MT, and CI hexamers are located at (number of H-bonds, Rg) values of (25, 1.0), (23, 1.4) and (27, 0.97), respectively. Compared with the mutant hexamer, the wild type hexamer and cross-interaction hexamer have an increased number of H-bonds and a decreased value of Rg, implying that the wild type can induce the mutant to more compact, ordered aggregates.
Fig 6.
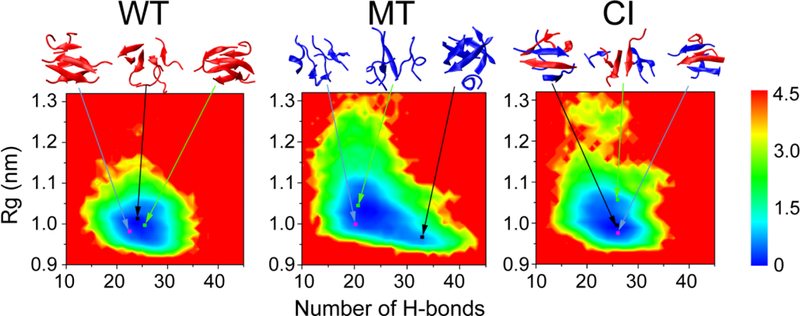
Free energy surfaces (in kcal/mol) of the wild type hexamer, mutant hexamer and cross-interaction hexamer, respectively at 310 K as functions of the total number of intra- and intermolecular H-bonds (Number of H-bonds) and radius of gyration (Rg). Their minimum energy basins are located at different (number of H-bonds, Rg) values of (25, 1.0 nm), (23, 1.4 nm) and (27, 0.97 nm), respectively, implying that the wild type can induce mutant to more compact, ordered aggregates. Representative structures are also given, along with their probabilities: 10.1% (black line), 4.1% (light blue line) and 2.8% (green line) at WT systems; 7.6% (black line), 3.5% (light blue line) and 2.2% (green line) at MT systems; 5.3% (black line), 4.9% (light blue line) and 3.6% (green line) at CI systems;
The origin of the synergistic effect between the wild type and the I254R mutant
To further investigate the molecular mechanism of the synergistic interaction between the wild type and the I254R mutant peptides, we calculated the contact probability of three different combinations of peptides (WT-WT, MT-MT and WT-MT) in the CI system. As shown in Fig. 7, WT-WT and MT-MT species still retain the same characteristics of MC-MC and SC-SC interactions as the WT and MT hexameric systems; however, the WT-MT species have the strongest MC-MC (Fig. 7C) and SC-SC (Fig. 7F) interactions, which suggest that the WT can cross-interact with its mutant. In the WT-MT peptide pairs, T253-R254 and I254-T253 pairs of main-chains have highest probabilities, also showing a preference for an antiparallel organization. As to side-chain interactions, the hydrophobic isoleucine plays an important role in cross-interaction between WT and MT: 26.5% in the I254-I255 pair and 21.1% in the I255-I255. The error bars of SC-SC contact probabilities in each probability map were calculated using the 150–200 ns and 200–250 ns data. Our calculations show that all the error bars are very small compared to the contact probabilities (Table S7-S12). The small errors reflect the reliability and good convergence of the REMD simulations.
Fig 7.
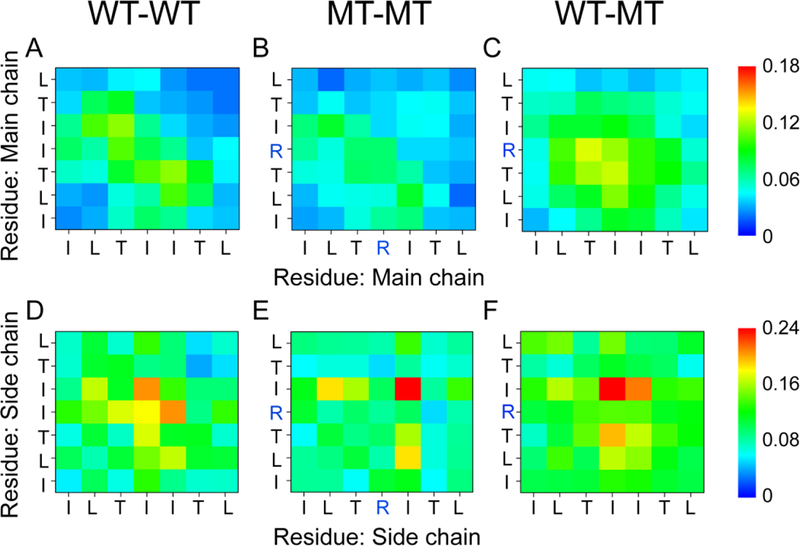
Contact probability map. Main-chain-main-chain (MC-MC) and side-chain -side-chain (SC-SC) contact probabilities in the cross-interaction system, WT-WT (A, D), MT-MT (B, E) and WT-MT (C, F). The wild-type species has a higher tendency to interact with the mutant than with itself.
To estimate the contribution of non-bonded interactions and reveal the prion-like behavior of the core fragment, we also calculated the inter-peptide binding free energy using the MM/PBSA method as shown in Tables 1 and 2. As can be seen in Table 1, both WT-WT and WT-MT peptide pairs have attractive interaction energy, while the I254R mutant has repulsive energy. The mixed, cross-interacting hexamer overcomes the repulsive energy and even has a small thermodynamic preference.
Table 1.
Partition of total energies (in kcal/mol). The mixed, cross-interacting hexamer overcomes the repulsive energy and even has a small thermodynamic preference.
| Systems | Eangle | Edih | Eelec | EvdW | Gpolar | Gnonpolar | Etotal |
|---|---|---|---|---|---|---|---|
| WT | 2126.1 ±4.9 |
518.3 ±2.0 |
−2651. ±10.2 |
−274.6 ±3.5 |
−221.7 ±5.4 |
25.0 ±0.5 |
−478.8 ±9.7 |
| MT | 2136.4 ±4.9 |
523.4 ±2.3 |
−1561.0 ±19.9 |
−285.2 ±5.5 |
−807.6 ±15.0 |
27.3 ±0.7 |
33.3 ±12.0 |
| CI | 2131.5 ±5.7 |
520.7 ±4.9 |
−2198.8 ±9.2 |
−277.6 ±3.4 |
−426.3 ±5.9 |
26.4 ±0.5 |
−224.1 ±9.0 |
| ΔH | 0.3 ±0.9 |
−0.2 ±0.6 |
−92.4 ±2.4 |
2.3 ±0.7 |
88.4 ±1.7 |
0.3 ±0.1 |
−1.3 ±1.8 |
Table 2.
Binding energies (in kcal/mol). The wild type species have a higher tendency to interact with its charged mutant due to strongest binding energy.
| ΔEvdw | ΔEelec | ΔGpolar | ΔGnonpolar | ΔGbinding | |
|---|---|---|---|---|---|
| WT-WT | −87.9±8.8 | −72.4± 12.4 | 85.6± 12.2 | −13.1 ±1.3 | −87.8± 10.3 |
| MT-MT | −97.0± 10.3 | 82.6± 18.6 | −55.3 ± 17.5 | −14.9± 1.6 | −84.6± 12.1 |
| WT-MT | −101.3 ±8.1 | −102.1 ±11.0 | 120.9± 11.4 | −15.4 ± 1.3 | −97.9±8.9 |
In Table 2 we calculated the binding free energy between three randomly selected peptides and three other peptide chains in the hexamers for both wild type and mutant systems. The positive value of electrostatic energy including both potential energy in vacuum (ΔEelec) and solvation energy (ΔGpolar) shows that introducing arginine, a positively charged residue, is unfavorable for aggregation. However, the WT-MT binding in the cross-interaction system weakens the negative effect. The large negative value of the van der Waals energy (ΔEvdw) and nonpolar solvation energy (ΔGnonpolar) reveal that the strong hydrophobicity of the peptides is an intrinsic driving force of aggregation. Comparison of the total binding free energies of the three models, when wild type and mutant peptides coexist, shows that the wild type species have a higher tendency to interact with its charged mutant due to strongest binding energy.
Recently, it has been shown experimentally that proximally immobilized ions can modulate hydrophobic interactions of conformationally stable β-peptides64. Our results of synergistic interaction between that wild type and the I254R mutant peptide illustrate a similar physical origin.
Conclusions
We have investigated the self-assembly of the aggregation-nucleating fragment 251ILTIITL257 of p53 protein and its I254R gatekeeper mutant, and their cross-interaction by performing three 250 ns atomistic REMD simulations starting from a random state. Structural analyses of the three systems show that both the wild type and cross-interaction systems mainly form a bilayer organization with three- and four-stranded antiparallel β-sheets with parallel β-strands in each sheet. The I254R mutation introduces a positive charge that is sufficient to suppress the aggregation tendency of the wild type peptides. The β-sheet rich structures are intrinsically stabilized by strong hydrophobic interaction, especially between the two isoleucines. Previously, it has been suggested that evolution tends to avoid aggregation from isoleucines65. Strikingly, we found that the wild type 251ILTIITL257 has a higher propensity to form β-sheet and interact with the I254R mutant, resulting in trapping the disordered peptides and extending the β-sheet rich ordered structure.
Our simulations provide insight into the difference between full length (or core domain) p53 aggregates and the classical nucleation-growth of amyloid fibrils20. Experimental studies of p53 aggregation in cells led to the suggestion that the I254R gatekeeper mutation alleviates the interaction of aggregating mutants with wild-type p53.23 At first sight, this conflicts with our observations of cross-interactions between p53 core fragment and its I254R mutant. However, the disagreement raises the question of what is the difference between the aggregation behaviors of an isolated peptide and when the fragment is highly buried in the protein core. Nature has evolved a way to seal aggregation prone peptide fragments in protein cores21. Mutations and unfolding perturbations often lead to exposure of amyloidogenic fragments resulting in aggregation-related diseases. p53 can constitute such an example. The first step in p53 aggregation is unfolding and exposing core fragments like 251ILTIITL257. Wild type aggregates can trap the already unfolded mutant p53, recruiting it into the aggregates20. Similarly, the Aβ peptides can seed tau protein also by stretching tau and exposing its hydrophobic core39. This is likely why the aggregation-disabled I254R mutant cannot trigger wild-type p53 aggregation12.
If we label the p53 core fragments like 251ILTIITL257as a generic “aggregating” species, and its gatekeeper I254R mutant as “non-aggregating” species, our study well demonstrates that the “aggregating” species can trigger and recruit “non-aggregating” species in a prion-like propagation fashion. Thus, when designing peptides to inhibit the protein aggregation, the designed peptide should not only be aggregation free on its own, it should also have the ability to resist being recruited and merged into an “aggregating” species. Our results indicated the gatekeeper mutation (I254R) does not completely eliminate the amyloid prone nature of the sequence ILTRITL. The mutation most likely eliminates or delayed the kinetic formation of nucleation core. Thus the I254R mutant could have the entropy barrier, in a similar way as some prion sequences with medium amyloid propensity 66. With the seeding of wild type p53 fragment, the wt p53 nucleus overcomes the entropy barrier of I254R mutant. The situation is also similar to the effects of an amyloid stretch within a long sequence to trigger and recruit otherwise non-amyloidogenic sequence into amyloid filaments67. Nucleation and seeding of prion protein aggregates is strongly influenced by dynamic interactions between the aggregate core forming domain and its flanking regions68. Most prion propagation among prion-like proteins with cross-seeding barriers.69 Still, it is recently found that prion resistant species (pig) can also be affected by bovine prions.70
Supplementary Material
Acknowledgments
G.W. acknowledges the financial support from the NSF of China (Grant No.: 91227102 and 11274075). R.N. and B.M thank the financial support from NCI, NIH, under contract number HHSN261200800001E. This research was supported (in part) by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. Simulations were performed at the NIH biowulf supercomputer cluster and the National High Performance Computing Center of Fudan University.
Footnotes
The authors declare no competing financial interest.
References
- 1.Wei G, Jewett AI and Shea J-E, Physical Chemistry Chemical Physics, 2010, 12, 3622–3629. [DOI] [PubMed] [Google Scholar]
- 2.Patel HR, Pithadia AS, Brender JR, Fierke CA and Ramamoorthy A, J Phys Chem Lett, 2014, 5, 1864–1870. [DOI] [PubMed] [Google Scholar]
- 3.Baram M, Atsmon-Raz Y, Ma B, Nussinov R and Miller Y, Physical Chemistry Chemical Physics, 2015, DOI: 10.1039/c5cp03338a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang M, Hu R, Chen H, Chang Y, Ma J, Liang G, Mi J, Wang Y and Zheng J, Physical Chemistry Chemical Physics, 2015, 17, 23245–23256. [DOI] [PubMed] [Google Scholar]
- 5.Larini L, Gessel MM, LaPointe NE, Do TD, Bowers MT, Feinstein SC and Shea J-E, Physical Chemistry Chemical Physics, 2013, 15, 8916–8928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miller Y, Ma B and Nussinov R, Chem Rev, 2010, 110, 4820–4838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nussbaum JM, Seward ME and Bloom GS, Prion, 2013, 7, 14–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dikiy I and Eliezer D, Biochim Biophys Acta, 2012, 1818, 1013–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cao P, Abedini A and Raleigh DP, Curr Opin Struct Biol, 2013, 23, 82–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Levy CB, Stumbo AC, Ano Bom AP, Portari EA, Cordeiro Y, Silva JL and De Moura-Gallo CV, Int JBiochem Cell Biol, 2011, 43, 60–64. [DOI] [PubMed] [Google Scholar]
- 11.Ano Bom AP, Rangel LP, Costa DC, de Oliveira GA, Sanches D, Braga CA, Gava LM, Ramos CH, Cepeda AO, Stumbo AC, De Moura Gallo CV, Cordeiro Y and Silva JL, J Biol Chem, 2012, 287, 28152–28162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vogelstein B, Lane D and Levine AJ, Nature, 2000, 408, 307–310. [DOI] [PubMed] [Google Scholar]
- 13.Vousden KH and Prives C, Cell, 2009, 137, 413–431. [DOI] [PubMed] [Google Scholar]
- 14.Riley T, Sontag E, Chen P and Levine A, Nat Rev Mol Cell Biol, 2008, 9, 402–412. [DOI] [PubMed] [Google Scholar]
- 15.Ishimaru D, Andrade LR, Teixeira LSP, Quesado P. a., Maiolino LM, Lopez PM, Cordeiro Y, Costa LT, Heckl WM, Weissmuller G, Foguel D and Silva JL, Biochemistry, 2003, 42, 9022–9027. [DOI] [PubMed] [Google Scholar]
- 16.Brown CJ, Lain S, Verma CS, Fersht AR and Lane DP, Nat Rev Cancer, 2009, 9, 862–873. [DOI] [PubMed] [Google Scholar]
- 17.Goh AM, Coffill CR and Lane DP, J Pathol, 2011, 223, 116–126. [DOI] [PubMed] [Google Scholar]
- 18.Freed-Pastor WA and Prives C, Genes Dev, 2012, 26, 1268–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ji X, Ma L, Huang Q, Li Z, Zhao J, Huang W, Ma B and Yu L, Current pharmaceutical design, 2014, 20, 1259–1267. [DOI] [PubMed] [Google Scholar]
- 20.Huang Q, Yu L, Levine AJ, Nussinov R and Ma B, Biochim Biophys Acta, 2014, 1844, 198–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rigacci S, Bucciantini M, Relini A, Pesce A, Gliozzi A, Berti A and Stefani M, Biophys J, 2008, 94, 3635–3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Higashimoto Y, Asanomi Y, Takakusagi S, Lewis MS, Uosaki K, Durell SR, Anderson CW, Appella E and Sakaguchi K, Biochemistry, 2006, 45, 1608–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu J, Reumers J, Couceiro JR, De Smet F, Gallardo R, Rudyak S, Cornelis A, Rozenski J, Zwolinska A, Marine JC, Lambrechts D, Suh YA, Rousseau F and Schymkowitz J, Nat Chem Biol, 2011, 7, 285–295. [DOI] [PubMed] [Google Scholar]
- 24.Silva JL, De Moura Gallo CV, Costa DC and Rangel LP, Trends Biochem Sci, 2014, 39, 260–267. [DOI] [PubMed] [Google Scholar]
- 25.Strano S, Rossi M, Fontemaggi G, Munarriz E, Soddu S, Sacchi A and Blandino G, FEBSLetters, 2001, 490, 163–170. [DOI] [PubMed] [Google Scholar]
- 26.Strano S, Fontemaggi G, Costanzo A, Rizzo MG, Monti O, Baccarini A, Del Sal G, Levrero M, Sacchi A, Oren M and Blandino G, J Biol Chem, 2002, 277, 18817–18826. [DOI] [PubMed] [Google Scholar]
- 27.Milner J and Medcalf EA, Cell, 1991, 65, 765–774. [DOI] [PubMed] [Google Scholar]
- 28.Chan WM, Siu WY, Lau A and Poon RYC, Molecular and Cellular Biology, 2004, 24, 3536–3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bullock a. N. and Fersht a. R., Nature reviews. Cancer, 2001, 1, 68–76. [DOI] [PubMed] [Google Scholar]
- 30.Ghosh S, Ghosh D, Ranganathan S, Anoop A, Jha SKP,NN, Padinhateeri R and Maji SK, Biochemistry, 2014, 53, 5995–6010. [DOI] [PubMed] [Google Scholar]
- 31.Wang G and Fersht AR, Proc Natl Acad Sci U S A, 2015, 112, 2443–2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reumers J, Maurer-Stroh S, Schymkowitz J and Rousseau F, Hum Mutat, 2009, 30, 431–437. [DOI] [PubMed] [Google Scholar]
- 33.Muller PA and Vousden KH, Nat Cell Biol, 2013, 15, 2–8. [DOI] [PubMed] [Google Scholar]
- 34.Qi R, Luo Y, Wei G, Nussinov R and Ma B, The Journal of Physical Chemistry Letters, 2015, 6, 3276–3282. [Google Scholar]
- 35.Wilcken R, Wang G, Boeckler FM and Fersht AR, Proc Natl Acad Sci U S A, 2012, 109, 13584–13589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang G and Fersht AR, Proc Natl Acad Sci U S A, 2012, 109, 13590–13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang G and Fersht AR, Proc Natl Acad Sci U S A, 2015, 112, 2437–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang M, Hu R, Chen H, Gong X, Zhou F, Zhang L and Zheng J, JChem Inf Model, 2015, 55, 1628–1639. [DOI] [PubMed] [Google Scholar]
- 39.Colby DW, Zhang Q, Wang S, Groth D, Legname G, Riesner D and Prusiner SB, Proc Natl Acad Sci U S A, 2007, 104, 20914–20919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Han W and Wu YD, Proteins, 2007, 66, 575–587. [DOI] [PubMed] [Google Scholar]
- 41.Zhou R, Methods Mol Biol, 2007, 350, 205–223. [DOI] [PubMed] [Google Scholar]
- 42.Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE and Berendsen HJ, J Comput Chem, 2005, 26, 1701–1718. [DOI] [PubMed] [Google Scholar]
- 43.Lindorff-Larsen K, Piana S, Palmo K, Maragakis P, Klepeis JL, Dror RO and Shaw DE, Proteins, 2010, 78, 1950–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Qi R, Luo Y, Ma B, Nussinov R and Wei G, Biomacromolecules, 2014, 15, 122–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xie L, Luo Y, Lin D, Xi W, Yang X and Wei G, Nanoscale, 2014, 6, 9752–9762. [DOI] [PubMed] [Google Scholar]
- 46.Zhang T, Nguyen PH, Nasica-Labouze J, Mu Y and Derreumaux P, J Phys Chem B, 2015, 119, 6941–6951. [DOI] [PubMed] [Google Scholar]
- 47.Do TD, Chamas A, Zheng X, Barnes A, Chang D, Veldstra T, Takhar H, Dressler N, Trapp B, Miller K, McMahon A, Meredith SC, Shea JE, Lazar Cantrell K and Bowers MT, Biochemistry, 2015, 54, 4050–4062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Patriksson A and van der Spoel D, Phys Chem Chem Phys, 2008, 10, 2073–2077. [DOI] [PubMed] [Google Scholar]
- 49.Garcia AE and Sanbonmatsu KY, Proteins, 2001, 42, 345–354. [DOI] [PubMed] [Google Scholar]
- 50.Wei G and Shea JE, Biophys J, 2006, 91, 1638–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu C and Shea JE, PLoS Comput Biol, 2010, 6, e1000998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Levine ZA, Larini L, LaPointe NE, Feinstein SC and Shea JE, Proc Natl Acad Sci U S A, 2015, 112, 2758–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tarus B, Nguyen PH, Berthoumieu O, Faller P, Doig AJ and Derreumaux P, Eur J Med Chem, 2015, 91, 43–50. [DOI] [PubMed] [Google Scholar]
- 54.Bussi G, Donadio D and Parrinello M, J Chem Phys, 2007, 126, 014101. [DOI] [PubMed] [Google Scholar]
- 55.Parrinello M and Rahman A, Journal of Applied Physics, 1981, 52, 7182. [Google Scholar]
- 56.Darden T, York D and Pedersen L, The Journal of Chemical Physics, 1993, 98, 10089. [Google Scholar]
- 57.Li H, Luo Y, Derreumaux P and Wei G, Biophys J, 2011, 101, 2267–2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lu Y, Derreumaux P, Guo Z, Mousseau N and Wei G, Proteins: Structure, Function, and Bioinformatics, 2009, 75, 954–963. [DOI] [PubMed] [Google Scholar]
- 59.Humphrey W, Dalke A and Schulten K, J Mol Graph, 1996, 14, 33–38, 27–38. [DOI] [PubMed] [Google Scholar]
- 60.Luo R, David L and Gilson MK, J Comput Chem, 2002, 23, 1244–1253. [DOI] [PubMed] [Google Scholar]
- 61.Smardova J, Liskova K, Ravcukova B, Kubiczkova L, Sevcikova S, Michalek J, Svitakova M, Vybihal V, Kren L and Smarda J, Pathol Oncol Res, 2013, 19, 421–428. [DOI] [PubMed] [Google Scholar]
- 62.Ponchel F and Milner J, British journal of cancer, 1998, 77, 1555–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Khoo KH, Andreeva A and Fersht AR, J Mol Biol, 2009, 393, 161–175. [DOI] [PubMed] [Google Scholar]
- 64.Ma CD, Wang C, Acevedo-Velez C, Gellman SH and Abbott NL, Nature, 2015, 517, 347–350. [DOI] [PubMed] [Google Scholar]
- 65.Ma B and Nussinov R, Curr Top Med Chem, 2007, 7, 999–1005. [DOI] [PubMed] [Google Scholar]
- 66.Sabate R, Rousseau F, Schymkowitz J, Batlle C and Ventura S, Prion, 2015, 9, 200–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hu L, Cui W, He Z, Shi X, Feng K, Ma B and Cai YD, PLoS One, 2012, 7, e39369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Krammer C, Kremmer E, Schatzl HM and Vorberg I, Prion, 2008, 2, 99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Aguzzi A and Lakkaraju AK, Trends CellBiol, 2016, 26, 40–51. [DOI] [PubMed] [Google Scholar]
- 70.Hammarstrom P and Nystrom S, Prion, 2015, 9, 266–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.